 Hemant K. Tiwari1*
Hemant K. Tiwari1* Amit Patki1 Jeffrey Lieberman2 T. Scott Stroup2
Amit Patki1 Jeffrey Lieberman2 T. Scott Stroup2 David B. Allison1,3,4 Rudolph L. Leibel5 and Wendy K. Chung5
David B. Allison1,3,4 Rudolph L. Leibel5 and Wendy K. Chung5
- 1 Section on Statistical Genetics, Department of Biostatistics, University of Alabama at Birmingham, Birmingham, AL, USA
- 2 Department of Psychiatry, Columbia University Medical Center, New York State Psychiatric Institute, New York, NY, USA
- 3 Department of Nutrition Sciences, University of Alabama at Birmingham, Birmingham, AL, USA
- 4 Nutrition Obesity Research Center, University of Alabama at Birmingham, Birmingham, AL, USA
- 5 Department of Pediatrics, Division of Molecular Genetics and the Naomi Berrie Diabetes Center of Columbia University Medical Center, Columbia University, New York, NY, USA
Antipsychotic drugs are widely used in treating schizophrenia, bipolar disorder, and other psychiatric disorders. Many of these drugs, despite their therapeutic advantages, substantially increase body weight. We assessed the association of alleles of 31 genes implicated in body weight regulation with weight gain among patients being treated with specific antipsychotic medications in the clinical antipsychotic trials in intervention effectiveness study, we found that rs2237988 in Potassium Channel Inwardly Rectifying Subfamily J Member 11 (KCNJ11), rs13269119 in Solute carrier family 30 member 8 (SLC30A8), and rs9922047 in fat mass and obesity associated (FTO) were associated with percent weight gain. We also observed the significant interaction of rs11643744 by treatment effect on the weight gain.
Introduction
Antipsychotic drugs (APDs) are effective treatments for several common psychiatric conditions, including schizophrenia and bipolar disorder. There has been a dramatic increase in the use of APDs in recent years. Some but not all APDs are associated with weight gain and metabolic dysregulation, a concern because many individuals with psychotic disorders are already metabolically vulnerable (Allison et al., 1999; Allison and Casey, 2001; Cope et al., 2005; Brecher et al., 2007; Strassnig et al., 2007; Correll et al., 2009; Varley and McClellan, 2009). Though possessing many advantages, many APDs have a major liability by virtue of their ability to promote substantiated weight gain and metabolic dysregulation in an already metabolically vulnerable population. During the past decade there have considerable research efforts to quantify, understand, prevent, and alleviate these adverse effects. Also, the marked increase in the use of these drugs, the importance of the mental illnesses they are used to treat, their profound effects on weight and metabolic functioning, and the known harms of obesity, diabetes, and metabolic dysregulation all underscore the importance of continued research in this area.
Even among patients taking any single APD, there is great variability in the amount of weight gained while taking the drug. These inter-individual differences could be due to genetic differences in the genes encoding the molecular targets for the drug in question and/or the genes encoding molecules affecting the pharmacokinetics of the drug (Cope et al., 2005). Although several investigators have examined genetic predictors of weight gain among persons taking APDs, few have tested for genetic mediators of weight gain response per se, or have examined a large number of candidate genes in large number of subjects (Need et al., 2009). In this study, therefore, we aimed to examine in the clinical antipsychotic trials in intervention effectiveness (CATIE) study by testing for genetic association of weight gain with single nucleotide polymorphisms (SNPs) in genes involved in body weight regulation.
Materials and Methods
Gene Selection and Study Data
We related 31 obesity-related genes to the effects of five APDs (perphenazine, olanzapine, quetiapine, risperidone, and ziprasidone) on the weight gain in 732 participants with genotypic data (out of 774 participants receiving treatment with at least one of these drugs) of the total 1493 subjects in CATIE study. The baseline antipsychotic medication at the time of randomization and other descriptive analysis such as distribution of age, sex, race, and baseline weight is given in Table 1. The CATIE study was conducted between January 2001 and December 2004 at 57 clinical sites in the United States, ascertaining individuals who were 18–65 years of age, had the DSM-IV diagnosis of schizophrenia, and were able to take appropriate oral medications. Patients were randomly assigned to oral olanzapine, perphenazine, quetiapine, risperidone under double-blinded treatment conditions in Phase 1, and followed for up to 18 months or until treatment was discontinued for any reason. Ziprasidone was added to the study in 2002. Information on the prior antipsychotic medications names and their generation are given in Table S1A in Supplementary Material and number of individuals randomized to a particular antipsychotic drug at base line is given in Table S1B in Supplementary Material. Note that the information provided in Tables S1A,B in Supplementary Material is restricted to individuals included for further genetic analysis.
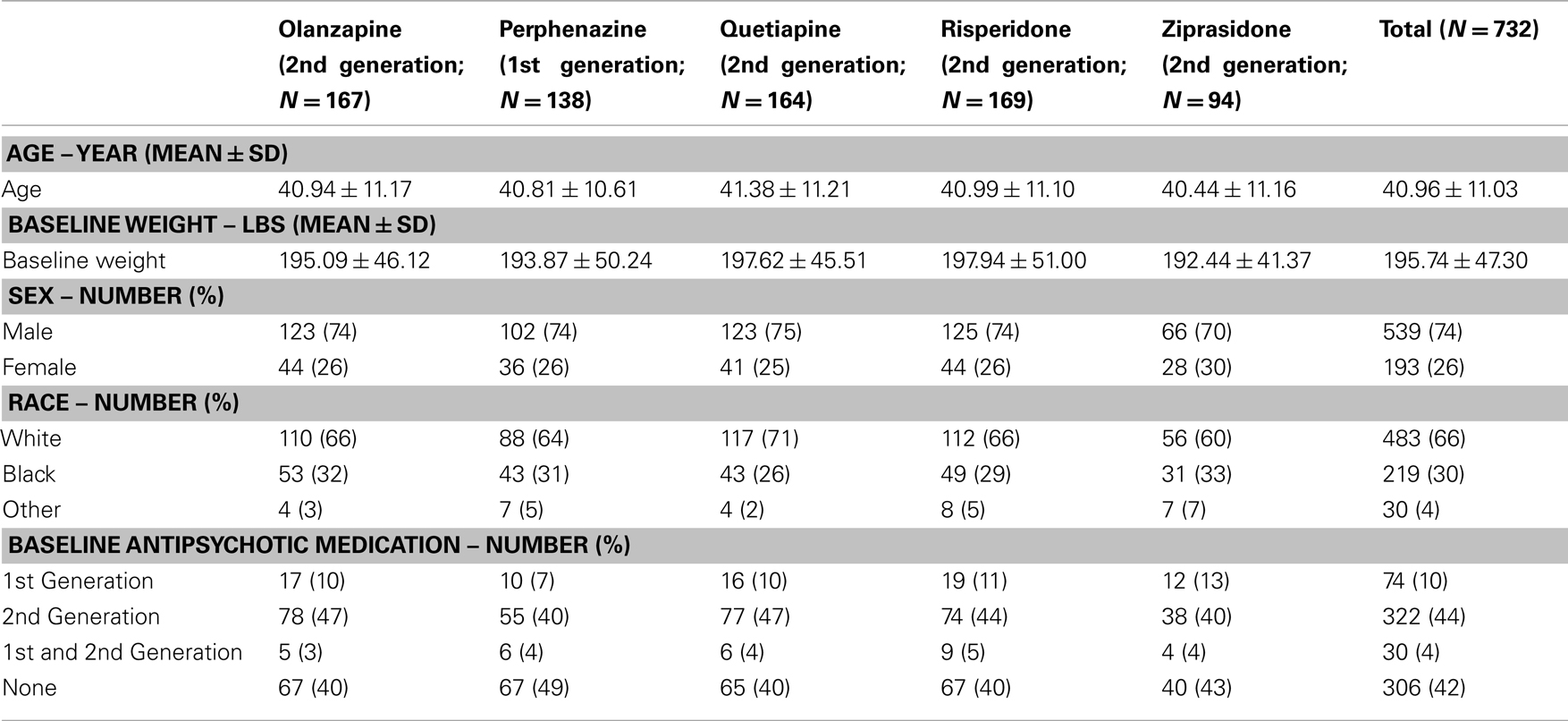
Table 1. Baseline covariates and antipsychotic drug history of patients with available genotypic data prior to randomization.
In addition to the primary psychiatric outcomes, patients were also monitored for metabolic effects of the drugs, including pulse, blood pressure, and body weight to evaluate the effect of antipsychotic treatments on weight gain, glucose and lipid metabolism, waist–hip ratio (WHR), and body mass index (BMI; Stroup et al., 2003 and Lieberman et al., 2005).
We investigated total weight gain in lbs within the duration of the study, weight gain per month of treatment, and weight gain > 7% in 732 individuals. The results are shown in the Table 2A (without any covariate adjustment) and Table 2B with covariates such as age, age squared, sex, race, baseline weight (only included for weight gain outcome variable), and treatment (olanzapine, quetiapine, risperidone, ziprasidone, and perphenazine). Our results given in Table 2A were similar to the results given in Table 3 of original study by Lieberman et al., 2005. Note that Lieberman et al., 2005 did not adjust for covariates in their analysis of weight measures.
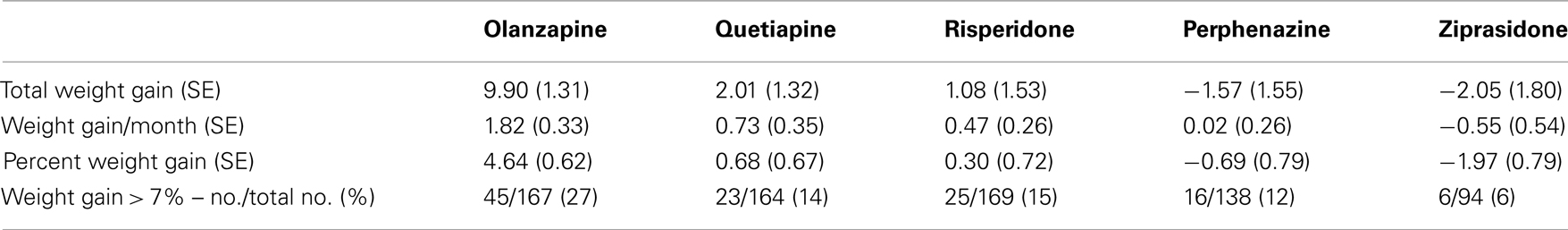
Table 2A. Weight measures without adjusting for covariates.
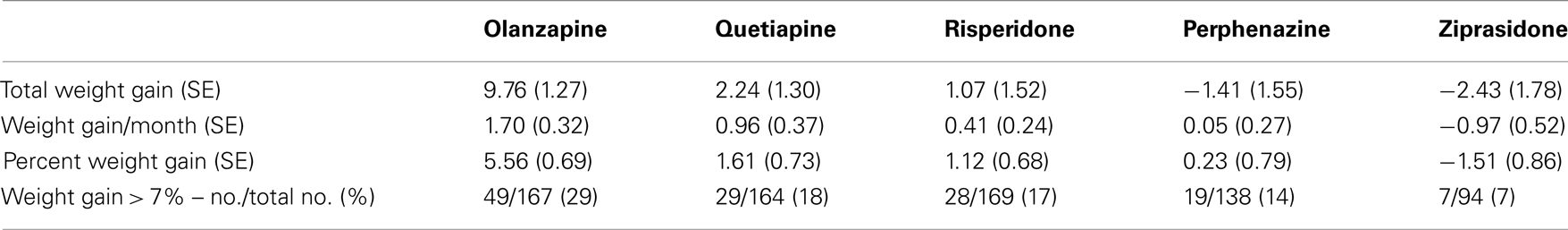
Table 2B. Weight measures after adjusting for covariates.
Genome-wide association analysis (GWAS) of the CATIE data was conducted by Perlegen Sciences by using the Affymetrix 500 K gene chip on 774 cases. Only 732 individuals had both clinical and genetic data available. The 1084 SNPs spanning the 31 selected candidate genes were used to perform the post hoc association analysis for effects of gene-by-drug interactions on weight gain during the course of treatment. Genes related to body weight regulation were selected, in part, because of their roles in the neural systems affected by psychotropic agents and included (Chung and Leibel (, 2008; Lindgren and McCarthy, 2008; van Vliet-Ostaptchouk et al 2009; Walley et al 2009). These included the dopamine receptors DRD2 and DRD4; Serotonin Transporter (SLC6A4), 5-Hydroxytryptamine Receptors (HTR1A, HTR2A, HTR2C, HTR1F, and HTR6); adrenergic receptors (ADRA1A, ADRA2A, ADRB3); Histamine receptors 1 and 2 (HRH1, HRH2); Cholinergic receptor, muscarinic 3 (CHRM3); synaptosomal associated protein SNAP25; genes associated with obesity including Leptin (LEP), Leptin receptor (LEPR), and fat mass and obesity associated (FTO); and genes associated with type 2 diabetes including peroxisome proliferator activated receptor gamma (PPARG); Transcription Factor 7 Like 2 (TCF7L2), Potassium Channel Inwardly Rectifying Subfamily J Member 11(KCNJ11), hematopoietically expressed homeobox (HHEX), Insulin like Growth Factor Binding Protein 2 (IGF2BP2), CDK5 Regulator Subunit-Associated Protein 1-Like (CDKAL1), Solute carrier family 30 member 8 (SLC30A8), Cyclin Dependent Kinase Inhibitors 2A and 2B (CDKN2A and CDKN2B), and Insulin degrading enzyme (IDE). We included intragenic SNPs and SNPs within or 50 kb 5′ or 3′ of these candidate genes. The list of all genes included in the study and number of SNPs within and flanking the genes are provided in Table S2 in Supplementary Material.
Body weight was measured at baseline and then repeatedly at the post-randomization visits. The timing of the visits varied by patient; hence, we did not have gain-in-weight measurements for all individuals at specific intervals following the start of treatment. With antipsychotics, the majority of weight gain (but by no means all) generally occurs by 120 days (Wetterling, 2001; Gentile, 2006; Gebhardt et al., 2009). This observation, coupled with the fact that we had considerable data around that time point, led to our choice of 120 days in the model. Hence, we predicted weight gain for each subject at the 120th day with a mixed model using weights measured at given time points as outcome, and both drug treatment duration and the square-root of drug treatment duration after randomization as predictors. We derived the prediction equation for weight gain at the 120th day by two ways, namely, Model 1 predicted weight gain estimated using combined data with all treatments as a non-linear function of time points only as described above; and Model 2 predicted weight gain estimated separately for each treatment as a non-linear function of time points and randomized treatment (i.e., we derived the five prediction equations for weight gain for each treatment by stratified sample with respect to treatments olanzapine, quetiapine, risperidone, perphenazine, and ziprasidone). Model 1 results are depicted in Tables, 3A with and without covariate adjustments, respectively. Similarly, we estimated weight gain, percent weight gain, and weight gain > 7% at 120th day, using model 2 and the results are depicted in Tables 4A without adjustment of covariates and with adjustment of covariates, respectively. Note that model 1 weight gain predictions were underestimating compared to other studies (Allison et al., 1999; Wirshing et al., 1999; Sicard et al., 2010; Cuerda et al., 2011; and Citrome et al., 2011), specifically for olanzapine-induced weight gain in schizophrenic patients. However, model 2 weight gain predictions were consistent with other studies. Both outcome variables, namely, weight gain and percent weight gain were checked for normality, and observations beyond four SD were considered to be outliers and were omitted from further analysis in the generalized linear model (GLM). However, we observed that both the analyses including outliers and the analyses excluding outliers gave similar results.
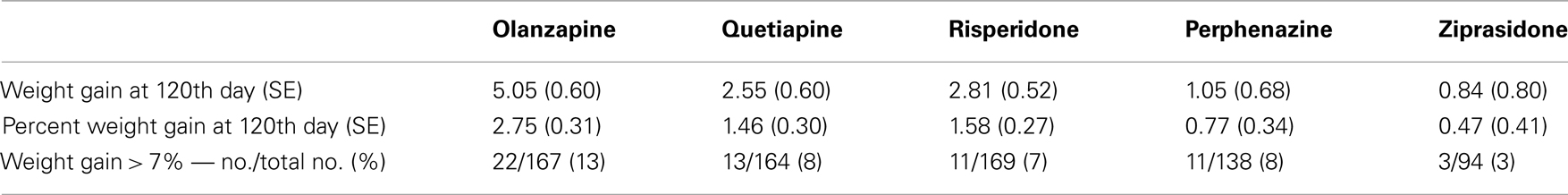
Table 3A. Weight measures without adjusting for covariates (Model 1: weight gain was estimated using combined data for all treatments).
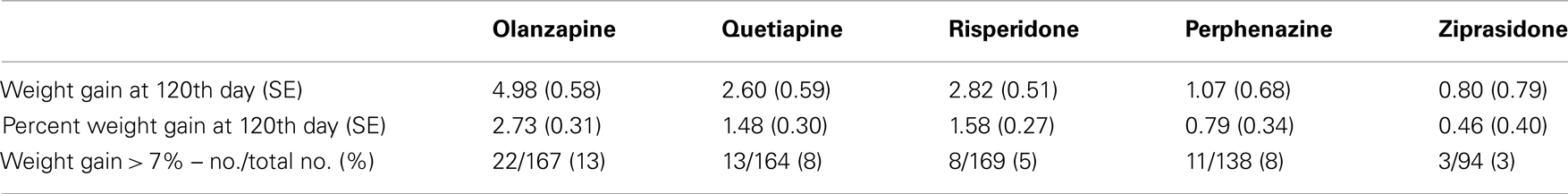
Table 3B. Weight measures after adjusting for covariates (Model 1: weight gain was estimated using combined data for all treatments).
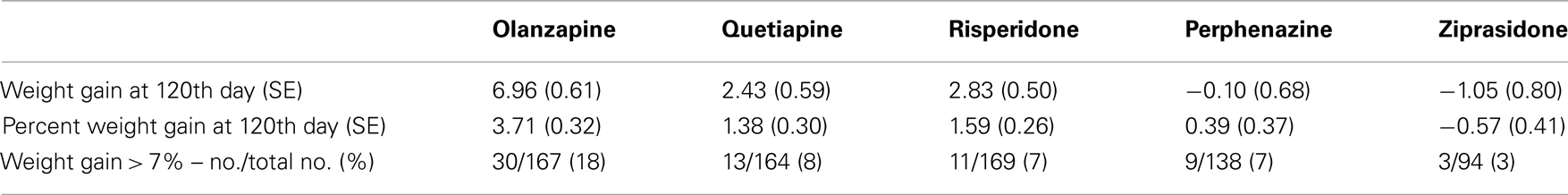
Table 4A. Weight measures without adjusting for covariates (Model 2: weight gain was estimated separately for each treatment).
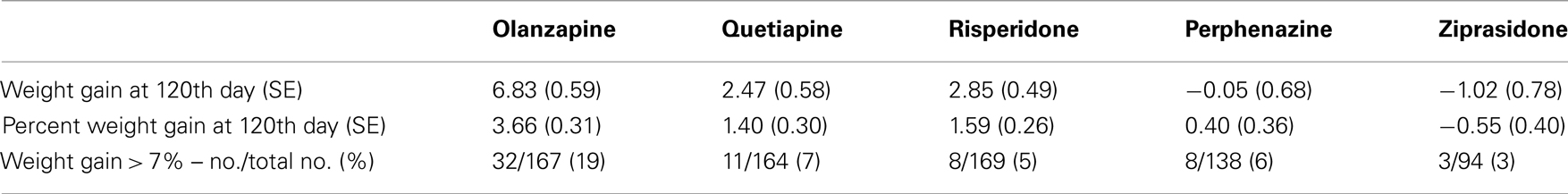
Table 4B. Weight measures after adjusting for covariates (Model 2: weight gain was estimated separately for each treatment).
We tested Hardy–Weinberg equilibrium (HWE) at each locus by using the chi-square with 1 degree of freedom (df) test (Weir, 1996). SNPs on the X chromosome were tested for HWE test by using data from females only. We also computed linkage disequilibrium (LD) among markers in the same gene by using r2 values. We tested the effect of each of 1084 SNPs on weight gain and percent weight gain at the 120th day by using a GLM framework.
We modeled age, age squared, sex, race, baseline weight (only included for weight gain outcome variable), and treatment (olanzapine, quetiapine, risperidone, ziprasidone, and perphenazine) as covariates in the linear model in addition to additive and dominance effects of the SNP. However, almost all of the individuals were off the prior drugs within 3 months after randomization. To control for extraneous variance due to antipsychotic drug history before randomization, and continual use of drugs (a cross-titration period) in addition to treatment drugs after randomization, we created a variable called “highest generation drug.” The highest generation drug was defined as the maximum of first and/or second generation drugs used before randomization to treatment drugs. We included two interaction terms in the linear model to control for extraneous variance due to prior drug use and are described as follows. The first interaction term in the linear model included highest generation drug used before randomization by its duration (i.e., how long the individual was on the highest generation drug before randomization). In cases in which the participant was taking multiple drugs of the same generation, we chose the drug with the longest duration of administration. Similarly, to control for extraneous variance due to some participants’ continual use of prior drugs in addition to randomized treatment drug in the titration period, we included the second interaction term in the model involving highest generation drug among prior drugs by its duration. Similar models were used for quantile regression (Redden et al., 2004) by using data at the 10th and 90th quantiles of the respective variables. Note that using these covariates will control for extraneous variance and will have very little effect on our association testing of primary outcome and SNPs. Multiple test correction for association testing was performed using a method proposed be Gao et al., 2008 for correlated SNPs within genes.
Results
Five treatment drugs were randomly assigned to the patients in the study: olanzapine, quetiapine, risperidone, ziprasidone, and perphenazine. After adjustment for covariates, treatment drug had a significant effect on percent weight gain (p-values < 0.0001). In a pair-wise comparison of each treatment drug, ziprasidone was used as a reference because it has been found to produce the lowest weight gain compared with the other drugs in this study (Allison et al., 1999; Brecher et al., 2007; Strassnig et al., 2007). Olanzapine had a significant effect on both weight gain and percent weight gain after Scheffe correction for multiple pair-wise comparisons (p-values < 0.0001). Means and SEs are in Tables 2–4. Tables 5A contain the significant p-values for main effects of SNPs and also SNP x Treatment interaction effects in genes FTO, KCNJ11, CDKL1, and SLC30A, using a GLM and a quantile regression method corresponding to phenotypes estimated using combined treatment groups (Model 1) and separately within each treatment group (Model 2), respectively. We included only FTO, KCNJ11, CDKL1, and SLC30A in Tables 5A because the five SNPs (rs11643744 in FTO (model 1 p-value = 0.024 and model 2 p-value = 0.02), rs9922047 in FTO (model 1 p-value = 0.002 and model 2 p-value = 0.05), and rs2237988 in KCNJ11 (model 1 p-value = 0.013), rs4712595 in CDKL1 (model 2 p-value = 0.03 and marginally significant (p = 0.051) for model 1, and rs13269119 in SLC30A8 (model 2 p-value = 0.04)) in these genes were found to be significant after a multiple tests correction. HWE test results and association results for all 1084 SNPs are provided in Tables S3A,B in Supplementary Material for weight gain when weight gain was estimated using all treatment groups together (model 1) and in Tables S4A,B in Supplementary Material for percent weight gain when weight gain was estimated separately within each treatment group (model 2). In the SNP association analysis using quantile regression at the 10th percentile, SNP rs2237988 5′ of KCNJ11 in an intron of ABCC8 (which encodes the Sulfonylurea Receptor) showed a significant association with percent weight gain even after multiple tests correction for multiple testing (Multiple tests corrected p-value 0.013, Table 5A for model 1). However, the significant association was not observed with percent weight gain when similar analysis was performed using estimated percent weight gain separately within each treatment group (p = 3.78 × 10−4 and multiple tests corrected p-value 0.30 in Table 5B). We further analyzed rs2237988 with adjustments for diabetes status since KCNJ11 and ABCC8 have been causally implicated in diabetes mellitus (Florez et al., 2003). Results are shown in Tables, 6A for model 1 (phenotype estimated using combined treatment groups) and model 2 (phenotype estimated using separately within each treatment group), respectively. We found that rs2237988 showed significant associations with percent weight gain when these parameters were adjusted for serum glucose concentration [p = 2.6 × 10−6 (multiple test corrected p-value = 0.003) in Table 6A) using model 1. Also, percent weight gain association with rs2237988 was significant after adjusting for serum glucose concentration in model 2 (p = 4.67 × 10−5 and multiple tests corrected p-value 0.04 in Table 6B). The SNPs rs4712595 of CDKL1 and rs13269119 of SLC30A8 showed significant associations with percent weight gain even after stringent multiple tests correction for multiple testing in the analysis of 10th percentile, when percent weight was analyzed using estimated percent weight gain separately within each treatment group (multiple tests corrected p-value 0.03 and 0.04, respectively, see Table 5B). Since both genes CDKL1 and SLC30A8 have been implicated in diabetes mellitus, we further analyzed rs4712595 and rs13269119 adjusting for diabetes status (see Results in Tables 6C). Only association between rs13269119 of SLC30A8 and percent weight gain remain significant after adjusting of glucose (p = 5.08 × 10−5, and p = 0.04 with multiple tests correction).
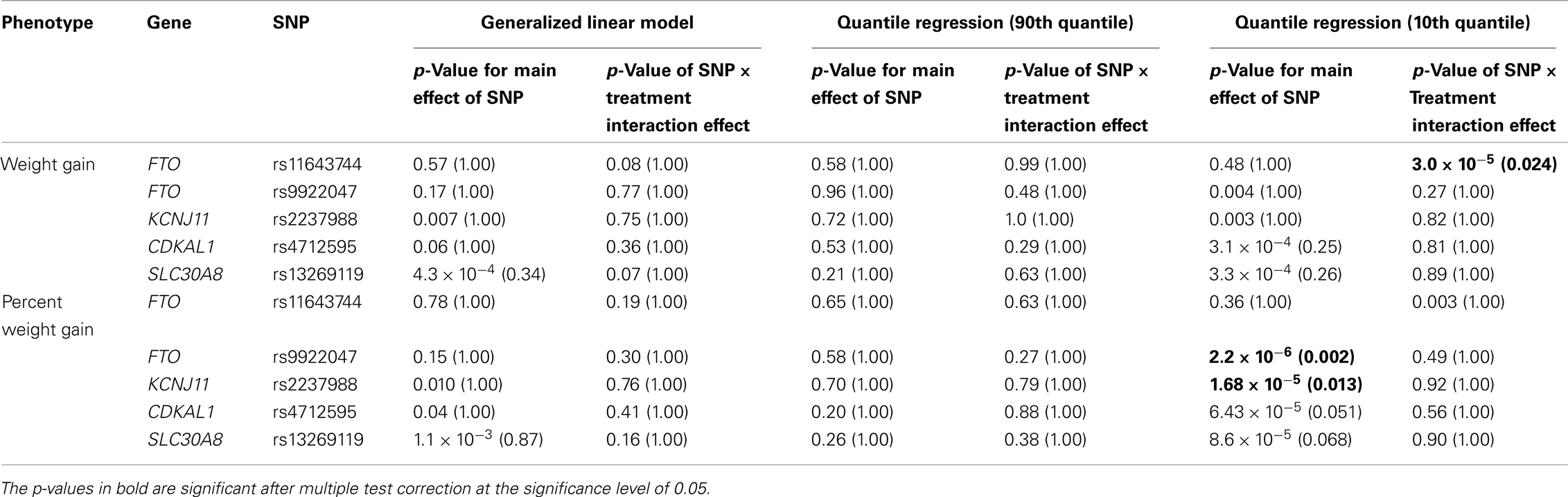
Table 5A. The p-values corresponding to the association results for significant markers with p-values corrected for multiple testing in parenthesis, using prediction model 1 (i.e., weight gain and percent weight gain at 120th day estimated with combined treatment groups).

Table 5B. The p-values corresponding to the association results for significant markers with p-values corrected for multiple testing in parenthesis, using prediction model 2 (i.e., weight gain and percent weight gain at 120th day estimated separately within each treatment group).
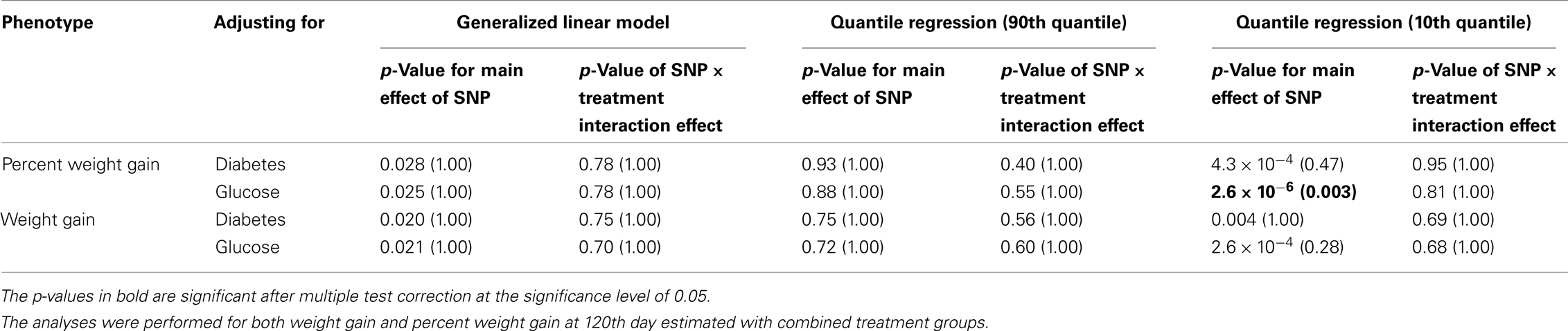
Table 6A. The p-values corresponding to the association results for rs2237988 marker in KCNJ11 after adjusting for diabetes status/glucose level with p-values corrected for multiple testing in parenthesis.
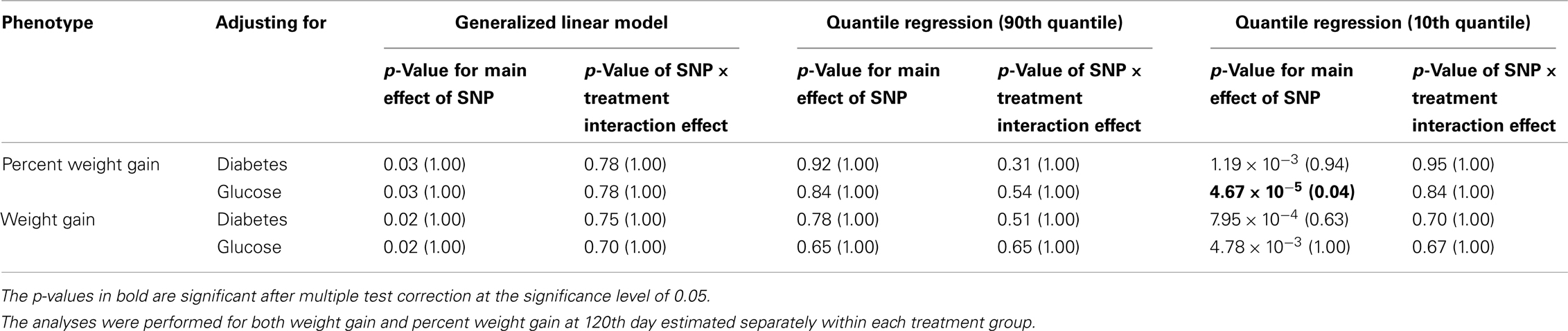
Table 6B. The p-values corresponding to the association results for rs2237988 marker on KCNJ11 after adjusting for diabetes status/glucose level with p-values corrected for multiple testing in parenthesis.
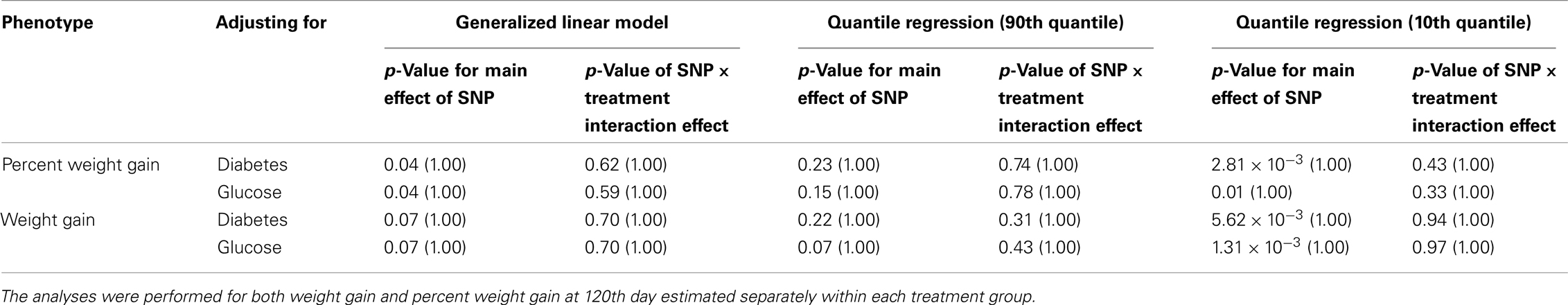
Table 6C. The p-values corresponding to the association results for rs4712595 marker on CDKAL1 after adjusting for diabetes status/glucose level with p-values corrected for multiple testing in parenthesis.
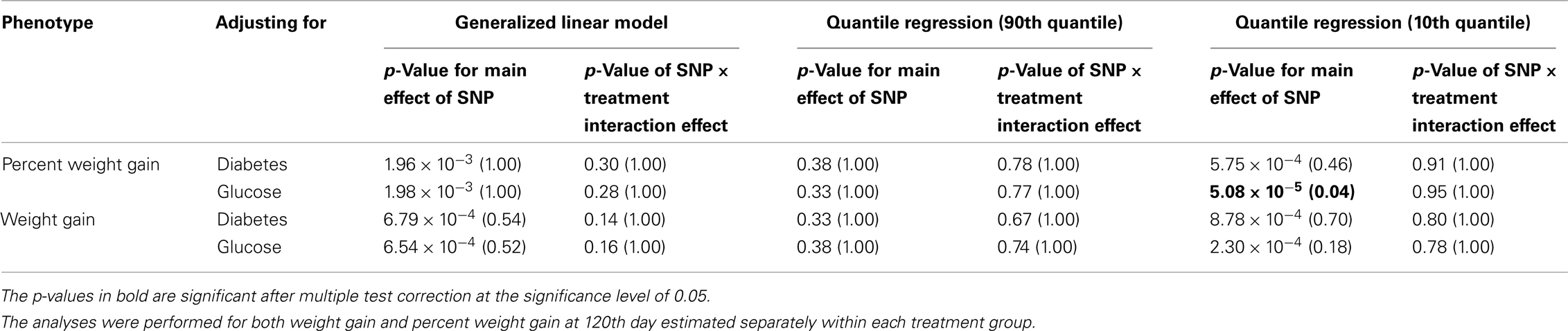
Table 6D. The p-values corresponding to the association results for rs13269119 marker on SLC30A8 after adjusting for diabetes status/glucose level with p-values corrected for multiple testing in parenthesis.
The SNP rs9922047 in FTO (a non-coding SNP in intron 1) was significantly associated with percent weight gain in the quantile regression at the 10th percentile using model 1 (multiple tests corrected p-value = 0.002) and also in model 2 (multiple tests corrected p-value = 0.05). An interaction between rs11643744 (another non-coding SNP in intron 1) in the FTO and treatment drug for weight gain was found to be significant after multiple tests correction in both models 1 (multiple tests corrected p-value = 0.024) and 2 (multiple tests corrected p-value = 0.02).
The adjusted mean values and SEs of weight gain for each genotype at rs2237988 (KCNJ11), rs4712595 (CDKAL1), rs13269119 (SLC30A8), rs9922047 (FTO) are shown in Tables 7A, respectively. The adjusted mean values and SEs from the quantile regression model for the treatment drug-by-genotype interaction effect at rs11643744 in FTO gene are shown in Table 8. We also investigated whether significant associations of KCNJ11, CDKAL1, SLC30A8, FTO SNPs with drug-related weight gain might be due to antecedent predisposition to adiposity, as reflected in baseline body weight. The association p-values of SNPs with baseline weight and BMI in all candidate genes are given in Tables S5A,B in Supplementary Material. There was no significant association detected for any candidate gene – including KCNJ11, CDKAL1, SLC30A8, FTO – with the baseline weight or BMI.
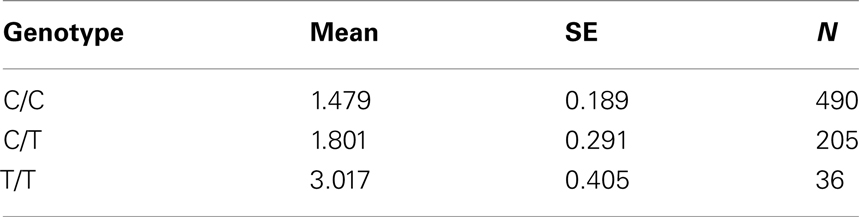
Table 7A. Adjusted mean percent weight gain corresponding to genotypes at rs2237988 (KCNJ11), using model 2.
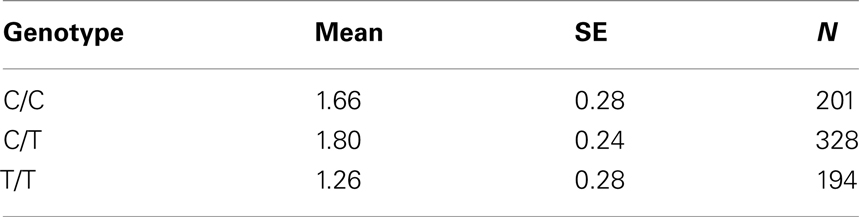
Table 7B. Adjusted mean percent weight gain corresponding to genotypes at rs4712595 (CDKAL1), using model 2.
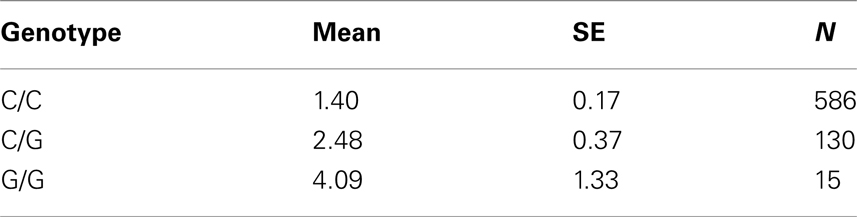
Table 7C. Adjusted mean percent weight gain corresponding to genotypes at rs13269119 (SLC30A8), using model 2.
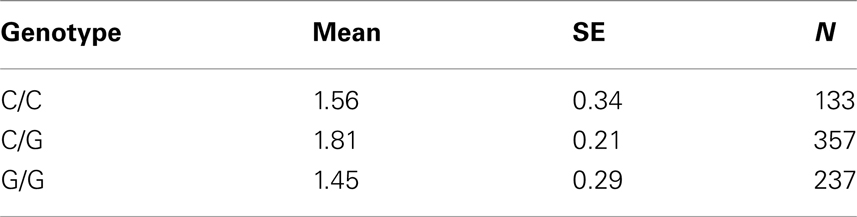
Table 7D. Adjusted mean percent weight gain corresponding to genotypes at rs9922047 (FTO), using model 2.
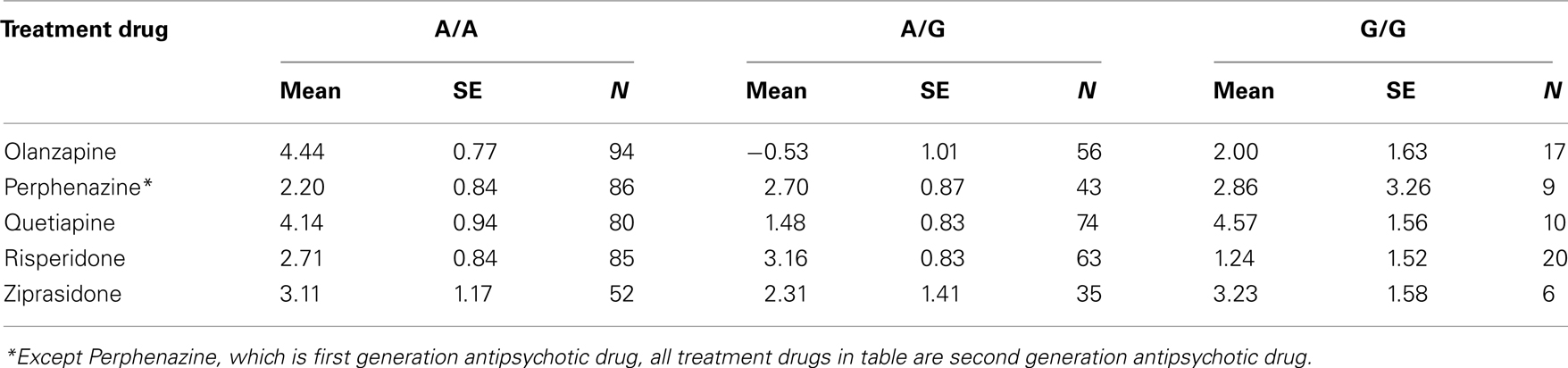
Table 8. Adjusted mean weight gain in lbs for treatment and genotypes at rs11643744 (FTO gene) in the interaction analyses.
Discussion
fillskip0pt We performed genetic analysis on predicted induced weight gain and percent weight gain at the 120th day in schizophrenic patients from CATIE study. The analyses were performed on outcome variable estimated using two ways, namely, predicted weight gain estimated using combined data with all treatments (model 1); and predicted weight gain estimated separately for each treatment (model 2). We found a significant association of rs2237988 in the KCNJ11 gene with percent weight gain estimated using combined data with all treatments. KCNJ11 encodes an ATP-sensitive potassium channel, Kir6.2, – that interacts with the SUR1 (ABCC8) subunit to determine insulin release (Inagaki et al., 1995). Both ABCC8 and KCNJ11 have been causally implicated in rare forms of diabetes, and ATP-sensitive potassium channels mediate some aspects of hypothalamic function related to food intake (Spanswick et al., 1997). In several studies type 2 diabetes prevalence among persons with schizophrenia has been found to be higher than in the general population (Lean and Pajonk, 2003; Gough and O’Donovan, 2005; Holt and Peveler, 2006; Allison et al., 2009). Also, the risk of diabetes in schizophrenia is increased by use of atypical APDs (Lean and Pajonk, 2003). The Consensus Statement, issued in 2004, indicated that drug-induced insulin resistance may occur because of weight gain or change in body fat distribution, or by direct effects on insulin-sensitivity target tissues (American Diabetes Association et al., 2004). We therefore further analyzed the genotypic data in KCNJ11 for association with percent weight gain with adjustment for baseline diabetes status (presence or absence), and serum glucose concentration at the 120th day after randomization. The serum glucose concentration was estimated for each subject at the 120th day with a mixed model by using the serum glucose concentration measured at given time points as the outcome. The association of rs2237988 in KCNJ11 with percent weight gain remained significant after multiple tests correction and with adjustment for the serum glucose concentration (p = 0.003). However, the significant association between rs2237988 and percent weight gain was not significant when using estimated percent weight gain separately within each treatment group. We also observed significant associations of rs4712595 of CDKL1 gene and rs13269119 of SLC30A8 with percent weight gain estimated at 120th day separately within each treatment. Both CDKL1 gene and SLC30A8 have been implicated for Type 2 diabetes. Thus, we further analyzed the genotypic data with percent weight gain with adjustment for baseline diabetes status (presence or absence), and serum glucose concentration at the 120th day after randomization. The association of rs13269119 in SLC30A8 with percent weight gain remained significant after multiple tests correction and with adjustment for the serum glucose concentration (p = 0.04). We found that rs4712595 of CDKAL1 association disappeared after correction for diabetes status; implying weight gain may be mediated by type 2 diabetes in the presence of rs4712595 in Schizophrenic patients. Genetic intervals containing SLC30A8 (8q24.11) and CDKL1 (6p22.3) have both been repeatedly implicated in type 2 diabetes (T2D) by association analyses (Lyssenko et al., 2008; McCarthy, 2010). SLC30A8 (solute carrier family 30 (zinc transporter), member 8) mediates zinc transport, specifically in beta cells. The gene is co-expressed in insulin-producing cells (Chimienti et al. 2004) where zinc participates in the processing of proinsulin to insulin and the release of insulin in response to glucose (Kirchhoff et al. 2008). CDKL1 (cdk5 regulatory subunit-associated protein 1-like 1) is associated with T2D and low birthweight, the latter possibly contributing to the risk of diabetes. In individuals in whom T2D is associated with allelic variation in CDKL1, T2D is not contingent upon obesity, as it is for many other diabetes-associated genes. The molecular physiology of this protein is not well understood, but it may play a role in proinsulin processing and/or mitochondrial oxidative metabolism related to beta cell release of insulin in response to glucose (Steinthorsdottir et al. 2007; Kirchhoff et al. 2008; Ohara-Imaizumi et al. 2010). Atypical antipsychotic medications may adversely affect beta cell function directly (Ader et al. 2005) – rendering allelic variation in genes such as SLC30 A8 and CDKL1 possible mediators of individual susceptibility to T2D as a consequence of exposure to these drugs. Increased circulating
insulin, or alterations of the timing and magnitude of endogenous release, may cause of increased body weight by effects on food intake (Mäkimattila et al. 1999), and might account for the association with weight gain detected here.
Fat mass and obesity associated has been associated with adiposity in several large genome-wide association studies (Dina et al. 2007; Frayling et al. 2007; Scuteri et al. 2007). The FTO SNP, rs9939609, which was previously associated with obesity, was not associated with weight gain in our study. Recently, Perez-Iglesias et al. 2010 investigated the association of FTO rs9939609 with weight gain in the context of treatment with haloperidol, Olanzapine, risperidone, ziprasidone, aripiprazole, or quetiapine. After 1 year of treatment with APDs, there was no significant effect on weight gain of the 3 genotypes defined by the rs9939609 variant. However, the two SNPs associated with weight gain in CATIE, rs9922047, and rs11643744 are within 10 kb, and 20 kb respectively, of rs9939609 and are in LD with rs9939609. Note that LD varies among populations and estimation of LD depends on the sample size. Therefore, it is possible that Perez-Iglesias et al. 2010 did not observed any association with rs9939609 due to difference between genetic make-up of the studies (Spanish sample in Perez-Iglesias and European American sample in our study) and small sample size (n = 143 in Perez-Iglesias and n = 732 in our study). FTO encodes a putative non-heme dioxygenase, but it is unclear how genetic variation at the FTO locus leads to increased adiposity (Han et al., 2010). The association may be conveyed – in part – by effects related to a nearby gene, RPGRP1L, that is a component of the ciliary basal body, an organelle implicated in the obesity of individuals with Bardet–Biedl syndrome (Stratigopoulos et al. 2008, 2011).
The genes implicated in this analysis play a role in energy homeostasis, presumably by hypothalamic mechanisms. These genes have also been implicated in aspects of glucose/insulin homeostasis. We endeavored to control for the latter because we were focused more on body weight effects, and the subjects were not fully phenotyped regarding diabetes. However, some antipsychotics have been implicated having direct adverse consequences for beta cell function (Best et al., 2005). The genes that we identified here may mediate effects on both body weight and diabetes susceptibility.
Conflict of Interest Statement
Dr. Allison has received funds from multiple pharmaceutical companies, litigators, and book publishers with interests in antipsychotic-induced weight gain.
Acknowledgments
This research was supported in part by NIH grant R01DK52431-16, and New York Obesity Research Center Grant P30 DK26687-25. We also thank Mihir Limdi, a summer intern in the Section on Statistical Genetics, Department of Biostatistics at UAB for his help in preparing the manuscript.
References
Ader, M., Kim, S. P., Catalano, K. J., Ionut, V., Hucking, K., Richey, J. M., Kabir, M., and Bergman, R. N. (2005). Metabolic dysregulation with atypical antipsychotics occurs in the absence of underlying disease: a placebo-controlled study of olanzapine and risperidone in dogs. Diabetes 54, 862–871.
Allison, D. B., and Casey, D. E. (2001). Antipsychotic-induced weight gain: a review of the literature. J Clin Psychiatry 62(Suppl. 7), 22–31. [Review].
Allison, D. B., Mentore, J. M., Heo, M., Chandler, L., Cappelleri, J. C., Infante, M., and Weiden, P. (1999). Antipsychotic-induced weight gain: a comprehensive research synthesis. Am. J. Psychiatry 156, 1686–1696.
Allison, D. B., Newcomer, J. W., Dunn, A. L., Blumenthal, J. A., Fabricatore, A. N., Daumit, G. L., Cope, M. B., Riley, W. T., Vreeland, B., Hibbeln, J. R., and Alpert, J. E. (2009). Obesity among those with mental disorders: a National Institute of Mental Health meeting report. Am. J. Prev. Med. 36, 341–350.
American Diabetes Association American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. (2004). Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 27, 596–601.
Best, L., Yates, A. P., and Reynolds, G. P. (2005). Actions of antipsychotic drugs on pancreatic ß-cell function: contrasting effects of clozapine and haloperidol. J Psychopharmacol. 19, 597–601.
Brecher, M., Leong, R. W., Stening, G., Osterling-Koskinen, L., and Jones, A. M. (2007). Quetiapine and long-term weight change: a comprehensive data review of patients with schizophrenia. J. Clin. psychiatry 68, 597.
Chimienti, F., Devergnas, S., Favier, A., and Seve, M. (2004). Identification and cloning of a beta-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes 53, 2330–2337.
Chung, W. K., and Leibel, R. L. (2008). “Energy and metabolism and obesity: research and clinical applications,” in Molecular Physiology of Monogenic and Syndromic Obesities in Human, ed. P. Donohoue (New Jersey: Humana Press), 1–22.
Citrome, L., Holt, R. I., Walker, D. J., and Hoffmann, V. P. (2011). Weight gain and changes in metabolic variables following olanzapine treatment in schizophrenia and bipolar disorder. Clin. Drug Investig. 31, 455–482.
Cope, M. B., Nagy, T. R., Fernández, J. R., Geary, N., Casey, D. E., and Allison, D. B. (2005). Antipsychotic drug-induced weight gain: development of an animal model. Int. J. Obes. (Lond.) 29, 607–614.
Correll, C. U., Manu, P., Olshanskiy, V., Napolitano, B., Kane, J. M., and Malhotra, A. K. (2009). Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA 302, 1765–1773; Erratum in: JAMA 302, 2322.
Cuerda, C., Merchan-Naranjo, J., Velasco, C., Gutierrez, A., Leiva, M., de Castro, M. J., Parellada, M., Giráldez, M., Bretón, I., Camblor, M., García-Peris, P., Dulín, E., Sanz, I., Desco, M., and Arango, C. (2011). Influence of resting energy expenditure on weight gain in adolescents taking second-generation antipsychotics. Clin. Nutr. doi: 10.1016/j.clnu.2011.03.007. [Epub ahead of print].
Dina, C., Meyre, D., Gallina, S., Durand, E., Korner, A., Jacobson, P., Carlsson, L. M., Kiess, W., Vatin, V., Lecoeur, C., Delplanque, J., Vaillant, E., Pattou, F., Ruiz, J., Weill, J., Levy-Marchal, C., Horber, F., Potoczna, N., Hercberg, S., Le Stunff, C., Bougneres, P., Kovacs, P., Marre, M., Balkau, B., Cauchi, S., Chevre, J. C., and Froguel, P. (2007). Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726.
Florez, J. C., Hirschhorn, J., and Altshuler, D. (2003). The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu. Rev. Genomics Hum. Genet. 4, 257–291.
Frayling, T. M., Timpson, N. J., Weedon, M. N., Zeggini, E., Freathy, R. M., Lindgren, C. M., Perry, J. R., Elliott, K. S., Lango, H., Rayner, N. W., Shields, B., Harries, L. W., Barrett, J. C., Ellard, S., Groves, C. J., Knight, B., Patch, A. M., Ness, A. R., Ebrahim, S., Lawlor, D. A., Ring, S. M., Ben-Shlomo, Y., Jarvelin, M. R., Sovio, U., Bennett, A. J., Melzer, D., Ferrucci, L., Loos, R. J., Barroso, I., Wareham, N. J., Karpe, F., Owen, K. R., Cardon, L. R., Walker, M., Hitman, G. A., Palmer, C. N., Doney, A. S., Morris, A. D., Smith, G. D., Hattersley, A. T., and McCarthy, M. I. (2007). A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894.
Gao, X., Starmer, J., and Martin, E. (2008). A multiple testing correction for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 21, 361–369.
Gebhardt, S., Haberhausen, M., Heinzel-Gutenbrunner, M., Gebhardt, N., Remschmidt, H., Krieg, J. C., Hebebrand, J., and Theisen, F. M. (2009). Antipsychotic-induced body weight gain: predictors and a systematic categorization of the long-term weight course. J. Psychiatr. Res. 43, 620–626.
Gentile, S. (2006). Long-term treatment with atypical antipsychotics and the risk of weight gain: a literature analysis. Drug Saf. 29, 303–319. [Review].
Gough, S. C., and O’Donovan, M. C. (2005). Clustering of metabolic comorbidity in schizophrenia: a genetic contribution? J. Psychopharmacol. 19(6 Suppl.), 47–55.
Han, Z., Niu, T., Chang, J., Lei, X., Zhao, M., Wang, Q., Cheng, W., Wang, J., Feng, Y., and Chai, J. (2010). Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 464, 1205–1209.
Holt, R. I., and Peveler, R. C. (2006). Association between antipsychotic drugs and diabetes. Diabetes Obes. Metab. 8, 125–135.
Inagaki, N., Gonoi, T., Clement, J. P. IV, Namba, N., Inazawa, J., Gonzalez, G., Aguilar-Bryan, L., Seino, S., and Bryan, J. (1995). Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270, 1166–1670.
Kirchhoff, K., Machicao, F., Haupt, A., Schäfer, S. A., Tschritter, O., Staiger, H., Stefan, N., Häring, H. U., and Fritsche, A. (2008). Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia 51, 597–601.
Lean, M. E., and Pajonk, F. G. (2003). Patients on atypical antipsychotic drugs: another high-risk group for type 2 diabetes. Diabetes Care 26, 1597–1605.
Lieberman, J. A., Stroup, T. S., McEvoy, J. P., Swartz, M. S., Rosenheck, R. A., Perkins, D. O., Keefe, R. S., Davis, S. M., Davis, C. E., Lebowitz, B. D., Severe, J., Hsiao, J. K., and Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. (2005). Effectiveness of antipsychotic drugs in patients with schizophrenia. N. Engl. J. Med. 353, 1209–1223.
Lindgren, C. M., and McCarthy, M. I. (2008). Mechanisms of disease: genetic insights into the etiology of type 2 diabetes and obesity. Nat. Clin. Pract. Endocrinol. Metab. 4, 156–163.
Lyssenko, V., Jonsson, A., Almgren, P., Pulizzi, N., Isomaa, B., Tuomi, T., Berglund, G., Altshuler, D., Nilsson, P., and Groop, L. (2008). Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 359, 2220–2232.
Mäkimattila, S., Nikkilä, K., and Yki-Järvinen, H. (1999). Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus. Diabetologia 42, 406–412.
Need, A. C., Keefe, R. S., Ge, D., Grossman, I., Dickson, S., McEvoy, J. P., and Goldstein, D. B. (2009). Pharmacogenetics of antipsychotic response in the CATIE trial: a candidate gene analysis. Eur. J. Hum. Genet. 17, 946–957.
Ohara-Imaizumi, M., Yoshida, M., Aoyagi, K., Saito, T., Okamura, T., Takenaka, H., Akimoto, Y., Nakamichi, Y., Takanashi-Yanobu, R., Nishiwaki, C., Kawakami, H., Kato, N., Hisanaga, S., Kakei, M., and Nagamatsu, S. (2010). Deletion of CDKAL1 affects mitochondrial ATP generation and first-phase insulin exocytosis. PLoS ONE 5, e15553. doi: 10.1371/journal.pone.0015553
Perez-Iglesias, R., Mata, I., Amado, J. A., Berja, A., Garcia-Unzueta, M. T., Martínez García, O., Arranz, M. J., Vazquez-Barquero, J. L., and Crespo-Facorro, B. (2010). Effect of FTO, SH2B1, LEP, and LEPR polymorphisms on weight gain associated with antipsychotic treatment. J. Clin. Psychopharmacol. 30, 661–666.
Redden, D. T., Fernández, J. R., and Allison, D. B. (2004). A simple significance test for quantile regression. Stat. Med. 23, 2587–2597.
Scuteri, A., Sanna, S., Chen, W. M., Uda, M., Albai, G., Strait, J., Najjar, S., Nagaraja, R., Orru, M., Usala, G., Dei, M., Lai, S., Maschio, A., Busonero, F., Mulas, A., Ehret, G. B., Fink, A. A., Weder, A. B., Cooper, R. S., Galan, P., Chakravarti, A., Schlessinger, D., Cao, A., Lakatta, E., and Abecasis, G. R. (2007). Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 3, e115. doi: 10.1371/journal.pgen.0030115
Sicard, M. N., Zai, C. C., Tiwari, A. K., Souza, R. P., Meltzer, H. Y., Lieberman, J. A., Kennedy, J. L., and Müller, D. J. (2010). Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics 11, 1561–1571.
Spanswick, D., Smith, M. A., Groppi, V. E., Logan, S. D., and Ashford, M. L. (1997). Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390, 521–525.
Steinthorsdottir, V., Thorleifsson, G., Reynisdottir, I., Benediktsson, R., Jonsdottir, T., Walters, G. B., Styrkarsdottir, U., Gretarsdottir, S., Emilsson, V., Ghosh, S., Baker, A., Snorradottir, S., Bjarnason, H., Ng, M. C., Hansen, T., Bagger, Y., Wilensky, R. L., Reilly, M. P., Adeyemo, A., Chen, Y., Zhou, J., Gudnason, V., Chen, G., Huang, H., Lashley, K., Doumatey, A., So, W. Y., Ma, R. C., Andersen, G., Borch-Johnsen, K., Jorgensen, T., van Vliet-Ostaptchouk, J. V., Hofker, M. H., Wijmenga, C., Christiansen, C., Rader, D. J., Rotimi, C., Gurney, M., Chan, J. C., Pedersen, O., Sigurdsson, G., Gulcher, J. R., Thorsteinsdottir, U., Kong, A., and Stefansson, K. (2007). A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 39, 770–775.
Strassnig, M., Miewald, J., Keshavan, M., and Ganguli, R. (2007). Weight gain in newly diagnosed first-episode psychosis patients and healthy comparisons. Schizophr. Res. 93, 90–98.
Stratigopoulos, G., LeDuc, C. A., Cremona, M. L., Chung, W. K., and Leibel, R. L. (2011). Cut-like homeobox 1 (CUX1) regulates expression of the fat mass and obesity-associated and retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) genes and coordinates leptin receptor signaling. J. Biol. Chem. 286, 2155–2170.
Stratigopoulos, G., Padilla, S. L., LeDuc, C. A., Watson, E., Hattersley, A. T., McCarthy, M. I., Zeltser, L. M., Chung, W. K., and Leibel, R. L. (2008). Regulation of Fto/Ftm gene expression in mice and humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1185–R1196.
Stroup, T. S., McEvoy, J. P., Swartz, M. S., Byerly, M. J., Glick, I. D., Canive, J. M., McGee, M. F., Simpson, G. M., Stevens, M. C., and Lieberman, J. A. (2003). The National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project: schizophrenia trial design and protocol development. Schizophr Bull. 29, 15–31.
van Vliet-Ostaptchouk, J. V., Hofker, M. H., van der Schouw, Y. T., Wijmenga, C., and Onland-Moret, N. C. (2009). Genetic variation in the hypothalamic pathways and its role on obesity. Obes. Rev. 10, 593–609.
Varley, C. K., and McClellan, J. (2009). Implications of marked weight gain associated with atypical antipsychotic medications in children and adolescents. JAMA 302, 1811–1812.
Walley, A. J., Asher, J. E., and Froguel, P. (2009). The genetic contribution to non-syndromic human obesity. Nat. Rev. Genet. 10, 431–142. [Review].
Wetterling, T. (2001). Bodyweight gain with atypical antipsychotics. A comparative review. Drug Saf. 24, 59–73. [Review].
Keywords: antipsychotic drugs, weight gain, schizophrenia, CATIE Study, candidate genes, energy homeostasis, KCNJ11 gene, FTO gene
Citation: Tiwari HK, Patki A, Lieberman J, Stroup TS, Allison DB, Leibel RL and Chung WK (2011) Association of allelic variation in genes mediating aspects of energy homeostasis with weight gain during administration of antipsychotic drugs (CATIE Study). Front. Gene. 2:56. doi: 10.3389/fgene.2011.00056
Received: 13 February 2011; Accepted: 13 August 2011;
Published online: 01 September 2011.
Edited by:
Colin Palmer, University of Dundee, UKReviewed by:
Naohiko Anzai, Dokkyo Medical University School of Medicine, JapanJatinder K. Lamba, University of Minnesota, USA
Copyright: © 2011 Tiwari, Patki, Lieberman, Stroup, Allison, Leibel and Chung. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Hemant K. Tiwari, Section on Statistical Genetics, Department of Biostatistics, University of Alabama at Birmingham, Ryals Public Health Building, Suite 327, Birmingham, AL 35294, USA. e-mail:aHRpd2FyaUB1YWIuZWR1