 Mark Steven Miller*
Mark Steven Miller* Lance D. Miller
Lance D. Miller
- Department of Cancer Biology, Comprehensive Cancer Center, Wake Forest School of Medicine, Winston-Salem, NC, USA
Mutation in RAS proteins is one of the most common genetic alterations observed in human and experimentally induced rodent cancers. In vivo, oncogenic mutations have been shown to occur at exons 12, 13, and 61, resulting in any 1 of 19 possible point mutations in a given tumor for a specific RAS isoform. While some studies have suggested a possible role of different mutant alleles in determining tumor severity and phenotype, no general consensus has emerged on the oncogenicity of different mutant alleles in tumor formation and progression. Part of this may be due to a lack of a single, signature pathway that shows significant alterations between different mutations. Rather, it is likely that subtle differences in the activation, or lack thereof, of downstream effectors by different RAS mutant alleles may determine the eventual outcome in terms of tumor phenotype. This paper reviews our current understanding of the potential role of different RAS mutations on tumorigenesis, highlights studies in model cell culture and in vivo systems, and discusses the potential of expression array and computational network modeling to dissect out differences in activated RAS genes in conferring a transforming phenotype.
Introduction
The RAS gene family consists of three small G proteins – Ha-, N-, and Ki-ras (with Ki-ras existing as the predominant Ki-ras4B and the alternatively spliced Ki-ras4A isoforms) – that play a central role in cell signaling (Barbacid, 1987; Malumbres and Barbacid, 2003). RAS proteins are anchored on the cytoplasmic side of the cell membrane, where they mediate signal transduction downstream from tyrosine kinase membrane receptors to a variety of effector molecules, stimulating a cascade of parallel phosphorylation reaction pathways that ultimately culminate with the activation of nuclear transcription factors. The three main effector pathways that are activated downstream of RAS – RAF/MEK/MAPK, PI3K/AKT, and RAL–GDS – play major roles in mediating signals relating to cell proliferation, cell survival, cell adhesion, and cell motility (Fan and Bertino, 1997; Campbell et al., 1998; Gille and Downward, 1999; Zuber et al., 2000; Bounacer et al., 2004). Each of the RAS isoforms appears to differentially regulate its downstream effectors in vivo, resulting in marked differences in the strength and type of signal produced (Hancock, 2003; Ehrhardt et al., 2004; Moon, 2006; Omerovic et al., 2008). This differential signaling appears to be mediated partly by the trafficking pathways used by each RAS isoform to reach the plasma membrane, as well as the location of each isoform in the plasma membrane itself (Chiu et al., 2002; Hancock, 2003; Moon, 2006); N- and Ha-ras associate with lipid rafts in the plasma membrane, whereas Ki-ras appears to be located in non-raft domains.
The RAS pathway is one of the most prevalent oncogenic alterations in both human and experimentally induced animal tumors (Bos, 1989; Conti, 1992; Malumbres and Barbacid, 2003). In vivo, oncogenic mutations have been shown to occur at exons 12, 13, and 61, resulting in any 1 of 19 possible point mutations for each RAS isoform. When stimulated by upstream signaling molecules, wild type RAS proteins interact with guanine nucleotide exchange factors to replace GDP with GTP, resulting in an activated protein conformation. RAS activity is terminated by interaction with GTPase activating protein, which stimulates the GTPase activity of the protein and converts GTP back to GDP, thereby restoring the inactive form of RAS. Mutations in RAS inhibit the GTPase activity and lock the protein in the active GTP bound conformation (Barbacid, 1987; Bos, 1989; Malumbres and Barbacid, 2003) In particular, mutations in the Ki-ras gene have been shown to play a key role in the pathogenesis of a variety of human tumors, with mutations occurring in 95% of pancreatic tumors, 50% of colon tumors, and 30% of lung adenocarcinomas (Barbacid, 1987; Bos et al., 1987; Conti, 1992; Malumbres and Barbacid, 2003). Of these cancers, lung and colon cancer are the first and second leading cause of cancer-related deaths in the U.S., respectively (Jemal et al., 2010). Among the candidate genes implicated in the initiation of these cancers, Ki-ras has received considerable attention as mutations in Ki-ras appear in early neoplastic lesions in both human and experimentally induced murine lung and colon tumors, and influence both tumor progression and drug resistance (Cerny et al., 1992; Reynolds et al., 1992; Hruban et al., 1993; Westra et al., 1993, 1996; Li et al., 1994a; Miller, 1994; Gryfe et al., 1997; Grady and Markowitz, 2002; Agbunag and Bar-Sagi, 2004; Fleming et al., 2005). The spectrum of RAS mutations differs by organ site and allele frequency, probably as a result of different environmental exposures and tissue specific differences in RAS expression. The Sanger Institute’s COSMIC database (Catalog of Somatic Mutations in Cancer; http://www.sanger.ac.uk/genetics/CGP/cosmic/add_info/) integrates data from the published literature on type and frequency of somatic mutations in human cancers. Using the database search tools, we examined the spectrum and frequency of Ki-ras mutations (Table 1). Consistent with the literature, Ki-ras mutations were observed most frequently in cancers of the lung, large intestine (including colon, rectal, and anal), pancreas, and biliary tract (including bile duct and gall bladder). Based on a collective mutational analysis involving >15,000 tumors, the most frequent alterations observed were point mutations at codons 12, 13, and 61. A spectrum of predominant mutant alleles were observed, and their relative frequencies are shown in Table 1 as the percentage of all mutant alleles observed for a given tumor type. Consistent with historical observations, ASP12, VAL12, and CYS12 emerged as the predominant mutant Ki-ras alleles. However, that the alleles distributed with large variation within each cancer type may reflect the non-redundant functions of the different alleles in tumorigenesis. Large variation across cancer types was also observed for some alleles, such as CYS12, ASP12, and ASP13, suggesting that mutant allele functionality may also depend, to some extent, on the tumor tissue of origin.
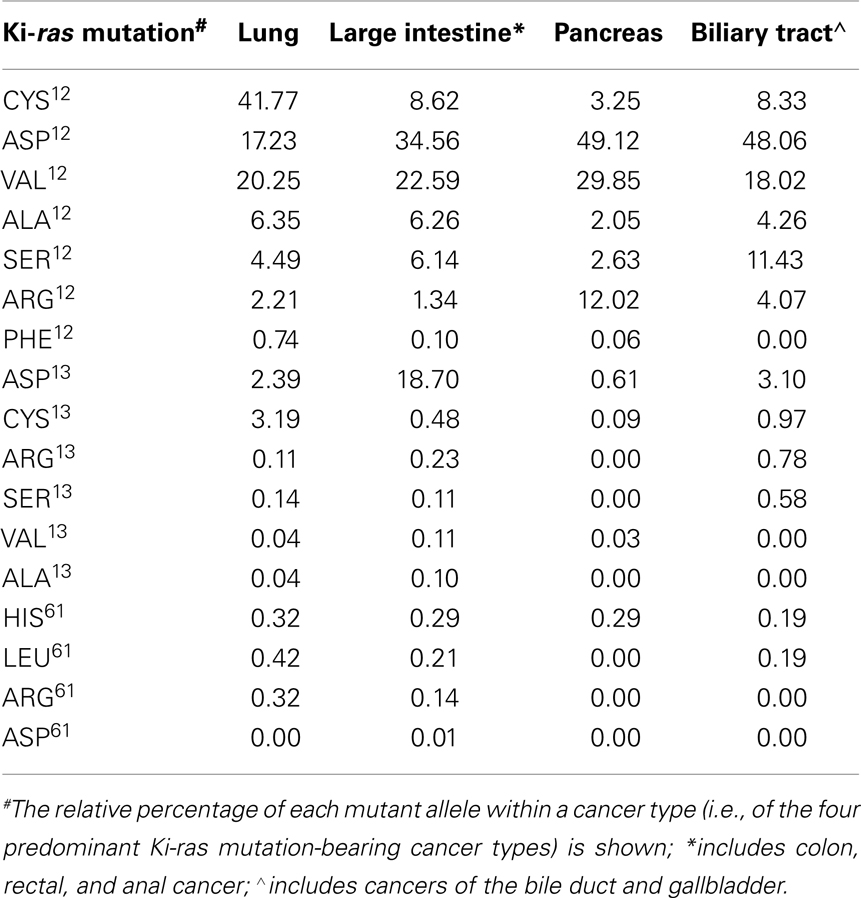
Table 1. Relative frequencies of the major Ki-ras mutations by cancer type: analysis of the Sanger COSMIC database.
Although some studies have provided evidence for mutation specific effects of different mutant RAS alleles, most studies and therapeutic approaches have treated RAS mutations as a single entity – the gene is either mutated or wild type. We believe that the different RAS mutations exhibit subtle differences in their ability to signal to their downstream effectors, which may impact their relative contribution to the carcinogenic process, their role as driver mutations, and tumor responsiveness to novel therapeutic agents that target RAS or its downstream effectors. Thus, this manuscript reviews the evidence obtained from biochemical, cell culture, and animal model data, as well as the limited number of human studies available, documenting the differential response of cells to different mutant RAS alleles.
In vitro Evidence for Differential Effects of Different Mutant RAS Alleles
Studies on the potential differences in the mutagenicity/oncogenicity of different RAS mutant alleles began shortly after the identification of RAS as a transforming oncogene. Initial studies demonstrated that different Ha-ras mutant alleles exhibited differences in their ability to transform mouse fibroblasts (Fasano et al., 1984; Seeburg et al., 1984; Der et al., 1986). Focusing on the Ha-ras gene, Fasano et al. (1984) found that the VAL12 mutation was the most potent in terms of the induction of focus formation in the NIH3T3 assay, with ARG12, ASP12, SER12, ASP13, and SER13 exhibiting transforming efficiencies that were 60, 50, 40, 20, and 0.1% relative to the VAL12 mutant. Seeburg et al. (1984), found somewhat similar results in Rat-1 cells with mutants to both Ki-ras and Ha-ras. Although not as quantitative as Fasano et al. (1984), these authors found that alleles of both Ki- and Ha-ras containing the VAL12 and ARG12 mutations exhibited greater transforming activity than alleles with the CYS12, ASP12, ASN12, and SER12 mutations. All of the mutant alleles exhibited growth in soft agar. Der et al. (1986) examined 17 different codon 61 mutations in the Ha-ras gene and found that the transforming activity in NIH 3T3 cells varied by more than 300-fold between the different mutant alleles.
Several groups (Gibbs et al., 1984a,b; McGrath et al., 1984; Sweet et al., 1984; Manne et al., 1985), using purified Ha-ras produced in E. coli, demonstrated that the valine 12 mutant exhibited 5 to 10-fold lower GTPase activity than the normal wild type allele. It was initially thought that the relative level of GTPase activity might account for the differences in transforming potential between the different mutant alleles. However, the studies by Der et al. (1986) and Colby et al. (1986) did not find any correlation between the transforming potency and GTPase activity of the different alleles. Subsequent to these early studies, the biochemical activity of RAS and its interactions with its downstream receptors has continued to be the focus of intensive investigations. Several laboratories have demonstrated that mutations in specific amino acids have very significant effects on both guanine nucleotide exchange factors and RAS GTPase activity (Nielsen et al., 2001; Zhang et al., 2005; Filchtinski et al., 2010; Lukman et al., 2010). However, many of these mutational analyses have been limited to examining amino acids that cause conformational changes in regions of the RAS genes associated with GTPase activity, such as the A59G variant (Lukman et al., 2010), rather than the common mutagenic variant alleles associated with tumorigenesis. More recently, redox agents such as reactive oxygen species and reactive nitrogen species have been shown to enhance the rate of guanine nucleotide exchange as a result of the formation of a thiyl radical on CYS118 Ha-ras (Del Villar et al., 1996; Kjeldgaard et al., 1996; Reuther and Der, 2000; Ford et al., 2002; Jourd’heuil et al., 2003; Schrammel et al., 2003; Williams et al., 2003; Heo et al., 2005, 2006; Davis et al., 2011). This raises the possibility that codon 12, 13, and 61 mutations could affect RAS GTPase activity by either (1) increasing oxidative stress and thereby increasing thiylation of CYS118 or (2) altering the accessibility of the CYS118 residue to modification by reactive oxygen species. As will be discussed below, the CYS12 allele has been shown in a variety of experimental systems to exhibit less potent tumorigenic activity than other mutations in codon 12. It is thus possible that the CYS12 variant could provide a new redox active cysteine motif in the RAS protein that differentially responds to the increased oxidative stress of the cancer cell environment in a different manner than other mutant RAS alleles. Clearly, none of these potential mechanisms are mutually exclusive and suggest new areas of inquiry into the mechanisms of the observed differential effects of RAS mutants on tumorigenicity.
In vivo Evidence for Differential Effects of Different Mutant RAS Alleles
The initial studies utilizing the NIH3T3 focus assay were confirmed using in vivo models. Studies from this and other laboratories have demonstrated an association between the histological stage of both murine and human lung tumors and the presence of specific mutant RAS alleles in tumor tissue. Early studies by Nuzum et al. (1990) demonstrated that, following treatment of adult A/J mice with urethane, mouse lung adenocarcinomas (ACs) had a high incidence of GLU61 → ARG61 (CAA → CGA) mutations relative to the smaller adenomas (ADs), which preferentially exhibited GLU61 → LEU61 (CAA → CTA) mutations. Li et al. (1994b) also demonstrated that lung ACs showed a higher incidence of mutations at the second base of codon 61 following treatment of newborn mice with nitrochrysene and its metabolites. Utilizing a transplacental treatment protocol, whereby mice are exposed in utero to a single dose of the chemical carcinogen 3-methylcholanthrene, we have demonstrated in three independent studies using different strains of mice that different RAS mutations are associated with tumor stage (Leone-Kabler et al., 1997; Gressani et al., 1999; Jennings-Gee et al., 2006). Treatment of pregnant mice with 3-methylcholanthrene resulted in a high incidence of lung tumors in the offspring 6–12 months after birth. Mice harboring a VAL12, ARG12, ASP12, or ARG13 mutant Ki-ras gene were more likely to contain later stage tumors than mice with the CYS12 or wild type allele, which exhibited mostly benign ADs and hyperplasias. Further work with this model also suggested that the type of mutation induced in Ki-ras following in utero exposure to the chemical carcinogen was associated with specific types of damage (hypermethylation vs. base pair mutations) at the Ink4a gene locus (Mizesko et al., 2001). Interestingly, the mutant Ki-ras alleles associated with progression to later stage tumors in our transplacental mouse studies were the same ones associated with a trend for poorer patient outcomes in a clinical study of human lung cancer (Keohavong et al., 1996). A clinical study examining the prognostic significance of Ki-ras mutations in lung cancer patients found that patients containing CYS, ARG, and ASP mutations at codon 12 appeared to have a poorer prognosis than those containing hydrophobic amino acid substitutions such as VAL or ALA (Siegfried et al., 1997). However, the sample sizes for this analysis were small and the authors did not find an overall association of Ki-ras mutations and poorer patient survival, as has been noted in several other studies (reviewed in Rodenhuis and Slebos, 1992). Similar observations have been made in colon cancer, as Finkelstein et al. (1993a,b) reported that the Ki-ras ASP12 mutant allele was associated with the metastatic properties of colon tumors.
In addition to the ability of different RAS mutant alleles to initiate tumor formation, the above mentioned results obtained in vivo also suggest differences in tumor progression imparted by the different RAS variant alleles. In the in vivo studies cited above, a consistent finding was that more aggressive tumors (i.e., ACs) were more prevalent with one type of RAS mutation, often the VAL12 allele, whereas benign lesions such as ADs or hyperplasias were more prevalent with other types of RAS variants, most notably the CYS12 mutation. These results suggest that specific RAS mutant alleles can impart a greater growth advantage than other alleles, and the often disparate results may be due to context and organ dependent effects of the different alleles (Guerra et al., 2003). Studies by Cespedes et al. (2006) have extended these findings and also confirmed the relatively weak transforming activity of the Ki-ras CYS12 allele observed in our studies. These authors transfected NIH3T3 cells with the ASP12, VAL12, and CYS12 alleles of Ki-ras. When the transforming properties of the alleles were assessed, the Ki-ras VAL12 allele exhibited a more aggressive tumorigenic phenotype than the Ki-ras ASP12 allele, which was attributed to the inability of the ASP12 allele to signal through the RAF/MEK/ERK pathway. When the transfected cells were injected into nude mice, the Ki-ras CYS12 containing cells failed to establish tumors.
Along the same lines, studies by this group utilizing NIH3T3 cells found that the Ki-ras VAL12 allele, in contrast to the wild type and Ki-ras VAL13 allele, exhibited increased glycolysis (Vizan et al., 2005). In addition, cells transfected with the VAL12 mutant allele were resistant to the induction of apoptosis induced by confluence and exhibited a much greater ability to grow in soft agar. The VAL12 mutant clones exhibited elevated levels of p-AKT, increased expression of Bcl-2, E-cadherin, β-catenin, and focal adhesion kinase, and decreased expression of RhoA (Guerrero et al., 2000). Similarly, Recktenwald et al. (2008) found that the VAL12 and ASP13 variants of Ki-ras enhanced cell survival and resistance to oxidative stress. Although they did not specifically address this in their paper, the VAL12 allele exhibited greater protection against formaldehyde- and H2O2-mediated toxicity and reduced caspase 3/7 activity relative to the ASP13 allele. Thus, a generally consistent finding across the animal studies, cell culture experiments, and human patient samples suggests that the CYS12 allele is associated with a relatively weak or no transforming activity, while VAL12 and ASP12 alleles were associated with more aggressive oncogenic properties. These transforming properties were not only associated with increased tumor formation, but seemed to also play a role in tumor progression.
The somewhat conflicting studies with human samples most likely result from the multitude of alterations that occur in human tumors by the time they are diagnosed. It is very likely that tumors with weakly transforming RAS alleles may require additional alterations at other oncogenic loci in order to drive tumorigenesis. Thus, in some cases, a RAS mutation such as the VAL12 allele could act as a driver mutation whereas in other cases, such as the CYS12 allele, mutations in other genes may be the key drivers of tumorigenesis. A major limitation of studies utilizing human tissue is the fact that human tumors contain several mutations in a variety of oncogenic loci. If a weakly oncogenic Ki-ras mutation occurs in a particular patient’s tumor, it is likely that by the time the tumor is isolated, mutations in other critical driver genes would have occurred. To our knowledge, no studies have been published which attempt to correlate specific mutations in Ki-ras with the presence or absence of other known driver mutations. Thus, attempts to associate specific RAS mutations with patient outcome or survival may not be able to distinguish the relative contribution of a specific Ki-ras mutation to the tumorigenic process.
In order to characterize the role of Ki-ras mutations in the initiation of lung tumorigenesis, several investigators have utilized inducible expression systems in transgenic mice. These included: (1) regulation of a murine Ki-ras ASP12 cDNA transgene via a doxycyline (DOX) regulated promoter (Fisher et al., 2001); (2) sporadic activation of a human Ki-ras VAL12 cDNA transgene via a Cre-lox mediated recombination construct (Meuwissen et al., 2001; Guerra et al., 2003); (3) spontaneous activation of the murine Ki-ras ASP12 gene via a somatic recombination system (Johnson et al., 2001); and (4) sporadic activation of the same murine Ki-ras ASP12 gene mutation via a Cre based lox-stop-lox construct (Jackson et al., 2001). Expression of these mutant Ki-ras alleles were shown to be a strong oncogenic stimulus for lung epithelial cells, resulting in significant increases in lung tumorigenicity. In each of these models, 100% of the mice developed aggressive ACs and died within 2–10 months from their lung tumor burden.
In contrast to these results our laboratory, utilizing a transgenic mouse in which the mutant human Ki-ras CYS12 allele is regulated in a DOX-inducible and lung-specific manner (Floyd et al., 2005), found that these mice developed hyperplasias and small, benign adenomas (ADs) after 12 months of DOX treatment that rarely progressed to the carcinoma stage. The CYS12 transgene appeared to signal to a subset of its downstream effectors, exhibiting increases in proliferative and both anti- and pro-apoptotic signals, as well as up-regulation of cell cycle inhibitory molecules. Mice harboring this mutant Ki-ras allele exhibited increased signaling through the RAS/RAF/ERK/cyclin D1, p38, and RAL/GDS pathways, but no alterations in signaling through the JNK and PI3K/AKT pathways (Floyd et al., 2006; Dance-Barnes et al., 2008). Interestingly, the relatively benign tumor phenotype observed in Ki-ras CYS12 mice was also observed in mice with activated and wild type RAF genes (Kerkhoff et al., 2000). These authors developed a strain of mice expressing an activated human c-raf-1 transgene specifically in the lung (SPC-c-raf-1-BXB mice) that also exhibited a relatively benign tumor phenotype, despite increased phosphorylation of downstream effector molecules in the ERK pathway. Because mutant RAF is downstream of RAS and thus signals to a specific subset of RAS effector molecules, these experiments provide further in vivo confirmation of the need for the activation of multiple RAS downstream effector molecules to mediate the full transformation of lung epithelial cells and provide the most definitive evidence to date of the potential for different Ki-ras mutations to exhibit differential effects on the carcinogenic process. With the limitations that these studies often involved different inducible gene systems, different strains of mice, and the unknowns of the potential effects of integration of the transgene in the mouse genome, future studies will need to take advantage of targeted knock-in technologies to create a series of transgenic mouse strains that replace the endogenous mouse Ki-ras gene with various mutated alleles and allow the mutants to be expressed from the natural murine Ki-ras promoter. These types of studies will be critical in dissecting out the effect of different RAS mutations on downstream signaling pathways and tumorigenicity.
Differential Expression of Ki-ras Mutants in Human Lung Tumors
Several studies using human tissue samples and cell lines (Bhattacharjee et al., 2001; Beer et al., 2002; Miura et al., 2002; Virtanen et al., 2002; Wikman et al., 2002; Creighton et al., 2005) have utilized RNA and cDNA microarray analyses to document the complex array of alterations in signaling pathways that accompany lung tumorigenesis. Yao et al. (2002) isolated lung tumors 6 and 14 months following treatment of A/J mice with a single injection of N-methylnitrosourea at 6 weeks of age. Using the Affymetrix Atlas Mouse™ cDNA Expression Array, consisting of 588 known mouse genes, they were able to identify 19 genes that showed differential expression between ADs and ACs, as well as 10 genes that exhibited similar alterations in expression levels between the two tumor types. Subsequent studies from the same group, using the more extensive Affymetrix Mu74Av2 chip, which can interrogate 36,000 full-length mouse genes and EST clusters from the Unigene Database, identified 50 genes that were either up-or down-regulated in ADs vs. ACs, including genes involved in cell cycle control, differentiation, and apoptosis (Bonner et al., 2004). In addition, when the murine tumors were compared with lung ACs isolated from human patients, the murine ADs and ACs clustered with two groups of human ACs that differed in their differentiation status, with murine ADs clustering with the well differentiated human ACs and murine ACs clustering with the less differentiated tumors. These investigators also identified 39 genes that were similarly regulated in murine and human lung ACs, further emphasizing the appropriateness of the mouse as a model for human lung cancer. Similarly, Jacks’ group (Sweet-Cordero et al., 2005) used the data obtained from this study in combination with their own analysis of mouse tumors from transgenic Ki-rasLA mice, which express the Ki-ras ASP12 transgene sporadically as a result of spontaneous recombination (Johnson et al., 2001), and compared the results obtained with the murine lung tumors with human arrays from a variety of sources, including their own analyses as well as those from Beer et al. (2002) and Bhattacharjee et al. (2001). These investigators were able to demonstrate patterns of gene expression that were common to both human and murine lung ACs (Sweet-Cordero et al., 2005). Most interesting, these researchers demonstrated that a Ki-ras expression signature in human ACs that was not identifiable by statistical analysis unless the mouse data was included in the integrated analyses, due to the high degree of variability inherent in human lung tumor tissues, results which have been confirmed by Creighton et al. (2005).
Interestingly, in all of these studies mutated RAS genes were treated as a single entity, as the comparison that was made was samples containing mutated RAS genes vs. those that did not. Thus, to date, it is unclear to what extent these signatures reflect the differential effects of different RAS mutant alleles on downstream signaling pathways. Accounting for this added complexity may allow a further refinement of the signatures down to the subset of RAS stimulated genes that are the most critical for tumor initiation and maintenance. Clearly, different RAS mutant alleles may engage different subsets of RAS downstream effector pathways, which may influence the gene expression profiles. This could be the reason that statistical significance for RAS mutated vs. wild type tumors could not be reached until the mouse data, which had a more homogenous Ki-ras mutational spectrum, were added.
Recent studies have utilized synthetic lethal and genome wide inhibitory RNA approaches to identify RAS effector molecules that are critical for tumorigenesis (Kassie et al., 2008; Barbie et al., 2009; Luo et al., 2009; Scholl et al., 2009; Singh et al., 2009; Vicent et al., 2010). These studies have clearly shown the dependency of tumor cells on a subset of RAS downstream effector pathways when mutated RAS genes are present. All of these studies have been conducted with the alleles that animal and in vitro studies have identified as the most oncogenic RAS mutations. Future studies will thus need to compare the use of synthetic lethal approaches with different RAS mutants. This will allow an understanding of which mutant RAS alleles signal to these critical pathways and will be an important approach in developing novel therapeutic agents.
Emerging Research
A number of studies have shown that mutations in Ki-ras are prognostic factors for poor patient outcome in lung cancer (Mitsudomi et al., 1991; Rodenhuis and Slebos, 1992; Rosell et al., 1993, 1996; Keohavong et al., 1996; Siegfried et al., 1997; Huncharek et al., 1999). Patients whose tumors contained Ki-ras mutations often exhibited poorer overall survival and reduced time to disease progression. As noted above, one study found that lung cancer patients whose tumors contained the CYS, ARG, or ASP mutations at codon 12 appeared to have a poorer prognosis than those containing hydrophobic amino acid substitutions such as VAL or ALA (Siegfried et al., 1997). However, a recent study in Stage III colon cancer patients failed to find an association with disease-free or overall survival (Ogino et al., 2009). A limitation of both of these studies was the fact that mutations in Ki-ras were treated as a single entity, so that all patients with mutations were compared to patients with wild type Ki-ras.
More recent studies have examined the effects of RAS mutations on drug sensitivity. Garassino et al. (2011) found that lung tumors of patients harboring the CYS12 mutations were less sensitive to cisplatin therapy but exhibited increased sensitivity to taxol and pemetrexed relative to the VAL12 and ASP12 mutant alleles. The ASP12 mutation demonstrated increased resistance to taxol and enhanced sensitivity to sorafenib. While none of the RAS mutants exhibited differential sensitivity to the tyrosine kinase inhibitors erlotinib, a study examining colorectal cancer patients with chemotherapy-refractory metastatic disease who harbored the Ki-ras ASP13 mutation found a small but statistically significant increase in overall survival and progression-free survival relative to patients harboring other Ki-ras mutations (De et al., 2010). These results clearly suggest that different RAS mutations may have a significant impact on patient response to therapeutic interventions, and that the RAS mutational profile may need to be considered in future clinical trials assessing the effects of novel agents in tumors harboring RAS mutations.
Recent studies have identified novel mutations in RAS genes, whose influence on tumorigenicity are first being assessed. Mutations in exon 4 of Ki-ras coding for LYS117 and ALA146 were associated with an increased probability of disease-free survival despite increases in RAS-GTP (Janakiraman et al., 2010). In addition, recent studies by To et al. (2008) demonstrated that the minor Ki-ras4A isoform may be the critical form of Ki-ras that is responsible for lung carcinogenesis. Smith et al. (2010) recently identified four additional Ki-ras mutations. The ASN117 and THR146 mutations, when transfected into NIH3T3 cells, produced a similar number of transformed foci as the ASP13 mutation but fewer colonies than the VAL12 and ASP12 mutant alleles. The PHE19 and GLN164 mutant alleles, similar to the wild type allele, had little transforming activity. When expression arrays were performed for cell lines harboring the different alleles, differential expression of a number of genes associated with tumor proliferation and cell survival were observed, consistent with the different alleles stimulating different gene expression programs. This work is among the first to document mutant-specific alterations in gene transcriptional programming, and may yield further insights into the effector pathways of allele-specific signaling.
Finally, a Ki-ras single nucleotide polymorphism in the 3′ untranslated region of the gene that disrupts binding of the let7 microRNA has been shown to be a potential prognostic marker of ovarian cancer risk, as 25% of unselected ovarian cancer cases and 61% of hereditary ovarian cancer patients lacking mutations in the BRCA genes harbored this RAS variant allele (Ratner et al., 2010). Interestingly, Hwang and Cohen (1997) found that deletion or inversion of a splicing enhancer region located in the 3′ untranslated region of the Ha-ras gene reduced the transcription of RAS mRNA and thus decreased the transforming activity of this variant allele. Thus, the list of mutant RAS alleles with the differential ability to influence tumorigenicity continues to grow. How each of these mutant alleles influences carcinogenicity and the mechanisms by which they do so are an area of research that should be a major emphasis for future studies.
Conclusion and Future Perspectives
Since the identification of mutant RAS genes as transforming oncogenes, investigators have attempted to identify a mechanistic basis for the allele-specific differences that could account for the observed differences in transforming ability observed in vitro and in vivo. These studies have so far failed to identify a specific gene pathway or pathways that account for the differential effects of different RAS mutants. The majority of evidence obtained to date and described above suggests that, in particular, the CYS12 mutation may exhibit less potent transforming properties than the VAL12 and ASP12 mutants, but a rigorous comparison of all possible RAS mutations of biological significance in the same experimental system has not been conducted. Most studies have examined only a limited number of mutations. In addition, the list of clinically relevant and biologically active RAS mutations continues to grow.
Some of the reason for the lack of a clear mechanistic basis for the observed phenotypic differences imparted by different mutant RAS alleles may be due to very subtle effects of the mutant RAS alleles on multiple gene pathways (either at the transcriptome or proteome level) and context as well as cell and tissue specific effects of mutant RAS genes. Recent advances in technology should enable us to determine the basis of these differences going forward. Biochemical and kinetic studies demonstrating the redox sensitivity of the CYS118 residue (Heo et al., 2005; Heo and Campbell, 2006; Raines et al., 2007) suggest that specific mutant forms of RAS could exhibit differential sensitivity to the redox state of the cell, which could influence tumor progression by either direct effects on RAS GTPase activity or indirect effects via alterations of protein interactions between RAS and its downstream effectors. As illustrated by the expression profiling studies described above (Smith et al., 2010), applications of transcriptomic and proteomic expression profiling hold much promise for delineating the allele-specific effects of different RAS mutant alleles. These studies need to be done in both human tumor samples as well as novel transgenic models.
The development of knock-in mouse models for each of the RAS mutant alleles would be a critically important advance in the field. Because of the tissue specific and dose related effects of RAS gene expression, it will be important to test these alleles in the same genetic background, driven from the endogenous RAS promoters, to eliminate some of the uncertainties associated with traditional transgenic constructs. While these studies should initially be done with Ki-ras, which is mutated more frequently than other RAS family members in human cancer, extending this research to other RAS family members will allow the development of a comprehensive understanding of RAS signaling networks. This will provide important information in terms of developing targeted therapies to the alleles most responsible for driving tumorigenesis as opposed to those that have much less significant contributions to maintenance of the tumorigenic phenotype.
It is likely that the answer will not be simple and that more than one mechanism will influence the behavior of different mutant RAS alleles. Since RAS is the most commonly mutated gene in human cancers and signals to a wide variety of molecules involved in proliferation, cell death, cell survival, oxidative stress, angiogenesis, inflammation, and drug resistance (Campbell et al., 1998; Hancock, 2003; Malumbres and Barbacid, 2003; Young et al., 2009), it is likely that patients whose tumors harbor specific RAS mutations will exhibit differences in survival, tumor aggressiveness, and response to chemotherapy agents. An understanding of the properties of each of the RAS mutants in specific tissues will aid in future attempts to personalize cancer treatment regimens and assure the best possible outcomes for patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors were supported by National Cancer Institute grant R01-CA136910 (Mark Steven Miller) and the Wake Forest University Cancer Center Support Grant P30-CA12197.
Abbreviations
AC, adenocarcinomas; AD, adenomas; CCSP, Clara cell secretory protein; DOX, doxycycline; IHC, immunohistochemistry; RT-PCR, reverse transcription-polymerase chain reaction; rtTA, reverse tetracycline trans-activator; tet, tetracycline.
References
Agbunag, C., and Bar-Sagi, D. (2004). Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res. 64, 5659–5663.
Barbie, D. A., Tamayo, P., Boehm, J. S., Kim, S. Y., Moody, S. E., Dunn, I. F., Schinzel, A. C., Sandy, P., Meylan, E., Scholl, C., Frohling, S., Chan, E. M., Sos, M. L., Michel, K., Mermel, C., Silver, S. J., Weir, B. A., Reiling, J. H., Sheng, Q., Gupta, P. B., Wadlow, R. C., Le, H., Hoersch, S., Wittner, B. S., Ramaswamy, S., Livingston, D. M., Sabatini, D. M., Meyerson, M., Thomas, R. K., Lander, E. S., Mesirov, J. P., Root, D. E., Gilliland, D. G., Jacks, T., and Hahn, W. C. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112.
Beer, D. G., Kardia, S. L., Huang, C. C., Giordano, T. J., Levin, A. M., Misek, D. E., Lin, L., Chen, G., Gharib, T. G., Thomas, D. G., Lizyness, M. L., Kuick, R., Hayasaka, S., Taylor, J. M., Iannettoni, M. D., Orringer, M. B., and Hanash, S. (2002) Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 8, 816–824.
Bhattacharjee, A., Richards, W. G., Staunton, J., Li, C., Monti, S., Vasa, P., Ladd, C., Beheshti, J., Bueno, R., Gillette, M., Loda, M., Weber, G., Mark, E. J., Lander, E. S., Wong, W., Johnson, B. E., Golub, T. R., Sugarbaker, D. J., and Meyerson, M. (2001). Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. U.S.A. 98, 13790–13795.
Bonner, A. E., Lemon, W. J., Devereux, T. R., Lubet, R. A., and You, M. (2004). Molecular profiling of mouse lung tumors: association with tumor progression, lung development, and human lung adenocarcinomas. Oncogene 23, 1166–1176.
Bos, J. L., Fearon, E. R., Hamilton, S. R., Verlaan-de Vries, M., van Boom, J. H., van der Eb, A. J., and Vogelstein, B. (1987). Prevalence of ras gene mutations in human colorectal cancers. Nature 327, 293–297.
Bounacer, A., McGregor, A., Skinner, J., Bond, J., Poghosyan, Z., and Wynford-Thomas, D. (2004). Mutant ras-induced proliferation of human thyroid epithelial cells requires three effector pathways. Oncogene 23, 7839–7845.
Campbell, S. L., Khosravi-Far, R., Rossman, K. L., Clark, G. J., and Der, C. J. (1998). Increasing complexity of Ras signaling. Oncogene 17, 1395–1413.
Cerny, W. L., Mangold, K. A., and Scarpelli, D. G. (1992). K-ras mutation is an early event in pancreatic duct carcinogenesis in the Syrian golden hamster. Cancer Res. 52, 4507–4513.
Cespedes, M. V., Sancho, F. J., Guerrero, S., Parreno, M., Casanova, I., Pavon, M. A., Marcuello, E., Trias, M., Cascante, M., Capella, G., and Mangues, R. (2006). K-ras Asp12 mutant neither interacts with Raf, nor signals through Erk and is less tumorigenic than K-ras Val12. Carcinogenesis 27, 2190–2200.
Chiu, V. K., Bivona, T., Hach, A., Sajous, J. B., Silletti, J., Wiener, H., Johnson, R. L., Cox, A. D., and Philips, M. R. (2002). Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 4, 343–350.
Colby, W. W., Hayflick, J. S., Clark, S. G., and Levinson, A. D. (1986). Biochemical characterization of polypeptides encoded by mutated human Ha-ras1 genes. Mol. Cell. Biol. 6, 730–734.
Conti, C. J. (1992). “Mutations of genes of the ras family in human and experimental tumors,” in Comparative Molecular Carcinogenesis, eds A. J. P. Klein-Szanto, M. W. Anderson, J. C. Barrett, and T. J. Slaga (New York: Wiley-Liss, Inc.), 357–378.
Creighton, C. J., Bromberg-White, J. L., Misek, D. E., Monsma, D. J., Brichory, F., Kuick, R., Giordano, T. J., Gao, W., Omenn, G. S., Webb, C. P., and Hanash, S. M. (2005). Analysis of tumor-host interactions by gene expression profiling of lung adenocarcinoma xenografts identifies genes involved in tumor formation. Mol. Cancer Res. 3, 119–129.
Dance-Barnes, S. T., Kock, N. D., Floyd, H. S., Moore, J. E., Mosley, L. J., D’Agostino, R. B. Jr., Pettenati, M. J., and Miller, M. S. (2008). Effects of mutant human Ki-ras(G12C) gene dosage on murine lung tumorigenesis and signaling to its downstream effectors. Toxicol. Appl. Pharmacol. 231, 77–84.
Davis, M. F., Vigil, D., and Campbell, S. L. (2011) Regulation of Ras proteins by reactive nitrogen species. Free Radic. Biol. Med. 51, 565–575.
De, R. W., Jonker, D. J., Di, N. F., Sartore-Bianchi, A., Tu, D., Siena, S., Lamba, S., Arena, S., Frattini, M., Piessevaux, H., Van, C. E., O’Callaghan, C. J., Khambata-Ford, S., Zalcberg, J. R., Simes, J., Karapetis, C. S., Bardelli, A., and Tejpar, S. (2010). Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304, 1812–1820.
Del Villar, K., Dorin, D., Sattler, I., Urano, J., Poullet, P., Robinson, N., Mitsuzawa, H., and Tamanoi, F. (1996). C-terminal motifs found in Ras-superfamily G-proteins: CAAX and C-seven motifs. Biochem. Soc. Trans. 24, 709–713.
Der, C. J., Finkel, T., and Cooper, G. M. (1986). Biological and biochemical properties of human rasH genes mutated at codon 61. Cell 44, 167–176.
Ehrhardt, A., David, M. D., Ehrhardt, G. R., and Schrader, J. W. (2004). Distinct mechanisms determine the patterns of differential activation of H-Ras, N-Ras, K-Ras 4B, and M-Ras by receptors for growth factors or antigen. Mol. Cell. Biol. 24, 6311–6323.
Fan, J., and Bertino, J. R. (1997). K-ras modulates the cell cycle via both positive and negative regulatory pathways. Oncogene 14, 2595–2607.
Fasano, O., Aldrich, T., Tamanoi, F., Taparowsky, E., Furth, M., and Wigler, M. (1984). Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 81, 4008–4012.
Filchtinski, D., Sharabi, O., Ruppel, A., Vetter, I. R., Herrmann, C., and Shifman, J. M. (2010). What makes Ras an efficient molecular switch: a computational, biophysical, and structural study of Ras-GDP interactions with mutants of Raf. J. Mol. Biol. 399, 422–435.
Finkelstein, S. D., Sayegh, R., Bakker, A., and Swalsky, P. (1993a). Determination of tumor aggressiveness in colorectal cancer by K- ras-2 analysis. Arch. Surg. 128, 526–531; discussion 531–532.
Finkelstein, S. D., Sayegh, R., Christensen, S., and Swalsky, P. A. (1993b). Genotypic classification of colorectal adenocarcinoma. Biologic behavior correlates with K-ras-2 mutation type. Cancer 71, 3827–3838.
Fisher, G. H., Wellen, S. L., Klimstra, D., Lenczowski, J. M., Tichelaar, J. W., Lizak, M. J., Whitsett, J. A., Koretsky, A., and Varmus, H. E. (2001). Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 15, 3249–3262.
Fleming, J. B., Shen, G. L., Holloway, S. E., Davis, M., and Brekken, R. A. (2005). Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: justification for K-ras-directed therapy. Mol. Cancer Res. 3, 413–423.
Floyd, H. S., Farnsworth, C. L., Kock, N. D., Mizesko, M. C., Little, J. L., Dance, S. T., Everitt, J., Tichelaar, J. W., Whitsett, J. A., and Miller, M. S. (2005). Conditional expression of the mutant Ki-rasG12C allele results in formation of benign lung adenomas: development of a novel mouse lung tumor model. Carcinogenesis 26, 2196–2206.
Floyd, H. S., Jennings-Gee, J. E., Kock, N. D., and Miller, M. S. (2006). Genetic and epigenetic alterations in lung tumors from bitransgenic Ki-ras(G12C) expressing mice. Mol. Carcinog. 45, 506–517.
Ford, E., Hughes, M. N., and Wardman, P. (2002). Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radic. Biol. Med. 32, 1314–1323.
Garassino, M. C., Marabese, M., Rusconi, P., Rulli, E., Martelli, O., Farina, G., Scanni, A., and Broggini, M. (2011). Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann. Oncol. 22, 235–237.
Gibbs, J. B., Sigal, I. S., Poe, M., and Scolnick, E. M. (1984a). Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci.U.S.A. 81, 5704–5708.
Gibbs, J. B., Ellis, R. W., and Scolnick, E. M. (1984b). Autophosphorylation of v-Ha-ras p21 is modulated by amino acid residue 12. Proc. Natl. Acad. Sci. U.S.A. 81, 2674–2678.
Gille, H., and Downward, J. (1999). Multiple ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem. 274, 22033–22040.
Grady, W. M., and Markowitz, S. D. (2002). Genetic and epigenetic alterations in colon cancer. Annu. Rev. Genomics Hum. Genet. 3, 101–128.
Gressani, K. M., Leone-Kabler, S. L., O’Sullivan, M. G., Case, L. D., Malkinson, A. M., and Miller, M. S. (1999). Strain-specific lung tumor formation in mice transplacentally exposed to 3-methylcholanthrene and post-natally exposed to butylated hydroxytoluene. Carcinogenesis 20, 2159–2165.
Gryfe, R., Swallow, C., Bapat, B., Redston, M., Gallinger, S., and Couture, J. (1997). Molecular biology of colorectal cancer. Curr. Probl. Cancer 21, 233–300.
Guerra, C., Mijimolle, N., Dhawahir, A., Dubus, P., Barradas, M., Serrano, M., Campuzano, V., and Barbacid, M. (2003). Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 4, 111–120.
Guerrero, S., Casanova, I., Farre, L., Mazo, A., Capella, G., and Mangues, R. (2000). K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 60, 6750–6756.
Hancock, J. F. (2003). Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4, 373–384.
Heo, J., and Campbell, S. L. (2006). Ras regulation by reactive oxygen and nitrogen species. Biochemistry 45, 2200–2210.
Heo, J., Prutzman, K. C., Mocanu, V., and Campbell, S. L. (2005). Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J. Mol. Biol. 346, 1423–1440.
Heo, J., Raines, K. W., Mocanu, V., and Campbell, S. L. (2006). Redox regulation of RhoA. Biochemistry 45, 14481–14489.
Hruban, R. H., van Mansfeld, A. D., Offerhaus, G. J., van Weering, D. H., Allison, D. C., Goodman, S. N., Kensler, T. W., Bose, K. K., Cameron, J. L., and Bos, J. L. (1993). K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 143, 545–554.
Huncharek, M., Muscat, J., and Geschwind, J. F. (1999). K-ras oncogene mutation as a prognostic marker in non-small cell lung cancer: a combined analysis of 881 cases. Carcinogenesis 20, 1507–1510.
Hwang, D. Y., and Cohen, J. B. (1997). A splicing enhancer in the 3′-terminal c-H-ras exon influences mRNA abundance and transforming activity. J. Virol. 71, 6416–6426.
Jackson, E. L., Willis, N., Mercer, K., Bronson, R. T., Crowley, D., Montoya, R., Jacks, T., and Tuveson, D. A. (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248.
Janakiraman, M., Vakiani, E., Zeng, Z., Pratilas, C. A., Taylor, B. S., Chitale, D., Halilovic, E., Wilson, M., Huberman, K., Ricarte Filho, J. C., Persaud, Y., Levine, D. A., Fagin, J. A., Jhanwar, S. C., Mariadason, J. M., Lash, A., Ladanyi, M., Saltz, L. B., Heguy, A., Paty, P. B., and Solit, D. B. (2010). Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 70, 5901–5911.
Jemal, A., Siegel, R., Xu, J., and Ward, E. (2010). Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300.
Jennings-Gee, J. E., Moore, J. E., Xu, M., Dance, S. T., Kock, N. D., McCoy, T. P., Carr, J. J., and Miller, M. S. (2006). Strain-specific induction of murine lung tumors following in utero exposure to 3-methylcholanthrene. Mol. Carcinog. 45, 676–684.
Johnson, L., Mercer, K., Greenbaum, D., Bronson, R. T., Crowley, D., Tuveson, D. A., and Jacks, T. (2001). Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116.
Jourd’heuil, D., Jourd’heuil, F. L., and Feelisch, M. (2003). Oxidation and nitrosation of thiols at low micromolar exposure to nitric oxide. Evidence for a free radical mechanism. J. Biol. Chem. 278, 15720–15726.
Kassie, F., Anderson, L. B., Higgins, L., Pan, Y., Matise, I., Negia, M., Upadhyaya, P., Wang, M., and Hecht, S. S. (2008). Chemopreventive agents modulate the protein expression profile of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone plus benzo[a]pyrene-induced lung tumors in A/J mice. Carcinogenesis 29, 610–619.
Keohavong, P., DeMichele, M. A., Melacrinos, A. C., Landreneau, R. J., Weyant, R. J., and Siegfried, J. M. (1996). Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin. Cancer Res. 2, 411–418.
Kerkhoff, E., Fedorov, L. M., Siefken, R., Walter, A. O., Papadopoulos, T., and Rapp, U. R. (2000). Lung-targeted expression of the c-Raf-1 kinase in transgenic mice exposes a novel oncogenic character of the wild-type protein. Cell Growth Differ. 11, 185–190.
Kjeldgaard, M., Nyborg, J., and Clark, B. F. (1996). The GTP binding motif: variations on a theme. FASEB J. 10, 1347–1368.
Leone-Kabler, S. L., Wessner, L. L., McEntee, M. F., D’Agostino, R. B., and Miller, M. S. (1997). Ki-ras mutations are an early event and correlate with tumor stage in transplacentally-induced murine lung tumors. Carcinogenesis 18, 1163–1168.
Li, Z. H., Zheng, J., Weiss, L. M., and Shibata, D. (1994a). c-k-ras and p53 mutations occur very early in adenocarcinoma of the lung. Am. J. Pathol. 144, 303–309.
Li, E. E., Heflich, R. H., Bucci, T. J., Manjanatha, M. G., Blaydes, B. S., and Delclos, K. B. (1994b). Relationships of DNA adduct formation, K-ras activating mutations and tumorigenic activities of 6-nitrochrysene and its metabolites in the lungs of CD-1 mice. Carcinogenesis 15, 1377–1385.
Lukman, S., Grant, B. J., Gorfe, A. A., Grant, G. H., and McCammon, J. A. (2010). The distinct conformational dynamics of K-Ras and H-Ras A59G. PLoS Comput. Biol. 6. e1000922. doi: 10.1371/journal.pcbi.1000922
Luo, J., Emanuele, M. J., Li, D., Creighton, C. J., Schlabach, M. R., Westbrook, T. F., Wong, K. K., and Elledge, S. J. (2009). A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835–848.
Malumbres, M., and Barbacid, M. (2003). RAS oncogenes: the first 30 years. Nat. Rev. Cancer 3, 459–465.
Manne, V., Bekesi, E., and Kung, H. F. (1985). Ha-ras proteins exhibit GTPase activity: point mutations that activate Ha-ras gene products result in decreased GTPase activity. Proc. Natl. Acad. Sci. U.S.A. 82, 376–380.
McGrath, J. P., Capon, D. J., Goeddel, D. V., and Levinson, A. D. (1984). Comparative biochemical properties of normal and activated human ras p21 protein. Nature 310, 644–649.
Meuwissen, R., Linn, S. C., van, d. V., Mooi, W. J., and Berns, A. (2001). Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene 20, 6551–6558.
Miller, M. S. (1994). Transplacental lung carcinogenesis: a pharmacogenetic mouse model for the modulatory role of cytochrome P450 1A1 on lung cancer initiation. Chem. Res. Toxicol. 7, 471–481.
Mitsudomi, T., Steinberg, S. M., Oie, H. K., Mulshine, J. L., Phelps, R., Viallet, J., Pass, H., Minna, J. D., and Gazdar, A. F. (1991). ras gene mutations in non-small cell lung cancers are associated with shortened survival irrespective of treatment intent. Cancer Res. 51, 4999–5002.
Miura, K., Bowman, E. D., Simon, R., Peng, A. C., Robles, A. I., Jones, R. T., Katagiri, T., He, P., Mizukami, H., Charboneau, L., Kikuchi, T., Liotta, L. A., Nakamura, Y., and Harris, C. C. (2002). Laser capture microdissection and microarray expression analysis of lung adenocarcinoma reveals tobacco smoking- and prognosis-related molecular profiles. Cancer Res. 62, 3244–3250.
Mizesko, M. C., Grewe, C., Grabner, A., and Miller, M. S. (2001). Alterations at the Ink4a locus in transplacentally induced murine lung tumors. Cancer Lett. 172, 59–66.
Moon, A. (2006). Differential functions of Ras for malignant phenotypic conversion. Arch. Pharm. Res. 29, 113–122.
Nielsen, K. H., Gredsted, L., Broach, J. R., and Willumsen, B. M. (2001). Sensitivity of wild type and mutant ras alleles to Ras specific exchange factors: identification of factor specific requirements. Oncogene 20, 2091–2100.
Nuzum, E. O., Malkinson, A. M., and Beer, D. G. (1990). Specific Ki-ras codon 61 mutations may determine the development of urethan-induced mouse lung adenomas or adenocarcinomas. Mol. Carcinog. 3, 287–295.
Ogino, S., Meyerhardt, J. A., Irahara, N., Niedzwiecki, D., Hollis, D., Saltz, L. B., Mayer, R. J., Schaefer, P., Whittom, R., Hantel, A., Benson, A. B. III, Goldberg, R. M., Bertagnolli, M. M., and Fuchs, C. S. (2009). KRAS mutation in stage III colon cancer and clinical outcome following intergroup trial CALGB 89803. Clin. Cancer Res. 15, 7322–7329.
Omerovic, J., Hammond, D. E., Clague, M. J., and Prior, I. A. (2008). Ras isoform abundance and signalling in human cancer cell lines. Oncogene 27, 2754–2762.
Raines, K. W., Bonini, M. G., and Campbell, S. L. (2007). Nitric oxide cell signaling: S-nitrosation of Ras superfamily GTPases. Cardiovasc. Res. 75, 229–239.
Ratner, E., Lu, L., Boeke, M., Barnett, R., Nallur, S., Chin, L. J., Pelletier, C., Blitzblau, R., Tassi, R., Paranjape, T., Hui, P., Godwin, A. K., Yu, H., Risch, H., Rutherford, T., Schwartz, P., Santin, A., Matloff, E., Zelterman, D., Slack, F. J., and Weidhaas, J. B. (2010). A KRAS-variant in ovarian cancer acts as a genetic marker of cancer risk. Cancer Res. 70, 6509–6515.
Recktenwald, C. V., Kellner, R., Lichtenfels, R., and Seliger, B. (2008). Altered detoxification status and increased resistance to oxidative stress by K-ras transformation. Cancer Res. 68, 10086–10093.
Reuther, G. W., and Der, C. J. (2000). The Ras branch of small GTPases: Ras family members don’t fall far from the tree. Curr. Opin. Cell Biol. 12, 157–165.
Reynolds, S. H., Wiest, J. S., Devereux, T. R., Anderson, M. W., and You, M. (1992). “Protooncogene activation in spontaneously occurring and chemically induced rodent and human lung tumors,” in Comparative Molecular Carcinogenesis, eds A. J. P. Klein-Szanto, M. W. Anderson, J. C. Barrett, and T. J. Slaga (New York: Wiley-Liss, Inc.), 303–320.
Rodenhuis, S., and Slebos, R. J. (1992). Clinical significance of ras oncogene activation in human lung cancer. Cancer Res. 52, 2665s–2669s.
Rosell, R., Li, S., Skacel, Z., Mate, J. L., Maestre, J., Canela, M., Tolosa, E., Armengol, P., Barnadas, A., and Ariza, A. (1993). Prognostic impact of mutated K-ras gene in surgically resected non-small cell lung cancer patients. Oncogene 8, 2407–2412.
Rosell, R., Monzo, M., Pifarre, A., Ariza, A., Sanchez, J. J., Moreno, I., Manuel, J., Lopez, M. P., Abad, A., and Maria de Anta, J. (1996). Molecular staging of non-small cell lung cancer according to K- ras genotypes. Clin. Cancer Res. 2, 1083–1086.
Scholl, C., Frohling, S., Dunn, I. F., Schinzel, A. C., Barbie, D. A., Kim, S. Y., Silver, S. J., Tamayo, P., Wadlow, R. C., Ramaswamy, S., Dohner, K., Bullinger, L., Sandy, P., Boehm, J. S., Root, D. E., Jacks, T., Hahn, W. C., and Gilliland, D. G. (2009). Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137, 821–834.
Schrammel, A., Gorren, A. C., Schmidt, K., Pfeiffer, S., and Mayer, B. (2003) S-nitrosation of glutathione by nitric oxide, peroxynitrite, and (*)NO/O(2)(*-). Free Radic. Biol. Med. 34, 1078–1088.
Seeburg, P. H., Colby, W. W., Capon, D. J., Goeddel, D. V., and Levinson, A. D. (1984). Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature 312, 71–75.
Siegfried, J. M., Gillespie, A. T., Mera, R., Casey, T. J., Keohavong, P., Testa, J. R., and Hunt, J. D. (1997). Prognostic value of specific KRAS mutations in lung adenocarcinomas. Cancer Epidemiol. Biomarkers Prev. 6, 841–847.
Singh, A., Greninger, P., Rhodes, D., Koopman, L., Violette, S., Bardeesy, N., and Settleman, J. (2009). A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500.
Smith, G., Bounds, R., Wolf, H., Steele, R. J., Carey, F. A., and Wolf, C. R. (2010). Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours – implications for personalised cancer medicine. Br. J. Cancer 102, 693–703.
Sweet, R. W., Yokoyama, S., Kamata, T., Feramisco, J. R., Rosenberg, M., and Gross, M. (1984). The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 311, 273–275.
Sweet-Cordero, A., Mukherjee, S., Subramanian, A., You, H., Roix, J. J., Ladd-Acosta, C., Mesirov, J., Golub, T. R., and Jacks, T. (2005). An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat. Genet. 37, 48–55.
To, M. D., Wong, C. E., Karnezis, A. N., Del, R. R., Di, L. R., and Balmain, A. (2008). Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat. Genet. 40, 1240–1244.
Vicent, S., Chen, R., Sayles, L. C., Lin, C., Walker, R. G., Gillespie, A. K., Subramanian, A., Hinkle, G., Yang, X., Saif, S., Root, D. E., Huff, V., Hahn, W. C., and Sweet-Cordero, E. A. (2010). Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J. Clin. Invest. 120, 3940–3952.
Virtanen, C., Ishikawa, Y., Honjoh, D., Kimura, M., Shimane, M., Miyoshi, T., Nomura, H., and Jones, M. H. (2002). Integrated classification of lung tumors and cell lines by expression profiling. Proc. Natl. Acad. Sci. U.S.A. 99, 12357–12362.
Vizan, P., Boros, L. G., Figueras, A., Capella, G., Mangues, R., Bassilian, S., Lim, S., Lee, W. N., and Cascante, M. (2005). K-ras codon-specific mutations produce distinctive metabolic phenotypes in NIH3T3 mice fibroblasts. Cancer Res. 65, 5512–5515.
Westra, W. H., Baas, I. O., Hruban, R. H., Askin, F. B., Wilson, K., Offerhaus, G. J., and Slebos, R. J. (1996). K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res. 56, 2224–2228.
Westra, W. H., Slebos, R. J., Offerhaus, G. J., Goodman, S. N., Evers, S. G., Kensler, T. W., Askin, F. B., Rodenhuis, S., and Hruban, R. H. (1993). K-ras oncogene activation in lung adenocarcinomas from former smokers. Evidence that K-ras mutations are an early and irreversible event in the development of adenocarcinoma of the lung. Cancer 72, 432–438.
Wikman, H., Kettunen, E., Seppanen, J. K., Karjalainen, A., Hollmen, J., Anttila, S., and Knuutila, S. (2002). Identification of differentially expressed genes in pulmonary adenocarcinoma by using cDNA array. Oncogene 21, 5804–5813.
Williams, J. G., Pappu, K., and Campbell, S. L. (2003). Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc. Natl. Acad. Sci. U.S.A. 100, 6376–6381.
Yao, R., Wang, Y., Lubet, R. A., and You, M. (2002). Differentially expressed genes associated with mouse lung tumor progression. Oncogene 21, 5814–5821.
Young, A., Lyons, J., Miller, A. L., Phan, V. T., Alarcon, I. R., and McCormick, F. (2009). Ras signaling and therapies. Adv. Cancer Res. 102, 1–17.
Zhang, B., Zhang, Y., Shacter, E., and Zheng, Y. (2005). Mechanism of the guanine nucleotide exchange reaction of Ras GTPase – evidence for a GTP/GDP displacement model. Biochemistry 44, 2566–2576.
Keywords: ras, cancer, mutation, tumorigenesis
Citation: Miller MS and Miller LD (2012) RAS mutations and oncogenesis: not all RAS mutations are created equally. Front. Gene. 2:100. doi: 10.3389/fgene.2011.00100
Received: 24 August 2011; Accepted: 13 December 2011;
Published online: 03 January 2012.
Edited by:
Ji Luo, National Institute of Health, USAReviewed by:
Ji Luo, National Institute of Health, USAMassimo Broggini, Istituto di Ricerche Farmacologiche Mario Negri, Italy
Copyright: © 2012 Miller and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mark Steven Miller, Department of Cancer Biology, Medical Center Blvd., Comprehensive Cancer Center, Wake Forest School of Medicine, Winston-Salem, NC 27157, USA. e-mail:bXNtaWxsZXJAd2FrZWhlYWx0aC5lZHU=