

- Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo, São Paulo, Brasil
Long non-coding RNAs (lncRNAs) transcribed from intergenic and intronic regions of the human genome constitute a broad class of cellular transcripts that are under intensive investigation. While only a handful of lncRNAs have been characterized, their involvement in fundamental cellular processes that control gene expression highlights a central role in cell homeostasis. Not surprisingly, aberrant expression of regulatory lncRNAs has been increasingly documented in different types of cancer, where they can mediate both oncogenic or tumor suppressor effects. Interaction with chromatin remodeling complexes that promote silencing of specific genes or modulation of splicing factor proteins seem to be two general modes of lncRNA regulation, but it is conceivable that additional mechanisms of action are yet to be unveiled. LncRNAs show greater tissue specificity compared to protein-coding mRNAs making them attractive in the search of novel diagnostics/prognostics cancer biomarkers in body fluid samples. In fact, lncRNA prostate cancer antigen 3 can be detected in urine samples and has been shown to improve diagnosis of prostate cancer. We suggest that an unbiased screening of the presence of RNAs in easily accessible body fluids such as serum and urine might reveal novel circulating lncRNAs as potential biomarkers in many types of cancer. Annotation and functional characterization of the lncRNA complement of the cancer transcriptome will conceivably provide new venues for early diagnosis and treatment of the disease.
Over the last decade, advances in genome-wide analyses of the eukaryotic transcriptome have revealed that most of the human genome is transcribed, generating a large repertoire of (>200 nt) long non-coding RNAs (lncRNAs) that map to intronic and intergenic regions (Birney et al., 2007; Dinger et al., 2009; Ponting et al., 2009; Kapranov et al., 2010). These include subsets of polyadenylated and non-polyadenylated transcripts that accumulate differently in the nucleus and cytoplasm of cells (Kapranov et al., 2007, 2010). The catalog of human lncRNAs has expanded dramatically just in the last several years; in fact, recently published deep RNA sequencing reveals that the range, depth, and complexity of the human transcriptome is far from fully characterized (Mercer et al., 2012) and expectations are that very soon the human lncRNA genes will outnumber protein-coding genes. The definition and naming of lncRNAs are currently evolving in the literature and different classes or categories of lncRNA have been described (Prensner and Chinnaiyan, 2011; Wright and Bruford, 2011). The categorization recently proposed by the HUGO Gene Nomenclature Committee (HGNC) is an ongoing project (Wright and Bruford, 2011), where lncRNAs were described as spliced, capped, and polyadenylated RNAs (Wright and Bruford, 2011); this clearly does not encompass all different lncRNAs that may be also unspliced and/or non-polyadenylated (Nakaya et al., 2007; Kapranov et al., 2010; Yang et al., 2011a). The rapid increase in the number of described lncRNAs along with the lack of uniform and systematic annotation nomenclature for the diverse and extensive amount of lncRNAs expressed in human tissues imposes a considerable limitation regarding the completeness of any database related to lncRNAs (Paschoal et al., 2012).
It is apparent that lncRNAs may act through diverse molecular mechanisms and play regulatory and structural roles in important biological processes (see Mattick, 2009 for a review). Presently, the mechanisms of action of only a few lncRNAs have been characterized in detail (Wang and Chang, 2011), and many of these lncRNAs have an altered expression in different types of human cancer (Huarte and Rinn, 2010; Gibb et al., 2011; Prensner and Chinnaiyan, 2011).
Cancers are the result of a process where somatic cells mutate and escape the controlled balance exerted by gene expression programs and cellular networks that maintain cellular homeostasis and normally prevent their unwanted expansion. Cancer cells differ from normal cells in many important characteristics, including loss of differentiation, increased invasiveness, and decreased drug sensitivity. Genes that affect these processes can be classified into two major groups: tumor suppressor genes and oncogenes. Tumor suppressor genes protect cells against mutations that initiate transformation. Conversely, oncogenes initiate the cellular transformation process when inappropriately activated. LncRNAs have been recently implicated as having tumor suppressor and oncogenic roles (Huarte and Rinn, 2010; Gibb et al., 2011; Prensner and Chinnaiyan, 2011).
General Mechanisms of lncRNA Function Implicated in Cancer
Long non-coding RNAs can activate cellular pathways that lead to tumorigenesis, in analogy to protein-coding oncogenes. The molecular mechanisms by which lncRNAs exert their biological functions has been extensively reviewed by (Wang and Chang, 2011). Here we highlight some examples of lncRNAs implicated in oncogenic functions.
One example of such an oncogenic lncRNA is metastasis-associated in lung adenocarcinoma transcript 1 (MALAT1), a nuclear-retained non-coding RNA that has been recently shown to regulate alternative splicing by modulating serine/arginine (SR) splicing factor phosphorylation (Tripathi et al., 2010). Increased expression of the lncRNA MALAT1 has been first observed in metastatic non-small cell lung cancer (Ji et al., 2003; see details in the next section), followed by endometrial stromal sarcoma of the uterus (Yamada et al., 2006), and more recently in six other types of cancer, including hepatocellular carcinoma, breast, pancreas, lung, colon, and prostate cancers (Lin et al., 2007). Recently, short hairpin RNA inhibition of MALAT1 in human cervical cancer cells was shown to suppress cell proliferation and invasion (Guo et al., 2010), whereas RNA interference-mediated silencing of MALAT1 reduced the in vitro migration of lung adenocarcinoma cells by influencing the expression of motility-related genes (Tano et al., 2010). Altogether, these findings reinforce the role of MALAT1 as an oncogenic lncRNA, and point to one of the possible modes of action of lncRNAs, namely through their interaction with and modulation of splicing factor proteins.
The mode of action of lncRNAs through the interaction with chromatin remodeling complexes may be a more general one, as it has been documented for two lncRNAs. One example is ANRIL (antisense non-coding RNA in the INK4 locus) that is altered in an estimated 30–40% of human tumors (Kim and Sharpless, 2006). Tumor suppressor p15INK4B is silenced by its antisense ANRIL transcript (Yu et al., 2008); lncRNA ANRIL is required for the recruitment of polycomb PRC1 and PRC2 complexes to the INK4B locus and for silencing of p15INK4B tumor suppressor gene (Yap et al., 2010; Kotake et al., 2011).
Another example is HOTAIR (HOX Antisense Intergenic RNA), a metastasis-associated gene located in the mammalian HOXC locus that reprograms chromatin state to promote cancer metastasis (Gupta et al., 2010). HOTAIR lncRNA interacts with Polycomb Repressor Complex PRC2, determining PRC2 localization and repression of the HOXD locus (Rinn et al., 2007). Recently it was found that HOTAIR serves as a scaffold for at least two distinct histone modification complexes. HOTAIR binds the PRC2 complex responsible for H3K27 methylation and also LSD1, a histone lysine demethylase that mediates enzymatic demethylation of H3K4Me2 (Tsai et al., 2010). Although the precise mechanism of HOTAIR activities remains to be elucidated, it is clear that HOTAIR participates in silencing of metastasis suppressor genes thus promoting metastasis, as discussed below.
Large-Scale Expression Profiling of lncRNAs in Patients
Cancer gene profiling studies have had an enormous impact on understanding of the biology of cancers, pointing to the biological heterogeneity of specific cancer types, providing identification of novel oncogenes and tumor suppressors, and defining pathways that interact to drive the growth of individual cancers (Cowin et al., 2010). Large-scale genomic studies are underway, such as The Cancer Genome Atlas project that aims to catalog in each cancer type the changes in DNA copy number and methylation, as well as in small (19–25 nt) non-coding microRNA (miRNA) and protein-coding mRNA expression (Cancer_Genome_Atlas_Research_Network, 2008, 2011). Noteworthy, changes in expression of lncRNAs have not been analyzed in these large cohort studies.
Identification of lncRNAs correlated to cancer has benefited in the past decade from the development of a number of effective high-throughput expression analyses technologies as well as from the increasing realization that lncRNAs may play important roles in physiological and pathological processes in the cell (see Table 1). Early efforts to identify molecular cancer markers based on the screening of cDNA libraries enriched in tumor-specific transcripts have identified lncRNAs whose expression levels correlate to cancer. Using a differential display approach, the lncRNA DD3, later named prostate cancer antigen 3 (PCA3), was initially identified as overexpressed in prostate tumors relative to benign prostate hyperplasia and normal epithelium (Bussemakers et al., 1999). Further studies later indicated that PCA3 is a very specific prostate cancer gene whose mechanism of action is still not identified (Marks and Bostwick, 2008; Shappell, 2008).
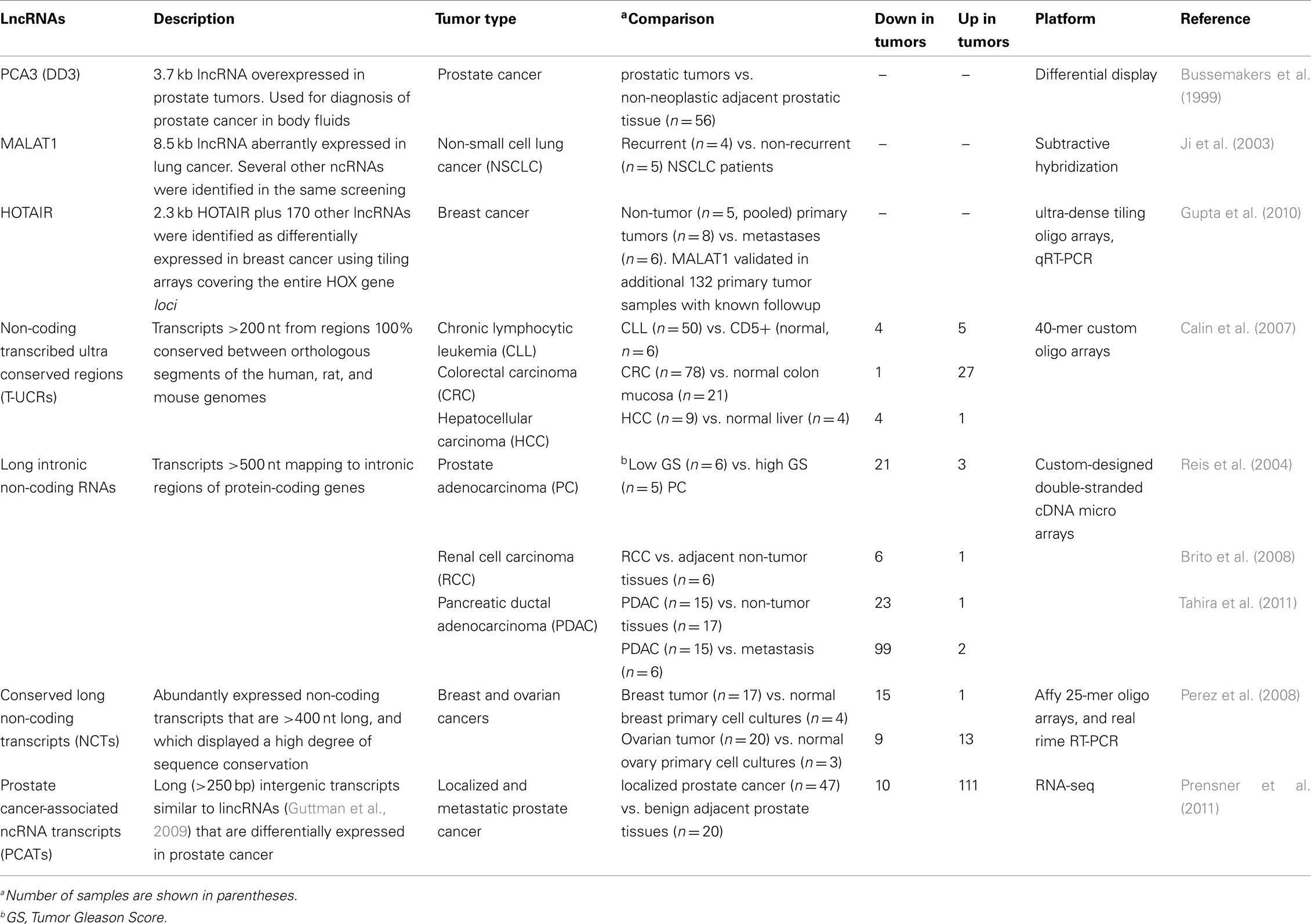
Table 1. Long non-coding RNAs differentially expressed in human cancer.
Another report that used large-scale transcriptome analysis to look for differential gene expression in cancer and gave attention to a differentially expressed lncRNA, namely MALAT1, employed a subtractive hybridization approach to determine differences in gene expression between primary non-small cell lung cancer tumors of five patients that were cured by surgery and tumors of four patients that subsequently metastasized (Ji et al., 2003). In all, 26 transcripts were found more than once, and among them the novel lncRNA named MALAT1. Subsequently, 31 samples from stage I patients suffering from adenocarcinoma or squamous cell carcinoma were analyzed by qPCR, and the expression levels of MALAT1 were significantly higher in metastasizing adenocarcinomas compared to non-metastasizing ones (p = 0.03); interestingly, no significant differences in gene expression were found for squamous cell carcinomas (n = 34; Ji et al., 2003). These data provided evidence that the association of lncRNA MALAT1 with metastasis depended on the lung tumor’s histology.
A large-scale gene expression approach specifically designed to look for lncRNAs correlated to cancer has employed hybridization of RNA derived from normal human breast epithelia, primary breast carcinomas, and distant metastases to ultra-dense tiling arrays covering the entire HOX gene loci (Gupta et al., 2010). The authors found that 233 transcribed regions in the HOX loci, comprising 170 lncRNAs and 63 HOX exons, were differentially expressed (Gupta et al., 2010), with a systematic variation in the expression of HOX lncRNAs among normal breast epithelia, primary tumor, and metastases. Dozens of HOX lncRNAs were expressed in normal breast but showed reduced expression in all cancer samples; conversely, a set of HOX lncRNAs was frequently expressed in primary tumors but not in metastases (Gupta et al., 2010). Notably, one such metastasis-associated lncRNA was HOTAIR, which had a unique association with patient prognosis (Gupta et al., 2010).
Oligoarrays were used to interrogate 481 ultra conserved regions (UCRs) in the human genome (Calin et al., 2007); UCRs are a subset of conserved sequences that are located in both intra- and intergenic regions and are absolutely conserved (100%) between orthologous regions of the human, rat, and mouse genomes (Bejerano et al., 2004). A total of 256 UCRs (53%) were identified as non-coding genomic regions (Bejerano et al., 2004). The authors investigated the expression of UCRs in a panel of 173 samples, including 133 human cancers [e.g., chronic lymphocytic leukemias (CLL), colorectal (CRC), and hepatocellular carcinomas (HCC)] and 40 corresponding normal tissues (Calin et al., 2007). Specific sets of UCRs were differentially expressed in distinct tumor types, and among them, 42 were non-coding UCRs (48% of the differentially expressed UCRs). This work demonstrated that the transcribed UCR expression profiles can be used to differentiate human cancers (Calin et al., 2007).
Custom-designed cDNA microarrays interrogating selected sets of protein-coding genes and lncRNAs from intronic/intergenic genomic regions were used for obtaining expression profiles from clinical samples of a number of cancer types (Reis et al., 2004; Brito et al., 2008; Tahira et al., 2011). In prostate cancer, RNA from 27 patient tumor samples with Gleason scores ranging from 5 to 10 were hybridized to these custom-designed microarrays (Reis et al., 2004). Among the 56 transcripts that were found to be significantly correlated to the degree of prostate tumor differentiation (Gleason score), 23 were lncRNAs mapping to intronic regions (Reis et al., 2004). Among the top twelve transcripts most significantly correlated to tumor differentiation, six were antisense intronic lncRNAs as shown by orientation-specific RT-PCR or northern blot analysis with strand-specific riboprobe (Reis et al., 2004).
Aberrant expression of intronic lncRNAs was studied in clear cell renal cell carcinoma (RCC) using matched samples of tumor and adjacent non-neoplastic tissue obtained from six patients (Brito et al., 2008). A set of 55 transcripts was significantly down-regulated in clear cell RCC relative to the matched non-tumor tissue; among the down-regulated transcripts, 49 mapped to untranslated or coding exons of protein-coding genes and 6 were lncRNAs mapped to intronic regions in genomic loci of protein-coding genes (Brito et al., 2008).
More recently, pancreatic ductal adenocarcinoma (PDAC) was studied, aiming at identifying gene expression profiles of protein-coding and lncRNAs correlated to pancreatic cancer and metastasis in 38 clinical samples of tumor and non-tumor pancreatic tissues (Tahira et al., 2011). Statistically significant expression signatures comprising protein-coding mRNAs and intronic/intergenic lncRNAs that correlate to PDAC or to pancreatic cancer metastasis were identified; interestingly, loci harboring intronic lncRNAs differentially expressed in PDAC metastases were enriched in genes associated to the MAPK pathway (Tahira et al., 2011).
Whole-genome tiling arrays were utilized to identify the expression of novel lncRNAs across the entire human genome (Perez et al., 2008). The authors hybridized RNA from normal lung cell cultures to the tiling arrays and found 495 transcriptionally active regions originated from non-protein-coding sequence (intergenic or intronic regions) and chose 15 candidate RNAs for subsequent real-time RT–PCR, northern blot, and sequencing experiments of which three were intronic lncRNAs (Perez et al., 2008). Altered expression of these lncRNAs was found in patient samples in both breast and ovarian cancers (Perez et al., 2008).
The first high-throughput sequencing of polyA+ RNA (RNA-seq) from a large cohort of 102 prostate tissues and cells lines has been recently reported (Prensner et al., 2011). The work has identified 121 unannotated prostate cancer-associated lncRNA transcripts (PCATs) and has characterized one lncRNA, PCAT-1, as a prostate-specific regulator of cell proliferation, showing that it is a target of PRC2 (Prensner et al., 2011). Patterns of PCAT-1 and PRC2 expression stratified patient tissues into molecular subtypes distinguished by expression signatures of PCAT-1-repressed target genes. These findings establish the utility of RNA-seq to identify disease-associated lncRNAs that may improve the stratification of cancer subtypes (Prensner et al., 2011).
Although the functional consequences of the deregulation of lncRNAs in cancer development are currently unknown, the studies discussed above indicate that this class of transcripts may play important functions in both normal and malignant tissues.
lncRNAs as a Diagnostic Test Tool
Molecular markers of malignancy are important diagnostic and prognostic tools that help patient management in the oncology clinics. Cancer is a multi-factorial disease and for most types of malignancies an increase in the number of available assessment and management tools is desirable. Expression of the lncRNA MALAT1 has been identified by Kaplan–Meier analyses as a prognostic parameter for patient survival in stage I non-small cell lung cancer (Ji et al., 2003). MALAT1 has been subsequently validated as a marker for endometrial stromal sarcoma of the uterus (Yamada et al., 2006) and for HCC and a spectrum of five other human carcinomas (Lin et al., 2007). In addition, increased expression of MALAT1 has been recently shown to be an independent prognostic factor for HCC following liver transplantation (Lai et al., 2011).
Increased expression of lncRNA HOTAIR was shown to be associated with metastasis in breast cancer patients, having a unique association with patient prognosis (Gupta et al., 2010). Subsequently, HOTAIR expression levels was found to correlate with metastasis in colorectal carcinoma (Kogo et al., 2011), and to predict tumor recurrence in hepatocellular carcinoma (Yang et al., 2011b).
At present, few lncRNAs have been characterized as potential biomarkers in human fluids. Measurement of lncRNA PCA3 in patient urine samples has been shown to allow more sensitive and specific diagnosis of prostate cancer than the widely used PSA (prostate-specific antigen) serum levels (Fradet et al., 2004; Tinzl et al., 2004; Shappell, 2008). The potential of PCA3 urine assay to aid prostate cancer diagnosis and minimize unnecessary biopsies has been extensively documented, highlighting its advantages over PSA and pointing to future challenges for this new diagnostic biomarker (Lee et al., 2011).
The lncRNA HULC (highly upregulated in liver cancer) is highly expressed in HCC patients and can be detected in the blood by conventional PCR methods (Panzitt et al., 2007). It has been later shown that HULC lncRNA expression is not confined to HCC, but is also expressed in colorectal carcinomas that metastasize to the liver (Matouk et al., 2009).
Diagnosis and treatment follow up of complex multi-factorial diseases such as cancer could conceivably be improved by screening of a larger number of molecular biomarkers in easily accessible sample specimens. In fact, highly stable cell-free circulating nucleic acids (cfCNA), both RNA and DNA species, have been discovered in the blood, plasma, and urine of humans (Tong and Lo, 2006). At present, there is evidence of a good correlation between tumor-associated changes in genomic, epigenetic, or transcriptional patterns and alterations in cfCNA levels (Schwarzenbach et al., 2011), strongly pointing to the utility of this blood biomarker class as promising clinical tools.
The release of nucleic acids into the blood is thought to be related to the apoptosis and necrosis of cancer cells in the tumor microenvironment and is also the result of secretion. Circulating RNAs are detectable in the serum and plasma of cancer patients, being surprisingly stable in spite of the fact that high amounts of RNases circulate in the blood of cancer patients. This implies that RNA may be protected from degradation by its packaging into microparticles, which include exosomes, microvesicles, apoptotic bodies, and apoptotic microparticles (Orozco and Lewis, 2010). Microparticles are small, membranous vesicles that can contain DNA, RNA, miRNA, intracellular proteins, and extracellular surface markers from the parental cells; they can be secreted from intracellular multivesicular bodies or released from the surface of blebbing membranes (Cocucci et al., 2009; Orozco and Lewis, 2010). The detection and identification of RNA in serum and plasma can be carried out using microarray technologies or reverse transcription quantitative real-time PCR (O’Driscoll et al., 2008). The reported RNA content of microvesicles and exosomes thus far includes primarily small miRNAs and long protein-coding mRNAs (Record et al., 2011).
Recent advances in small non-coding miRNA expression profiling in human cancer and their potential as therapeutic targets and novel biomarkers have been reviewed (Farazi et al., 2011; Munker and Calin, 2011). The presence of small non-coding miRNAs in serum of cancer patients was first described for diffuse large B-cell lymphoma patients (Lawrie et al., 2008). Circulating miRNAs were subsequently used in assessing patients with prostate cancer (Mitchell et al., 2008) and at present circulating miRNAs have been characterized as potential biomarkers in over ten different cancers (Kosaka et al., 2010). Despite being promising biomarkers for cancer diagnosis and prognosis, there have been conflicting findings about circulating miRNAs from the same tumor reported from different studies (Kosaka et al., 2010). These discrepancies might be due to the lack of an established endogenous miRNA control to normalize for circulating miRNA levels, and also to the different extraction and quantification methods used among the studies (Kosaka et al., 2010). An effort to standardize results is warranted by putting forward recommendations for controlling pre-analytical variables, including the reduction of contaminant cellular miRNAs of hematopoietic origin in the isolation and quantization of cell-free circulating RNAs (Duttagupta et al., 2011).
We speculate that in addition to miRNAs and mRNAs the human serum might contain a considerable amount of lncRNAs that will eventually be detected by the use of unbiased high-throughput technologies such as genome tiling expression microarrays or RNA-seq deep-sequencing of serum samples. Such approaches should be subjected to the same controls regarding pre-analytical variables (Duttagupta et al., 2011), including the reduction of contaminant hematopoietic cells in the isolation and quantization of cell-free circulating lncRNAs.
Comparative studies of lncRNAs in serum from cancer patient large cohorts and from normal subjects will possibly reveal novel circulating lncRNAs as potential biomarkers in many types of cancers.
Conclusion and Perspectives
Long non-coding RNA expression profiles in human cancers have highlighted the potential value of this class of non-coding RNAs as tumor markers in patient diagnosis and prognosis. The rapidly expanding catalog of lncRNAs holds promises that in the near future lncRNAs will become ever more important in cancer patient management. An analogy can be made with the impact of small miRNA profiling in many types of cancer (Braconi et al., 2011; Ferracin et al., 2011; Schetter and Harris, 2011; Wang and Sen, 2011), which has provided different experimental lines of evidence that deregulation of miRNAs not only results as consequence of cancer progression but also directly affects gene networks that promote tumor initiation and progression in a cause-effect manner (Lovat et al., 2011). As the catalog of lncRNAs grows, it will become important to elucidate the genetic networks and pathways regulated by the abnormally expressing lncRNAs in cancer cells as a means to understanding the role of these lncRNAs in the induction of malignant transformation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by grants from Fundação de Amparo a Pesquisa do Estado de São Paulo, and from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brasil. Eduardo M. Reis and Sergio Verjovski-Almeida received established investigator fellowships from CNPq.
References
Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S., and Haussler, D. (2004). Ultraconserved elements in the human genome. Science 304, 1321–1325.
Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigo, R., Gingeras, T. R., Margulies, E. H., Weng, Z., Snyder, M., Dermitzakis, E. T., Thurman, R. E., Kuehn, M. S., Taylor, C. M., Neph, S., Koch, C. M., Asthana, S., Malhotra, A., Adzhubei, I., Greenbaum, J. A., Andrews, R. M., Flicek, P., Boyle, P. J., Cao, H., Carter, N. P., Clelland, G. K., Davis, S., Day, N., Dhami, P., Dillon, S. C., Dorschner, M. O., Fiegler, H., Giresi, P. G., Goldy, J., Hawrylycz, M., Haydock, A., Humbert, R., James, K. D., Johnson, B. E., Johnson, E. M., Frum, T. T., Rosenzweig, E. R., Karnani, N., Lee, K., Lefebvre, G. C., Navas, P. A., Neri, F., Parker, S. C., Sabo, P. J., Sandstrom, R., Shafer, A., Vetrie, D., Weaver, M., Wilcox, S., Yu, M., Collins, F. S., Dekker, J., Lieb, J. D., Tullius, T. D., Crawford, G. E., Sunyaev, S., Noble, W. S., Dunham, I., Denoeud, F., Reymond, A., Kapranov, P., Rozowsky, J., Zheng, D., Castelo, R., Frankish, A., Harrow, J., Ghosh, S., Sandelin, A., Hofacker, I. L., Baertsch, R., Keefe, D., Dike, S., Cheng, J., Hirsch, H. A., Sekinger, E. A., Lagarde, J., Abril, J. F., Shahab, A., Flamm, C., Fried, C., Hackermuller, J., Hertel, J., Lindemeyer, M., Missal, K., Tanzer, A., Washietl, S., Korbel, J., Emanuelsson, O., Pedersen, J. S., Holroyd, N., Taylor, R., Swarbreck, D., Matthews, N., Dickson, M. C., Thomas, D. J., Weirauch, M. T., Gilbert, J., Drenkow, J., Bell, I., Zhao, X., Srinivasan, K. G., Sung, W. K., Ooi, H. S., Chiu, K. P., Foissac, S., Alioto, T., Brent, M., Pachter, L., Tress, M. L., Valencia, A., Choo, S. W., Choo, C. Y., Ucla, C., Manzano, C., Wyss, C., Cheung, E., Clark, T. G., Brown, J. B., Ganesh, M., Patel, S., Tammana, H., Chrast, J., Henrichsen, C. N., Kai, C., Kawai, J., Nagalakshmi, U., Wu, J., Lian, Z., Lian, J., Newburger, P., Zhang, X., Bickel, P., Mattick, J. S., Carninci, P., Hayashizaki, Y., Weissman, S., Hubbard, T., Myers, R. M., Rogers, J., Stadler, P. F., Lowe, T. M., Wei, C. L., Ruan, Y., Struhl, K., Gerstein, M., Antonarakis, S. E., Fu, Y., Green, E. D., Karaöz, U., Siepel, A., Taylor, J., Liefer, L. A., Wetterstrand, K. A., Good, P. J., Feingold, E. A., Guyer, M. S., Cooper, G. M., Asimenos, G., Dewey, C. N., Hou, M., Nikolaev, S., Montoya-Burgos, J. I., Löytynoja, A., Whelan, S., Pardi, F., Massingham, T., Huang, H., Zhang, N. R., Holmes, I., Mullikin, J. C., Ureta-Vidal, A., Paten, B., Seringhaus, M., Church, D., Rosenbloom, K., Kent, W. J., Stone, E. A., NISC Comparative Sequencing Program, Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children’s Hospital Oakland Research Institute, Batzoglou, S., Goldman, N., Hardison, R. C., Haussler, D., Miller, W., Sidow, A., Trinklein, N. D., Zhang, Z. D., Barrera, L., Stuart, R., King, D. C., Ameur, A., Enroth, S., Bieda, M. C., Kim, J., Bhinge, A. A., Jiang, N., Liu, J., Yao, F., Vega, V. B., Lee, C. W., Ng, P., Shahab, A., Yang, A., Moqtaderi, Z., Zhu, Z., Xu, X., Squazzo, S., Oberley, M. J., Inman, D., Singer, M. A., Richmond, T. A., Munn, K. J., Rada-Iglesias, A., Wallerman, O., Komorowski, J., Fowler, J. C., Couttet, P., Bruce, A. W., Dovey, O. M., Ellis, P. D., Langford, C. F., Nix, D. A., Euskirchen, G., Hartman, S., Urban, A. E., Kraus, P., Van Calcar, S., Heintzman, N., Kim, T. H., Wang, K., Qu, C., Hon, G., Luna, R., Glass, C. K., Rosenfeld, M. G., Aldred, S. F., Cooper, S. J., Halees, A., Lin, J. M., Shulha, H. P., Zhang, X., Xu, M., Haidar, J. N., Yu, Y., Ruan, Y., Iyer, V. R., Green, R. D., Wadelius, C., Farnham, P. J., Ren, B., Harte, R. A., Hinrichs, A. S., Trumbower, H., Clawson H, Hillman-Jackson, J., Zweig, A. S., Smith, K., Thakkapallayil, A., Barber, G., Kuhn, R. M., Karolchik, D., Armengol, L., Bird, C. P., de Bakker, P. I., Kern, A. D., Lopez-Bigas, N., Martin, J. D., Stranger, B. E., Woodroffe, A., Davydov, E., Dimas, A., Eyras, E., Hallgrímsdóttir, I. B., Huppert, J., Zody, M. C., Abecasis, G. R., Estivill, X., Bouffard, G. G., Guan, X., Hansen, N. F., Idol, J. R., Maduro, V. V., Maskeri, B., McDowell, J. C., Park, M., Thomas, P. J., Young, A. C., Blakesley, R. W., Muzny, D. M., Sodergren, E., Wheeler, D. A., Worley, K. C., Jiang, H., Weinstock, G. M., Gibbs, R. A., Graves, T., Fulton, R., Mardis, E. R., Wilson, R. K., Clamp, M., Cuff, J., Gnerre, S., Jaffe, D. B., Chang, J. L., Lindblad-Toh, K., Lander, E. S., Koriabine, M., Nefedov, M., Osoegawa, K., Yoshinaga, Y., Zhu, B., and de Jong, P. J. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816.
Braconi, C., Henry, J. C., Kogure, T., Schmittgen, T., and Patel, T. (2011). The role of microRNAs in human liver cancers. Semin. Oncol. 38, 752–763.
Brito, G. C., Fachel, A. A., Vettore, A. L., Vignal, G. M., Gimba, E. R., Campos, F. S., Barcinski, M. A., Verjovski-Almeida, S., and Reis, E. M. (2008). Identification of protein-coding and intronic noncoding RNAs down-regulated in clear cell renal carcinoma. Mol. Carcinog. 47, 757–767.
Bussemakers, M. J., Van Bokhoven, A., Verhaegh, G. W., Smit, F. P., Karthaus, H. F., Schalken, J. A., Debruyne, F. M., Ru, N., and Isaacs, W. B. (1999). DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 59, 5975–5979.
Calin, G. A., Liu, C. G., Ferracin, M., Hyslop, T., Spizzo, R., Sevignani, C., Fabbri, M., Cimmino, A., Lee, E. J., Wojcik, S. E., Shimizu, M., Tili, E., Rossi, S., Taccioli, C., Pichiorri, F., Liu, X., Zupo, S., Herlea, V., Gramantieri, L., Lanza, G., Alder, H., Rassenti, L., Volinia, S., Schmittgen, T. D., Kipps, T. J., Negrini, M., and Croce, C. M. (2007). Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12, 215–229.
Cancer_Genome_Atlas_Research_Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068.
Cancer_Genome_Atlas_Research_Network. (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615.
Cocucci, E., Racchetti, G., and Meldolesi, J. (2009). Shedding microvesicles: artefacts no more. Trends Cell Biol. 19, 43–51.
Cowin, P. A., Anglesio, M., Etemadmoghadam, D., and Bowtell, D. D. (2010). Profiling the cancer genome. Annu. Rev. Genomics Hum. Genet. 11, 133–159.
Dinger, M. E., Amaral, P. P., Mercer, T. R., and Mattick, J. S. (2009). Pervasive transcription of the eukaryotic genome: functional indices and conceptual implications. Brief. Funct. Genomic. Proteomic. 8, 407–423.
Duttagupta, R., Jiang, R., Gollub, J., Getts, R. C., and Jones, K. W. (2011). Impact of cellular miRNAs on circulating miRNA biomarker signatures. PLoS ONE 6, e20769. doi:10.1371/journal.pone.0020769
Farazi, T. A., Spitzer, J. I., Morozov, P., and Tuschl, T. (2011). miRNAs in human cancer. J. Pathol. 223, 102–115.
Ferracin, M., Querzoli, P., Calin, G. A., and Negrini, M. (2011). MicroRNAs: toward the clinic for breast cancer patients. Semin. Oncol. 38, 764–775.
Fradet, Y., Saad, F., Aprikian, A., Dessureault, J., Elhilali, M., Trudel, C., Masse, B., Piche, L., and Chypre, C. (2004). uPM3, a new molecular urine test for the detection of prostate cancer. Urology 64, 311–315; discussion 315–316.
Gibb, E. A., Brown, C. J., and Lam, W. L. (2011). The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 10, 38.
Guo, F., Li, Y., Liu, Y., Wang, J., and Li, G. (2010). Inhibition of metastasis-associated lung adenocarcinoma transcript 1 in CaSki human cervical cancer cells suppresses cell proliferation and invasion. Acta Biochim. Biophys. Sin. (Shanghai) 42, 224–229.
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., Tsai, M. C., Hung, T., Argani, P., Rinn, J. L., Wang, Y., Brzoska, P., Kong, B., Li, R., West, R. B., Van De Vijver, M. J., Sukumar, S., and Chang, H. Y. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076.
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., Huarte, M., Zuk, O., Carey, B. W., Cassady, J. P., Cabili, M. N., Jaenisch, R., Mikkelsen, T. S., Jacks, T., Hacohen, N., Bernstein, B. E., Kellis, M., Regev, A., Rinn, J. L., and Lander, E. S. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227.
Huarte, M., and Rinn, J. L. (2010). Large non-coding RNAs: missing links in cancer? Hum. Mol. Genet. 19, R152–R161.
Ji, P., Diederichs, S., Wang, W., Boing, S., Metzger, R., Schneider, P. M., Tidow, N., Brandt, B., Buerger, H., Bulk, E., Thomas, M., Berdel, W. E., Serve, H., and Muller-Tidow, C. (2003). MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22, 8031–8041.
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., Stadler, P. F., Hertel, J., Hackermuller, J., Hofacker, I. L., Bell, I., Cheung, E., Drenkow, J., Dumais, E., Patel, S., Helt, G., Ganesh, M., Ghosh, S., Piccolboni, A., Sementchenko, V., Tammana, H., and Gingeras, T. R. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488.
Kapranov, P., St. Laurent, G., Raz, T., Ozsolak, F., Reynolds, C. P., Sorensen, P. H., Reaman, G., Milos, P., Arceci, R. J., Thompson, J. F., and Triche, T. J. (2010). The majority of total nuclear-encoded non-ribosomal RNA in a human cell is “dark matter” un-annotated RNA. BMC Biol. 8, 149. doi:10.1186/1741-7007-8-149
Kim, W. Y., and Sharpless, N. E. (2006). The regulation of INK4/ARF in cancer and aging. Cell 127, 265–275.
Kogo, R., Shimamura, T., Mimori, K., Kawahara, K., Imoto, S., Sudo, T., Tanaka, F., Shibata, K., Suzuki, A., Komune, S., Miyano, S., and Mori, M. (2011). Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326.
Kosaka, N., Iguchi, H., and Ochiya, T. (2010). Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 101, 2087–2092.
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., and Xiong, Y. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962.
Lai, M. C., Yang, Z., Zhou, L., Zhu, Q. Q., Xie, H. Y., Zhang, F., Wu, L. M., Chen, L. M., and Zheng, S. S. (2011). Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med. Oncol. doi: 10.1007/s12032-011-0004-z. [Epub ahead of print].
Lawrie, C. H., Gal, S., Dunlop, H. M., Pushkaran, B., Liggins, A. P., Pulford, K., Banham, A. H., Pezzella, F., Boultwood, J., Wainscoat, J. S., Hatton, C. S., and Harris, A. L. (2008). Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 141, 672–675.
Lee, G. L., Dobi, A., and Srivastava, S. (2011). Prostate cancer: diagnostic performance of the PCA3 urine test. Nat. Rev. Urol. 8, 123–124.
Lin, R., Maeda, S., Liu, C., Karin, M., and Edgington, T. S. (2007). A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 26, 851–858.
Lovat, F., Valeri, N., and Croce, C. M. (2011). MicroRNAs in the pathogenesis of cancer. Semin. Oncol. 38, 724–733.
Marks, L. S., and Bostwick, D. G. (2008). Prostate cancer specificity of PCA3 gene testing: examples from clinical practice. Rev. Urol. 10, 175–181.
Matouk, I. J., Abbasi, I., Hochberg, A., Galun, E., Dweik, H., and Akkawi, M. (2009). Highly upregulated in liver cancer noncoding RNA is overexpressed in hepatic colorectal metastasis. Eur. J. Gastroenterol. Hepatol. 21, 688–692.
Mattick, J. S. (2009). The genetic signatures of noncoding RNAs. PLoS Genet. 5, e1000459. doi:10.1371/journal.pgen.1000459
Mercer, T. R., Gerhardt, D. J., Dinger, M. E., Crawford, J., Trapnell, C., Jeddeloh, J. A., Mattick, J. S., and Rinn, J. L. (2012). Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 30, 99–104.
Mitchell, P. S., Parkin, R. K., Kroh, E. M., Fritz, B. R., Wyman, S. K., Pogosova-Agadjanyan, E. L., Peterson, A., Noteboom, J., O’Briant, K. C., Allen, A., Lin, D. W., Urban, N., Drescher, C. W., Knudsen, B. S., Stirewalt, D. L., Gentleman, R., Vessella, R. L., Nelson, P. S., Martin, D. B., and Tewari, M. (2008). Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. U.S.A. 105, 10513–10518.
Nakaya, H. I., Amaral, P. P., Louro, R., Lopes, A., Fachel, A. A., Moreira, Y. B., El-Jundi, T. A., Da Silva, A. M., Reis, E. M., and Verjovski-Almeida, S. (2007). Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol. 8, R43.
O’Driscoll, L., Kenny, E., Mehta, J. P., Doolan, P., Joyce, H., Gammell, P., Hill, A., O’Daly, B., O’Gorman, D., and Clynes, M. (2008). Feasibility and relevance of global expression profiling of gene transcripts in serum from breast cancer patients using whole genome microarrays and quantitative RT-PCR. Cancer Genomics Proteomics 5, 94–104.
Orozco, A. F., and Lewis, D. E. (2010). Flow cytometric analysis of circulating microparticles in plasma. Cytometry A 77, 502–514.
Panzitt, K., Tschernatsch, M. M., Guelly, C., Moustafa, T., Stradner, M., Strohmaier, H. M., Buck, C. R., Denk, H., Schroeder, R., Trauner, M., and Zatloukal, K. (2007). Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132, 330–342.
Paschoal, A. R., Maracaja-Coutinho, V., Setubal, J. C., Simões, Z. L. P., Verjovski-Almeida, S., and Durham, A. M. (2012). Non-coding transcription characterization and annotation: a guide and web resource for non-coding RNA databases. RNA Biol. 9. [Epub ahead of print].
Perez, D. S., Hoage, T. R., Pritchett, J. R., Ducharme-Smith, A. L., Halling, M. L., Ganapathiraju, S. C., Streng, P. S., and Smith, D. I. (2008). Long, abundantly expressed non-coding transcripts are altered in cancer. Hum. Mol. Genet. 17, 642–655.
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641.
Prensner, J. R., and Chinnaiyan, A. M. (2011). The emergence of lncRNAs in cancer biology. Cancer Discov. 1, 391–407.
Prensner, J. R., Iyer, M. K., Balbin, O. A., Dhanasekaran, S. M., Cao, Q., Brenner, J. C., Laxman, B., Asangani, I. A., Grasso, C. S., Kominsky, H. D., Cao, X., Jing, X., Wang, X., Siddiqui, J., Wei, J. T., Robinson, D., Iyer, H. K., Palanisamy, N., Maher, C. A., and Chinnaiyan, A. M. (2011). Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 29, 742–749.
Record, M., Subra, C., Silvente-Poirot, S., and Poirot, M. (2011). Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 81, 1171–1182.
Reis, E. M., Nakaya, H. I., Louro, R., Canavez, F. C., Flatschart, A. V., Almeida, G. T., Egidio, C. M., Paquola, A. C., Machado, A. A., Festa, F., Yamamoto, D., Alvarenga, R., Da Silva, C. C., Brito, G. C., Simon, S. D., Moreira-Filho, C. A., Leite, K. R., Camara-Lopes, L. H., Campos, F. S., Gimba, E., Vignal, G. M., El-Dorry, H., Sogayar, M. C., Barcinski, M. A., Da Silva, A. M., and Verjovski-Almeida, S. (2004). Antisense intronic non-coding RNA levels correlate to the degree of tumor differentiation in prostate cancer. Oncogene 23, 6684–6692.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., Goodnough, L. H., Helms, J. A., Farnham, P. J., Segal, E., and Chang, H. Y. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323.
Schetter, A. J., and Harris, C. C. (2011). Alterations of microRNAs contribute to colon carcinogenesis. Semin. Oncol. 38, 734–742.
Schwarzenbach, H., Hoon, D. S., and Pantel, K. (2011). Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437.
Shappell, S. B. (2008). Clinical utility of prostate carcinoma molecular diagnostic tests. Rev. Urol. 10, 44–69.
Tahira, A. C., Kubrusly, M. S., Faria, M. F., Dazzani, B., Fonseca, R. S., Maracaja-Coutinho, V., Verjovski-Almeida, S., Machado, M. C., and Reis, E. M. (2011). Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol. Cancer 10, 141.
Tano, K., Mizuno, R., Okada, T., Rakwal, R., Shibato, J., Masuo, Y., Ijiri, K., and Akimitsu, N. (2010). MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 584, 4575–4580.
Tinzl, M., Marberger, M., Horvath, S., and Chypre, C. (2004). DD3PCA3 RNA analysis in urine – a new perspective for detecting prostate cancer. Eur. Urol. 46, 182–186; discussion 187.
Tong, Y. K., and Lo, Y. M. (2006). Diagnostic developments involving cell-free (circulating) nucleic acids. Clin. Chim. Acta 363, 187–196.
Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., Freier, S. M., Bennett, C. F., Sharma, A., Bubulya, P. A., Blencowe, B. J., Prasanth, S. G., and Prasanth, K. V. (2010). The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938.
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., Shi, Y., Segal, E., and Chang, H. Y. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693.
Wang, J., and Sen, S. (2011). MicroRNA functional network in pancreatic cancer: from biology to biomarkers of disease. J. Biosci. 36, 481–491.
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914.
Wright, M. W., and Bruford, E. A. (2011). Naming “junk”: human non-protein coding RNA d gene nomenclature. Hum. Genomics 5, 90–98.
Yamada, K., Kano, J., Tsunoda, H., Yoshikawa, H., Okubo, C., Ishiyama, T., and Noguchi, M. (2006). Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Sci. 97, 106–112.
Yang, L., Duff, M. O., Graveley, B. R., Carmichael, G. G., and Chen, L. L. (2011a). Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 12, R16.
Yang, Z., Zhou, L., Wu, L. M., Lai, M. C., Xie, H. Y., Zhang, F., and Zheng, S. S. (2011b). Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 18, 1243–1250.
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., Gil, J., Walsh, M. J., and Zhou, M. M. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674.
Keywords: long non-coding RNA, cancer, diagnostics, expression signature
Citation: Reis EM and Verjovski-Almeida S (2012) Perspectives of long non-coding RNAs in cancer diagnostics. Front. Gene. 3:32. doi: 10.3389/fgene.2012.00032
Received: 21 December 2011; Paper pending published: 14 January 2012;
Accepted: 20 February 2012; Published online: 08 March 2012.
Edited by:
Philipp Kapranov, St. Laurent Institute, USAReviewed by:
Georges St. Laurent, St. Laurent Institute, USAElai Davicioni, GenomeDx Biosciences Inc., Canada
Copyright: © 2012 Reis and Verjovski-Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Eduardo M. Reis and Sergio Verjovski-Almeida, Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo, 05508-900 São Paulo, Brasil. e-mail:ZW1yZWlzQGlxLnVzcC5icg==;dmVyam9AaXEudXNwLmJy