
- Brudnick Neuropsychiatric Research Institute, University of Massachusetts Medical School, Worcester, MA, USA
Background
Interactions between endogenous factors, e.g., gene allelic variants, and exogenous factors, e.g., exposure to specific environments, can be reasonably suspected as the cause of the most common chronic communication disorders. However, the elucidation of genetic factors and their interactions with non-genetic factors in behavioral diseases has been challenged. To date, two main strategies have been used for the identification of genes for diseases of unknown molecular etiology. Over two decades, linkage analysis and positional cloning of the disease gene were effective in the identification of genes for Mendelian or monogenic single-gene disorders (Botstein and Risch, 2003; McKusick, 2007). The methodological and statistical threshold criteria, applied for linkage analysis in families, provides robust evidence for identification of chromosomal locus and the gene mutation characterized by high penetrance. Among common neuropsychiatric diseases, the approach was successful in the identification of genes for familial early onset Alzheimer’s disease (AD) carrying autosomal-dominant mutations in PSEN1, PSEN2 genes (Rogaev et al., 1995; Sherrington et al., 1995). Another genetic approach, genome-wide association (GWA), or direct genetic association analysis in candidate-genes, employs relatively common genetic variations across the genome or in selected genes for comparative analysis in case–control groups. GWA has been applied for hundreds phenotype traits, including neuropsychiatric conditions (Wellcome Trust Case Control Consortium, 2007; Donnelly, 2008). The genetic association methodology generated much more controversial data in interpretation (Pearson and Manolio, 2008). In most cases, the GWA studies identified only a minor proportion of genetic contribution to common illnesses with little medical diagnostic value. Nonetheless, the genetic association method revealed some convincing data. For example, as confirmed in numerous studies, ApoE ε4 allelic isoform is a common risk factor for AD in ethnic groups of Caucasian and Asian origin. Analysis of two large Caucasian cohorts stratified by age showed that the ApoE-genotype dependent lifetime risks for AD are similar to those of Mendelian-disease genes with major risk effect (Genin et al., 2011). Unlike AD, however, no mutant gene convincingly inherited as a Mendelian trait has yet been described for schizophrenia and affective disorders, despite the fact that many mutigenerational pedigrees are available. GWA employs relatively common single-nucleotide polymorphisms (SNPs) across the genome. The recently emerged concept that rare genetic variations, rather than common population variations, underlie common diseases challenges the standard genetic association approach in neuropsychiatric genetics.
Rare and Non-Coding Variations
Indeed, rare copy-number variations (CNVs) in a few chromosomal loci have been found in a small proportion of schizophrenia samples (Karayiorgou et al., 1995; International Schizophrenia Consortium, 2008; Mulle et al., 2010; Vacic et al., 2011). Among numerous genes implicated for schizophrenia, a few candidate-genes, such as DISC or NPAS3, with disrupted structure due to chromosomal translocations in single patients, can be noted (Millar et al., 2000; Kamnasaran et al., 2003).
Emerging new generation sequencing technology, termed deep or massively parallel sequencing (MPS), may transform the field. Direct sequencing of all genes, or preferably whole-genome sequences, will provide complete genetic information of the patient. However, from our current knowledge on population genetic variability, we can expect >3.5 million SNPs and >1,000 CNVs of genomic sequences per individual in comparison to reference genome sequence. Thus, excluding genetic “background” and identification of disease-related variations in the individual genomes remains a challenge in study of behavior diseases.
Currently available MPS platforms (e.g., Illumina or Complete Genomics) produce biases to different types of errors in nucleotide sequences (Lam et al., 2011). Another challenge is a biological interpretation of genetic variations found via direct genome sequencing analysis. Application of high-throughput functional assays in cellular models and in animals testing the biological significance of suspected mutations or polymorphisms in selected genes may be one possible solution.
de novo Mutations
Another recently proposed approach is the identification of genes via analysis of de novo mutations in exome. Despite reduced reproductive fitness, the rate of incidence for schizophrenia and autism is relatively stable (0.4–1%) in worldwide populations (Bassett et al., 1996; Saha et al., 2005; Center for Disease Control and Prevention, 2009). The Hardy–Weinberg concept could explain, in part, the long-term maintenance of recessive mutant alleles in populations. Another explanation is that spontaneous de novo mutation process contributes to the relatively frequent population occurrence of schizophrenia and autism. In this event, we may anticipate the increased risk for disease with increased parental age due to age-dependent increased mutation frequency in gametogenesis. Indeed, the effect of paternal (but not maternal) age on the risk for schizophrenia has been reported. Meta-analysis studies suggest that there is an increased relative risk for schizophrenia from 1.84 to 4.62 in children of fathers with an older age of fatherhood (Hubert et al., 2011). Instead of searching for large families and pedigrees, the small nuclear families with one proband and no other schizophrenia cases in the family history are a subject of interest. Recently, putative causative de novo mutations were found in an analysis of genomic exomes of 4 of 20 analyzed probands with autism (O’Roak et al., 2011). Two studies employed sequencing of exomes for schizophrenia. In summary, in 35 probands from family trios, several different genes bearing de novo mutations were found. With one exception for a DGCR2 gene located on schizophrenia-associated 22q11.2 microdeletion region, the mutant genes are not presented in putative schizophrenia-associated chromosomal loci or pathways predicted in previous studies (Girard et al., 2011; Xu et al., 2011). There was no match between the genes with de novo mutations found in independent families. Thus, the pathogenic significance of the identified mutations for schizophrenic trait must be further verified.
Estimates made from preliminary analysis of parent–child genome sequences suggest a rate of de novo spontaneous germline mutation ∼1.1 × 10−8 bp for human haploid genome (Roach et al., 2010). It would correspond to ∼60–70 new mutations per diploid genome in each individual, which are non-inherited from the parents. Because only a small portion of the genome, <1%, is represented by exome, most of the spontaneous germline mutations must be located in non-coding genomic regions. Therefore, the complete genome sequencing in search of de novo mutations in neuropsychiatric diseases is a promising strategy. The search for mutations in sequences for enhancers, insulators, and repressors, which are often physically distant (e.g., >1 Mb) from the gene transcriptional start, along with 5′-and 3′-UTRs (untranslated gene regions) and promoter gene regions must be analyzed.
Epigenomics
Unlike genetics, epigenomic variations are generally reversible alterations that can be identified by analysis of the state of DNA methylation or histone modifications specific for active and non-active chromatin (Park, 2008).
On one hand, the genomic regions which are targets for the epigenomic regulations are under pressure of negative selection. These regions are characterized by a relative deficiency in SNPs (Tolstorukov et al., 2011). On the other hand, the mutations in these regions, if they occur, may directly affect the chromatin structure or DNA methylation, thereby affecting the capacity of these regions for either epigenomic stress-induced or cell specific (e.g., neuronal) regulations.
I believe it would be reasonable to hypothesize that the genetic–epigenomic interactions (GEI) play a significant role in common human illness, particularly in behavioral diseases, including major psychiatric diseases, such as schizophrenia, affective disorders, autism, or even in age-related dementias. Any SNP or structural genetic variation in cis-position located at a distant regulatory region or nearby gene can alter the constitutive or tissue-specific state of active chromatin and regulation of the gene (Figure 1). Certain epigenetic mutations can be silent, but will manifest due to programmed epigenomic transformations during development, the aging process, or triggered under specific conditions, e.g., hypothalamus – axilatory mediated stress conditions, or exposure to infection or chemical compounds. We could speculate that the silent variations affecting the epigenomic state play a role in some psychiatric disorders with reversible clinical manifestation (schizophrenia and bipolar disorder) or stress-induced and age-related diseases.
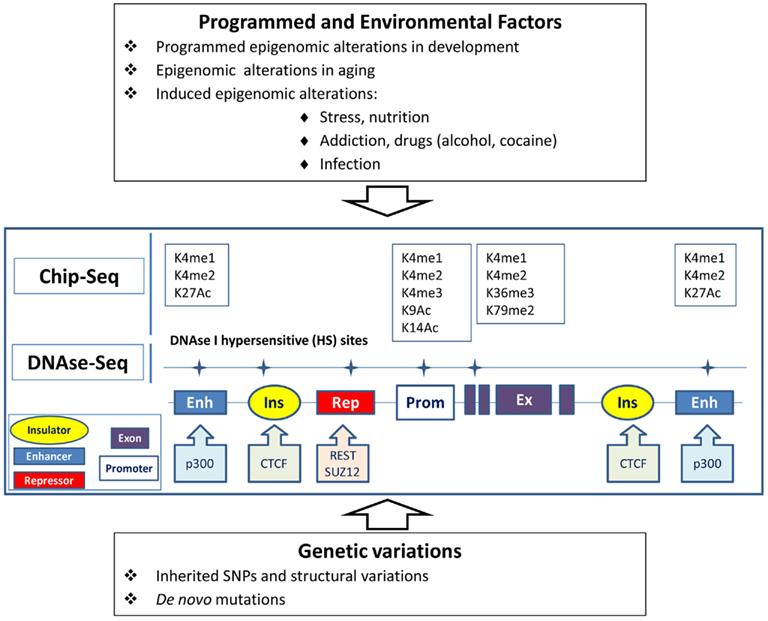
Figure 1. Epigenomic and genetic interactions in regulation of chromatin and gene activity underlying behavior traits. Genes can be regulated via genomic elements (enhancers, repressors, insulators) in tissue-specific manner. Epigenomic modification of chromatin in brain cells can be affected by programmed transformation, environmental factors, and genetic variations in the regulatory regions. Alteration of the transcription of the gene can also be caused by mutation in the regulatory elements without obvious changes in chromatin. Open or active chromatin structures are identified by the mapping of DNAse-hypersensitive sites and by Chip-seq technology, which tracks the genomic regions forming complexes with differentially modified histones (K) or the sequences binding with transcription regulatory proteins.
Despite the fact that the genetic role of schizophrenia is well-established, and that multiple informative schizophrenia families have been available for a long time, there is, as yet, no robust evidence for mutations in genes altering the protein structure in any significant number of schizophrenia cases as observed, e.g., in AD. Classical twin analysis showed that the contribution of non-genetic factors to schizophrenia is at least 50%. The number of autism spectrum disorder (ASD) cases continues to increase. Currently, ASD is diagnosed in 1 of 110 children (Center for Disease Control and Prevention, 2009). The cause of the rising rate for ASD is unknown. Recent re-evaluation of large cohorts of twins also demonstrated that there is a significant role of non-genetic factors in ASDs, and up to 37–38% of genetic factors in contrast to the previous conception of an ∼90% heritability in autism. A surprisingly significant role of shared twin environment, evidenced by high dizygotic twin concordance, was observed (Hallmayer et al., 2011). The hypothesis of GEI seems to be very relevant to ASD and schizophrenia.
Modified genomic DNA and chromatin complexes can be extracted from neuronal cells separately from glial cell populations from postmortem brain specimens (Matevossian and Akbarian, 2008). We can attempt to determine whether structural or single-nucleotide variations in individual genomes (genetic variations) correlate with individual variations in DNA methylation or methylation/acetylation histone modifications (epigenomic variations) in the same loci in neuronal cells. Genetic variations can be identified by whole-genome MPS re-sequencing. Epigenomic variations in the same individual genomes can be detected by MPS using rapidly progressing methodologies for analysis of DNA methylation with single-nucleotide resolution, DNAse-I hypersensitive sites, or Chip-seq data for transcriptional start sites or transcriptional regulatory elements. For example, Chip-seq can track transcriptional start sites via detection of sites for histone H3 trimethylated at lysine 4 (H3K4me3) and other transcriptional regulatory elements including enhancers enriched with histone H3 acetylated at lysine 27 (H3K27Ac) and histone H3 monomethylated at lysine 4 (H3K4me1). DNAse-I hypersensitive sites (at least ∼1–2% of genome) mark open or active chromatins associated with majority of regulatory and transcriptional start sites.
Beyond neuropsychiatric illnesses, I suggest that GEIs may also underlie changes in non-pathogenic behavioral traits and that the interplay between genetic variations and epigenomic modifications could be identified through the study of non-conventional animal models. Genomic sequencing, coupled with epigenomic studies, provides perspective in the identification of alterations in genome correlated with rapid behavior changes under certain selection process in rodents (e.g., in rats) or, in follow-up artificial selection in domesticated animals. Rapid changes in behavior, from native aggressive defending reaction to tolerant or even to “the man’s best friend” behavior in Canidae species, can be achieved in just a few generations, as demonstrated in the domestication experiments of silver foxes selected for tameability (Spady and Ostrander, 2008; Trut et al., 2009; Parker et al., 2010). The patterns of genetic alterations underlying changes in this behavior paradigm have yet to be identified. Perhaps it is not an exaggeration to speculate that elucidation of such a mechanism may also contribute to understanding the evolution of normal and abnormal social behavior in humans, and even of our own tolerance or intolerance of each other.
Acknowledgments
Supported, in part, by NIH/NINDS NS045854, NARSAD Distinguished Investigator Award, NIH/NIA AG029360, EU FP7 HEALTH-RF (ADAMS project), Rostok Group. I would like to thank A. Grigorenko for her assistance with figure preparation.
References
Bassett, A. S., Bury, A., Hodgkinson, K. A., and Honer, W. G. (1996). Reproductive fitness in familial schizophrenia. Schizophr. Res. 21, 151–160.
Botstein, D., and Risch, N. (2003). Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat. Genet. 33(Suppl.), 228–237.
Center for Disease Control Prevention. (2009). Autism and Developmental Disabilities Monitoring (ADDM) Network. Available at: http://www.cdc.gov/ncbddd/autism/addm.html
Donnelly, P. (2008). Progress and challenges in genome-wide association studies in humans. Nature 456, 728–731.
Genin, E., Hannequin, D., Wallon, D., Sleegers, K., Hiltunen, M., Combarros, O., Bullido, M. J., Engelborghs, S., De, D. P., Berr, C., Pasquier, F., Dubois, B., Tognoni, G., Fievet, N., Brouwers, N., Bettens, K., Arosio, B., Coto, E., Del, Z. M., Mateo, I., Epelbaum, J., Frank-Garcia, A., Helisalmi, S., Porcellini, E., Pilotto, A., Forti, P., Ferri, R., Scarpini, E., Siciliano, G., Solfrizzi, V., Sorbi, S., Spalletta, G., Valdivieso, F., Vepsalainen, S., Alvarez, V., Bosco, P., Mancuso, M., Panza, F., Nacmias, B., Bossu, P., Hanon, O., Piccardi, P., Annoni, G., Seripa, D., Galimberti, D., Licastro, F., Soininen, H., Dartigues, J. F., Kamboh, M. I., Van, B. C., Lambert, J. C., Amouyel, P., and Campion, D. (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907.
Girard, S. L., Gauthier, J., Noreau, A., Xiong, L., Zhou, S., Jouan, L., Dionne-Laporte, A., Spiegelman, D., Henrion, E., Diallo, O., Thibodeau, P., Bachand, I., Bao, J. Y., Tong, A. H., Lin, C. H., Millet, B., Jaafari, N., Joober, R., Dion, P. A., Lok, S., Krebs, M. O., and Rouleau, G. A. (2011). Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863.
Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., Miller, J., Fedele, A., Collins, J., Smith, K., Lotspeich, L., Croen, L. A., Ozonoff, S., Lajonchere, C., Grether, J. K., and Risch, N. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68, 1095–1102.
Hubert, A., Szoke, A., Leboyer, M., and Schurhoff, F. (2011). Influence of paternal age in schizophrenia. Encephale 37, 199–206.
International Schizophrenia Consortium. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241.
Kamnasaran, D., Muir, W. J., Ferguson-Smith, M. A., and Cox, D. W. (2003). Disruption of the neuronal PAS3 gene in a family affected with schizophrenia. J. Med. Genet. 40, 325–332.
Karayiorgou, M., Morris, M. A., Morrow, B., Shprintzen, R. J., Goldberg, R., Borrow, J., Gos, A., Nestadt, G., Wolyniec, P. S., and Lasseter, V. K. (1995). Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc. Natl. Acad. Sci. U.S.A. 92, 7612–7616.
Lam, H. Y., Clark, M. J., Chen, R., Chen, R., Natsoulis, G., O’Huallachain, M., Dewey, F. E., Habegger, L., Ashley, E. A., Gerstein, M. B., Butte, A. J., Ji, H. P., and Snyder, M. (2011). Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 30, 78–82.
Matevossian, A., and Akbarian, S. (2008). Neuronal nuclei isolation from human postmortem brain tissue. J.Vis.Exp. 20, e914.
McKusick, V. A. (2007). Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 80, 588–604.
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., Devon, R. S., St Clair, D. M., Muir, W. J., Blackwood, D. H., and Porteous, D. J. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423.
Mulle, J. G., Dodd, A. F., McGrath, J. A., Wolyniec, P. S., Mitchell, A. A., Shetty, A. C., Sobreira, N. L., Valle, D., Rudd, M. K., Satten, G., Cutler, D. J., Pulver, A. E., and Warren, S. T. (2010). Microdeletions of 3q29 confer high risk for schizophrenia. Am. J. Hum. Genet. 87, 229–236.
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., Karakoc, E., Mackenzie, A. P., Ng, S. B., Baker, C., Rieder, M. J., Nickerson, D. A., Bernier, R., Fisher, S. E., Shendure, J., and Eichler, E. E. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589.
Parker, H. G., Shearin, A. L., and Ostrander, E. A. (2010). Man’s best friend becomes biology’s best in show: genome analyses in the domestic dog. Annu. Rev. Genet. 44, 309–336.
Pearson, T. A., and Manolio, T. A. (2008). How to interpret a genome-wide association study. JAMA 299, 1335–1344.
Roach, J. C., Glusman, G., Smit, A. F., Huff, C. D., Hubley, R., Shannon, P. T., Rowen, L., Pant, K. P., Goodman, N., Bamshad, M., Shendure, J., Drmanac, R., Jorde, L. B., Hood, L., and Galas, D. J. (2010). Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328, 636–639.
Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K., Tsuda, T., Mar, L., Sorbi, S., Nacmias, B., Piacentini, S., Amaducci, L., Chumakov, I., Cohen, D., Lannfelt, L., Fraser, P. E., Rommens, J. M., and St George-Hyslop, P. H. (1995). Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778.
Saha, S., Chant, D., Welham, J., and McGrath, J. (2005). A systematic review of the prevalence of schizophrenia. PLoS Med. 2, e141. doi: 10.1371/journal.pmed.0020141
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J. F., Bruni, A. C., Montesi, M. P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sanseau, P., Polinsky, R. J., Wasco, W., Da Silva, H. A., Haines, J. L., Perkicak-Vance, M. A., Tanzi, R. E., Roses, A. D., Fraser, P. E., Rommens, J. M., and St George-Hyslop, P. H. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760.
Spady, T. C., and Ostrander, E. A. (2008). Canine behavioral genetics: pointing out the phenotypes and herding up the genes. Am. J. Hum. Genet. 82, 10–18.
Tolstorukov, M. Y., Volfovsky, N., Stephens, R. M., and Park, P. J. (2011). Impact of chromatin structure on sequence variability in the human genome. Nat. Struct. Mol. Biol. 18, 510–515.
Trut, L., Oskina, I., and Kharlamova, A. (2009). Animal evolution during domestication: the domesticated fox as a model. Bioessays 31, 349–360.
Vacic, V., McCarthy, S., Malhotra, D., Murray, F., Chou, H. H., Peoples, A., Makarov, V., Yoon, S., Bhandari, A., Corominas, R., Iakoucheva, L. M., Krastoshevsky, O., Krause, V., Larach-Walters, V., Welsh, D. K., Craig, D., Kelsoe, J. R., Gershon, E. S., Leal, S. M., Dell, A. M., Morris, D. W., Gill, M., Corvin, A., Insel, P. A., McClellan, J., King, M. C., Karayiorgou, M., Levy, D. L., Delisi, L. E., and Sebat, J. (2011). Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471, 499–503.
Wellcome Trust Case Control Consortium. (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678.
Citation: Rogaev EI (2012) Genomics of behavioral diseases. Front. Gene. 3:45. doi: 10.3389/fgene.2012.00045
Received: 02 February 2012; Accepted: 12 March 2012;
Published online: 02 April 2012.
Copyright: © 2012 Rogaev. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence:ZXZnZW55LnJvZ2FldkB1bWFzc21lZC5lZHU=