 April K. Marrone
April K. Marrone Halyna R. Shcherbata*
Halyna R. Shcherbata*- Max Planck Research Group of Gene Expression and Signaling, Max Planck Institute for Biophysical Chemistry, Goettingen, Germany
Mammalian musculature is a very robust and dynamic tissue that goes through many rounds of degeneration and regeneration in an individual’s lifetime. There is a biological program that maintains muscle progenitor cells that, when activated, give rise to intermediate myoblast progeny that consequently differentiate into mature muscle cells. Recent works have provided a picture of the role that microRNAs (miRNAs) play in maintaining aspects of this program. Intriguingly, a subset of these miRNAs is de-regulated in muscular dystrophies (MDs), a group of fatal inherited neuromuscular disorders that are often associated with deficiencies in the Dystrophin (Dys) complex. Apparently, transcriptional expression of many of the muscle specific genes and miRNAs is dependent on chromatin state regulated by the Dys–Syn–nNOS pathway. This puts Dystrophin at the epicenter of a highly regulated program of muscle gene expression in which miRNAs help to coordinate networking between multiple phases of muscle maintenance, degeneration, and regeneration. Therefore, understanding the role of miRNAs in physiology of normal and diseased muscle tissue could be useful for future applications in improving the MD therapies and could open new clinical perspectives.
Function of the Dystrophin Glycoprotein Complex
Muscular dystrophies (MDs) are a group of fatal inherited diseases (~30), associated with progressive muscle wasting caused by muscle membrane fragility, abnormal metabolic control, increased oxidative and energetic stress and abnormal proliferation of satellite cells (Campbell, 1995; Cohn and Campbell, 2000; Constantin et al., 2006; Vercherat et al., 2009; Wallace and McNally, 2009). The most prevalent and severe Duchenne MD (DMD) and the milder Becker MD (BMD) both result from mutations in the Dystrophin (Dys) gene. Dys, the largest gene in the human genome (1.2 Mb, 79 exons), encodes a structural cytoplasmic protein that is the key component of the larger membrane-associated multiprotein dystrophin glycoprotein complex (DGC; Kunkel et al., 1986; Hoffman et al., 1987). The DGC classically consists of dystrophin, dystroglycans α and β, sarcoglycans α, β, Γ, and δ, sarcospan, and other proteins, such as α-dystrobrevin, syntrophins α1, β1, β2, Γ1, Γ2, calveolin-3, Growth factor receptor–bound protein-2 (Grb2), and neuronal Nitric Oxide Synthase (nNOS) have been shown to be associated with the DGC (Durbeej and Campbell, 2002).
The DGC has multiple roles in muscle: mechanical stabilization of the muscle sarcolemma via anchoring the extracellular matrix (ECM) to the cytoskeleton, signal transduction between the internal and external environments of the muscle cell and provides a scaffold responsible for the membrane localization of signaling proteins (Ervasti and Sonnemann, 2008). The DGC, via Syntrophin (Syn) anchors a variety of signaling molecules to their functional sites at the membrane. For example, it localizes nNOS at the sarcolemma consequently regulating intramuscular generation of nitric oxide (NO; Wehling et al., 2001). nNOS signaling nitrosylates the histone deacetylases (HDACs) that regulate gene transcription in muscle progenitor cells by controlling the activity of myogenic (MyoD, Myf5, myogenin, and MRF4) and MEF2 family proteins (McKinsey et al., 2000; Puri et al., 2001). Upon myoblast differentiation, HDACs are displaced from the chromatin that then becomes hyperacetylated leading to transcriptional activation. Since Dys deficiency leads to downregulation of nNOS signaling (Brenman et al., 1995), it indirectly plays a role in control of the balance between acetylation and deacetylation of muscle differentiation genes and has an impact on the regenerative potential of muscle stem cells. It is not clear if the DGC is just a scaffold for Syn–nNOS or, since it has been shown that the DGC is also a signal-transducing module involved in cross talk between the internal and external environments of the muscle cell (Moore and Winder, 2010), it can also actively control epigenetic characteristics of muscle cells via the nNOS signaling pathway.
Muscle Maintenance and Regeneration
Muscles can withstand rigorous mechanical stress and, if damaged, be repaired by the progeny of satellite cells (Figure 1). Satellite cells are quiescent muscle progenitor cells that reside along the basal lamina of the muscle fiber and proliferate only in response to specific signals, for example muscle injury (Charge and Rudnicki, 2004). For continuous and sufficient muscle regeneration, satellite cells must be easily activated to proliferate but also should return to quiescence to maintain their stem cell characteristics (Dhawan and Rando, 2005).
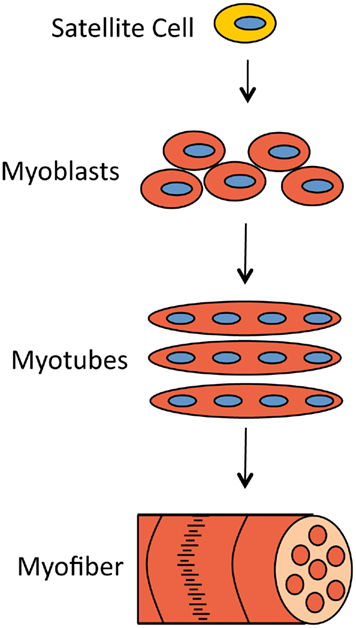
Figure 1. Muscle damage can be repaired by the progeny of satellite cells. When muscle fibers need to be replaced, signals are sent to otherwise quiescent satellite cells, which activates their proliferation and generates proliferating myoblasts. Myoblasts later differentiate and fuse to form myotubes. Myotubes cluster into myofibers, the structures that muscle cells consist of.
The progeny of satellite progenitor cells are primary myoblasts. These myoblasts proliferate to increase in number and upon activation differentiate and fuse to form myotubes. These myotubes then cluster into myofibers which again cluster to form muscle cells or myocytes. This program must be tightly regulated to ensure that muscle is replenished as necessary and that satellite cells are not lost in an untimely manner (Buckingham, 2006).
In order for a cell to be a myocyte it must express specific transcription factors, including Mef2, MyoD, and myogenin among others (Sabourin and Rudnicki, 2000). Before myoblasts express these factors they are in a proliferative state that requires repression of terminal differentiation genes and cell cycle inhibitors. In order for differentiation of myoblasts to occur, proliferation must halt. This requires the coordinated agreement of cell cycle inhibition and initiation of muscle specific gene expression.
In dystrophic patients, Dys insufficiency causes the muscle sarcolemma to become fragile leading to damage that cannot be easily repaired due to inadequate response of satellite cells. This results in chronic inflammation of muscle tissue by penetration of immune cells and eventually leads to the replacement of myofibers with adipose and fibrotic tissue (Porter et al., 2002).
Age-dependent muscle degeneration in MD patients is associated with defects in multiple processes that occur in different cell types such as activation of satellite cell proliferation, progenitor cell maintenance, myoblast differentiation, muscle cell homeostasis, immune cell recruitment, etc. These processes are also required during normal muscle development and growth and the transcriptional circuitry and signaling pathways controlling these events have been extensively studied using many model organisms. In this review we will discuss how recent studies have also revealed a new level of regulation of muscle gene expression that is mediated by miRNAs. Since miRNAs have great potential for therapeutics, understanding their basic biology and specific functions in healthy and diseased muscle tissue is of great importance.
The Dys–Syn–nNOS Pathway and miRNA Levels are Reciprocally Regulated
The DGC through the Dys–Syn–nNOS pathway regulates the expression of miRNAs required for muscle tissue maintenance and regeneration (Cacchiarelli et al., 2010; Figure 2). Indeed, it has been found that mRNA levels are altered in the human DMD pathology, which correlates with mis-expressed miRNAs (Eisenberg et al., 2007). Specifically nNOS signaling controls modification of HDAC2, which in normal situations prevents binding to promoter regions that would lead to transcriptional activation. When Dys is absent this signaling is disturbed, which also results in miRNA dysregulation (Figure 3). Interestingly it was found that most of the mis-expressed miRNAs found in DMD patients are returned to normal expression levels when HDACs were inhibited by restoring NO signaling (Cacchiarelli et al., 2010). This is in accord with a previous study that showed that class I HDAC inhibitors and NO-inhibited HDACs effectively ameliorate MD in mouse models (Colussi et al., 2008).
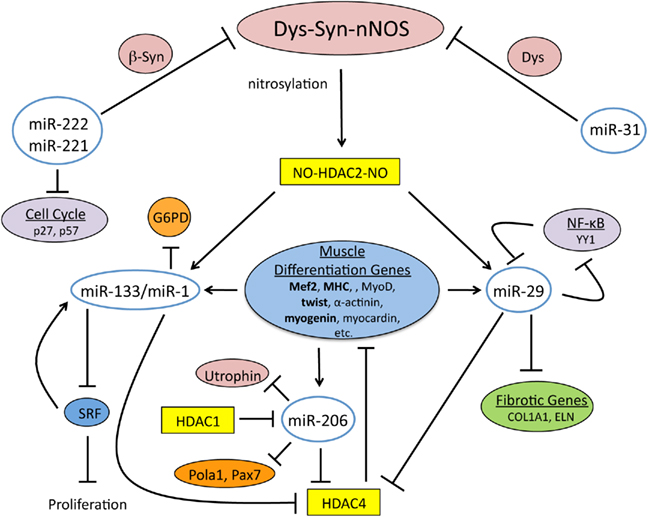
Figure 2. The Dys–Syn–nNOS pathway and miRNAs coordinate the muscle differentiation program. Dys–Syn–nNOS signaling regulates the epigenetic profile of muscle cells via nitrosylation of HDAC2 that influences gene expression by altering the acetylation status of histones. miR-221 and miR-222 share the same seed sequence and both target cell cycle inhibitors, p27 and p57. miR-222 also targets β1-Syn implying an impact on nNOS signaling. miR-31 temporally targets terminal differentiation genes including Dys. miR-1 and miR-133 are transcriptionally controlled by the nitrosylation state of HDAC2, which is regulated by nNOS. miR-133 targets SRF during proliferation, which in a self regulatory manner promotes expression of miR-133. miR-1 targets G6PD and HDAC4 during differentiation, and since HDAC4 regulates the transcription of muscle differentiation genes, Dys–Syn–nNOS signaling indirectly promotes differentiation. In a self-promoting regulatory loop, the expression of muscle differentiation genes results in miR-1 expression. miR-29 is also transcriptionally controlled by the nitrosylation state of HDAC2. Additional regulation of miR-29 comes from YY1, a member of NF-κB signaling. The skeletal muscle specific miRNA, miR-206 is also upregulated by muscle gene expression and aids in cell cycle inhibition by targeting DNA Pola1 and Pax7. Utrophin is another target of miR-206 providing another link between it and the DGC.
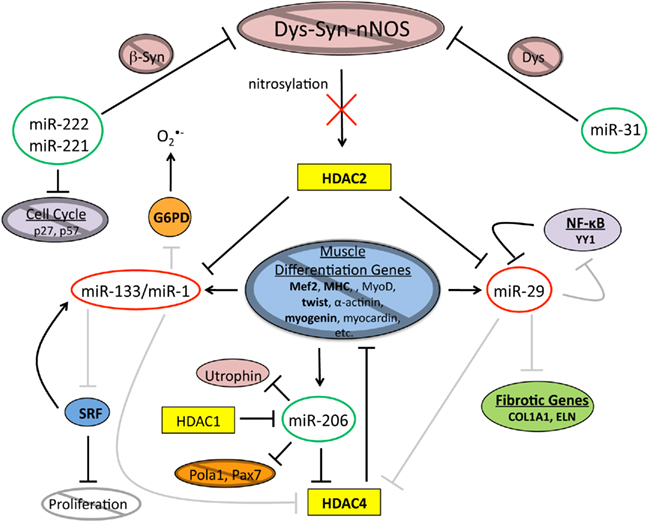
Figure 3. In dystrophic muscle the epigenetic network of muscle differentiation is disrupted. When Dys is absent, Dys–Syn–nNOS signaling is disrupted inhibiting nitrosylation of HDAC2. Consequently, levels of miR-133/miR-1 and miR-29, all of which are repressed by non-nitrosylated HDAC2, are down. The regulatory roles of miR-133/miR-1 are disturbed leading to inhibition of muscle differentiation gene expression and an increase in oxidative cellular stress. Muscle tissue generated under imbalanced conditions will contribute to the procession of adipogenesis. The decrease in miR-29 levels correlates with an increase in the amount of Collagen and Elastin that are indicative of fat and fibrotic tissue. Due to reasons that are not quite clear at this time, levels of miR-206 and miR-31 are increased altering the equilibrium between the proliferation and differentiation states. miRNAs circled in green are upregulated and miRNAs circled in red are down regulated, bold letters indicate upregulated protein levels, gray circles with diagonal lines and gray lines indicate deficiencies.
Not only does the DGC play a role in miRNA regulation, but it is also regulated via miRNAs. For example, miR-222 regulates the Dys–Syn–nNOS pathway possibly by targeting the 3′-UTR of β1-Syntrophin (De Arcangelis et al., 2010). Since Syntrophins bind to Dys and act as a scaffold for nNOS, miR-222 could control the timing of NO signaling during differentiation. miR-222 has the same seed sequence as miR-221, both are controlled by the Ras–MAPK pathway and are downregulated upon differentiation of myoblasts (Cardinali et al., 2009). Additionally, miR-221 and miR-222 promote cell cycle progression by down-regulating the cyclin-dependent kinase inhibitors p27 (Cdkn1b/Kip1) and p57 (Cdkn1c/Kip2) that are essential for the maintenance of the proliferative state (le Sage et al., 2007; Medina et al., 2008). Since both miR-221 and miR-222 are significantly upregulated in MDs (Eisenberg et al., 2007; Greco et al., 2009; De Arcangelis et al., 2010), the cell cycle kinetics of muscle progenitor cells could also be affected.
Another miRNA that is known to regulate muscle terminal differentiation genes is miR-31, which directly targets the 3′-UTR or Dys consequently inactivating the Dys–Syn–nNOS pathway (Cacchiarelli et al., 2011). In normal muscle, miR-31 activity is detected in early myoblasts to suppress precocious expression of late differentiation markers, while in dystrophic muscles miR-31 levels are elevated probably due to activation of the regenerative program in differentiating satellite cells and myoblasts. Increased miR-31 levels in these cells subsequently reduce the amount of Dys, the lack of which correlates with a differentiation delay (Greco et al., 2009; Cacchiarelli et al., 2011). Importantly, in dystrophic conditions, when Dys synthesis is rescued through exon skipping, inhibition of miR-31 significantly improved Dys production (Cacchiarelli et al., 2011). As a result, downregulation of miR-31 most likely promotes generation of NO via Dys–Syn–nNOS signaling and subsequent HDAC2 nitrosylation. Once nitrosylated, HDAC2 no longer inhibits expression of select miRNAs (Cacchiarelli et al., 2010), for example muscle specific miR-1 that marks differentiating myoblasts (Chen et al., 2006).
miRNAs Balance the Epigenetic Network of Dystrophic Muscle
miR-1 is evolutionarily conserved from invertebrates to vertebrates and via targeting another histone deacetylase, HDAC4 (Chen et al., 2006) it can modulate epigenetic profiles of muscle genes. Multiple HDAC4 transcriptionally regulated genes promote muscle differentiation, including Mef2 (Lu et al., 2000; McKinsey et al., 2000; Chen et al., 2006). Insights from research in Drosophila showed that miR-1 is necessary for post-mitotic growth but not establishment of muscles (Sokol and Ambros, 2005). miR-1 expression is regulated initially by Twist, a mesoderm fate determining transcription factor, then in later stages by Mef2 (Sokol and Ambros, 2005), a central regulator of myogenesis. This puts miR-1 in a positive feedback loop where its expression is initiated and promoted by the muscle differentiation program (myogenic factors and Dys–Syn–nNOS signaling) and then, miR-1 acts to reassure terminal differentiation through downregulation of HDAC4, a repressor of myogenic genes.
In mdx mouse and human DMD patients, when Dys is absent and NO signaling is perturbed, miR-1 is downregulated in differentiating myoblasts (McCarthy and Esser, 2007; Greco et al., 2009; Cacchiarelli et al., 2010). Myofibers generated under such conditions are not robust and can be lost as part of the disease pathology. Transcriptomic analysis carried out on the dystrophic mouse revealed an altered gene expression profile that might affect proper myofiber differentiation (Ghahramani Seno et al., 2010). Additionally, miR-1 targets Glucose-6-phosphate dehydrogenase (G6PD) that controls oxidative stress by maintaining levels of oxidized and reduced glutathione, a major antioxidant (Cacchiarelli et al., 2010). Since dystrophic muscle is sensitive to different stresses and has altered levels of reactive oxygen/nitrogen species (Martins et al., 2008; Shkryl et al., 2009; Kucherenko et al., 2011; Marrone et al., 2011), finding new ways to protect against oxidative stress is of a great significance. Importantly, miRNAs have been shown in many organisms to be involved in stress response (Biggar and Storey, 2011; Dorn, 2011).
Once muscle differentiation genes start to be expressed they in turn can regulate expression of miRNAs. miR-1 and miR-133 are transcribed together from a single primary transcript (Chen et al., 2006); however, they perform antagonistic functions in muscle regeneration. For instance, during proliferation miR-133 is upregulated to target serum response factor (SRF), which is important during muscle differentiation (Gauthier-Rouviere et al., 1996; Wang et al., 2002; Li et al., 2005; Chen et al., 2006). In a negative feedforward loop SRF also promotes the expression of miR-133 (Chen et al., 2006). In addition, induction of miR-1/133 and miR-206 (discussed later) transcription is regulated by other factors that drive myogenesis including MyoD and Mef2 (Kim et al., 2006; Rao et al., 2006; Rosenberg et al., 2006); however, the amount of miR-133 decreases during differentiation. It is interesting that miR-133, an enhancer of proliferation, and miR-1, an enhancer of differentiation, are involved in temporally separated developmental processes but are transcribed together. This supports the idea that there is posttranscriptional regulation of these miRNAs by an as yet unknown mechanism. For example, primary miRNA (pri-miRNA) could be processed by auxiliary factors. It has been shown that an RNA processing protein (hnRNP A1) promotes production of one miRNA over the other members of the cluster via binding to the terminal loop of the pri-miRNA (Michlewski et al., 2008). Despite initial beliefs that the terminal loops of pri-miRNAs are unimportant, a large number of pri-miRNAs including transcripts of miR-1 and miR-133 have conserved terminal loops, which suggests that miR-1/133 in muscle could be regulated posttranscriptionally. Nonetheless, similar to miR-1, in dystrophic muscle miR-133 is downregulated (McCarthy and Esser, 2007; Cacchiarelli et al., 2010) supporting that proliferation and differentiation of satellite cells are disrupted in DMD patients.
When early muscle transcription factors start to be expressed they promote expression of several miRNAs, which in turn fine-tune signals required to reprogram cells to differentiate and fuse into myotubes. Skeletal muscle specific miR-206 is upregulated in activated myoblasts to aid differentiation and halt proliferation via cell cycle arrest (Kim et al., 2006). This is accomplished by inhibiting DNA synthesis by targeting Pola1, a DNA polymerase (Kim et al., 2006). Additionally, miR-206 targets the satellite cell self renewal factor, Pax7 (Cacchiarelli et al., 2010), and the muscle differentiation inhibitor HDAC4 (Williams et al., 2009). Though miR-206 can be positively regulated by muscle differentiation genes similar to miR-1/133, in dystrophic muscle the levels of this miRNA are higher than normal (McCarthy et al., 2007; Yuasa et al., 2008; Greco et al., 2009; Cacchiarelli et al., 2010). Interestingly, Utrophin, a Dys homolog, which due to a compensatory mechanism is upregulated in the mdx mouse, is another target of miR-206 (Rosenberg et al., 2006). However, increased levels of miR-206 as a part of the DMD pathology do not agree with increased levels of Utrophin (McCarthy et al., 2007), suggesting that compensatory mechanisms act independent from miR-206. Unlike miR-1/133, miR-206 is not repressed by HDAC2, but by HDAC1 (Cacchiarelli et al., 2010). This indicates that miR-206 is not regulated by the Dys–Syn–nNOS pathway; however, miR-206 contributes to the imbalance of the regeneration program in dystrophic muscle.
miRNAs Regulate Tissue Replacement in Compromised Dystrophic Muscle
Even though different types of MDs have specific mechanisms of muscle degeneration, inflammation is a hallmark symptom of dystrophic muscle. Accordingly, an increase in miR-222 and miR-223 levels in dystrophic muscle is believed to be due to inflammation (Eisenberg et al., 2007; Greco et al., 2009). Inflammation is associated with fat and fibrotic replacement of muscle fibers and degeneration (Porter et al., 2002). To elucidate the differences seen in different types of muscular disorders a comprehensive study was carried out in which muscle specimens were analyzed representing 10 different human conditions (Eisenberg et al., 2007). What was learned from this study is that (1) there is a large amount of variation in miRNA levels in disease states, (2) there are shared misregulated miRNAs across muscular disorders (miR-146b and miR-miR-221) and (3) only a small number of miRNAs are unique to an individual disorder such as DMD (miR-95, miR-30d, miR-486, miR-331, miR-193b, miR-30a_5p, miR-30e_5p, and miR-26a).
Previously it was thought that improper fate determination of satellite cell progeny generated extra fibrotic tissue in dystrophic muscle (Shefer et al., 2004; Brack et al., 2007). Fibrosis therefore ought to occur at the expense of muscle regeneration. However, recently it has been shown that in addition to satellite muscle progenitor cells, there are also mesenchymal progenitor cells living among myofibers that give rise to adipocytes and fibroblasts which generate fat/fibrotic tissue (Joe et al., 2010; Uezumi et al., 2010). These cells have been termed fibro/adipogenic progenitors (Natarajan et al., 2010). What is amazing about these cells is that they are activated during muscle regeneration similar to satellite cells, but in a non-autonomous manner receive signals from the surrounding muscle environment. If muscle regeneration is producing normal healthy muscle fibers then adipogenesis is inhibited. However, if muscle regeneration is compromised then adipogenesis proceeds and fibrotic tissue will arise in the interstitial space between myofibers (Figure 4). Certain proteins are indicative of fibrotic tissue such as collagen and elastin. Recently it has been shown that their mRNAs are targeted by the miR-29 family, which helps to modify ECM organization (van Rooij et al., 2008). miR-29 is an enhancer of differentiation and is repressed in myoblasts by Yin Yang 1 (YY1), a protein that is positively regulated by NF-κB and is a member of the Polycomb group that acts as a transcriptional silencer (Wang et al., 2008). During myogenesis expression of miR-29 is upregulated by SRF and Mef2, and in a self regulatory manner, miR-29 suppresses YY1 and HDAC4 by targeting their 3′-UTRs (Wang et al., 2008; Li et al., 2009). Interestingly, the expression of miR-29 is also controlled by the Dys–Syn–nNOS pathway where, similar to miR-1, nitrosylation of HDAC2 allows for its transcription (Cacchiarelli et al., 2010). In mdx mouse and human DMD patients there is therefore an expected decrease in miR-29 levels which promotes an increase in fibrotic tissue (Eisenberg et al., 2007; Greco et al., 2009; Cacchiarelli et al., 2010).
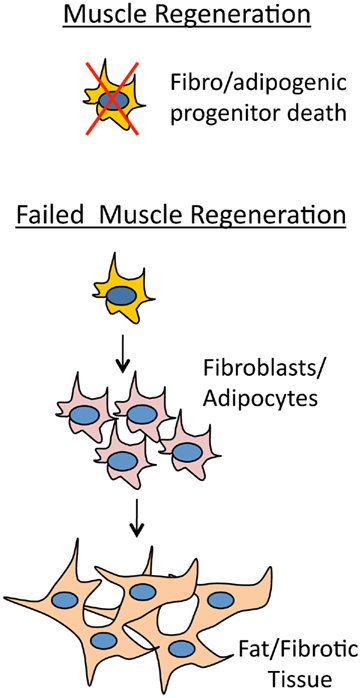
Figure 4. Myotube health regulates adipogenesis. If muscle is regenerating normally then fibro/adipogenic progenitor cells will receive a non-cell autonomous signal from the muscle environment and die. The signal from the degenerative muscle environment stimulates fibro/adipogenic progenitor cells to give rise to adipocites and fibroblasts that will differentiate into fat and fibrotic tissue.
All of these findings lead to the conclusion that in dystrophic muscle the alteration in miRNA levels is the result of an imbalanced regeneration program, where not only activation of satellite cell proliferation but also myoblast differentiation are impaired (Figure 3). The overall result is that damaged muscle fibers do not properly regenerate resulting in fat and fibrotic tissue replacement with accompanying inflammation. Each of these processes is accompanied by misregulation of certain miRNAs that can be considered in a multistep treatment of these fatal neuromuscular disorders.
New Options for miRNAs in Muscular Dystrophy Therapies
As discussed above, miRNA dysregulation contributes to a variety of MD pathologies and the miRNA regulatory network is disturbed in dystrophic muscle. In order to return the diseased tissue to normal mode, multiple processes have to be adjusted, and miRNAs are important functional units controlling different aspects of muscle regeneration and maintenance. In addition, miRNAs have been shown to be perfect players in fine-tuning different developmental processes: stem cell division and maintenance, differentiation, cell cycle control, etc (Shcherbata et al., 2006; Inui et al., 2010). Therefore re-establishing the normal miRNA levels may allow for improvement of misregulated protein synthesis. The proteomic fingerprint can be altered by variation of miRNA levels, which can be achieved in a positive or a negative manner. miRNAs can be inhibited by in vivo silencing via use of chemically modified antisense nucleotides locked nucleid acids (LNA; Kumar et al., 1998), 2′-O-methyl-RNA oligonucleotides (2′-O-me-RNA; Matera and Ward, 1993), microRNA sponges (Ebert et al., 2007), or cholesterol conjugated RNA analogs (antagomirs; Krutzfeldt et al., 2005). Inversely miRNA levels can be increased by in vivo injection or viral delivery resulting in reduced expression of upregulated disease genes (Fasanaro et al., 2010; Pfeifer and Lehmann, 2010).
An exemplary result of miRNA targeting in DMD therapy comes from Cacchiarelli et al. (2011). In mdx mice symptoms of muscle degeneration can be reversed by the introduction of viral vectors that allow for transcriptional exon skipping that removes a non-sense mutation in the Dys gene. However, these therapies have not been able to restore full muscle function due to low Dys levels. Dys is a target for miR-31 and any replacement of Dys via genetic manipulation could potentially be inhibited by the presence of this miRNA. The authors were able to significantly increase Dys levels in exon skipping treated DMD myoblasts by downregulation of miR-31 using a miR-31 sponge and an anti-miR-31 LNA oligonucleotide. These results set a precedent for the use of miRNAs as therapeutic targets to alleviate muscular dystrophy. It is unknown at this time if there are other mRNA targets of miR-31, alteration of which would give rise to deleterious side effects of this treatment.
Fibrotic degeneration, which is characteristic in mdx mice, was also successfully decreased by increasing miR-29 levels (Cacchiarelli et al., 2010). Electroporation of mdx muscles with miR-29 had a therapeutic effect resulting in significant reduction of collagen deposition and an increase in both collagen and elastin mRNAs. These data prove that miRNAs are crucial regulators of the ECM that can possibly regulate the signals sent from the muscle environment to fibro/adipogenic progenitors, and targeting/promoting such miRNAs could inhibit fibrosis allowing for more successful muscle regeneration.
Though it seems obvious from the discussion presented thus far that Dys–Syn–nNOS signaling plays a key role in balancing the epigenetic network of muscle regeneration, it must be considered that therapies restoring NO signaling alone may not be fully curative. Patients with the milder BMD in some cases are missing the domain of Dys essential for Dys–Syn–nNOS signaling (Wells et al., 2003; Lai et al., 2009). Since these patients do not succumb to their illness, the malfunction of nNOS signaling is still compatible with life. Key muscle regenerative miRNAs regulated by nNOS signaling are probable therapeutic targets; however other isolated miRNAs should be given attention for disease treatment too. Screening for misregulated miRNAs has been done by several groups in dystrophic animal models and DMD patients which allowed to compile a comprehensive list of differentially regulated miRNAs that can be clustered intro three functional categories: regeneration, degeneration, and inflammation (Eisenberg et al., 2007; Greco et al., 2009).
Regenerative miRNAs are upregulated during embryonic development and post ischemic regeneration (Greco et al., 2009; Suh and Blelloch, 2011), supporting their role in proliferation and differentiation of muscle cell progenitors. This indicates that functional tissue replacement is based on satellite cell populations, and for miRNA-based therapies to be effective progenitor cell maintenance and division should be taken into consideration. Muscle satellite cells are quiescent until they become proliferative upon response to muscle damage or heavy use. It will be of interest to determine what role miRNAs have in the initial activation of satellite cells, since it has been shown that miRNA signatures differ from self-renewing to quiescent stem cells and from proliferative to differentiating progenitor cells (Arnold et al., 2011).
Initially it was believed that the role of the DGC was to simply provide structural support at costameres and without this support the muscle cell would tear and waste away. However, that is obviously not the only avenue with which disease progresses. The DGC is right in the middle of the muscle differentiation program, since without the Dys–Syn–nNos pathway muscle maintenance and regeneration is disrupted. This means that formed myotubes are compromised and contribute to tissue replacement by fat and fibrosis. The plethora of miRNAs implicated in the DMD pathology present a substantial and complex level of regulation that opens diverse avenues for future research and therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Gerd Vorbrüggen and all members of Shcherbata Lab for comments on the manuscript and Max Planck Society for financial support.
References
Arnold, C. P., Tan, R., Zhou, B., Yue, S. B., Schaffert, S., Biggs, J. R., Doyonnas, R., Lo, M. C., Perry, J. M., Renault, V. M., Sacco, A., Somervaille, T., Viatour, P., Brunet, A., Cleary, M. L., Li, L., Sage, J., Zhang, D. E., Blau, H. M., Chen, C., and Chen, C. Z. (2011). MicroRNA programs in normal and aberrant stem and progenitor cells. Genome Res. 21, 798–810.
Biggar, K. K., and Storey, K. B. (2011). The emerging roles of microRNAs in the molecular responses of metabolic rate depression. J. Mol. Cell Biol. 3, 167–175.
Brack, A. S., Conboy, M. J., Roy, S., Lee, M., Kuo, C. J., Keller, C., and Rando, T. A. (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810.
Brenman, J. E., Chao, D. S., Xia, H., Aldape, K., and Bredt, D. S. (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752.
Buckingham, M. (2006). Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr. Opin. Genet. Dev. 16, 525–532.
Cacchiarelli, D., Incitti, T., Martone, J., Cesana, M., Cazzella, V., Santini, T., Sthandier, O., and Bozzoni, I. (2011). miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 12, 136–141.
Cacchiarelli, D., Martone, J., Girardi, E., Cesana, M., Incitti, T., Morlando, M., Nicoletti, C., Santini, T., Sthandier, O., Barberi, L., Auricchio, A., Musaro, A., and Bozzoni, I. (2010). MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 12, 341–351.
Campbell, K. P. (1995). Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80, 675–679.
Cardinali, B., Castellani, L., Fasanaro, P., Basso, A., Alema, S., Martelli, F., and Falcone, G. (2009). Microrna-221 and microrna-222 modulate differentiation and maturation of skeletal muscle cells. PLoS ONE 4, e7607. doi: 10.1371/journal.pone.0007607
Charge, S. B., and Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238.
Chen, J. F., Mandel, E. M., Thomson, J. M., Wu, Q., Callis, T. E., Hammond, S. M., Conlon, F. L., and Wang, D. Z. (2006). The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38, 228–233.
Cohn, R. D., and Campbell, K. P. (2000). Molecular basis of muscular dystrophies. Muscle Nerve 23, 1456–1471.
Colussi, C., Mozzetta, C., Gurtner, A., Illi, B., Rosati, J., Straino, S., Ragone, G., Pescatori, M., Zaccagnini, G., Antonini, A., Minetti, G., Martelli, F., Piaggio, G., Gallinari, P., Steinkuhler, C., Clementi, E., Dell’aversana, C., Altucci, L., Mai, A., Capogrossi, M. C., Puri, P. L., and Gaetano, C. (2008). HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. U.S.A. 105, 19183–19187.
Constantin, B., Sebille, S., and Cognard, C. (2006). New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: implications in DMD. J. Muscle Res. Cell Motil. 27, 375–386.
De Arcangelis, V., Serra, F., Cogoni, C., Vivarelli, E., Monaco, L., and Naro, F. (2010). Beta1-syntrophin modulation by miR-222 in mdx mice. PLoS ONE 5, 12098. doi: 10.1371/journal.pone.0012098
Dhawan, J., and Rando, T. A. (2005). Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 15, 666–673.
Durbeej, M., and Campbell, K. P. (2002). Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev. 12, 349–361.
Ebert, M. S., Neilson, J. R., and Sharp, P. A. (2007). MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4, 721–726.
Eisenberg, I., Eran, A., Nishino, I., Moggio, M., Lamperti, C., Amato, A. A., Lidov, H. G., Kang, P. B., North, K. N., Mitrani-Rosenbaum, S., Flanigan, K. M., Neely, L. A., Whitney, D., Beggs, A. H., Kohane, I. S., and Kunkel, L. M. (2007). Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. U.S.A. 104, 17016–17021.
Ervasti, J. M., and Sonnemann, K. J. (2008). Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 265, 191–225.
Fasanaro, P., Greco, S., Ivan, M., Capogrossi, M. C., and Martelli, F. (2010). microRNA: emerging therapeutic targets in acute ischemic diseases. Pharmacol. Ther. 125, 92–104.
Gauthier-Rouviere, C., Vandromme, M., Tuil, D., Lautredou, N., Morris, M., Soulez, M., Kahn, A., Fernandez, A., and Lamb, N. (1996). Expression and activity of serum response factor is required for expression of the muscle-determining factor MyoD in both dividing and differentiating mouse C2C12 myoblasts. Mol. Biol. Cell 7, 719–729.
Ghahramani Seno, M. M., Trollet, C., Athanasopoulos, T., Graham, I. R., Hu, P., and Dickson, G. (2010). Transcriptomic analysis of dystrophin RNAi knockdown reveals a central role for dystrophin in muscle differentiation and contractile apparatus organization. BMC Genomics 11, 345. doi: 10.1186/1471-2164-11-345
Greco, S., De Simone, M., Colussi, C., Zaccagnini, G., Fasanaro, P., Pescatori, M., Cardani, R., Perbellini, R., Isaia, E., Sale, P., Meola, G., Capogrossi, M. C., Gaetano, C., and Martelli, F. (2009). Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 23, 3335–3346.
Hoffman, E. P., Brown, R. H. Jr., and Kunkel, L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928.
Inui, M., Martello, G., and Piccolo, S. (2010). MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 11, 252–263.
Joe, A. W., Yi, L., Natarajan, A., Le Grand, F., So, L., Wang, J., Rudnicki, M. A., and Rossi, F. M. (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163.
Kim, H. K., Lee, Y. S., Sivaprasad, U., Malhotra, A., and Dutta, A. (2006). Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 174, 677–687.
Krutzfeldt, J., Rajewsky, N., Braich, R., Rajeev, K. G., Tuschl, T., Manoharan, M., and Stoffel, M. (2005). Silencing of microRNAs in vivo with “antagomirs.” Nature 438, 685–689.
Kucherenko, M. M., Marrone, A. K., Rishko, V. M., Magliarelli Hde, F., and Shcherbata, H. R. (2011). Stress and muscular dystrophy: a genetic screen for dystroglycan and dystrophin interactors in Drosophila identifies cellular stress response components. Dev. Biol. 352, 228–242.
Kumar, R., Singh, S. K., Koshkin, A. A., Rajwanshi, V. K., Meldgaard, M., and Wengel, J. (1998). The first analogues of LNA (locked nucleic acids): phosphorothioate-LNA and 2(′-thio-LNA. Bioorg. Med. Chem. Lett. 8, 2219–2222.
Kunkel, L. M., Hejtmancik, J. F., Caskey, C. T., Speer, A., Monaco, A. P., Middlesworth, W., Colletti, C. A., Bertelson, C., Muller, U., Bresnan, M., Shapiro, F., Tantravahi, U., Speer, J., Latt, S. A., Bartlett, R., Pericak-Vance, M. A., Roses, A. D., Thompson, M. W., Ray, P. N., Worton, R. G., Fischbeck, K. H., Gallano, P., Coulon, M., Duros, C., Boue, J., Junien, C., Chelly, J., Hamard, G., Jeanpierre, M., Lambert, M., Kaplan, J. C., Emery, A., Dorkins, H., Mcglade, S., Davies, K. E., Boehm, C., Arveiler, B., Lemaire, C., Morgan, G. J., Denton, M. J., Amos, J., Bobrow, M., Benham, F., Boswinkel, E., Cole, C., Dubowitz, V., Hart, K., Hodgson, S., Johnson, L., Walker, A., Roncuzzi, L., Ferlini, A., Nobile, C., Romeo, G., Wilcox, D. E., Affara, N. A., Ferguson-Smith, M. A., Lindolf, M., Kaariainen, H., De La, C. A., Ionasescu, V., Searby, C., Ionasescu, R., Bakker, E., Van Ommen, G. J., Pearson, P. L., Greenberg, C. R., Hamerton, J. L., Wrogemann, K., Doherty, R. A., Polakowska, R., Hyser, C., Quirk, S., Thomas, N., Harper, J. F., Darras, B. T., and Francke, U. (1986). Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature 322, 73–77.
Lai, Y., Thomas, G. D., Yue, Y., Yang, H. T., Li, D., Long, C., Judge, L., Bostick, B., Chamberlain, J. S., Terjung, R. L., and Duan, D. (2009). Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 119, 624–635.
le Sage, C., Nagel, R., Egan, D. A., Schrier, M., Mesman, E., Mangiola, A., Anile, C., Maira, G., Mercatelli, N., Ciafre, S. A., Farace, M. G., and Agami, R. (2007). Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 26, 3699–3708.
Li, S., Czubryt, M. P., Mcanally, J., Bassel-Duby, R., Richardson, J. A., Wiebel, F. F., Nordheim, A., and Olson, E. N. (2005). Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. U.S.A. 102, 1082–1087.
Li, Z., Hassan, M. Q., Jafferji, M., Aqeilan, R. I., Garzon, R., Croce, C. M., Van Wijnen, A. J., Stein, J. L., Stein, G. S., and Lian, J. B. (2009). Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J. Biol. Chem. 284, 15676–15684.
Lu, J., Mckinsey, T. A., Zhang, C. L., and Olson, E. N. (2000). Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 6, 233–244.
Marrone, A. K., Kucherenko, M. M., Wiek, R., Gopfert, M. C., and Shcherbata, H. R. (2011). Hyperthermic seizures and aberrant cellular homeostasis in Drosophila dystrophic muscles. Sci. Rep. 1. doi: 10.1038/srep00047.
Martins, A. S., Shkryl, V. M., Nowycky, M. C., and Shirokova, N. (2008). Reactive oxygen species contribute to Ca2+ signals produced by osmotic stress in mouse skeletal muscle fibres. J. Physiol. (Lond.) 586, 197–210.
Matera, A. G., and Ward, D. C. (1993). Nucleoplasmic organization of small nuclear ribonucleoproteins in cultured human cells. J. Cell Biol. 121, 715–727.
McCarthy, J. J., and Esser, K. A. (2007). MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J. Appl. Physiol. 102, 306–313.
McCarthy, J. J., Esser, K. A., and Andrade, F. H. (2007). MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am. J. Physiol. Cell Physiol. 293, C451–C457.
McKinsey, T. A., Zhang, C. L., Lu, J., and Olson, E. N. (2000). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111.
Medina, R., Zaidi, S. K., Liu, C. G., Stein, J. L., Van Wijnen, A. J., Croce, C. M., and Stein, G. S. (2008). MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 68, 2773–2780.
Michlewski, G., Guil, S., Semple, C. A., and Caceres, J. F. (2008). Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol. Cell 32, 383–393.
Moore, C. J., and Winder, S. J. (2010). Dystroglycan versatility in cell adhesion: a tale of multiple motifs. Cell Commun. Signal. 8, 3.
Natarajan, A., Lemos, D. R., and Rossi, F. M. (2010). Fibro/adipogenic progenitors: a double-edged sword in skeletal muscle regeneration. Cell Cycle 9, 2045–2046.
Pfeifer, A., and Lehmann, H. (2010). Pharmacological potential of RNAi – focus on miRNA. Pharmacol. Ther. 126, 217–227.
Porter, J. D., Khanna, S., Kaminski, H. J., Rao, J. S., Merriam, A. P., Richmonds, C. R., Leahy, P., Li, J., Guo, W., and Andrade, F. H. (2002). A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 11, 263–272.
Puri, P. L., Iezzi, S., Stiegler, P., Chen, T. T., Schiltz, R. L., Muscat, G. E., Giordano, A., Kedes, L., Wang, J. Y., and Sartorelli, V. (2001). Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 8, 885–897.
Rao, P. K., Kumar, R. M., Farkhondeh, M., Baskerville, S., and Lodish, H. F. (2006). Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. U.S.A. 103, 8721–8726.
Rosenberg, M. I., Georges, S. A., Asawachaicharn, A., Analau, E., and Tapscott, S. J. (2006). MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 175, 77–85.
Sabourin, L. A., and Rudnicki, M. A. (2000). The molecular regulation of myogenesis. Clin. Genet. 57, 16–25.
Shcherbata, H. R., Hatfield, S., Ward, E. J., Reynolds, S., Fischer, K. A., and Ruohola-Baker, H. (2006). The MicroRNA pathway plays a regulatory role in stem cell division. Cell Cycle 5, 172–175.
Shefer, G., Wleklinski-Lee, M., and Yablonka-Reuveni, Z. (2004). Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway. J. Cell Sci. 117, 5393–5404.
Shkryl, V. M., Martins, A. S., Ullrich, N. D., Nowycky, M. C., Niggli, E., and Shirokova, N. (2009). Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 458, 915–928.
Sokol, N. S., and Ambros, V. (2005). Mesodermally expressed Drosophila microRNA-1 is regulated by twist and is required in muscles during larval growth. Genes Dev. 19, 2343–2354.
Suh, N., and Blelloch, R. (2011). Small RNAs in early mammalian development: from gametes to gastrulation. Development 138, 1653–1661.
Uezumi, A., Fukada, S., Yamamoto, N., Takeda, S., and Tsuchida, K. (2010). Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 12, 143–152.
van Rooij, E., Liu, N., and Olson, E. N. (2008). MicroRNAs flex their muscles. Trends Genet. 24, 159–166.
Vercherat, C., Chung, T. K., Yalcin, S., Gulbagci, N., Gopinadhan, S., Ghaffari, S., and Taneja, R. (2009). Stra13 regulates oxidative stress mediated skeletal muscle degeneration. Hum. Mol. Genet. 18, 4304–4316.
Wallace, G. Q., and McNally, E. M. (2009). Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 71, 37–57.
Wang, D., Passier, R., Liu, Z. P., Shin, C. H., Wang, Z., Li, S., Sutherland, L. B., Small, E., Krieg, P. A., and Olson, E. N. (2002). Regulation of cardiac growth and development by SRF and its cofactors. Cold Spring Harb. Symp. Quant. Biol. 67, 97–105.
Wang, H., Garzon, R., Sun, H., Ladner, K. J., Singh, R., Dahlman, J., Cheng, A., Hall, B. M., Qualman, S. J., Chandler, D. S., Croce, C. M., and Guttridge, D. C. (2008). NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14, 369–381.
Wehling, M., Spencer, M. J., and Tidball, J. G. (2001). A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 155, 123–131.
Wells, K. E., Torelli, S., Lu, Q., Brown, S. C., Partridge, T., Muntoni, F., and Wells, D. J. (2003). Relocalization of neuronal nitric oxide synthase (nNOS) as a marker for complete restoration of the dystrophin associated protein complex in skeletal muscle. Neuromuscul. Disord. 13, 21–31.
Williams, A. H., Valdez, G., Moresi, V., Qi, X., Mcanally, J., Elliott, J. L., Bassel-Duby, R., Sanes, J. R., and Olson, E. N. (2009). MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326, 1549–1554.
Keywords: muscular dystrophy, Dys–Syn–nNOS, epigenetic regulation, dystrophin, miRNAs
Citation: Marrone AK and Shcherbata HR (2011) Dystrophin orchestrates the epigenetic profile of muscle cells via miRNAs. Front. Gene. 2:64. doi: 10.3389/fgene.2011.00064
Received: 12 July 2011; Paper pending published: 29 July 2011;
Accepted: 26 August 2011; Published online: 13 September 2011.
Edited by:
Michael Rossbach, Genome Institute of Singapore, SingaporeReviewed by:
Francesca Fanini, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, ItalyKin Fai Au, Stanford University, USA
Hui Ling, MD Anderson Cancer Center, USA
Copyright: © 2011 Marrone and Shcherbata. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Halyna R. Shcherbata, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Goettingen, Germany. e-mail: halyna.shcherbata@mpibpc. mpg.de