 Paris S. Salazar-Hamm1*
Paris S. Salazar-Hamm1* Kyana N. Montoya1
Kyana N. Montoya1 Liliam Montoya2
Liliam Montoya2 Kel Cook1
Kel Cook1 Schuyler Liphardt1
Schuyler Liphardt1 John W. Taylor2
John W. Taylor2 Joseph A. Cook1,3
Joseph A. Cook1,3 Donald O. Natvig1
Donald O. Natvig1- 1Department of Biology, University of New Mexico, Albuquerque, NM, United States
- 2Department of Plant and Microbial Biology, University of California, Berkeley, Berkeley, CA, United States
- 3Museum of Southwestern Biology, University of New Mexico, Albuquerque, NM, United States
Human lung mycobiome studies typically sample bronchoalveolar lavage or sputum, potentially overlooking fungi embedded in tissues. Employing ultra-frozen lung tissues from biorepositories, we obtained fungal ribosomal RNA ITS2 sequences from 199 small mammals across 39 species. We documented diverse fungi, including common environmental fungi such as Penicillium and Aspergillus, associates of the human mycobiome such as Malassezia and Candida, and others specifically adapted for lungs (Coccidioides, Blastomyces, and Pneumocystis). Pneumocystis sequences were detected in 83% of the samples and generally exhibited phylogenetic congruence with hosts. Among sequences from diverse opportunistic pathogens in the Onygenales, species of Coccidioides occurred in 12% of samples and species of Blastomyces in 85% of samples. Coccidioides sequences occurred in 14 mammalian species. The presence of neither Coccidioides nor Aspergillus fumigatus correlated with substantial shifts in the overall mycobiome, although there was some indication that fungal communities might be influenced by high levels of A. fumigatus. Although members of the Onygenales were common in lung samples (92%), they are not common in environmental surveys. Our results indicate that Pneumocystis and certain Onygenales are common commensal members of the lung mycobiome. These results provide new insights into the biology of lung-inhabiting fungi and flag small mammals as potential reservoirs for emerging fungal pathogens.
Introduction
Human lungs are estimated to inhale 500 to 100,000 fungal spores per day depending on local environmental conditions (Hasleton, 1972; Hamm et al., 2020b). Substantial attention has been paid to mechanisms fungi use to avoid the immune response that lungs can mount against pathogens (e.g. Kwon-Chung and Sugui, 2013; Wiesner and Klein, 2017; Ward et al., 2021). Only recently, however, has it been suggested that healthy lungs may harbor as many fungi as lungs diagnosed with overt infections (Richardson et al., 2019), but it remains unclear which fungi in healthy lung tissues present transiently as a result of spore inhalations and which fungi survive as commensals, pathogens, or mutualists.
Once thought to represent a sterile tissue, the lung is now known to possess a microbiome, including a fungal component, the mycobiome (Hamm et al., 2020b). Studies of the human lung mycobiome have been limited, with samples typically derived from sputum or bronchoalveolar lavage rather than from actual tissues. Previous studies typically focused on diseased lungs (Nguyen et al., 2015; Weaver et al., 2019), so knowledge of the human lung mycobiome and more broadly the mammalian lung mycobiome remains limited.
Museum collections provide opportunities to study animal-microbe interactions across broad spatial scales and temporal archives (Cheng et al., 2011; Carvalho et al., 2017; Dunnum et al., 2017; Schindel and Cook, 2018). The goal of the current study was to explore the value of ultra-frozen lung tissues from museum collections to characterize the lung mycobiome of wild small mammals using molecular and cultivation methods. Lung samples were chosen to represent the species diversity from arid environments across the southwestern United States, in part to determine whether such frozen tissues would help define the range and host species of Coccidioides, the causative agent of coccidioidomycosis (Valley fever).
Long before the advent of modern molecular methods there were indications in the 1940s and 1950s that apparently healthy mammals frequently harbor living fungi in their lung tissues. The evidence for this arose in part from attempts by Dr. C. W. Emmons (Emmons and Ashburn, 1942; Emmons, 1943) to determine whether rodents serve as natural reservoirs for coccidioidomycosis. Those studies demonstrated a relatively high incidence of Coccidioides among specific rodent hosts at certain localities (15% of pocket mice and 17% kangaroo rats at San Carlos Indian Reservation, San Carlos, Arizona; Emmons and Ashburn, 1942). Moreover, these and other studies demonstrated that a related fungus, now generally known as Blastomyces parvus (previously Haplosporangium parvum or Emmonsia parva), was detected even more frequently in mammalian lung tissues of a wide variety of mammalian hosts (Emmons and Ashburn, 1942; Jellison, 1950; Bakerspigel, 1956).
Species of Pneumocystis represent a special case in the context of the lung mycobiome, because they are obligately symbiotic lung fungi. They are broadly distributed across mammals, and it has been suggested that each mammalian species has at least one host-specific species of Pneumocystis (Danesi et al., 2020). Molecular surveys for Pneumocystis in domesticated animals and wildlife have used targeted PCR approaches, and success rates have been mixed depending on the animal species targeted (Demanche et al., 2001; Akbar et al., 2012; Danesi et al., 2020). We report that high-throughput sequencing targeting fungal ribosomal internal transcribed spacer (ITS) sequences identifies this group at high frequency (83%) among diverse and widely distributed small mammals. This approach also provides preliminary information regarding specific fungal-host associations and coevolutionary histories for this group.
Our molecular study also expands the results of early lung studies with respect to Coccidioides and Blastomyces and demonstrates that mammalian lung tissues host a diverse mycobiome, with many species either known or now appearing to be adapted to the lung environment. Our results further indicate that, along with Pneumocystidales, members of the Onygenales, Malasseziales, and Saccharomycetales are common constituents of the natural lung mycobiome. We hypothesize that members of all four orders are typically benign commensals rather than aggressive opportunistic pathogens and that the lung mycobiome provides an excellent opportunity to better understand the evolution of commensalism or pathogenicity.
Results
Tissue acquisition
A total of 199 ultra-frozen lung tissues were obtained through formal request from the University of New Mexico Museum of Southwestern Biology (MSB) and the University of California Berkeley Museum of Vertebrate Zoology (MVZ). Samples represented 39 species from six mammalian families (Heteromyidae, Cricetidae, Muridae, Sciuridae, Geomyidae, and Leporidae) (Supplementary Table 1). Samples spanned 45 localities within 19 counties across California, Arizona, and New Mexico. Museum specimen collection dates ranged from 1994 to 2019.
Confirmation of host identities
Because species designations that accompany most museum specimens are initially morphology based, we sequenced a region of the mitochondrial cytochrome b (cyt b) gene to test mammalian host identities. We successfully captured mitochondrial cyt b sequences, a common molecular barcode, from 156 of 199 samples to confirm or correct host identifications. We found that 22 of the frozen tissue samples had incorrect initial species designations. The analyses presented here reflect species designations based on cyt b sequences (Supplementary Table 1).
Lung mycobiome community analysis
We employed Illumina sequencing that targeted the fungal nuclear ribosomal RNA ITS2 region for fungal community analyses. Processing of ITS2 sequences with UPARSE of 199 small-mammalian lung samples produced a total of 16,515,699 sequences clustered into 762 operational taxonomic units (OTUs; Blaxter et al., 2005). The average number of fungal OTUs per sample was 43.7 (Supplementary Data Sheet 2). Ascomycota (48%) and Basidiomycota (20%) were the dominant phyla with only a few OTUs from Mucoromycota (3%). Twenty-nine percent of the OTUs could not be identified to phylum using either NCBI or UNITE databases. Among the most abundant and frequent OTUs were members of the Eurotiomycetes (Aspergillus, Penicillium, and Blastomyces), Sordariomycetes (Sordariaceae), Pneumocystidomycetes (Pneumocystis), Dothideomycetes (Alternaria, Curvularia, and Aureobasidium), Saccharomycetes (Candida and Geotrichum) and Malasseziomycetes (Malassezia) (Table 1). There was a strong correlation between OTU frequency (percentage of samples with a given OTU) and abundance (sequence read numbers for a given OTU) (Figure 1).
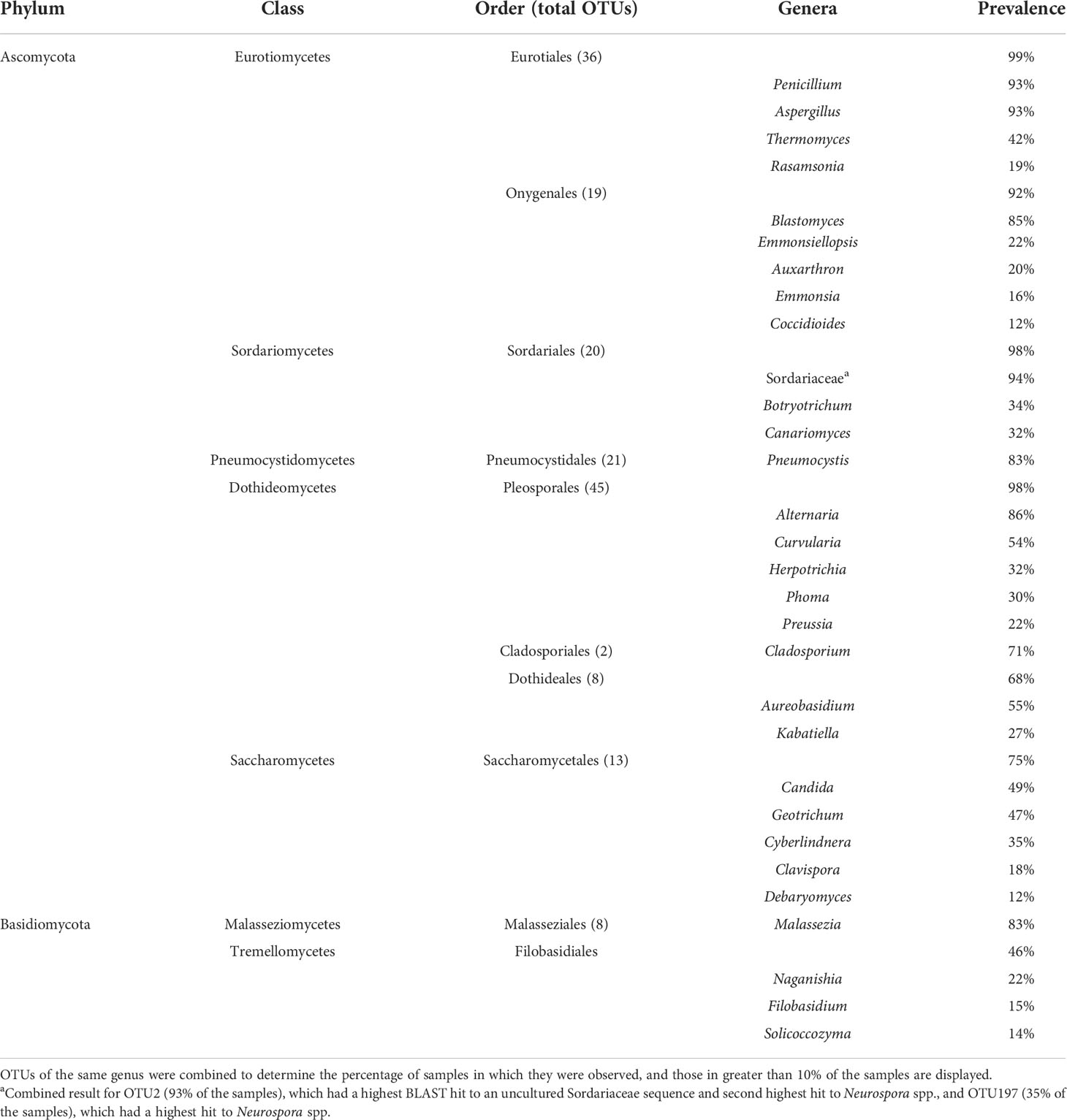
Table 1 Prevalence of major fungal taxa from ITS2 Illumina sequencing of 199 small-mammal lung samples.
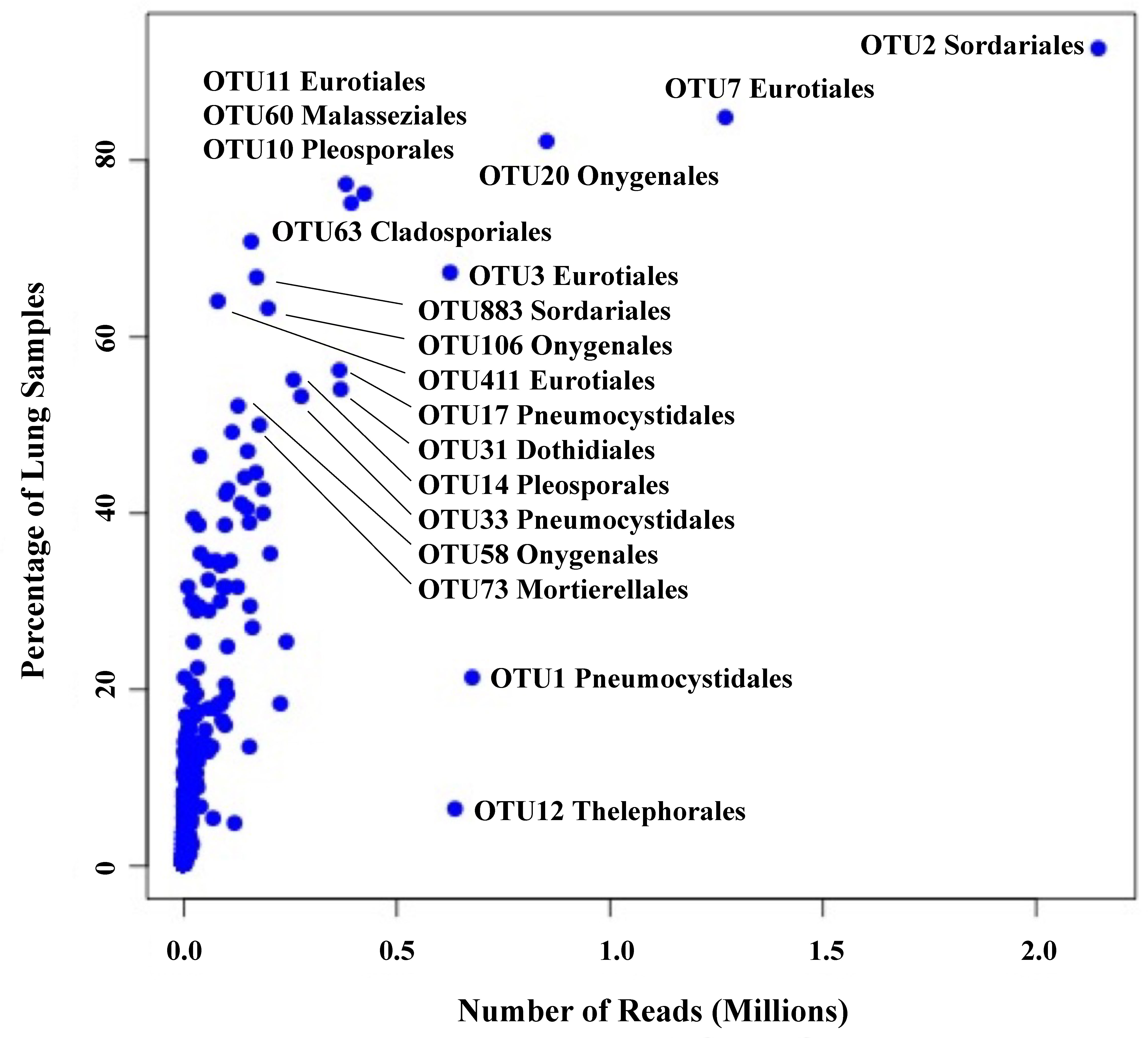
Figure 1 Relationship between frequency (percent of lung samples) and total reads for 762 OTUs. Taxonomic orders are given for those OTUs with a frequency of ≥50% plus two OTUs with high read counts, OTU1 (Pneumocystidales) and OTU12 (Thelephorales).
We focused special attention on the frequency, abundance, and diversity within the orders Onygenales, Pneumocystidales, Malasseziales, and Saccharomycetales because of previous reports regarding the lung mycobiome. The Onygenales accounted for 19 OTUs and one or more of these OTUs were in 183 (92%) samples (Table 1; Figure 2A). The Malasseziales accounted for eight OTUs (all in Malassezia) and were present in 166 (83%) of the samples (Table 1). The Pneumocystidales were represented by 21 OTUs (all in Pneumocystis) and were in 165 (83%) of the samples (Table 1; Figure 2B). Members of the Saccharomycetales accounted for 13 OTUs and occurred in 75% of the samples.
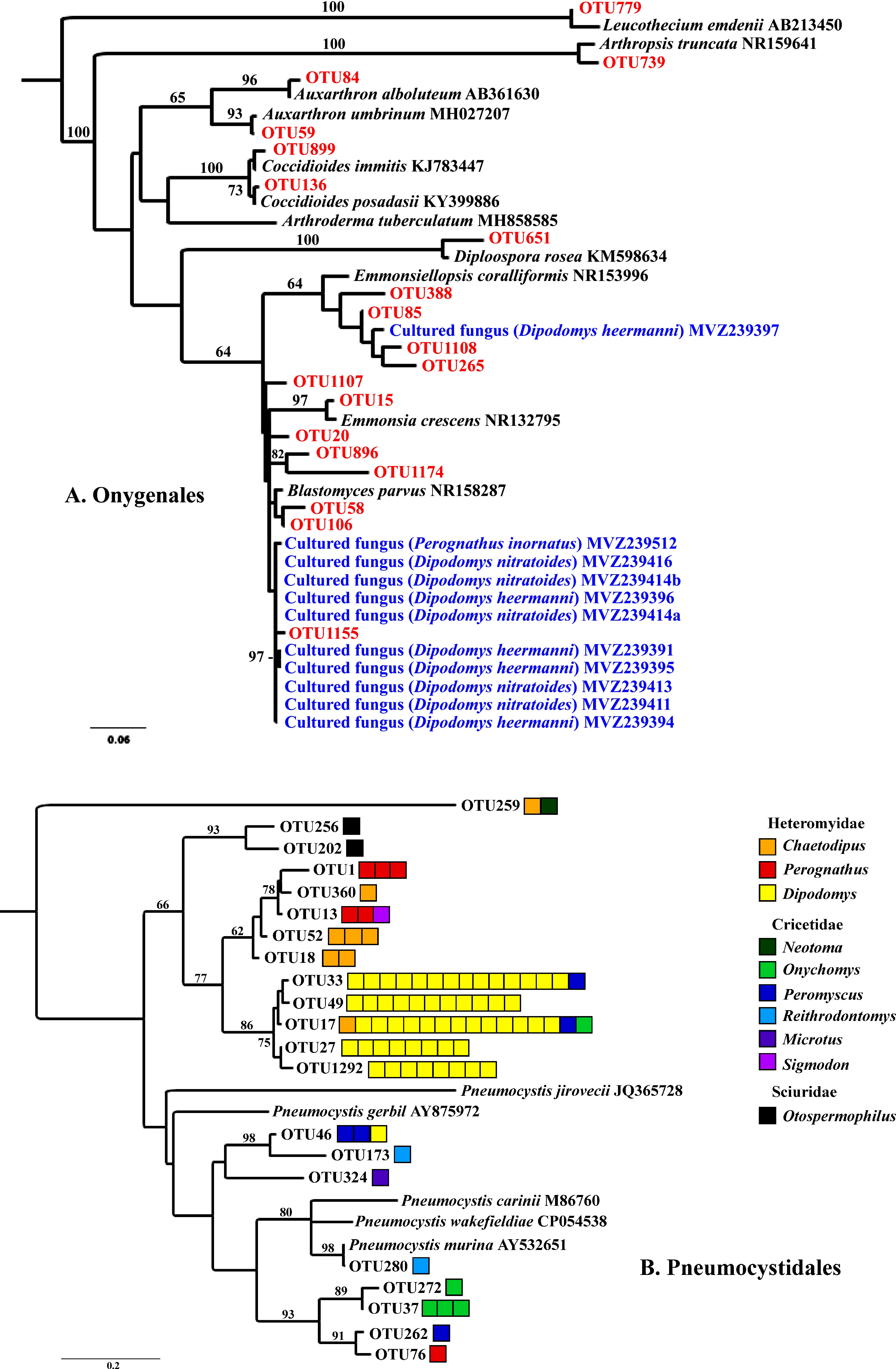
Figure 2 RaxML maximum likelihood phylogenies for Onygenales (A) and Pneumocystidales (B). Both trees have a mid-point root and bootstrap values (1000 replicates) are shown for branches with greater than 60% support. (A) Onygenalean fungi obtained through culturing (blue) and Illumina ITS2 sequencing (red) of lung tissues. Host species for which lung fungal cultures were obtained are in parentheses. Sequences in black type were acquired from GenBank. (B) Pneumocystidales OTUs were obtained from Illumina ITS2 sequencing. Each box to the right of an OTU represents one sample with >1,000 sequence reads for a given OTU. Boxes are color coded by genus with warm colors (red, orange, yellow) within Heteromyidae, cool colors (green, blue, purple) within Cricetidae, and black for Sciuridae. Branches with species names and accession numbers represent sequences obtained from GenBank. These trees are presented to illustrate the diversity of fungi from these two groups that were obtained from lung samples, together with associations among cultured fungi, ITS sequences, and mammalian species. Based on ITS sequences alone we were not able to resolve deeper evolutionary relationships unambiguously.
We note that 13 OTUs had top hits to species of Aspergillus, and members of this genus occurred in 93% of the lung samples (Table 1). The most frequent and abundant of these, OTU3, had top BLAST GenBank hits to A. fumigatus (the primary cause of aspergillosis) and occurred in 67% of the lung samples (Figure 1). The difficulty in assessing the significance of sequences from A. fumigatus and other members of the Eurotiales in the context of the lung mycobiome is discussed below.
Many of the fungi inhabiting lung tissues have been designated as pathotrophs (Supplementary Figure 1A). When functional guild designations of animal pathogens and parasites were pooled, it was evident that most samples had a high abundance of known fungal animal symbionts, but there remains a large unclassified component (Supplementary Figure 1B).
Alpha diversity calculated by OTU richness, the Simpson index, and the Shannon index showed no differences among host family, state, or collection year (Supplementary Figure 2).
Rarefaction curves indicated substantial coverage of OTU diversity, but coverage varied among samples (Supplementary Figure 3).
Differences in fungal community composition among samples were evaluated using nonmetric multidimensional scaling (NMDS) ordinations with the Bray-Curtis dissimilarity metric. There were no clear trends (p>0.05) in the fungal communities across time (collection year or month), location (state), or host (genus or family) although limited sample size may have dampened visible patterns (Supplementary Figure 4). Subsets of the data based on well sampled host families were analyzed to explore small spatial scale patterns. PERMANOVA was used to test for statistical differences between groups. In Kern County, California, 40 samples collected from four localities within a 50 km radius showed a clear separation existed in fungal community composition between the host genera of Dipodomys and Perognathus (Figure 3A, R2=0.22905, p=0.001). A similar pattern was not observed for Sierra County, New Mexico; while there was clustering of Chaetodipus samples, Dipodomys and Perognathus sampling was not sufficient to make direct comparisons with Kern County (Figure 3B, R2=0.07634, p=0.188). The lung communities in samples with Coccidioides present were not significantly different from those without Coccidioides sequences (Figure 3C, R2=0.00539, p=0.351). In NMDS analyses, lung samples with OTU3 (top BLAST hits to strains of Aspergillus fumigatus) substantially overlapped those without this OTU even though a PERMANOVA analysis suggested differences between the two sample classes (R2=0.00883, p=0.005). When only samples with >10,000 OTU3 reads were considered there was evidence of clustering for these samples (Figure 3D).
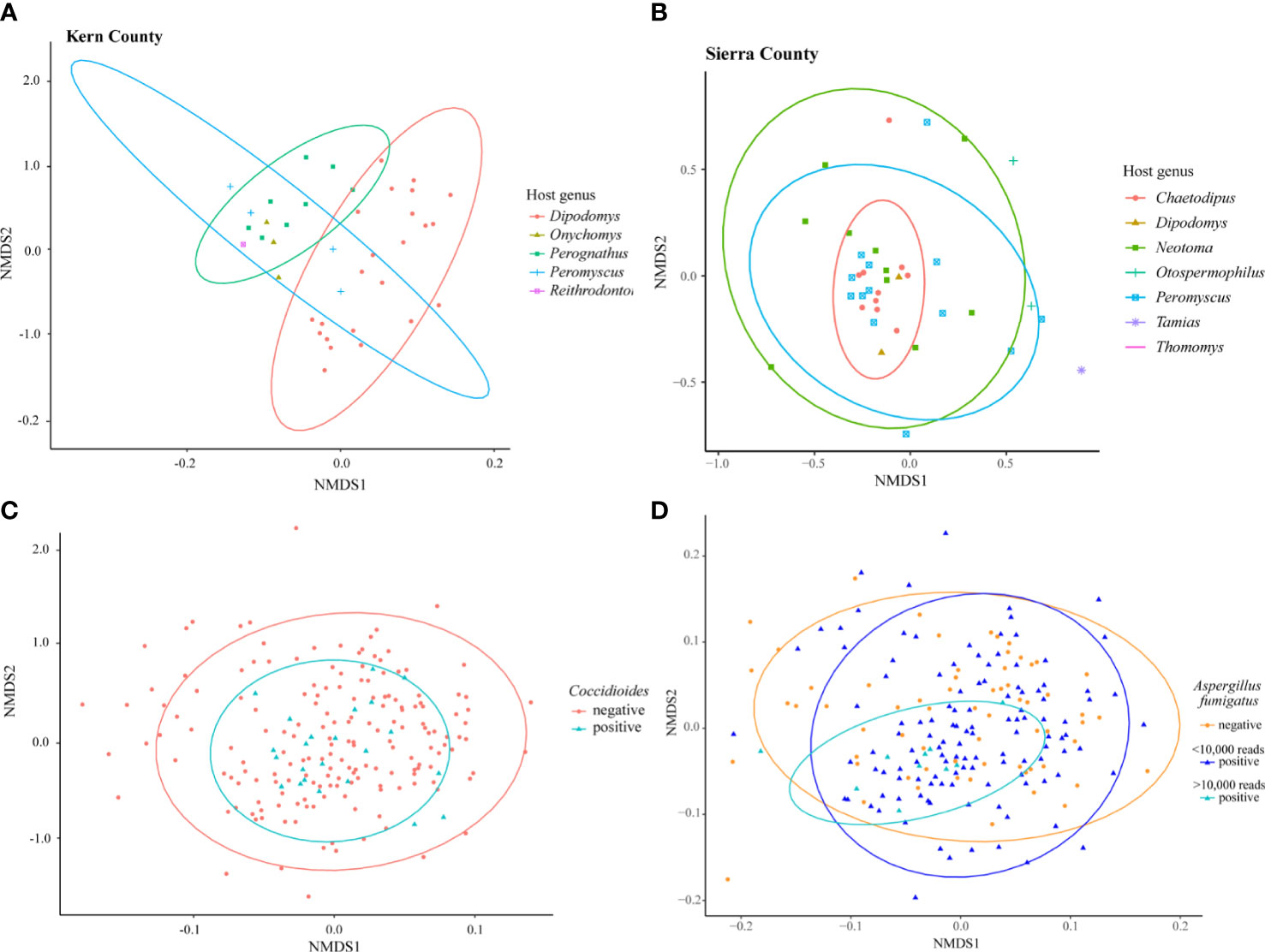
Figure 3 NMDS ordination of small-mammal samples from Kern County California only (A), Sierra County New Mexico only (B), and all 199 small-mammalian lung samples. (A) Samples collected within a 50 km radius in Kern County California suggest a difference in the lung mycobiome communities of species of Dipodomys and Perognathus (R2= 0.22905, p=0.001). (B) Samples within a 50 km radius in Sierra County New Mexico suggest a clustering of the lung mycobiome community within species of Chaetodipus but not differences from Neotoma or Peromyscus species (R2=0.07634, p=0.188). (C) Analysis of all 199 samples reveals no fungal community differences between those samples with Coccidioides and those without (R2=0.00539, p=0.351). (D) Analysis of all 199 samples demonstrated potential differences but also substantial overlap between those samples with OTU3 (top BLAST hits to strains of Aspergillus fumigatus) and those without (R2= 0.00883, p=0.005). A degree of clustering of samples with higher numbers of OTU3 reads (>10,000) was observed, however (indicated by the smaller turquoise oval). For the analysis shown, A. fumigatus sequence reads were removed to prevent the effect of abundant OTU3 on Bray-Curtis dissimilarity values. Results obtained when A. fumigatus reads were included in the analyses were in substance the same (R2= 0.01037, p=0.009, results not presented). Additional NMDS ordination results are presented for collection year, location by state, and host family and genus in Supplementary Figure 3.
A Mantel test revealed significant positive correlation between geographic distance and fungal community dissimilarity (r=0.1132, p=0.001). Positive spatial autocorrelation was seen at two distance classes, less than 44.8 km and between 135 km and 224 km indicating that samples collected closer together tend to have more similar fungal communities (Supplementary Figure 5A). Other distance classes had no positive or negative autocorrelation. Although lung fungal communities in general exhibited low Bray-Curtis similarity in pairwise comparisons, distance-decay analysis indicated significant decrease in similarity with distance (p<0.001; Supplementary Figure 5).
Coccidioides, Blastomyces parvus, and other Onygenales
Of the 199 small mammalian lungs for which we obtained Illumina ITS2 sequences, 24 (12%) produced Coccidioides sequence reads. These Coccidioides-positive samples fell within 14 mammalian species (Ammospermophilus harrisii, Chaetodipus intermedius, Cheatodipus penicillatus, Dipodomys heermanni, Dipodomys merriami, Neotoma albigula, Neotoma stephensi, Onychomys torridus, Otospermophilus variegatus, Perognathus ampulus, Peromyscus boylii, Peromyscus maniculatus, Sylvilagus audubonii, and Thomomys bottae) representing 10 genera and 5 families (Cricetidae, Heteromyidae, Sciuridae, Geomyidae, and Leporidae) (Figure 4; Supplementary Table 2). Positive Coccidioides samples were found in California (Kern County), Arizona (Cochise and Maricopa Counties) and New Mexico (Catron, Sierra, and Socorro Counties) (Figure 5; Supplementary Table 2). Coccidioides positive rates were highest in Maricopa County, Arizona (27%) and Sierra County, New Mexico (20%). UPARSE analysis produced two Coccidioides OTUs (OTU136 and OTU899) that were 97.38% similar. OTU136 shared a closer sequence similarity to C. posadasii and OTU899 shared a closer sequence similarity to C. immitis (Figure 2A).
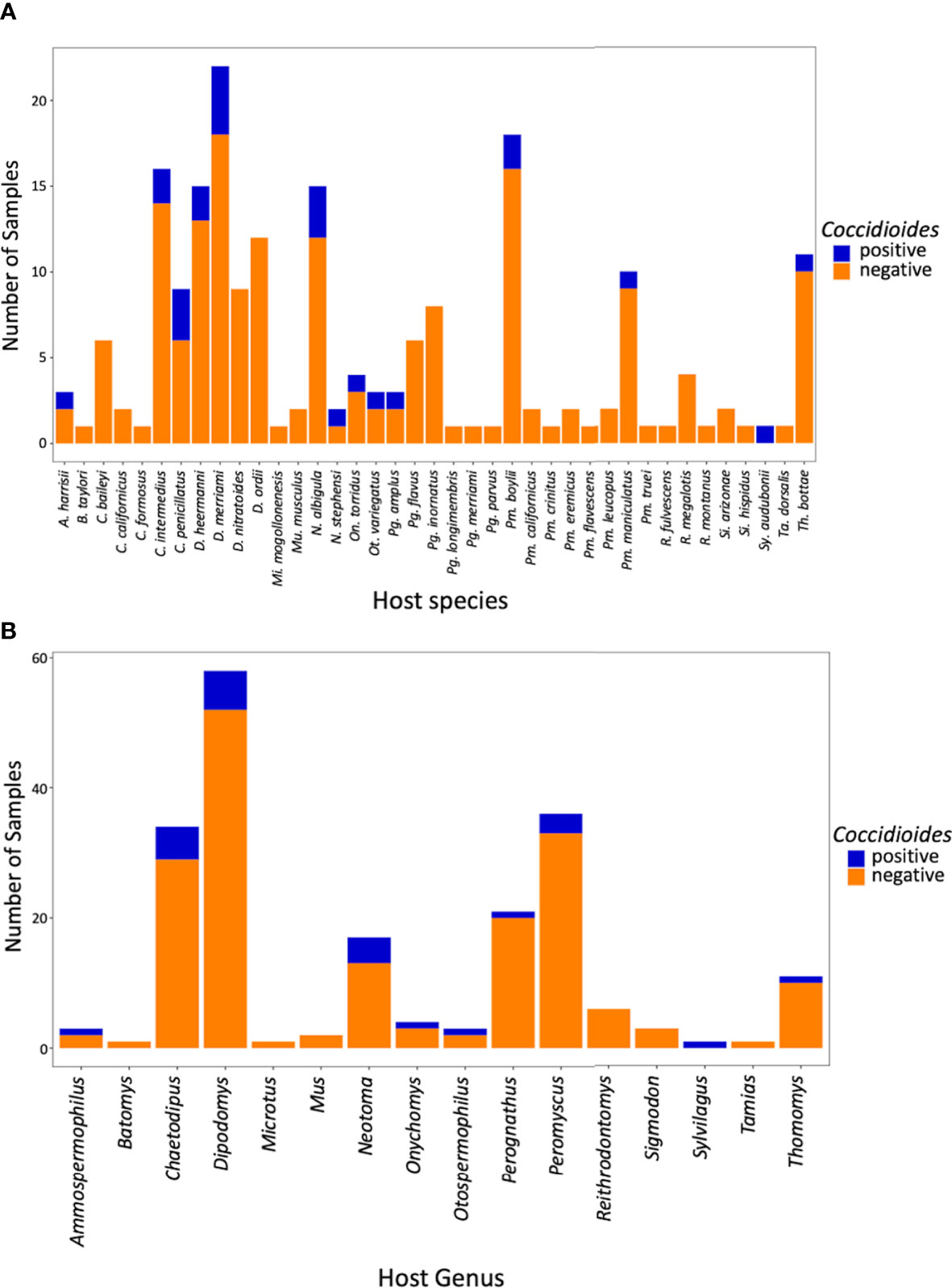
Figure 4 Bar plot of Coccidioides positive and negative samples by species (A) and by genus (B).
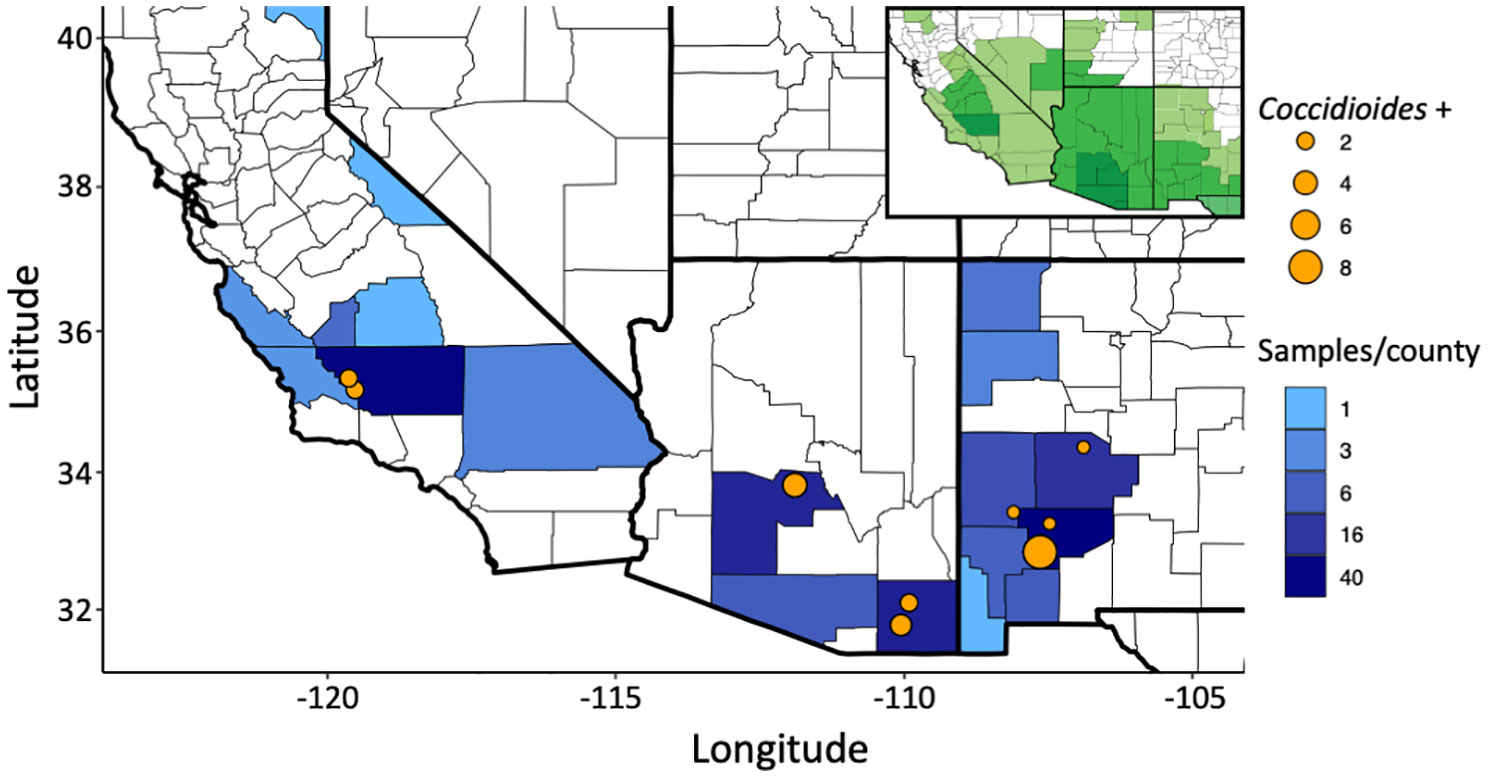
Figure 5 Sampling of small-mammalian lungs across the southwestern U.S. detect Coccidioides in wild animals. The number of samples sequenced per county is depicted in blue. Coccidioides positive trapping locations are shown with an orange dot with increasing size based on the number of positive samples in that specific location. GPS locations less than 1 km apart were merged. The insert indicates the endemic range for the genus Coccidioides, with darker shades of green indicating higher levels of incidence, as recognized by the U.S. Centers for Disease Control and Prevention (see Gorris et al., 2019).
Seven OTUs exhibited best hits to Blastomyces parvus in GenBank BLAST searches (Figure 2A). One or more of these OTUs occurred in 85% of the lungs sampled. Blastomyces OTU20 occurred in more than 80% of the samples and was the third most frequent OTU in the community data (Figure 1). Two other variants with top BLAST hits to B. parvus, OTU106 and OTU20, each occurred in more than half of the lungs sampled (Figure 1). Co-occurrence analyses revealed no significant correlation between the occurrence of members of the B. parvus group and species of Coccidioides (rho= -0.0257, p=0.6351, Supplementary Table 3).
Cultured members of the Onygenales
Although for most lung samples we did not have sufficient tissue for culture plating, we plated tissues from the lungs of 25 animals that were collected in 2019 during this study. This was done in part to test whether we could recover B. parvus and other members of the Onygenales, after preliminary sequencing efforts suggested the frequent presence of these fungi. Isolates whose top BLAST hits were to B. parvus were the most frequent among isolates obtained by direct culturing of lung tissues (11 of 29 isolates, Figure 2A; Supplementary Table 4).
Several OTUs had best BLAST GenBank hits to species in other Onygenalean genera, including Emmonsia, Emonsiellopsis, Auxarthron, Diploospora, Arthropsis, and Leucothecium (Figure 2A). Among these latter genera, only a species of Emmonsiellopsis was obtained in culture from mammal lungs (Figure 2A).
Pneumocystidales diversity
The 21 OTUs assigned to Pneumocystis were present in rodents of the families Heteromyidae, Cricetidae, and Sciuridae with patterns suggesting substantial but not complete host specificity (Figure 2B). Phylogenetic relationships among fungal OTUs from the Heteromyidae were substantially congruent with established relationships for the three mammalian genera, namely showing a closer relationship between Chaetodipus and Perognathus relative to Dipodomys (Alexander and Riddle, 2005). Pneumocystis sequences were especially prevalent in species of Dipodomys (32 of 57 samples, 56%), and were less frequently observed in species of Peromyscus (4 of 36 samples, 11%). Of the 165 samples containing Pneumocystis, 23 (14%) had multiple OTUs with >1000 reads each.
Additional fungi cultured from lung tissues
A total of 29 fungal isolates were obtained from five species of rodents from Kern County, California (Supplementary Table 4). In addition to isolates of the Onygenales referenced above, isolates included Aspergillus fumigatus, Aspergillus sp., several isolates of Penicillium sp., two members of the Chaetomiaceae, and two members of the Mucorales. As mentioned above, sequences from A. fumigatus were both frequent and abundant in the ITS2 Illumina data.
Discussion
Our Illumina sequencing results produced an average of just over 40 fungal OTUs per lung sample and more than 700 OTUs across the 199 samples examined (Supplementary Data Sheet 2). The following discussion assumes minimal contamination of samples from natural and laboratory environments. As described in Materials and Methods, all PCR-based experiments included negative controls. In addition, while the lung samples employed were collected over a period of twenty-five years by multiple scientists, they were obtained using sterile protocols designed to minimize contamination (Yates, 1996; Yates et al., 1996; Galbreath et al., 2019). The problem of potential environmental and laboratory contamination has received special interest in the context of species of Malassezia, once thought to be restricted to skin but now recognized as common in diverse microbiome and environmental samples unrelated to contamination (Spatz and Richard, 2020). While the potential for contamination by species of Malassezia and other environmental fungi provide a caution for interpreting results of microbiome studies in general, we note that the fungi we have focused on here, especially the Onygenales and Pneumocystidales, are not common in environmental samples.
We recognize four groups of fungi that occur in lung tissues: 1) those present because of incidental inhalation of spores but not truly colonizing lung surfaces or tissues; 2) fungi that are capable of colonizing lung tissue as commensals or pathogens transiently or long-term but are not specifically adapted to the lung; 3) fungi that are common members of the mammalian mycobiome that can exist in lung tissues, but which can also be associated with other tissues such as skin or gut, and 4) fungi adapted to the lung, either obligately (Pneumocystis species) or facultatively (Coccidioides species, Aspergillus fumigatus). These categories will likely be fluid depending on the fungi and mammals in question, for example because of potential differential susceptibility of mammalian species to pathogenicity.
The two most difficult mycobiome groups to distinguish between are those representing incidental inhalation (group 1) and transient colonization (group 2). Fungi present from incidental inhalation might be expected to occur in fewer samples and/or exhibit low read numbers, and their sequences would likely be common in environmental surveys. Such fungi could include sporulating members of the Sordariales, Helotiales, Pezizales, Dothidiales, Eurotiales, Hypocreales, Agaricales, and Pleosporales (Klich, 2002; Fröhlich-Nowoisky et al., 2009; Porras-Alfaro et al., 2011; Hamm et al., 2020a). A study by Kramer et al. of the airway mycobiome of cystic fibrosis patients found that sequences from many fungal species had high fluctuations both among different patients and over time when patients were sampled multiple times, suggesting the pulmonary mycobiome was dominated by species present temporarily because of inhalation of environmental spores (Kramer et al., 2015). Similarly, Rubio-Portillo et al. reported substantial variation among the mycobiomes of human bronchoalveolar lavage samples from different individuals, while in contrast they observed correlations between fungal OTUs from lavage samples and air samples from the home environments of test subjects (Rubio-Portillo et al., 2020). This further supports the potential relevance to lung mycobiome studies of incidental inhalation of spores from the local environment.
The importance of transient colonization (group 2), though difficult to separate from simple incidental inhalation in any specific instance, can be inferred from the fact that common environmental fungi not generally thought of as human pathogens occasionally cause disease, especially in immunocompromised individuals. Such fungi include species of Fusarium, Penicillium, Alternaria, and many others including members of the Mucoromycota (Skiada et al., 2017). And although the immune system of healthy individuals efficiently works against such infections, the triggering of defensive responses may in fact depend on transient colonization (Hernández-Chávez et al., 2017; Gago et al., 2019). With respect to our results, the strong correlation between read abundance and frequency (Figure 1) might support the argument that certain fungi, for example members of the Eurotiales not generally recognized as pathogens, are capable of colonization of healthy lung tissue beyond simply being present passively because of inhalation. This could include those species of Aspergillus that only rarely cause serious infections (Tsang et al., 2020).
We also acknowledge that A. fumigatus, sequences of which were common in our dataset (based on BLAST hits), is special in that it causes the most frequently diagnosed lung mycosis and is at the same time a common environmental saprotroph. Aspergillus fumigatus has multiple adaptations that permit colonization of lung tissues, including small spore size, thermotolerance, and molecular mechanisms that allow avoidance of immune responses (Robert and Casadevall, 2009; Casadevall, 2012; Kwon-Chung and Sugui, 2013; Raffa and Keller, 2019); and in that sense it might fit into either group 4 or group 2. Currently, it is difficult to know for certain whether the apparent frequent occurrence of A. fumigatus in the lungs of small mammals reflects the large number of spores in the environment or colonization of lung tissues. However, the fact that in NMDS analyses samples with high read counts for OTU3 (presumed A. fumigatus) formed a cluster relative to samples with low and no OTU3 reads (Figure 3D) may provide support for colonization in such samples.
Taken as a whole, our results have similarities and differences with respect to results obtained for the lung mycobiome of humans. Fungi commonly detected in healthy human lungs have included Candida (Saccharomycetales) Malassezia (Malasseziales), Cladosporium (Cladosporiales), Penicillium and Aspergillus (Eurotiales), and Pneumocystis (Tipton et al., 2017). Similarly, OTUs representing the Malasseziales and Saccharomycetales (including Candida), commonly associated with the human mycobiome (group 3), were well represented in our data (83% and 49% of the samples, respectively; Figure 1; Table 1). The relatively high diversity of Malasseziales fungi (eight OTUs) in our samples is similar to results reported previously for both healthy and diseased human lungs (van Woerden et al., 2013; Willger et al., 2014; Fraczek et al., 2018).
In our results, many OTUs were both abundant in sequence reads and frequency across samples despite being from different small mammals and various locations (Figure 1). This contrasts with results reported for humans that indicated high inter-individual variability for many OTUs (Kramer et al., 2015; Rubio-Portillo et al., 2020). In addition, sequences from members of the Onygenales were frequent in our samples but have been rare or non-existent in human samples. One potential contributing factor to these differences between our study and human studies is that we sampled lung tissues as opposed to bronchoalveolar lavage or sputum, and as a result our samples have the potential to detect fungi more deeply in tissues than would be obtained with lavage or sputum sampling. Another potential difference could reflect the fact that small, fossorial mammals spend substantial amounts of time in close contact with the soil environment, which could increase exposure to fungi in the soil environment, thereby reflecting more intense sampling of fungi in soil.
The most compelling results from our study relate to the Pneumocystidales and Onygenales, offering new insights into the biology of these organisms. Species of Pneumocystis have long been reported to be common obligate lung fungi with substantial host specificity, and most species are yet to be formally described (Mazars et al., 1997; Demanche et al., 2001; Laakkonen et al., 2001; Akbar et al., 2012). Members of this group possess genomic and physiological adaptations to be obligate biotrophs of mammals (Ma et al., 2016), and they clearly belong to the group of fungi adapted to the lung (group 4). Surveys for species of Pneumocystis in the lungs of non-human primates and bats using specific Pneumocystis-directed PCR produced positive results in 30-40% of the lung tissues examined (Demanche et al., 2001; Akbar et al., 2012), although one study reported a high incidence in shrews (80% of 83 samples; Danesi et al., 2020). Our Illumina approach resulted in 83% of the samples (165 of 199) containing sequences from members of this group. This percentage is almost certainly an underestimate, given that we examined only small amounts of lung tissue from each animal. Our results suggest that in some instances a single lung can harbor multiple Pneumocystis lineages, similar to results reported for P. carinii and P. wakefieldiae (Cushion et al., 2004; Latinne et al., 2021). For example, several different individuals of Dipodomys heermanni had more than 1000 reads assigned to both OTUs 27 and 1292. Moreover, in some instances our data suggest the possibility of lineage spillover across rodent families (Figure 2B). These are congruent with the findings of Latinne et al. (2021) obtained for Southeast Asian rodents indicating that certain Pneumocystis lineages may not be restricted to a single host. Nevertheless, host specificity of Pneumocystis species and their animal hosts is apparent for multiple genera of Heteromyidae especially Dipodomys (Figure 2B). The evolution of Pneumocystis host niche is complex and not well understood (Babb-Biernacki et al., 2020). Our study suggests that high-throughput non-targeted sequencing can be valuable to gain information toward understanding Pneumocystis diversity and host specificity.
Our results support the hypothesis that certain Onygenalean fungi, including species of Coccidioides and members of the Blastomyces parvus group, are adapted to infect and persist in lung tissues. Taylor and Barker have argued that species of Coccidioides are adapted to be endozoan inhabitants of small-mammal lungs rather than soil fungi with the ability to cause opportunistic infections (Taylor and Barker, 2019). This argument is based in part on genomic studies of Coccidioides species and other members of the Onygenales that showed a reduction of genes involved in plant decomposition and an increase in genes involved in the degradation of animal proteins (Sharpton et al., 2009). Moreover, sequences from species of Coccidioides and Blastomyces are rare in environmental surveys including studies that sample sites where species of Coccidioides are endemic (Porras-Alfaro et al., 2011; Bates et al., 2012), and the detection of Coccidioides species in soils is sporadic even with robust methods (Greene et al., 2000; Barker et al., 2012). Further, OTUs from the Onygenales typically do not have close relatives among sequences from environmental samples (uncultured fungi) in GenBank. This paucity of representation in environmental surveys contrasts with the diversity (19 OTUs) and prevalence (92%) of Onygenalean OTUs in the lung tissues examined here. An illustration of this disparity is found in a comparison of BLAST hits at GenBank using the eight most frequent Eurotiales OTUs in our dataset with those obtained with the eight most frequent Onygenales OTUs (Supplementary Table 5). The Eurotiales had equally good hits to GenBank entries from environmental samples and sequences obtained from known organisms. In contrast, the Onygenales sequences produced close matches to sequences from known organisms but not to environmental sequences.
Although coccidioidomycosis has been reported in diverse animal species (del Rocío Reyes-Montes et al., 2016), there are only two studies reporting the direct isolation of Coccidioides species from wild small mammals. One reported cultured Coccidioides from pocket mice (Chaetodipus intermedius, C. penicillatus, and C. baileyi, reported as Perognathus in the literature but later reassigned to Chaetodipus; Hafner and Hafner, 1983), the southern grasshopper mouse (Onychomys torridus), and a Merriam’s kangaroo rat (Dipodomys merriami) trapped in Arizona (Emmons and Ashburn, 1942). In Baja Mexico, a serology assay detected coccidioidal antibodies in a deer mouse (Peromyscus maniculatus) and desert woodrat (Neotoma lepida; Catalán-Dibene et al., 2014). Our ITS2 data represent the first high-throughput sequencing approach to attempt detection of Coccidioides species in wild small-mammal lungs. Here, we report 14 species of small mammals with sequences from Coccidioides, nine of which (Ammospermophilus harrisii, Dipodomys heermanni, Neotoma albigula, Neotoma stephensi, Otospermophilus variegatus, Perognathus ampulus, Peromyscus boylii, Sylvilagus audubonii, and Thomomys bottae) are newly documented host species (Figure 4A).
Early research from Emmons and colleagues suggested a higher correlation with heteromyid rodents and Coccidioides in the environment (Emmons and Ashburn, 1942; Emmons, 1943). Within the Heteromyidae rodents in our study, the genus Chaetodipus had a 15% positivity rate and Dipodomys had a 10% positivity rate, but they did not greatly differ from our overall average Coccidioides detection frequency (12%) (Figure 4B). In terms of future studies, Neotoma (woodrats) may be of particular interest with a Coccidioides positive rate of 24%, two-fold higher than our overall average (Figure 4B). An overlap analysis between the distribution models of rodents and species of Coccidioides has suggested that Neotoma lepida is a predominant co-occurring species (Ocampo-Chavira et al., 2020).
We were limited by sample size in terms of the Leporidae, Sciuridae, and Muridae. Notwithstanding, one Sylvilagus audubonii (desert cottontail) sample was positive for Coccidioides (Figure 4A). Sylvilagus audubonii DNA was found in association with Coccidioides positive burrows in Arizona (Kollath et al., 2019). Despite having only five samples of sciurids (squirrels), we detected Coccidioides in two genera (Ammospermophilus and Otospermophilus) corresponding to 40% of the samples. Only two murid samples were obtained, and neither were positive for Coccidioides.
With the expected expansion of coccidioidomycosis endemic regions in the United States (Lauer, 2017; Gorris et al., 2019), efforts have increased to model the factors affecting the distribution of the disease and predict its expansion under climate change scenarios. These efforts have, however, focused on environmental factors (climate, soil, elevation, and land cover variables) to predict ecological niches for C. immitis and C. posadasii (Baptista-Rosas et al., 2007; Gorris et al., 2018; Weaver et al., 2020; Dobos et al., 2021). Our study supports the prediction that small mammals play important roles as host reservoirs of Coccidioides species (del Rocío Reyes-Montes et al., 2016; Taylor and Barker, 2019), suggesting that the ecology, distribution, and taxonomy of small mammals should be incorporated into Coccidioides modeling efforts.
The presence of Coccidioides sequences does not appear to disrupt the lung mycobiome (Figure 3C). Studies with chytridiomycosis in frogs (Jani and Briggs, 2014) and snake fungal disease (Allender et al., 2018) suggest that these infections cause a disruption of the microbiome and a decrease in both fungal and bacterial diversity with fungal pathogen introduction. In sea star wasting disease, changes in microbial community composition occurred during disease progression, with decreasing species richness only in the late stages of the disease (Lloyd and Pespeni, 2018). The presence of Coccidioides without mycobiome disruption might imply a primarily commensal relationship with its host until a change in host conditions favoring a pathogenic state.
Emmons suggested a correlation between the presence of Blastomyces parvus (previously Haplosporangium parvum) and Coccidioides stating, “The isolation of H. parvum from rodents of a given area is taken to indicate that C. immitis is probably also present in that area” (Emmons, 1943). Our ITS2 sequencing revealed Blastomyces sequences in 85% of the samples suggesting that it is more common in small-mammal lungs than are species of Coccidioides (12%). When abundances were rarified, Blastomyces was present without Coccidioides in 77 samples, but only co-occurred with Coccidioides in 6 samples. A Spearman correlation and a permutation test found no statistical correlation between the two Onygenalean genera (Supplementary Table 3). Even without correlation, it is notable that Blastomyces parvus is in high frequency in small-mammal lungs despite its ability to cause adiaspiromycosis (Emmons and Ashburn, 1942). Emmonsia crescens (OTU15), detected in 16% of the samples, is an additional etiological agent of adiaspiromycosis (Table 1, Figure 2A) (Emmons and Jellison, 1960). Adiaspiromycosis often presents as a pulmonary infection common to fossorial mammals like rodents but is much rarer in humans (Anstead et al., 2012).
Lung samples employed in this study were chosen to reflect the geographic range of human coccidioidomycosis from low to high incidence to determine if we could detect sequences from species of Coccidioides. One of the most revealing aspects of our results, however, had to do with the high frequency of sequences from B. parvus and other members of the Onygenales among lung samples (92%). In addition, B. parvus was the most common fungal species cultured from lung tissue. We argue that the endozoan, small mammal reservoir hypothesis (Taylor and Barker, 2019) should be expanded beyond species of Coccidioides to encompass multiple Ajellomycetaceae (Onygenales) fungi including Blastomyces, Emergomyces, Emmonsia, and Emmonsiellopsis, an argument with significant clinical relevance. The phylogenetic diversity within this group is becoming ever more important to the medical field as over the last four decades there has been an increase in reports of novel Emmonsia-like human pathogens (Schwartz et al., 2015). Our study and others (Danesi et al., 2020) demonstrate the commonality of these Emmonsia-like fungi globally in wild animals. Host jumps have allowed and will likely continue to allow these fungi to evolve and diversify followed by radiation, specialization, and speciation which in some cases could increase their virulence (Thines, 2019).
This study was made possible by the frozen tissue collections of the UNM Museum of Southwestern Biology and the UCB Museum of Vertebrate Zoology. While acknowledging the value of the availability of diverse, frozen tissue collections, we also note that molecular barcoding of specimens to confirm host identifications is best practice (Dunnum et al., 2017). About 11% of the samples in this study showed incongruence between host identification based on cyt b sequences and that reported in museum databases. Nine of these frozen samples can be attributed to tissues that were apparently mislabeled at the time of collection, while 13 samples represent misidentification of closely related cryptic species for which morphology alone is inconclusive. Misidentification based on morphological features alone has been reported to be as high as 10% in some pathogen studies (Müller et al., 2013); however, without the ability to both test the host identity with a molecular approach and return to a physical voucher, these kinds of incongruencies would have been impossible to rectify. Directly linking genetic data to a physical specimen should become a necessary component of infectious disease studies (Thompson et al., 2021).
Materials and methods
Tissue acquisition
Ultra-frozen lung tissues were obtained from the University of New Mexico Museum of Southwestern Biology (MSB) and the University of California Berkeley Museum of Vertebrate Zoology (MVZ) by formal request. Sampling ranged from 1994 to 2019. Archival tissues loaned to this project included 39 species within five families (Heteromyidae, Cricetidae, Muridae, Sciuridae, and Geomyidae) of rodents and one rabbit species in Leporidae (Supplementary Table 1). Field sampling procedures followed established, sterile protocols developed to avoid cross-contamination and maximize the utility of these collections (Yates, 1996; Yates et al., 1996; Galbreath et al., 2019). Typically, specimens were preserved under a series of three-year protocols approved through the Institutional Animal Care and Use Committees (IACUC). The current protocol (Animal Welfare Assurance # D16-00565; A4023-01) is enforced under the United States Department of Agriculture Registration # 85-R-0002. Upon sacrifice of the mammal specimen following approved guidelines for animal care and use (Sikes et al., 2016), lung samples were collected into cryovials, immediately flash frozen in liquid nitrogen, and then archived and data-based at the ultrafrozen facilities at MSB and MVZ. Samples were obtained from 45 sampling localities from 19 counties in California, Arizona, and New Mexico within the known distribution of coccidioidomycosis (Figure 5).
DNA purification from lung tissues
Approximately 0.025 g of lung tissue was lyophilized for 24 hours followed by DNA extraction using the following CTAB procedure. Tissue was resuspended and ground in 500 µL cetyltrimethylammonium bromide (CTAB) lysis buffer (2% CTAB, 1.4 M NaCl, 20 mM EDTA, 100 mM Tris–HCl) plus β-mercaptoethanol (final concentration 0.2%) and 10 µL protease K (10 mg/mL) followed by an hour at 65°C. An isoamyl alcohol/chloroform extraction was performed by adding 500µL isoamyl alcohol-chloroform (1:24) followed by 20 minutes of gentle shaking and then centrifugation at 16,000g for 5 minutes. The upper aqueous phase was transferred to a fresh tube. DNA was precipitated by addition of 15 µL 3M sodium acetate (pH 5.2) and 500µL ice-cold isopropanol. Samples were inverted and incubated at -20°C for 10 minutes. After centrifugation, the pellet was washed twice, first with 500µL ice-cold 70% ethanol and then with 500µL ice-cold 100% ethanol. Ethanol was discarded and the pellet dried prior to resuspension in 50µL sterile H2O. DNA was further purified with Agencourt AMPure beads (Agencourt Bioscience Corporation, Beverly, MA, USA) following instructions from the manufacturer.
Host identifications
Host species were tentatively identified in the field using morphological parameters including but not limited to total length, tail length, hind foot length, ear length, reproductive data, total weight, and coat coloration. Geographic locality and field guides were additionally used for verification. To check species designations, we partially sequenced the mitochondrial cyt b region. Purified DNA extracted from lung tissue was used to amplify the cyt b region with primers targeting specific host groups. Cricetids (Neotoma, Peromyscus, Onychomys, Reithrodontomys, Baiomys, and Sigmodon), and murids (Mus) were amplified with the MSB05 and MSB14 modified primers as described in (Hope et al., 2010). Heteromyids (Chaetodipus, Dipodomys, and Perognathus), geomyids (Thomomys), and sciurids (Ammospermophilus, Tamias, and Otospermophilus) were amplified with the MVZ05 and MVZ14 primer pair (Smith and Patton, 1993). If the initial amplification was unsuccessful, we employed primer pair MVZ05 and MVZ04 which amplifies a shorter 426 bp segment (Smith et al., 1992). Polymerase chain reaction (PCR) conditions were as follows: initial denaturation at 95°C for 10 min, followed by 34 cycles of 95°C for 15 sec, annealing at 52°C for 30 sec, and extension at 72°C for 1 min, with a final extension at 72°C for 5 min, and holding at 4°C. PCR was followed by gel electrophoresis confirmation. Products were purified with ExoSAP-IT (Affymetrix, Santa Clara, CA) according to the manufacturer’s recommendations followed by Sanger sequencing using BigDye Terminator v3.1(Applied Biosystems, Foster City, CA) employing the forward and reverse primers. Identification was performed using BLAST searches. Cyt b sequences were deposited in GenBank (Sayers et al., 2021) under accession numbers OK134972 to OK135142 (Supplementary Table 1). For 43 samples that could not be verified with molecular methods, we utilized the archived host identification (https://arctos.database.museum).
Illumina library preparation and sequencing
The preparation of amplicon libraries was preceded by a PCR amplification using primers ITS1-F and ITS4 (White et al., 1990; Gardes and Bruns, 1993) to amplify the fungal nuclear ribosomal ITS region from whole-lung DNA. These reactions were performed in 15 µL reactions with 7.5 µL Premix ExTaq polymerase (Takara, Mountain View, CA), 1 µL of each (5 µM) primer, 2 µL of 1 mg/mL bovine serum albumin, 2.5 µL of sterile water and 1 µL of template DNA. PCR products were checked by gel electrophoresis. DNA concentration of successful amplifications were measured with a Qubit dsDNA HS kit (Life Technologies Inc., Gaithersburg, MD) and serial dilutions were performed to dilute inhibitors. A second PCR was preformed to contain a 29 (forward) or 25 (reverse) base linker, a 12 base barcode, a 29 (forward) or 34 (reverse) base pad, a 0-8 base heterogeneity spacer, and the fungal ITS2 5.8S-FUN and ITS4-FUN primers (Taylor et al., 2016), following the procedure in (Gao et al., 2019). Negative controls were included with all PCR procedures to ensure that amplified fragments did not result from laboratory contamination. PCR products were purified using AMPure magnetic beads (Beckman Coulter Inc., Brea, CA) following the manufacturer’s instructions and pooled to create DNA libraries following the Illumina MiSeq DNA library preparation protocol for paired-end reads. Libraries were quality checked for concentration and amplicon size using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) at the Vincent J. Coates Genomics Sequencing Laboratory (GSL, University of California, Berkeley, CA). Pyrosequencing was performed on the Illumina MiSeq PE300 sequencing platform at the GSL. All raw reads were deposited in the NCBI Short Read Archive (SRA) under BioProject ID PRJNA769405.
Sequence processing
Illumina sequences were processed initially with USEARCH v11 (Edgar, 2013). Sequences less than 250 bases in length were removed, as were adapter and primer sequences, and presumed chimeras. Sequences were clustered into OTUs at 99% similarity with UPARSE implemented in USEARCH (Edgar, 2010). Reports of nearly identical outcomes for community analyses utilizing OTUs and ASVs (Glassman and Martiny, 2018; Joos et al., 2020) support our choice of OTUs to avoid overestimating the number of fungal lineages due to intragenomic and intraspecific variation among ribosomal DNA repeats in the ITS2 region (Schoch et al., 2012; Estensmo et al., 2021). Representative OTU sequences were assigned taxonomy initially using SINTAX (Edgar, 2016) and the UNITE database v8.2 (Nilsson et al., 2019), supplemented with GenBank BLAST searches for taxonomic confirmation. The results presented in Figures 1, 2 and Table 1 reflect taxonomy confirmed with searches at GenBank. Fungal sequences from representative OTUs were deposited in GenBank under the accession numbers OK078030 to OK078530 (Supplementary Data Sheet 2 links OTUs and accession numbers). Code can be found at https://github.com/p-salazarhamm/mammal_lung_mycobiome.
Community and spatial analyses
We visualized differences in lung mycobiome composition among small mammals across time, space, and host using two-dimensional NMDS ordinations, a method commonly employed in microbial ecology to evaluate dissimilarities among communities (Legendre and Legendre, 2012). We transformed the data to relative abundance by dividing OTU read counts by the total of all reads within a sample. Ordinations were then performed on the relative abundance transformed fungal community data using the Bray-Curtis dissimilarity metric in the phyloseq package v1.30.0 (McMurdie and Holmes, 2013). We created subsets of the data and generated ordinations for intensely sampled counties to assess differences in fungal community by host on small spatial scales.
Spatial structure was examined by using a Mantel test in the vegan package v2.5-7 (Oksanen et al., 2019) in R v3.5.1 (R Core Team, 2018) to investigate a correlation between geographic distance and sample similarity. To assess the spatial distances at which communities were more or less similar than expected due to chance, we used a Mantel correlogram also in the vegan package. Furthermore, we plotted distance decay of similarity by plotting spatial distance between each pair of samples against the Bray-Curtis similarity between those samples.
Functional analyses
We used FUNGuild v1.1 (Nguyen et al., 2016) to assign trophic modes and guilds (functional groups) to fungal OTUs. Animal-fungal symbionts per sample were pooled by determining the percent of reads in each sample that belonged to OTUs whose guild assignments included animal pathogen and animal parasite.
Alpha diversity
OTU richness, the Simpson index, and the Shannon index were calculated to assess differences in alpha diversity of samples by location (state), host family, and collection date. To account for differing sequencing depths among samples, expected richness at 1000 reads was calculated using the rrarefy function in the vegan package (Oksanen et al., 2019). We used 1000 reads as a cutoff to retain reads from species of Coccidioides, which were in low abundance. We additionally computed the rarefaction curve to determine OTU richness at varying sequencing depths.
Co-occurrence analysis
Abundance data was rarified at three different read depths using the rrarefy.perm() function for computing 1000 rarefactions using EcolUtils R package (https://github.com/GuillemSalazar/EcolUtils). The patterns held consistent at the various depths (Supplementary Table 3). Seven OTUs (OTU20, OTU58, OTU1155, OTU1107, OTU106, OTU1174, and OTU896) identified as Blastomyces were grouped together to determine their co-occurrence with the two OTUs of Coccidioides (OTU136 and OTU899). The observed Spearman correlation was calculated. To test for significance, a permutation test was run for 10000 random draws and the observed rho statistic was compared to the corresponding null distribution.
Phylogenetic analyses
Illumina sequences representative of OTUs from the Onygenales and Pneumocystidales along with representative sequences from GenBank were subjected to phylogenetic analyses. Sequences were aligned with MAFFT using default parameters (gap open penalty 1.53, gap extension penalty 0.123) (Katoh and Standley, 2013), and alignments (Supplementary Data Sheets 3, 4) were subjected to maximum likelihood analysis with RAxML (Stamatakis, 2006) in each case employing 1000 bootstrap replicates (GTRCAT substitution model).
Culture isolation
If available, a portion of the lung tissue was used in culturing on a yeast glucose media (1% yeast extract, 2% glucose, 1.5% agar) with the addition of tetracycline (10 mg/L) and chloramphenicol (50 mg/L). Typically, 3-4 small lung fragments (approximately 0.25 cm each) were plated onto a single 10-cm agar plate. Plates were incubated at 25°C until fungal growth was visible. Colonies arising from these segments were transferred as hyphal tips to fresh individual plates. Colony and microscopic characteristics were assessed before selecting isolates for ITS Sanger sequencing. After 2-7 days growth, tissue was collected, and DNA was extracted using the above CTAB procedure with the reduction of the heating step to 30 min at 65°C. PCR was performed as above with primers ITS1-F and ITS4 (White et al., 1990; Gardes and Bruns, 1993) primers followed by confirmation with gel electrophoresis confirmation. Products were purified with ExoSAP-IT (Affymetrix, Santa Clara, California) according to the manufacturer’s recommendations. The entire ITS region was targeted for Sanger sequencing using BigDye Terminator v3.1 (Applied Biosystems, Foster City, CA) and sequences were identified using BLAST (Altschul et al., 1990). Sequences from lung fungal cultures were deposited in GenBank under accession numbers MW652389-MW652417 (Supplementary Table 4).
Data availability statement
The datasets presented in this study are available in online repositories. Access to the datasets is at https://www.ncbi.nlm.nih.gov/ with the accession number: PRJNA769405. Further inquiries should be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the animal study because this study involved frozen lung tissues from museum collections deposited over several decades by multiple researchers. No live animals were employed or sacrificed for the study. Although not directly related to our study, typical animal handling and current Animal Welfare Assurance protocols employed by scientists at the University of New Mexico Museum of Southwestern Biology are presented in Materials and Methods under the subheading “Tissue acquisition.”
Author contributions
PS-H, DN, JT, and JC conceptualized the project. PS-H, KM, LM, SL, and JC collected data. PS-H, KC, SL, and DN analyzed the data. PS-H and DN prepared the initial draft of the manuscript, and all authors participated in revisions. PS-H, JT, and DN provided funding. All authors contributed to the article and approved the submitted version.
Funding
PS-H was supported in part by a UNM Sevilleta LTER Summer Graduate Student Fellowship (NSF awards DEB 1655499 and DEB 1440478) and the UNM Sevilleta Field Station endowment fund, with additional support from research awards from the UNM Graduate and Professional Student Association and the Department of Biology Graduate Research Allocations Committee. PS-H was also supported by the Mycological Society of America Graduate Research Fellowship and the John W. Rippon Research Award. JT acknowledges the Valley Fever Research Initiative at the University of California, VFR-19-633952 and grant R01 AI148336 from the NIAID. JC acknowledges NSF support (NSF1561342, 2033482). Support was also provided by the UNM Department of Biology’s Molecular Biology Facility, supported by the UNM Center for Evolutionary & Theoretical Immunology (CETI) under National Institutes of Health grant P30GM110907.
Acknowledgments
This project was made possible by frozen tissue collections preserved at the Museum of Southwestern Biology at the University of New Mexico (UNM) and the Museum of Vertebrate Zoology at University of California Berkeley. The authors would like to thank the field crews that collected these tissues, in particular Dr. James L. Patton. We additionally would like to thank the UNM Center for Advanced Research Computing, supported in part by the National Science Foundation (NSF), for providing high performance computing resources used in this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffunb.2022.996574/full#supplementary-material
Supplementary Data Sheet 1 | Supplementary Figures and Tables.
Supplementary Data Sheet 2 | ITS2 OTU frequency and abundance, including taxonomic classifications designated by UNITE database v8.2 (Nilsson et al., 2019) and BLAST searches.
Supplementary Data Sheet 3 | Onygenales sequence alignment (NEXUS format).
Supplementary Data Sheet 4 | Pneumocystidales sequence alignment (NEXUS format).
References
Akbar H., Pinçon C., Aliouat-Denis C. M., Derouiche S., Taylor M. L., Pottier M., et al. (2012). Characterizing Pneumocystis in the lungs of bats: understanding Pneumocystis evolution and the spread of Pneumocystis organisms in mammal populations. Appl. Environ. Microbiol. 78, 8122–8136. doi: 10.1128/AEM.01791-12
Alexander L. F., Riddle B. R. (2005). Phylogenetics of the new world rodent family heteromyidae. J. Mammal. 86, 366–379. doi: 10.1644/BER-120.1
Allender M. C., Baker S., Britton M., Kent A. D. (2018). Snake fungal disease alters skin bacterial and fungal diversity in an endangered rattlesnake. Sci. Rep. 8, 12147. doi: 10.1038/s41598-018-30709-x
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anstead G. M., Sutton D. A., Graybill J. R. (2012). Adiaspiromycosis causing respiratory failure and a review of human infections due to Emmonsia and Chrysosporium spp. J. Clin. Microbiol. 50, 1346–1354. doi: 10.1128/JCM.00226-11
Babb-Biernacki S. J., Esselstyn J. A., Doyle V. P. (2020). Rethinking host range in Pneumocystis. PloS Pathog. 16, e1008824. doi: 10.1371/journal.ppat.1008824
Bakerspigel A. (1956). Haplosporangium in Saskatchewan rodents. Mycologia 48, 568–572. doi: 10.1080/00275514.1956.12024567
Baptista-Rosas R. C., Hinojosa A., Riquelme M. (2007). Ecological niche modeling of coccidioides spp. in western north American deserts. Ann. NY Acad. Sci. 1111, 35–46. doi: 10.1196/annals.1406.003
Barker B. M., Tabor J. A., Shubitz L. F., Perrill R., Orbach M. J. (2012). Detection and phylogenetic analysis of Coccidioides posadasii in Arizona soil samples. Fungal Ecol. 5, 163–176. doi: 10.1016/j.funeco.2011.07.010
Bates S. T., Nash III T.H., Garcia-Pichel F. (2012). Patterns of diversity for fungal assemblages of biological soil crusts from the southwestern united states. Mycologia 104, 353–361. doi: 10.3852/11-232
Blaxter M., Mann J., Chapman T., Thomas F., Whitton C., Floyd R., et al. (2005). Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc B. Biol. Sci. 360, 1935–1943. doi: 10.1098/rstb.2005.1725
Carvalho T., Becker C. G., Toledo L. F. (2017). Historical amphibian declines and extinctions in Brazil linked to chytridiomycosis. Proc. R. Soc B. Biol. Sci. 284, 20162254. doi: 10.1098/rspb.2016.2254
Casadevall A. (2012). Fungi and the rise of mammals. PloS Pathog. 8, e1002808. doi: 10.1371/journal.ppat.1002808
Catalán-Dibene J., Johnson S. M., Eaton R., Romero-Olivares A. L., Baptista-Rosas R. C., Pappagianis D., et al. (2014). Detection of coccidioidal antibodies in serum of a small rodent community in Baja California, Mexico. Fungal Biol. 118, 330–339. doi: 10.1016/j.funbio.2014.01.006
Cheng T. L., Rovito S. M., Wake D. B., Vredenburg V. T. (2011). Coincident mass extirpation of neotropical amphibians with the emergence of the infectious fungal pathogen Batrachochytrium dendrobatidis. proc. Nat. Acad. Sci. 108, 9502–9507. doi: 10.1073/pnas.1105538108
Cushion M. T., Keely S. P., Stringer J. R. (2004). Molecular and phenotypic description of pneumocystis wakefieldiae sp. nov., a new species in rats. Mycologia 96, 429–438. doi: 10.1080/15572536.2005.11832942
Danesi P., Falcaro C., Dukik K., Jiang Y., Rizzoli A. P., Allavena R., et al. (2020). Molecular diagnosis of Emmonsia-like fungi occurring in wild animals. Mycopathologia 185, 51–65. 10.1007/s11046-019-00353-8
del Rocío Reyes-Montes M., Pérez-Huitrón M. A., Ocaña-Monroy J. L., Frías-De-León M. G., Martínez-Herrera E., Arenas R., et al. (2016). The habitat of coccidioides spp. and the role of animals as reservoirs and disseminators in nature. BMC Infect. Dis. 16, 1–8. doi: 10.1186/s12879-016-1902-7
Demanche C., Berthelemy M., Petit T., Polack B., Wakefield A. E., Dei-Cas E., et al. (2001). Phylogeny of Pneumocystis carinii from 18 primate species confirms host specificity and suggests coevolution. J. Clin. Microbiol. 39, 2126–2133. doi: 10.1128/JCM.39.6.2126-2133.2001
Dobos R. R., Benedict K., Jackson B. R., McCotter O. Z. (2021). Using soil survey data to model potential Coccidioides soil habitat and inform valley fever epidemiology. PLoS One 16, e0247263. doi: 10.1371/journal.pone.0247263
Dunnum J. L., Yanagihara R., Johnson K. M., Armien B., Batsaikhan N., Morgan L., et al. (2017). Biospecimen repositories and integrated databases as critical infrastructure for pathogen discovery and pathobiology research. PLoS Negl. Trop. Dis. 11, e000513. doi: 10.1371/journal.pntd.0005133
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar R. C. (2016). SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv, 074161. doi: 10.1101/074161
Emmons C. W. (1943). Coccidioidomycosis in wild rodents: A method of determining the extent of endemic areas. Pub. Health Rep. 58, 1–5. doi: 10.2307/4584326
Emmons C. W., Ashburn L. L. (1942). The isolation of Haplosporangium parvum n. sp. and Coccidioides immitis from wild rodents. their relationship to coccidioidomycosis. Pub. Health Rep. 57, 1715–1727. doi: 10.2307/4584276
Emmons C. W., Jellison W. L. (1960). Emmonsia crescens sp. n. and adiaspiromycosis (haplomycosis) in mammals. Ann. NY Acad. Sci. 89, 91–101. doi: 10.1111/j.1749-6632.1960.tb20133.x
Estensmo E. L. F., Maurice S., Morgado L., Martin-Sanchez P. M., Skrede I., Kauserud H. (2021). The influence of intraspecific sequence variation during DNA metabarcoding: A case study of eleven fungal species. Mol. Ecol. Res. 21, 1141–1148. doi: 10.1111/1755-0998.13329
Fraczek M. G., Chishimba L., Niven R. M., Bromley M., Simpson A., Smyth L., et al. (2018). Corticosteroid treatment is associated with increased filamentous fungal burden in allergic fungal disease. J. Allergy Clin. Immunol. 142, 407–414. doi: 10.1016/j.jaci.2017.09.039
Fröhlich-Nowoisky J., Pickersgill D. A., Després V. R., Pöschl U. (2009). High diversity of fungi in air particulate matter. Proc. Nat. Acad. Sci. U.S.A. 10612814–12819. doi: 10.1073/pnas.0811003106
Gago S., Denning D. W., Bowyer P. (2019). Pathophysiological aspects of Aspergillus colonization in disease. Med. Mycol. 57, S219–S227. doi: 10.1093/mmy/myy076
Galbreath K. E., Hoberg E. P., Cook J. A., Armién B., Bell K. C., Campbell M. L., et al. (2019). Building an integrated infrastructure for exploring biodiversity: field collections and archives of mammals and parasites. J. Mammal. 100, 382–393. doi: 10.1093/jmammal/gyz048
Gao C., Montoya L., Xu L., Madera M., Hollingsworth J., Purdom E., et al. (2019). Strong succession in arbuscular mycorrhizal fungal communities. ISME J. 13, 214–226. doi: 10.1038/s41396-018-0264-0
Gardes M., Bruns T. D. (1993). ITS primers with enhanced specificity for basidiomycetes application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Glassman S. I., Martiny J. B. H. (2018). Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. mSphere 3, e00148–e00118. doi: 10.1128/mSphere.00148-18
Gorris M. E., Cat L. A., Zender C. S., Treseder K. K., Randerson J. T. (2018). Coccidioidomycosis dynamics in relation to climate in the southwestern united states. GeoHealth 2, 6–24. doi: 10.1002/2017GH000095
Gorris M. E., Cat L. A., Zender C. S., Treseder K. K., Randerson J. T. (2019). Expansion of coccidioidomycosis endemic regions in the united states in response to climate change. GeoHealth 3, 308–327. doi: 10.1029/2019GH000209
Greene D. R., Koenig G., Fisher M. C., Taylor J. W. (2000). Soil isolation and molecular identification of Coccidioides immitis. Mycologia 92, 406–410. doi: 10.1080/00275514.2000.12061175
Hafner J., Hafner M. S. (1983). Evolutionary relationships of heteromyid rodents. Great Basin Nat. Memoirs. 7, 3–29. Available at: https://scholarsarchive.byu.edu/gbnm/vol7/iss1/2.
Hamm P. S., Mueller R. C., Kuske C. R., Porras-Alfaro A. (2020a). Keratinophilic fungi: specialized fungal communities in a desert ecosystem identified using cultured-based and illumina sequencing approaches. Microbiol. Res. 239, 126530. doi: 10.1016/j.micres.2020.126530
Hamm P. S., Taylor J. W., Cook J. A., Natvig D. O. (2020b). Decades-old studies of fungi associated with mammalian lungs and modern DNA sequencing approaches help define the nature of the lung mycobiome. PloS Pathog. 16, e1008684. doi: 10.1371/journal.ppat.1008684
Hasleton P. S. (1972). The internal surface area of the adult human lung. J. Anat. 112, 391–400. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1271180/pdf/janat00322-0079.pdf.
Hernández-Chávez M. J., Luis A., Pérez-García L. A., Niño-Vega G. A., Mora-Montes H. M. (2017). Fungal strategies to evade the host immune recognition. J. Fungi 3, 51. doi: 10.3390/jof3040051
Hope A. G., Waltari E., Dokuchaev N. E., Abramov S., Dupal T., Tsvetkova A., et al. (2010). High-latitude diversification within Eurasian least shrews and Alaska tiny shrews (Soricidae). J. Mammal. 91, 1041–1057. doi: 10.1644/09-MAMM-A-402.1
Jani A. J., Briggs C. J. (2014). The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Nat. Acad. Sci. U.S.A. 111, E5049–E5058. doi: 10.1073/pnas.1412752111
Jellison W. L. (1950). Haplomycosis in Montana rabbits, rodents, and carnivores. Pub. Health Rep. 65, 1057–1063. doi: 10.2307/4587440
Joos L., Beirinckx S., Haegeman A., Debode J., Vandecasteele B., Baeyen S., et al. (2020). Daring to be differential: metabarcoding analysis of soil and plant-related microbial communities using amplicon sequence variants and operational taxonomical units. BMC Genom. 21, 1–17. doi: 10.1186/s12864-020-07126-4
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Klich M. A. (2002). Biogeography of Aspergillus species in soil and litter. Mycologia 94, 21–27. doi: 10.1080/15572536.2003.11833245
Kollath D. R., Miller K. J., Barker B. M. (2019). The mysterious desert dwellers: Coccidioides immitis and Coccidioides posadasii, causative fungal agents of coccidioidomycosis. Virulence 10, 222–233. doi: 10.1080/21505594.2019.1589363
Kramer R., Sauer-Heilborn A., Welte T., Guzman C. A., Abraham W. R., Höfle M. G. (2015). Cohort study of airway mycobiome in adult cystic fibrosis patients: differences in community structure between fungi and bacteria reveal predominance of transient fungal elements. J. Clin. Microbiol. 53, 2900–2907. doi: 10.1128/JCM.01094-15
Kwon-Chung K. J., Sugui J. A. (2013). Aspergillus fumigatus–what makes the species a ubiquitous human fungal pathogen? PloS Pathog. 9, e1003743. doi: 10.1371/journal.ppat.1003743
Laakkonen J., Fisher R. N., Case T. J. (2001). Pneumocystosis in wild small mammals from California. J. Wildl. Dis. 37, 408–412. doi: 10.7589/0090-3558-37.2.408
Latinne A., Chen H. W., Kuo C. C., Lorica R., Singleton G., Stuart A., et al. (2021). Revisiting the Pneumocystis host specificity paradigm and transmission ecology in wild southeast Asian rodents. Infect. Genet. Evol. 93, 104978. doi: 10.1016/j.meegid.2021.104978
Lauer A. (2017). “Coccidioidomycosis: Increasing incidence of an “orphan“ disease in response to environmental changes,” in Modeling the transmission and prevention of infectious disease. Ed. Hurst C. J. (Springer: Cham), 151–185.
Lloyd M. M., Pespeni M. H. (2018). Microbiome shifts with onset and progression of Sea star wasting disease revealed through time course sampling. Sci. Rep. 8, 16476. doi: 10.1038/s41598-018-34697-w
Ma L., Chen Z., Huang D. W., Kutty G., Ishihara M., Wang H., et al. (2016). Genome analysis of three Pneumocystis species reveals adaptation mechanisms to life exclusively in mammalian hosts. Nat. Commun. 7, 10740. doi: 10.1038/ncomms10740
Mazars E. D., Guyot K. A., Fourmaintraux S. O., Renaud F. R., Petavy F. R., Camus D. A., et al. (1997). Detection of Pneumocystis in European wild animals. J. Eukaryot. Microbiol. 44, 39S. doi: 10.1111/j.1550-7408.1997.tb05763.x
McMurdie P. J., Holmes S. (2013). Phyloseq: an r package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Müller L., Goncalves G. L., Cordeiro-Estrela P., Marinho J. R., Althoff S. L., Testoni A. F., et al. (2013). DNA Barcoding of sigmodontine rodents: identifying wildlife reservoirs of zoonoses. PloS One 8, e80282. doi: 10.1371/journal.pone.0080282
Nguyen N. H., Song Z., Bates S. T., Branco S., Tedersoo L., Menke J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nguyen L. D. N., Viscogliosi E., Delhaes L. (2015). The lung mycobiome: an emerging field of the human respiratory microbiome. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00089
Nilsson R. H., Larsson K. H., Taylor A. F., Bengtsson-Palme J., Jeppesen T. S., Schigel D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nuc. Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Ocampo-Chavira P., Eaton-Gonzalez R., Riquelme M. (2020). Of mice and fungi: Coccidioides spp. distribution models. J. Fungi. 6, 320. doi: 10.3390/jof6040320
Oksanen J., Blanchet F. G., Friendly M., Kindt R., Legendre P., McGlinn D., et al. (2019). Vegan: Community ecology package. r package version 2.5-6. The Comprehensive R Archive Network.
Porras-Alfaro A., Herrera J., Natvig D. O., Lipinski K., Sinsabaugh R. L. (2011). Diversity and distribution of soil fungal communities in a semiarid grassland. Mycologia 103, 10–21. doi: 10.3852/09-297
Raffa N., Keller N. (2019). A call to arms: Mustering secondary metabolites for success and survival of an opportunistic pathogen. PloS Pathog. 15, e1007606. doi: 10.1371/journal.ppat.1007606
R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at: (http://www.R-project.org/).
Richardson M., Bowyer P., Sabino R. (2019). The human lung and Aspergillus: you are what you breathe in? Med. Mycol. 57, S145–SS54. doi: 10.1093/mmy/myy149
Robert V. A., Casadevall A. (2009). Vertebrate endothermy restricts most fungi as potential pathogens. J. Infect. Dis. 200, 1623–1626. doi: 10.1086/644642
Rubio-Portillo E., Orts D., Llorca E., Fernández C., Antón J., Ferrer C., et al. (2020). The domestic environment and the lung mycobiome. Microorganism 8, 1717. doi: 10.3390/microorganisms8111717
Sayers E. W., Cavanaugh M., Clark K., Pruitt K. D., Schoch C. L., Sherry S. T., et al. (2021). GenBank. Nuc. Acids Res. 49, D92–D96. doi: 10.1093/nar/gkaa1023
Schindel D. E., Cook J. A. (2018). The next generation of natural history collections. PLoS Biol. 16, e2006125. doi: 10.1371/journal.pbio.2006125
Schoch C. L., Seifert K. A., Huhndorf S., Robert V., Spouge J. L., Levesque C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Nat. Acad. Sci. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Schwartz I. S., Kenyon C., Feng P., Govender N. P., Dukik K., Sigler L., et al. (2015). 50 years of Emmonsia disease in humans: the dramatic emergence of a cluster of novel fungal pathogens. PloS Path. 11, e1005198. doi: 10.1371/journal.ppat.1005198
Sharpton T. J., Stajich J. E., Rounsley S. D., Gardner M. J., Wortman J. R., Jordar V. S., et al. (2009). Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res. 19, 1722–1731. doi: 10.1101/gr.087551.108
Sikes R. S. and the Animal Care and Use Committee of the American Society of Mammalogists. (2016). 2016 Guidelines of the American society of mammalogists for the use of wild mammals in research and education. J. Mammal. 97, 663–688. doi: 10.1093/jmammal/gyw078
Skiada A., Pavleas I., Drogari-Apiranthitou M. (2017). Rare fungal infectious agents: a lurking enemy. F1000Res 6, 1917. doi: 10.12688/f1000research.11124.1
Smith M. F., Patton J. L. (1993). The diversification of south American murid rodents: evidence from mitochondrial DNA sequence data for the akodontine tribe. Biol. J. Linn. Soc 50, 149–177. doi: 10.1111/j.1095-8312.1993.tb00924.x
Smith M. F., Thomas W. K., Patton J. L. (1992). Mitochondrial DNA-like sequence in the nuclear genome of an akodontine rodent. Mol. Biol. Evol. 9, 204–215. doi: 10.1093/oxfordjournals.molbev.a040714
Spatz M., Richard M. L. (2020). Overview of the potential role of Malassezia in gut health and disease. Front. Cell. Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.00201
Stamatakis A. (2006). RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Taylor J. W., Barker B. M. (2019). The endozoan, small- mammal reservoir hypothesis and the life cycle of Coccidioides species. Med. Mycol. 57, S16–S20. doi: 10.1093/mmy/myy039
Taylor D. L., Walters W. A., Lennon N. J., Bochicchio J., Krohn A., Caporaso J. G., et al. (2016). Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for illumina amplicon sequencing. Appl. Environ. Microbiol. 82, 7217–7226. doi: 10.1128/AEM.02576-16
Thines M. (2019). An evolutionary framework for host shifts–jumping ships for survival. New Phytol. 224, 605–617. doi: 10.1111/nph.16092
Thompson C. W., Phelps K. L., Allard M. W., Cook J. A., Dunnum J. L., Ferguson A. W., et al. (2021). Preserve a voucher specimen! the critical need for integrating natural history collections in infectious disease studies. MBio 12, 1–20. doi: 10.1128/mBio.02698-20
Tipton L., Ghedin E., Morris A. (2017). The lung mycobiome in the next generation sequencing era. Virulence 8, 334–341. doi: 10.1080/21505594.2016.1235671
Tsang C. C., Tang J. Y. M., Ye H., Xing F., Lo S. K. F., Xiao C., et al. (2020). Rare/cryptic Aspergillus species infections and importance of antifungal susceptibility testing. Mycoses 63, 1283–1298. doi: 10.1111/myc.13158
van Woerden H. C., Gregory C., Brown R., Marchesi J. R., Hoogendoorn B., Matthews I. P. (2013). Differences in fungi present in induced sputum samples from asthma patients and non-atopic controls: a community-based case control study. BMC Infect. Dis. 13, 1–6. doi: 10.1186/1471-2334-13-69
Ward R. A., Thompson G. R., Villani A. C., Li B., Mansour M. K., Wuethrich M., et al. (2021). The known unknowns of the immune response to Coccidioides. J. Fungi 7, 377. doi: 10.3390/jof7050377
Weaver D., Gargo S., Bromley M., Bowyer P. (2019). The human lung mycobiome in chronic respiratory disease: limitations of methods and our current understanding. Curr. Fungal Infect. Rep. 13, 109–119. doi: 10.1007/s12281-019-00347-5
Weaver E., Kolivras K. N., Thomas R. Q., Thomas V. A., Abbas K. M. (2020). Environmental factors affecting ecological niche of Coccidioides species and spatial dynamics of valley fever in the united states. Spat. Spatio-temporal Epidemiol. 32, 100317. doi: 10.1016/j.sste.2019.100317
White T. J., Bruns T., Lee S., Taylor J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR protocols: A guide to methods and applications. Eds. Innis M. A., Gelfand D. H., Sninsky J. J., White T. J. (Academic Press, Orlando, Florida, USA), 315–322.
Wiesner D. L., Klein B. S. (2017). Lung epithelium: barrier immunity to inhaled fungi and driver of fungal-associated allergic asthma. Curr. Opin. Microbiol. 40, 8–13. doi: 10.1016/j.mib.2017.10.007
Willger S. D., Grim S. L., Dolben E. L., Shipunova A., Hampton T. H., Morrison H. G., et al. (2014). Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome 2, 1–15. doi: 10.1186/2049-2618-2-40
Yates T. L. (1996). “Tissues, cell suspensions and chromosomes,” in Measuring and monitoring biological diversity: Standard methods for mammals. Eds. Wilson D. E., Cole F. R., Nichols J. D., Rudran R., Foster M. S. (Washington D.C.: Smithsonian Institution Press), 275–278.
Keywords: Coccidioides, Valley fever, Onygenales, Blastomyces, Aspergillus fumigatus, Pneumocystis
Citation: Salazar-Hamm PS, Montoya KN, Montoya L, Cook K, Liphardt S, Taylor JW, Cook JA and Natvig DO (2022) Breathing can be dangerous: Opportunistic fungal pathogens and the diverse community of the small mammal lung mycobiome. Front. Fungal Biol. 3:996574. doi: 10.3389/ffunb.2022.996574
Received: 17 July 2022; Accepted: 24 August 2022;
Published: 26 September 2022.
Edited by:
Timothy Yong James, University of Michigan, United StatesReviewed by:
Lotus Lofgren, Duke University, United StatesSeung-Yoon Oh, Changwon National University, South Korea
Copyright © 2022 Salazar-Hamm, Montoya, Montoya, Cook, Liphardt, Taylor, Cook and Natvig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paris S. Salazar-Hamm, cHNoMTAyQHVubS5lZHU=