Abstract
Epigenetic information can be passed on from one generation to another via DNA methylation, histone modifications, and changes in small RNAs, a process called epigenetic memory. During a mammal’s lifecycle epigenetic reprogramming, or the resetting of most epigenetic marks, occurs twice. The first instance of reprogramming occurs in primordial germ cells and the second occurs following fertilization. These processes may be both passive and active. In order for epigenetic inheritance to occur the epigenetic modifications must be able to escape reprogramming. There are several examples supporting this non-Mendelian mechanism of inheritance including the prepacking of early developmental genes in histones instead of protamines in sperm, genomic imprinting via methylation marks, the retention of CenH3 in mammalian sperm and the inheritance of piwi-associated interfering RNAs. The ability of mammals to pass on epigenetic information to their progeny provides clear evidence that inheritance is not restricted to DNA sequence and epigenetics plays a key role in producing viable offspring.
Introduction
Epigenetic information is encrypted in genetic sequences, and includes DNA methylation, histone modifications and small RNA changes (Bonasio et al., 2010). Epigenetic memory is the ability to transfer epigenetic information from one generation to the next. Epigenetic information uses patterns of inheritance, which are not determined by DNA sequence alone and may result in an epigenetic memory, which like genetic memory can be stably inherited and passed onto progeny through meiosis, although epigenetic inheritance mainly defies Mendelian laws.
Epigenetic modifications can also be propagated through mitotic cell divisions, a process referred to as epigenetic somatic inheritance, resulting in somatic genetic memories. Both mitotic and meiotic epigenetic inheritance involve similar epigenetic mechanisms such as histones and methylation patterns. However the focus of this review will be on a process that is less common: transgenerational inheritance of epigenetic states through meiotic cell divisions (Whitelaw and Whitelaw, 2008).
In most cells in the body epigenetic marks are relatively stable and become fixed once cells differentiate or exit the cell cycle (Morgan et al., 2005). However there are two key situations in which epigenetic information undergoes extensive reprogramming and becomes erased and re-established (Feng et al., 2010).
Epigenetic reprogramming, which occurs first during gametogenesis and again in early embryogenesis, are events characterized by extensive, changes in DNA methylation and chromatin modifications (Reik, 2007; Lange and Schneider, 2010). During early stages of development, genes that are required later in development are temporarily repressed using reversible histone modifications, which allow for the expression of these genes when needed (Reik, 2007). These cells are in a pluripotent state that gives them the ability to differentiate into many different cell types as they develop and their fate becomes more defined and restricted (Reik, 2007).
Epigenetic Reprogramming
The germ line cells are unique as they undergo meiosis and are alone responsible for transmitting both genetic, and possibly epigenetic, information to the next generation (Combes and Whitelaw, 2010). However major reprogramming takes place in primordial germ cells (PGCs) in which parental imprints are erased and totipotency is restored.
Primordial germ cells are derived from epiblast cells and in females they enter meiotic arrest in prophase of meiosis I, while in males PGCs enter mitotic arrest until about birth when mitosis of spermatogonial stem cells begins (Morgan et al., 2005). Early PGCs have epigenetic marks similar to somatic cells, for example random X chromosome inactivation and imprinted genes. In somatic cells imprinted genes are those where only a single copy is expressed according to the parent-of-origin. However in E11.5 (embryonic day) PGCs as well as later stage germ cells, these same genes are expressed biallelically, indicating that the imprints have been largely erased from the gametes by this stage – a sign of epigenetic reprogramming. Using flow cytometry on allele-specific expression of imprinted genes it has been determine that the imprints are largely intact in migrating PGCs; out of four imprinted genes analyzed, Snrpn, Igf2, H19 were expressed monoallelically, whereas Igf2r was expressed biallelically (Szabo et al., 2002).
However by the time the PGCs arrive at the genital ridges, shown in Figure 1, imprinted genes are biallelically expressed and methylation in imprinted genes and single copy genes has been erased, with the majority of this demethylation occurring between E11.5 and E12.5 (Hajkova et al., 2008). In addition to the rapid loss of DNA methylation, several other reprogramming activities are known to occur including the loss of the repressive dimethylation of histone 3 lysine 9 and many histone modifications (Hajkova et al., 2008). The extent of demethylation over these few cell cycles occurs despite the presence of Dnmt1, a DNA methyltransferase, in the nucleus of the PGCs indicating that this process is likely to be active and it completes the first round of reprogramming (Hajkova et al., 2008). It is important to note however, that reprogramming does not occur universally and certain epigenetic marks present are not erased (Hajkova et al., 2008). However it has been determined that following the first round of reprogramming, male PGCs have approximately 60% less methylation, while female PGCs have approximately 70% less (Popp et al., 2010).
Figure 1
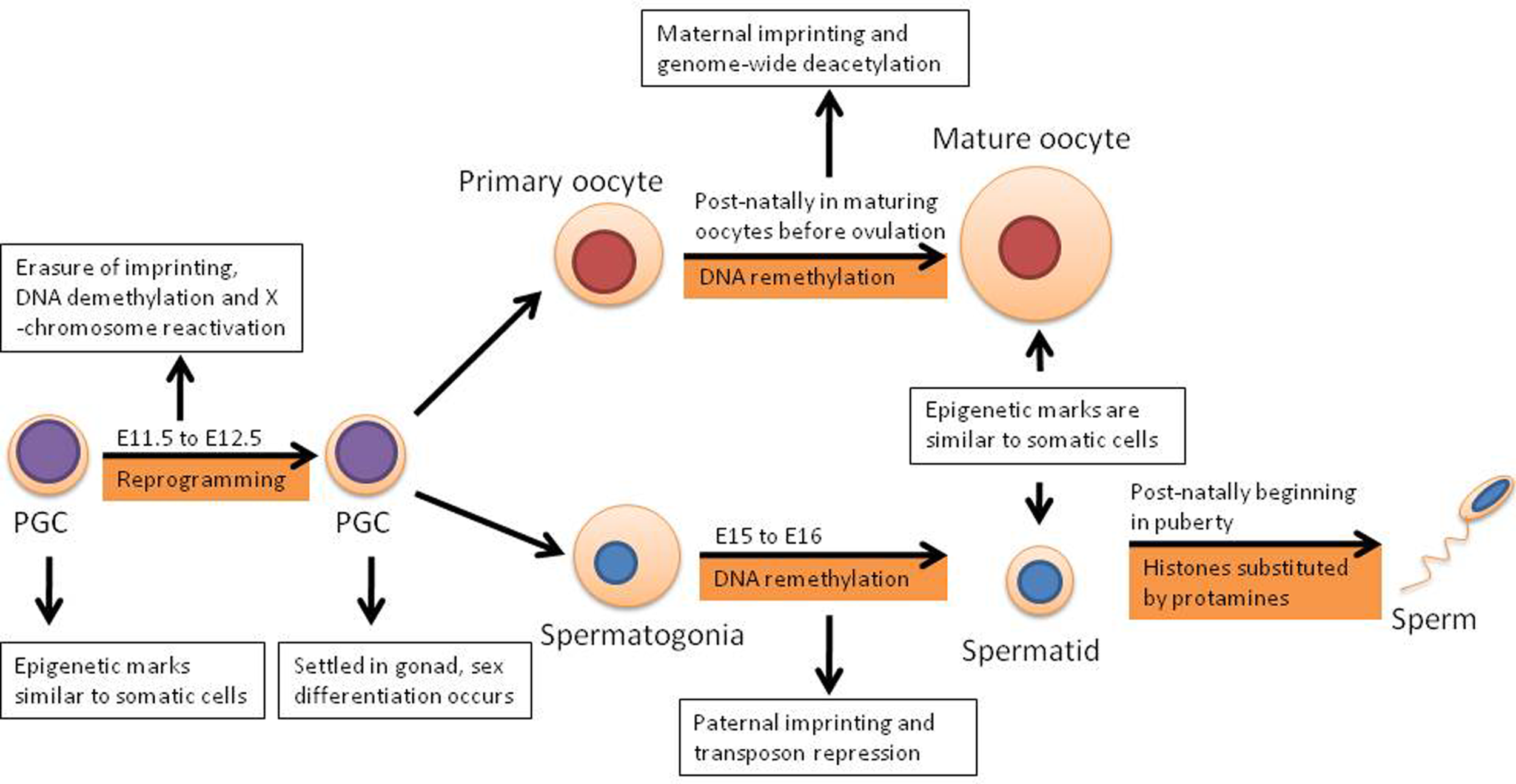
The first round of epigenetic reprogramming occurs in the germline according to embryonic day (E) of mammals prior to the maturity of gametes and is distinct in males and females.
DNA remethylation occurs several days later in the male germ line, between E15–E16. However in the female germ line the process occurs postnatally during oocyte growth (Sasaki and Matsui, 2008). The resulting DNA has a similar methylation state to somatic cells, although there are important differences including methylation patterns at regions which parental imprinting control regions (ICRs; Hemberger et al., 2009).
The second time reprogramming occurs is following fertilization, shown in Figure 2. Fertilization occurs between two parental genomes of extremely different epigenetic states, due to the fact that they are in different parts of the cell cycle, and considerable processing has to occur before they are able to begin transcription and cell division. The maternal genome is packaged in abundantly modified histones, while the paternal genome is densely wrapped in histone substitutes called protamines (Puri et al., 2010). It is important to note however that although the majority of histones are replaced during spermatogenesis, the transition is not complete (Puri et al., 2010).
Figure 2
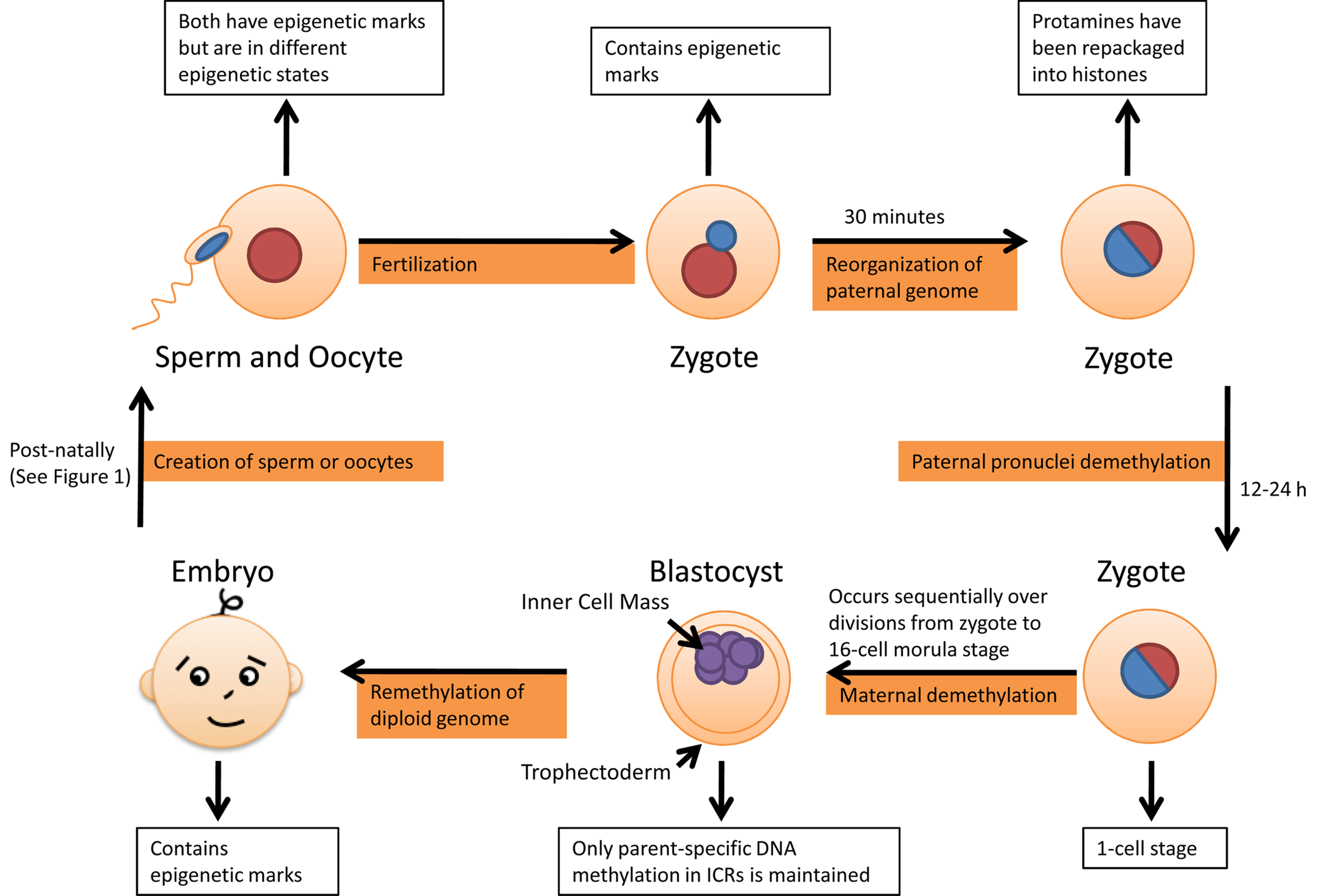
Second round of epigenetic reprogramming occurs following fertilization sequentially from zygote to morula stage, remethylation occurs only in inner cell mass, the mass of cells which eventually gives rise to the definitive structures of the fetus.
When the sperm arrives in the cytoplasm of the oocyte, a specialized cell with considerable chromatin remodeling capacity, the paternal genome is reorganized. The paternal genome is first decondensed and then repackaged into nucleosomes containing histone octamers (Puri et al., 2010). The replacement of protamines with histones generates chromatin structures that are compatible with somatic cellular processes, such as transcription (Ooi and Henikoff, 2007). Chromatin remodeling of the paternal genome occurs extremely quickly, with studies done in mice indicating that most protamines were removed within 30 min following fertilization (van der Heijden et al., 2005). The histones, which associate with the paternal chromatin, are more acetylated than those already present in the maternal chromatin (Lange and Schneider, 2010). It is possible this imbalance is passive due to the presence of non-acetylated histones from oocyte cytoplasm. It may also be an active process of incorporation of a particular acetylated histone variant (Morgan et al., 2005). At this point the paternal DNA undergoes DNA demethylation of almost the entire genome, erasing most of the epigenetic marks. The demethylation process occurs within the first 12–24 h following fertilization and is thought to be active because it occurs before DNA replication begins in the paternal pronucleus (Morgan et al., 2005; Combes and Whitelaw, 2010). However it is not known if there are any physical similarities between reprogramming in PGCs and reprogramming in zygotes.
The sperm activates the oocyte and the maternal genome completes its second meiotic division (Deng and Li, 2009). During the first cleavage division in the zygote, the maternal and paternal genomes combine so each of the daughter blastomeres inherits nearly equal amounts of methylated DNA. DNA methylation levels of the maternal genome in blastomeres sequentially decrease during each subsequent division. Reprogramming in the early embryo seems to be susceptible to environmental stress, and errors in this spatially and temporally highly coordinated process provide one important explanation for the high rate of embryo loss after fertilization (Haaf, 2006).
Like paternal demethylation, maternal demethylation leaves certain sites untouched and is accompanied by histone modifications. DNA methyltransferases Dnmt1, Dnmt3a, and Dnmt3b are enzymes that catalyze the transfer of a methyl group to DNA, thereby maintaining the methylation of CpG sites within the mammalian genome which allows for gene silencing as well as maintaining genome integrity (Tsumura et al., 2006). Dnmt1 is the most abundant key maintenance methyltransferase which operates at unmethylated cytosines of hemimethylated CpGs after DNA replication (Xu et al., 2010). Dnmt3a and Dnmt3b are responsible for de novo methylation at unmethylated CpGs, methylation of satellite repeats, methylation of H19 and Dlk1/Gtl2 differentially methylated regions (DMRs) and methylation of imprinted gene Rasgrf1 (Kato et al., 2007; Taberlay and Jones, 2011).
During maternal demethylation, Dnmt1 is excluded from the nucleus and so it is thought that this results in failure to maintain DNA methylation patterns during replication. These steps allow the cells to gain pluripotency and occur during the transition from the 1-cell zygote to the 16-cell morula stage, with the embryo remaining the same size despite cell division (Sasaki and Matsui, 2008; Combes and Whitelaw, 2010). In comparison to reprogramming in PGCs, parent-specific DNA methylation is maintained in ICRs during this stage (Edwards and Ferguson-Smith, 2007).
The process of epigenetic reprogramming prepares the early embryo for the development of the first two cell lineages in the blastocyst, resulting in differentiation between the trophoectoderm and the inner cell mass from the morula, a decision involving the relationship between epigenetic marks, cell position and transcription factors (Combes and Whitelaw, 2010). Not long after implantation, de novo genome-wide DNA methylation occurs in cells committed to the inner cell mass and is carried out by Dnmt3a and Dnmt3b (Watanabe et al., 2004). This results in an epigenetic asymmetry between the trophoectoderm and the inner cell mass, which is thought to occur due to the fact that the inner cell mass will give rise to the embryonic and adult tissues, whereas the trophectoderm forms tissues which are discarded at birth (Hemberger et al., 2009).
These DNA methylation patterns are maintained from that point on through mitotic divisions in somatic lineages (Bonasio et al., 2010). Both stages of reprogramming play a key role in the removal of inherited epigenetic modifications, but they do not remove all epigenetic marks.
Escaping Reprogramming: Epigenetic Inheritance
If the entire genome were reprogrammed in the germline it would be impossible for epigenetic modifications to be inherited. However there are epigenetic markers that can escape both incidences of reprogramming resulting in epigenetic modifications that persist in the somatic cells of the individual.
Protamine-mediated inheritance
One way that epigenetic inheritance can happen is due to the incomplete replacement of histones by small basic nuclear proteins called protamines during gametogenesis. Although the majority of DNA in sperm chromatin is bound by protamines, a small percentage, only 1% in mice for example, remains bound to nucleosomes (Wykes and Krawetz, 2003). In humans 4% (Hammoud et al., 2009) to 15% (Gatewood et al., 1987) of histones are retained in mature sperm. Studies indicate that the remaining histones are not spread randomly across the genome, and in fact their retention may be linked to gene activity in developmental processes (Hammoud et al., 2009).
A possible reason for the prepacking of very early developmental genes with histones in sperm is that it might prevent their tight condensation by protamines and thus facilitate their transcription in early embryos (Ooi and Henikoff, 2007). A study by Gardiner-Garden et al. (1998), detected histone-associated regions in ε and γ-globin gene in humans, both of which are transcribed in primitive erythroblasts in the embryonic yolk sac. The embryonic yolk sac differentiates at 3 weeks of gestation, and γ-globin predominates during the fetal period (Gardiner-Garden et al., 1998). In contrast, no histone-associated regions were found at β-globin genes, which have a minor involvement in the fetus but predominant after birth in bone marrow, or δ-globin, which is produced after birth (Peschle et al., 1985; Gardiner-Garden et al., 1998). The results indicate that the presence of histones may mark ε and γ-globin genes for early expression in the embryo (Gatewood et al., 1987; Gardiner-Garden et al., 1998). There is also evidence of enrichment of histones not only at genes expressed during early embryogenesis but also at specific imprinted genes. Histones are ideal candidates for this method of inheritance due to their influence on chromatin structure, which regulates access to genes (Ooi and Henikoff, 2007).
Methylation-mediated inheritance
When global genome reprogramming occurs in PGCs, intracisternal A particles (IAPs) are almost entirely protected from demethylation resulting in a significant fraction of all IAPs not being reprogrammed (Popp et al., 2010). In the second round of reprogramming following fertilization, IAPs as well as certain imprinted genes are once again left untouched (Feng et al., 2010). The shared protection from reprogramming may explain the parental imprinting effects observed with some endogenous genes whose expression are influenced by nearby IAP insertions (Rakyan et al., 2002). The most famous example of epigenetic inheritance comes from experiments focusing on agouti locus in mice that controls coat color and is linked to the epigenetic status of IAPs (Morgan et al., 1999).
In mice which carry the Avy allele, an IAP retrotransposon has inserted upstream of the agouti locus resulting in ectopic expression of agouti protein, resulting in hair follicle melanocytes to switch from the synthesis of black to yellow (Morgan et al., 1999). In addition to the yellow coat color, expression of the agouti genes is linked to obesity, diabetes, and increased susceptibility to tumors (Yen et al., 1994). Avy/A mice that have a hypomethylated Avy epiallele have a yellow coat, indicating that Avy is dominant over A. However when the cryptic promoter in the IAP is silenced, such as in the case of hypermethylation, Avy/A mice have a black coat because the epiallele is silenced, essentially reverting to wild type, and in this case Avy is no longer dominant resulting in mice indistinguishable from wild-type (A/A) mice.
The linkage between the coat color of the mother and offsring is due to the incomplete erasure of an epigenetic mark in the female germline, and is a clear example of epigenetic inheritance; however the probability of passing on an epiallele to the next generation is never 100% (Morgan et al., 1999). The insertion of IAP that acts as a controlling element of agouti expression has an unstable epigenetic state in the germline, resulting in the possibility of different cells of the same individual having a different epigenetic status (Morgan et al., 1999; Cropley et al., 2010).
In addition to IAPs, imprinted genes occasionally evade the second round of reprogramming and remain marked with their parental origin (Bartolomei, 2009). Imprints established in the germline must not only escape reprogramming but also the subsequent wave of de novo methylation that occurs (Morgan et al., 2005). DMRs are crucial to escaping reprogramming and many imprinted genes contain them. DMRs indicate areas that are methylated differently between parental alleles and some, which are derived from sperm and oocytes, behave as ICRs (Morgan et al., 2005). Genomic imprints are maintained through cis- and trans-acting DNA-binding factors that specifically recognize ICRs and prevent active demethylation on the paternally methylated alleles as well as facilitating maintenance DNA methylation and differential chromatin modifications (Bartolomei, 2009). One such factors is the protein PGC7/STELLA. Although Stella-/- eggs have normal ICR methylation, zygotes derived from these eggs are hypomethylated at multiple loci with maternally or paternally methylated ICRs. Zygotes also exhibit premature global loss of DNA methylation on the maternal pronucleus, indicating that the maternal genome must be protected from the demethylation that occurs on the paternal genome immediately after fertilization, and that the role for STELLA is widespread (Nakamura et al., 2007). Other proteins have been identified that are involved in the stability of imprints, such as RBBP1/ARID4A and RBBP1L1/ARID4B, which are involved in the maintenance of imprinting at the Snprn locus (Wu et al., 2006; Bartolomei, 2009).
Epigenetic marks must also be placed on newly replicated DNA in order for epigenetic inheritance to occur. Dnmt1 which has an established role in maintaining DNA methylation at imprinted loci is present in pre-implantation embryos (Li et al., 1993). Although the details are unclear it is likely DNA methylation is maintained through a combination of the oocyte-specific form of Dnmt1 and the somatic form, which is the form of the enzyme most often observed in mammalian cells (Hirasawa et al., 2008). A study using H19 and Dlk1/Gtl2 loci indicated that neither Dnmt3a nor Dnmt3b are required for the maintenance of methylation imprints and indicated that Dnmt1 of both the maternal and zygotic isoforms is sufficient for maintaining methylation imprints in the pre-implantation embryo (Hirasawa et al., 2008).
One widely studied imprinted gene in mice is the maternally expressed H19 gene. The mouse gene H19 encodes an RNA that is highly expressed in embryonic tissues of endodermal and mesodermal origin, and has been shown to be hypermethylated on the inactivated paternal allele in both somatic tissues and sperm. The differential methylation present in the somatic tissues is inherited from the gametes and preserved through embryogenesis, providing an example of epigenetic memory (Tremblay et al., 1997). H19 has also been shown to be hypermethylated on the paternal allele in humans, indicating that it is also escaping reprogramming.
Histone-mediated inheritance
The inheritance of histone marks and of small RNAs potentially through both oocyte and sperm might also contribute to epigenetic inheritance as well as to reprogramming across generations. Histones are the architectural proteins that package DNA into nucleosomal particles (Heinkoff et al., 2004). Modified histone variants may control barriers to transcription and perpetuate active chromatin states. Specifically, histone retention in mature sperm as previously described, occurs non-randomly resulting in the sperm genome providing not only paternal DNA sequence, but also molecular regulatory factors. If certain histones were not protected from becoming protamines, the embryo would not be able to develop correctly (Ooi and Henikoff, 2007; Ward, 2010).
Another example of epigenetic memory mediated by histones is by the retention of CENP-A (mammalian special histone H3 variant, CenH3) in mammalian sperm (Palmer et al., 1990). The role of CenH3 in higher eukaryotes is to assemble centromeric nucleosomes, whereby determining the location of the centromere (Amor et al., 2004). CENP-A, the core histone that replaces ordinary histone H3 in centromeric nucleosomes, is crucial because it ensures correct chromosome segregation during mitosis and meiosis (Dubin et al., 2010). In this way, centromere identity does not rely on DNA sequence but rather on the presence of CenH3, which has been proposed to act as an epigenetic mark of the centromere (Bernad et al., 2009). Incorporation of newly synthesized CENP-A has been shown to occur in telophase/early G1 in human cells (Dala, 2009). Kinetochore location is specified when CenH3-containing nucleosomes are segregated between daughter DNA strands which also contain new nucleosomes which do not contain CenH3. The result is a dilution of the CenH3 signal during S phase which leads to a self-propagating epigenetic state, marking centromere location as well as providing the optimal chromatin configuration for kinetochore formation and function (Gieni et al., 2008).
Small RNA-mediated inheritance
In addition to chromatin marks like histones, it is also possible for epigenetic inheritance to occur by inheritance of certain RNAs through the germline. In mammals, piwi-associated interfering RNAs (piRNAs) associate with an animal-specific subclass of Argonaute family called piwi proteins (Houwing et al., 2007). piRNAs are specifically expressed in germ cells and are important for their proper maturation (Kim, 2006). Transposons are nucleic acid parasites that are able to both move and propagate within a host genome (Kazazian, 2004). piRNAs are 24–30 nucleotides in length, and are responsible for transposon silencing in animals (Girard and Hannon, 2007; Brennecke et al., 2008). Approximately 50% of the genome in humans is made up of transposons and derepression of transposable elements occurs during epigenetic reprogramming, so piRNAs are crucial defense which ensure that transposons are not reactivated (Bernstein et al., 2007; Aravin et al., 2009). Reactivation of transposons in germ cells has extremely negative effects, as due to their high copy numbers as well as their ability to move around the genome, active transposons have the potential to be highly disruptive to their host, since integration of a transposon near or in a gene can disturb its coding sequences or expression pattern resulting in sterility (Aravin et al., 2009). Inheritance of piRNAs plays an important role of defending the germline against activation of transposons (O’Donnell and Boeke, 2007).
Previous research on the inheritance of epigenetic memory through piRNAs has been done in Drosophila. When female flies that have not been exposed to a specific active transposon are crossed with males that have established control over the same active transposon, the result is sterility in the progeny, a phenomenon referred to as hybrid dysgenesis. However the reverse cross does not result in the same outcome, supporting the hypothesis that maternally deposited piRNAs are responsible for epigenetic inheritance in this instance (Brennecke et al., 2008). piRNAs have also been shown to be required for male fertility in mice, with mutations resulting in the degeneration of the germline but somatic cells appearing to be relatively unaffected (Costa, 2008; Klattenhoff and Theurkauf, 2008). In addition, recent research in a Chinese population has indicated that genetic variants in piRNAs confer susceptibility to spermatogenic failure. This research indicates that piRNAs, although non-coding, have an important role in successful development as well as the ability to be passed on through epigenetic inheritance in humans (Jablonka and Raz, 2009; Gu et al., 2010). Thus, piRNAs are a crucial form of epigenetic memory due to their ability to silence transposons, thus ensuring the fertility of the progeny (Aravin et al., 2009).
Epigenetic Inheritance of Disease
Epigenetic memory is not only crucial to insuring viable offspring, but has already begun to be shown to play an important role in disease. Specifically, transgenerational epigenetic inheritance in humans may result in illnesses due to epimutations that are then passed on to progeny. One known case of epigenetic inheritance is increased risk for hereditary non-polyposis colorectal cancer (HNPCC), a sporadic colorectal cancer with mismatch repair deficiency due to genetic mutations or hypermethylation, particularly of MLH1 or MSH2 mismatch repair genes (Hitchins et al., 2005; Hesson et al., 2010). Mutations of MSH6, PMS2, and MLH3 have also been associated with a small number of HNPCC cases (Rustgi, 2007).
Hypermethylation of MLH1 is not limited to neoplastic cells but can also originate from the germline and therefore become widespread in normal somatic cells (Hitchins et al., 2007). Individuals possessing the germline epimutation have only one functional copy of the MLH1 gene from the conception and therefore cancers typical of HNPCC syndrome develop (Hesson et al., 2010). The presence of an MLH1 epimutation in the germline indicates potential for inheritance from parents to progeny. Studies have indicated that such inheritance is possible, with one family showing maternal transmission of the epimutation to the son, although the mutation was erased in his spermatozoa. In this case, the MLH1 epimutation that caused a predisposition to HNPCC in the mother was also present in the son, indicating he also had an increased risk of cancer. However, in her other children the epimutation was shown to revert to its normal state, indicating that the mutation was erased during reprogramming. These results indicated that germline transmission of an epigenetic state that confers disease susceptibility such as in the case of hypermethylation of MLH1 is possible. Overall, studies thus far have indicated that although epimutations are usually erased in the germline, they may be retained at a low frequency (Hitchins et al., 2007).
In addition to HNPCC, Prader–Willi (PWS) and Angelman (AS) syndromes may be caused by epigenetic inheritance in certain subgroups of patients. Both diseases are caused by the loss of function of imprinted genes 15q11-q13 in humans. Analysis of patients with PWS and AS has indicated that although in some cases the defect is caused by an imprinting center (IC) deletion; out of 51 patients with PWS 32 did not have the deletion, and out of 85 patients with AS, 66 patients did not have any mutation in IC elements either. However, in 27% of cases when the patient had AS it was indicated that there was an imprinting defect which occurred after fertilization. In such AS patients the imprinting defect occurred on the chromosome inherited from either maternal or paternal grandmother, while in PWS patients it occurred in the chromosome inherited from the paternal grandmother. These results indicate that the failure to erase the maternal imprint during spermatogenesis, or incomplete reprogramming, is one cause of both PWS and AS (Buiting et al., 2003; Santos-Reboucas and Pimentel, 2007).
Hereditary non-polyposis colorectal cancer, PWS, and AS have all shown that epigenetic inheritance may also be partially responsible for the inheritance of disease. Although human studies are still very limited, they have already begun to show the importance of epigenetic memory when it comes to illness and disease (Gluckman et al., 2007).
Concluding Remarks
There are many ways for a mammal to pass on epigenetic marks, such as inheritance of histones, piRNAs, and methylation signatures. What all of these epigenetic modifications with the ability to be passed on as epigenetic memory have in common is their importance in maintaining proper development and their ability to escape the near-genome-wide reprogramming that occurs in the germline. Epigenetic inheritance plays a crucial role in important functions such as expression of genes in early embryo development, imprinting, and the silencing of transposons and without epigenetic factors like DNA methylation and histone modifications development cannot proceed. Epigenetic inheritance may also lead to inheritance of epimutations, which increase risk of disease. As we begin to understand the importance of epigenetic memory it quickly becomes clear that inheritance is not simply a matter of Mendelian genetics and that our understanding of the complex interactions which generate life are far from complete.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Amor D. J. Kalitsis P. Sumer H. Choo A. (2004). Building the centromere: from foundation proteins to 3D organization. Trends Cell Biol.14, 359–368.10.1016/j.tcb.2004.05.009
2
Aravin A. A. van der Heijden G. W. Castaneda J. Vagin V. V. Hannon G. J. Bortvin A. (2009). Cytoplasmic compartmentalization of the fetal piRNA pathway in mice. PLoS Genet.5, e10000764.10.1371/journal.pgen.1000764
3
Bartolomei M. S. (2009). Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev.23, 2124–2133.10.1101/gad.1841409
4
Bernad R. Sanchez P. Losada A. (2009). Epigenetic specification of centromeres by CENP-A. Exp. Cell Res.315, 3233–3241.10.1016/j.yexcr.2009.07.023
5
Bernstein B. E. Meissner A. Lander E. S. (2007). The mammalian epigenome. Cell23, 669–681.10.1016/j.cell.2007.01.033
6
Bonasio R. Tu S. Reinberg D. (2010). Molecular signals of epigenetic states. Science330, 612–616.10.1126/science.1191078
7
Brennecke J. Malone C. D. Aravin A. A. Sachidanandam R. Stark A. Hannon G. J. (2008). An epigenetic role for maternally inherited piRNAs in transposon silencing. Science322, 1387–1392.10.1126/science.1165171
8
Buiting K. Grob S. Lich C. Gillessen-Kaesbach El-Maarri O. Horsthemke B. (2003). Epimutations in Prader-Willi and Angelman with an imprinting defect. Am. J. Hum. Genet.72, 571–577.
9
Combes A. Whitelaw E. (2010). Epigenetic reprogramming: enforcer or enabler of developmental fate?Dev. Growth Differ.52, 483–491.10.1111/j.1440-169X.2010.01185.x
10
Costa F. F. (2008). Non-coding RNAs, epigenetics and complexity. Gene410, 9–17.10.1016/j.gene.2007.12.008
11
Cropley J. E. Suter C. M. Beckman K. B. Martin D. I. K. (2010). CpG methylation of a silent controlling element in the murine avy allele is incomplete and unresponsive to methyl donor supplementation. PLoS ONE5, e9055.10.1371/journal.pone.0009055
12
Dala Y. (2009). Epigenetic specification of centromeres. Biochem. Cell Biol.87, 273–282.
13
Deng M. Li R. (2009). Sperm chromatin-induced ectopic polar body extrusion in mouse eggs after ICSI and delayed egg activation. PLoS ONE4, e7171.10.1371/journal.pone.0007171
14
Dubin M. Fuchs J. Graf R. Schubert I. Nellen W. (2010). Dynamics of a novel centromeric histone variant CenH3 reveals the evolutionary ancestral timing of centromere biogenesis. Nucleic Acids Res.1–12.
15
Edwards C. A. Ferguson-Smith A. C. (2007). Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol.19, 281–289.
16
Feng S. Jacobsen S. E. Reik W. (2010). Epigenetic reprogramming in plant and animal development. Science330, 622–627.10.1126/science.1190614
17
Gardiner-Garden M. Ballesteros M. G. Tam P. P. L. (1998). Histone- and Protamine-DNA Association: conservation of different patterns within B-globin domain in human sperm. Mol. Cell. Biol.18, 3350–3356.
18
Gatewood J. M. Cook G. R. Balhorn R. Bradbury E. M. Schmid C. W. (1987). Sequence-specific packaging of DNA in human sperm chromatin. Science236, 962–964.10.1126/science.3576213
19
Gieni R. S. Chan G. K. T. Henzel M. J. (2008). Epigenetics regulate centromere formation and kinetochore function. J. Cell. Biochem.104, 2027–2039.
20
Girard A. Hannon G. J. (2007). Conserved themes in small-RNA-mediated transposon control. Trends Cell Biol.18, 136–148.10.1016/j.tcb.2008.01.004
21
Gluckman P. Hanson M. Beedle A. (2007). Non-genomic transgenerational inheritance of disease risk. Bioessays29, 145–154.10.1002/bies.20522
22
Gu A. Ji G. Shi X. Long Y. Xia Y. Song L. Wang S. Wang X. (2010). Genetic variants in Piwi-interacting RNA pathway genes confer susceptibility to spermatogenic failure in a Chinese population. Hum. Reprod.25, 2955–2961.
23
Haaf T. (2006). Dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr. Top. Microbiol. Immunol.310, 13–22.
24
Hajkova P. Ancelin K. Waldmann T. Lacoste N. Lange U. C. Cesari F. Lee C. Almouzni G. Schneider R. Surani M. A. (2008). Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature452, 877–881.10.1038/nature06714
25
Hammoud S. S. Nix D. A. Zhang H. Purwar J. Carrell D. T. Cairns B. R. (2009). Distinctive chromatin in human sperm packages genes for embryo development. Nature460, 473–478.
26
Heinkoff S. Furuyama T. Ahmad K. (2004). Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet.20, 320–326.10.1016/j.tig.2004.05.004
27
Hemberger M. Dean W. Reik W. (2009). Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat. Rev. Mol. Cell Biol.10, 526–537.
28
Hesson L. Hitchins M. Ward R. (2010). Epimutations and cancer predisposition: importance and mechanisms. Curr. Opin. Genet. Dev.20, 290–298.
29
Hirasawa R. Chiba H. Kaneda M. Tajima S. Li E. Jaenisch R. Sasaki H. (2008). Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev.22, 1607–1616.10.1101/gad.1667008
30
Hitchins M. Williams R. Cheong K. Halani N. Lin V. A. Packham D. Ku S. Buckle A. Hawkins N. Burn J. Gallinger S. Goldblatt J. Kirk J. Tomlinson I. Scott R. Spigelman A. Suter C. Martin D. Suthers G. Ward R. (2005). MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastreoenterology129, 1392–1399.10.1053/j.gastro.2005.09.003
31
Hitchins M. Wong J. Suthers G. Suter C. Martin D. Hawkins N. Ward R. (2007). Inheritance of a cancer-associated MLH1 germ-line epimutation. N. Engl. J. Med.356, 697–705.
32
Houwing S. Kamminga L. M. Berezikov E. Cronembold D. Girard A. van den Else H. Filippov D. V. Blaser H. Raz E. Moens C. B. Plasterk R. H. A. Hannon G. J. Draper B. W. Ketting R. F. (2007). A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in zebrafish. Cell129, 69–82.10.1016/j.cell.2007.03.026
33
Jablonka E. Raz G. (2009). Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol.84, 131–176.
34
Kato Y. Masahiro K. Hata H. Kumaki K. Hisano M. Kohara Y. Okano M. Li E. Nozaki M. Sasaki H. (2007). Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum. Mol. Genet.16, 2272–2280.
35
Kazazian H. H. Jr. (2004). Mobile elements: drivers of genome evolution. Science303, 1626–1636.10.1126/science.1089670
36
Kim V. N. (2006). Small RNAs just got bigger: piwi-interacting RNAs (piRNAs) in mammalian testes. Genes Dev.20, 1993–1997.10.1101/gad.1456106
37
Klattenhoff C. A. Theurkauf W. E. (2008). Biogenesis and germline functions of piRNAs. Development135, 3–9.10.1242/dev.006486
38
Lange U. C. Schneider R. (2010). What an epigenome remembers. Bioessays32, 659–668.10.1002/bies.201000030
39
Li E. Beard C. Jaenisch R. (1993). Role for DNA methylation in genomic imprinting. Nature366, 362–365.10.1038/366362a0
40
Morgan H. D. Santos F. Green K. Dean W. Reik W. (2005). Epigenetic reprogramming in mammals. Hum. Mol. Genet.14(Suppl 1), R47–R58.
41
Morgan H. D. Sutherland H. G. Martin D. I. K. Whitelaw E. (1999). Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet.23, 314–318.
42
Nakamura T. Arai Y. Umebara H. Masuhara M. Kimura T. Taniguchi H. Sekimoto T. Ikawa M. Yoneda Y. Okabe Tanaka, S. Shiota K. Nakano T. (2007). PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol.9, 64–71.
43
O’Donnell K. A. Boeke J. D. (2007). Mighty piwis defend the germline against genome intruders. Cell129, 37–44.
44
Ooi S. L. Henikoff S. (2007). Germline histone dynamics and epigenetics. Curr. Opin. Cell Biol.19, 257–265.
45
Palmer D. K. O’Day K. Margolis R. L. (1990). The centromere specific histone CENP-A is selectively retained in discrete loci in mammalian sperm nuclei. Chromosoma100, 32–36.
46
Peschle C. Mavillo F. Care A. Migliaccio G. Migliaccio A. R. Salvo G. Samoggia P. Petti S. Guerriero R. Marinucci M. Lazzaro D. Russo G. Mastroberardino G. (1985). Haemoglobin switching in human embryos: asynchrony of zeta-alpha and epsilon-gamma-globin switches in primitive and definite erythropoietic lineage. Nature313, 235–238.10.1038/313235a0
47
Popp C. Dean W. Feng S. Cokus S. J. Andrews S. Pellegrini M. Jacobsen S. E. Reik W. (2010). Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature463, 1101–1105.10.1038/nature08829
48
Puri D. Dhawan J. Mishra R. K. (2010). The paternal hidden agenda: epigenetic inheritance through sperm chromatin. Epigenetics5, 386–391.10.4161/epi.5.5.12005
49
Rakyan V. Blewitt M. E. Drunker R. Preis J. I. Whitelaw E. (2002). Metastable epialleles in mammals. Trends Genet.18, 348–351.10.1016/S0168-9525(02)02709-9
50
Reik W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature447, 425–432.10.1038/nature05918
51
Rustgi A. K. (2007). The genetics of hereditary colon cancer. Genes Dev.21, 2525–2538.10.1101/gad.1593107
52
Santos-Reboucas C. B. Pimentel M. M. G. (2007). Implication of abnormal epigenetic patterns for human diseases. Eur. J. Hum. Genet.15, 10–17.
53
Sasaki H. Matsui Y. (2008). Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat. Rev. Genet.9, 129–140.
54
Szabo P. E. Hubner K. Scholer H. Mann J. R. (2002). Allele-specific expression of imprinted genes in mouse migratory primordial germ cells. Mech. Dev.115, 157–160.
55
Taberlay P. C. Jones P. A. (2011). DNA methylation and cancer. Prog. Drug. Res.67, 1–23.
56
Tremblay K. D. Duran K. L. Bartolomei M. S. (1997). A 5’ 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol. Cell. Biol.17, 4322–4329.
57
Tsumura A. Hayakawa T. Kumaki Y. Takebayashi S. Sakaue M. Matsuoka C. Shimotohno K. Ishikawa F. Li E. Ueda H. R. Nakayama J. Okano M. (2006). Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells11, 805–814.10.1111/j.1365-2443.2006.00984.x
58
van der Heijden M. S. Brody J. R. Dezentje D. A. Gallmeier E. Cunningham S. C. Swartz M. J. DeMarzo A. M. Offerhaus J. H. Isacoff W. H. Hruban R. H. Kern S. E. (2005). In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin. Cancer Res.11, 7508–7515.10.1158/1078-0432.CCR-05-1048
59
Ward W. S. (2010). Function of sperm chromatin structural elements in fertilization and development. Mol. Hum. Reprod.16, 30–36.
60
Watanabe D. Suetake I. Tajima S. Hanaoka K. (2004). Expression of Dnmt3b in mouse hematopoietic progenitor cells and spermatogonia at specific stages. Gene Expr. Patterns5, 43–49.10.1016/j.modgep.2004.06.008
61
Whitelaw N. C. Whitelaw E. (2008). Transgenerational epigenetic inheritance in health and disease. Curr. Opin. Genet. Dev.18, 273–279.
62
Wu M. Y. Tsai T. F. Beaudet A. L. (2006). Defiency of Rbbp1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Dev.20, 2859–2870.10.1101/gad.1452206
63
Wykes S. M. Krawetz S. A. (2003). The structural organization of sperm chromatin. J. Biol. Chem.278, 29471–29477.
64
Xu F. Mao C. Ding Y. Rui C. Wu L. Shi A. Zhang H. Zhang L. Xu Z. (2010). Molecular and enzymatic profiles of mammalian DNA methyltransferases: structures and targets for drugs. Curr. Med. Chem.17, 4052–4071.
65
Yen T. T. Gill A. M. Frigeri L. G. Barsh G. S. Wol G. L. (1994). Obesity, diabetes, and neoplasia in yellow A(vy)-mice: ectopic expression of the agouti gene. FASEB J.8, 479–488.
Summary
Keywords
epigenetic memory, germ line reprogramming, genomic imprinting, epigenetic inheritance, epigenetic disease inheritance
Citation
Migicovsky Z and Kovalchuk I (2011) Epigenetic Memory in Mammals. Front. Gene. 2:28. doi: 10.3389/fgene.2011.00028
Received
28 April 2011
Accepted
26 May 2011
Published
08 June 2011
Volume
2 - 2011
Edited by
Jaap Joles, University Medical Center Utrecht, Netherlands
Reviewed by
Rachel Giles, University Medical Center Utrecht, Netherlands; Alyson MacInnes, The Hubrecht Institute of Developmental Biology and Stem Cell Research, Netherlands
Copyright
© 2011 Migicovsky and Kovalchuk.
This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Igor Kovalchuk, Department of Biological Sciences, University of Lethbridge, 4401 University Drive, Lethbridge, Canada AB T1K 3M4. e-mail: igor.kovalchuk@uleth.ca
This article was submitted to Frontiers in Epigenomics, a aspecialty of Frontiers in Genetics.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.