
- 1 Department of Psychiatry, The University of Iowa, Iowa City, IA, USA
- 2 Neuroscience and Genetics Programs, The University of Iowa, Iowa City, IA, USA
- 3 Department of Psychological and Brain Sciences, Dartmouth College, Hanover, NH, USA
- 4 Center for Family Research, The University of Georgia, Athens, GA, USA
Chronic alcohol intake is associated with a wide variety of adverse health outcomes including depression, diabetes, and heart disease. Unfortunately, the molecular mechanisms through which these effects are conveyed are not clearly understood. To examine the potential role of epigenetic factors in this process, we examined the relationship of recent alcohol intake to genome wide methylation patterns using the Illumina 450 Methylation Bead Chip and lymphoblast DNA derived from 165 female subjects participating in the Iowa Adoption Studies. We found that the pattern of alcohol use over the 6-months immediately prior to phlebotomy was associated with, severity-dependent changes in the degree of genome wide methylation that preferentially hypermethylate the central portion of CpG islands with methylation at cg05600126, a probe in ABR, and the 5′ untranslated region of BLCAP attaining genome wide significance in two point and sliding window analyses of probe methylation data, respectively. We conclude that recent alcohol use is associated with widespread changes in DNA methylation in women and that further study to confirm these findings and determine their relationship to somatic function are in order.
Introduction
Together, alcohol use and dependence affect 8% of the adult United States each year and cause over 200 billion dollars of economic damage annually (Harwood, 2000; Grant et al., 2004). The mechanism(s) through which alcohol exerts this toll varies. During acute intoxication, much of the economic damage and personal injury results from the increased rate of accidental injury (Harwood, 2000). But after returning to sobriety, the risk for further damage from accidental injury markedly diminishes. However, in the case of the sustained heavy use of alcohol, the risk for increased morbidity does not remit after return to sobriety and the individual remains at increased risk for a large number of medical conditions including hypertension, heart disease, and impaired executive function in the absence of acute intoxication (Harwood, 2000; Sesso et al., 2008). At the microscopic level, this increased risk can be directly linked to adverse impact on tissue and organ damage (Pace et al., 2009). However, at the molecular level, the direct effects of long term alcohol use seem more complex with chronic changes in a number of biochemical pathways being noted (Köhnke, 2008).
Some of these cellular changes may be legacies of altered protein folding and trafficking bequeathed to the cell from periods of intoxication (Esteban-Pretel et al., 2011). However, most cellular proteins have limited lifetimes before they are intracellularly recycled. Hence, they are unlikely to be directly responsible for some of the chronic dysfunction seen in cells prepared from abstinent alcoholics (Burim et al., 2004; Welsh et al., 2011). Instead, some of these alterations may result from alcohol induced changes in genomic tone, which is defined as the stable transcriptional repertoire of a cell (Alter and Hen, 2009).
The factors that control the “genomic tone” or transcriptional repertoire of the given cell are diverse but can be generally categorized as genetic variation, tissue specific transcriptional activators/repressors, and epigenetic factors. Conceivably, chronic alcohol use could affect the type and distribution of both transcriptional and epigenetic factors thus changing the genomic tone of the given cell. Unfortunately, systematic methods for assessing tissue specific transcription factors are not commonly available. In contrast, recent advances in DNA methylation assessment technologies have made genome wide assessment of DNA methylation more accessible. Using these technologies, using both lymphoblast and primary tissues, we have recently demonstrated that smoking is associated with genome wide changes in DNA methylation and that those smoking associated changes are coordinated across tissues are cell preparations including lymphoblast, lymphocytes, and pulmonary macrophages (Monick et al., 2012).
These advancements are particularly welcome because in prior work using more restricted approaches, we and others have presented evidence that alterations in DNA methylation may be in part responsible for altered genomic tone observed in peripheral blood cells from subjects who chronically use alcohol (Bonsch et al., 2005; Bleich et al., 2006; Philibert et al., 2008; Hillemacher et al., 2009a). However, these studies were limited by the low number of genes and the limited number of subjects surveyed. In this communication, using the Illumina HumanMethylation450 BeadChip, which interrogates over 485,000 CpG residues, we examine the relationship between alcohol consumption and degree of DNA methylation in lymphoblast DNA prepared from 165 female subjects from the Iowa Adoption Studies, the largest case and control adoption studies of substance use in the world (Philibert, 2006).
Materials and Methods
The protocols and procedures used in the Iowa Adoptions Studies (IAS) have been described in detail elsewhere (Yates et al., 1998). In brief, the IAS is a case and control adoption study of the effects of genetic, environmental, and gene–environment interactions in the etiology of substance use and antisocial personality. The sample is a high risk cohort. Half of the 940 adoptees in the original study had at least one biological parent with a history of severe antisocial personality, alcoholism, or depression while the other half of the subjects had no biological history of illness. After birth, they were placed in adoptive homes without evidence of bias with respect to biological birth status.
The data used in the current study is derived from interviews with the Semi-Structured Interview for the Assessment of the Genetics of Alcoholism, Version II (Bucholz et al., 1994), during each of the last two waves of the IAS study (1999–2004 and 2005–2009). Using this data, subjects were classified on the basis of the frequency of their alcohol use in the past 6 months prior to assessment into four categories: (1) abstinent (no use in the past 6 months); (2) mild users (use of alcohol in between 1 and 8 weeks in the past 6 months); (3) moderate users (use of alcohol in between 9 and 25 weeks in the past 6 months); and (4) heavy users (alcohol use in every week in the past 6 months). The lymphoblast DNA was derived by Epstein Barr virus mediated transformation (Caputo et al., 1991) of lymphocytes obtained from blood donated by 165 female subjects during the last wave of the study.
The lymphoblast DNA used in this study was prepared from growth-entrained lymphoblast cell lines using our standard procedures (Philibert et al., 2008). In brief, on the day before DNA preparation, one-half of the cell media for each culture flask was exchanged. DNA was then prepared from the cell lines 24 h later using cold protein precipitation (Lahiri and Schnabel, 1993). After quantification and purity assessment using a Nanodrop (Thermo Scientific, USA) spectrophotometer, DNA was stored at −20°C and RNA was −80°C until use.
Genome wide DNA methylation of the DNA was assessed using the Illumina HumanMethylation450 BeadChip under contract by the University of Minnesota Genome Center using the protocol specified by the manufacturer and the contractor. The resulting microarray data were inspected for complete bisulfite conversion of the DNA, and average beta values (i.e., average methylation) for each CpG residue were determined using the GenomeStudio V2009.2; Methylation module Version 1.5.5., Version 3.2 (Illumina, San Diego). The resulting beta values were exported into Microsoft Excel and JMP (SAS Institute, USA) for data analysis. The HumanMethylation450 BeadChip contains 485,577 probes that recognize at least 20216 transcripts, potential transcripts, or isolated CpG islands. With respect to this sample, >99.7% of the 485,577 probes yielded statistically reliable data.
The methylation status of the serotonin transporter (SLC6A4) promoter region was previously assessed for 163 of the 165 samples using MALDI-TOF mass spectroscopy by Sequenom (San Diego, CA, USA) as described previously (Philibert et al., 2008, 2010). Using the sequence annotation files from both the current and the prior studies, we identified CpG residues that were assessed using both technologies. The methylation values for each residue were compared using Least Squares regression (Fleiss, 1981).
After logarithmic conversion, data were inspected for outliers then the initial data analyses were conducted using genome wide t-tests. Subsequently, beta values for each of the probes were aligned according to their physical location and the data re-analyzed using paired t-tests over a 11-probe sliding window in order to more adroitly capture methylation signatures over larger regions (Farthing et al., 2008; Dindot et al., 2009). All genome wide comparisons were corrected for multiple comparisons using the method of Benjamini and Hochberg (1995). For select loci, data were analyzed with respect to alcohol use status using ANOVA (Fleiss, 1981).
Pathway analysis of differentially methylated genes was conducted using GoMiner™ using default settings (Zeeberg et al., 2003). All values reported include nominal and FDR corrected values.
Results
The demographic and clinical characteristics of the 165 female subjects is shown in Table 1. Overall, the subjects were largely white and tended to be their mid-to-late 40s. Based on the analytical model and the distribution of daily drinking patterns among the subjects over the 6 months prior to phlebotomy, we divided the subjects into four groups. Consistent with enrichment of the sample for the diathesis of substance use, the majority of these 165 subjects in the study reported the use of alcohol in the past 6 months with the 28 subjects in the “heavy” use group reporting alcohol use every week for the past 26 weeks while the 50 “moderate” drinkers and the 47 “mild” drinkers reported drinking in 9–25 weeks, and 1–8 weeks in the past 26 weeks, respectively. Their current drinking pattern was reflective of their lifetime history of drinking alcohol. Only 5 of the 40 individuals who reported recent abstinence also reported one or more symptoms of lifetime alcohol dependence. In contrast, 15 of 28 of the heavy drinking reported one or more symptoms (p < 0.01 as compared to the abstinent group) with four of them meeting criteria for a lifetime diagnosis of alcohol dependence (three or more symptoms). Approximately 50% of the subjects also reported a past history of smoking with 27% continuing to smoke at the time of phlebotomy. However, despite the strong epidemiological associations of smoking and drinking behaviors (John et al., 2003), in this cohort of 165 subjects, there were no significant differences in the rates of smoking between the three groups.

Table 1. Clinical characteristics of the 165 female Iowa Adoptions Studies probands.
In our initial analyses, we contrasted the methylation values for the 40 abstinent individuals with the values for the 47 mild, 50 moderate, and 28 heavy drinkers using genome wide t-tests. The results of those analyses are shown in Table 2. The overall degree methylation was less in the abstinent group (44.26%) than in mild (44.64%), moderate (44.65%), or heavy (44.62%) drinking groups (all comparisons vs abstinent p < 0.0001). Despite this overall difference, as the table indicates, although some of the values show strong consistency across several partially independent comparisons, by themselves none of the comparisons between individual groups (e.g., heavy drinker vs mild) are statistically significant after genome wide comparison (best p-value after correction is p < 0.25) to values for the abstinent group. However, when the moderate and heavy drinkers are pooled together and compared to the abstinent group, the comparison at cg05600126, a probe in ABR, a gene known to be involved in vestibular function (Kaartinen et al., 2002), reaches genome wide significance after genome wide correction (p < 0.05; Table 3) with several other probes nearly reaching significance.
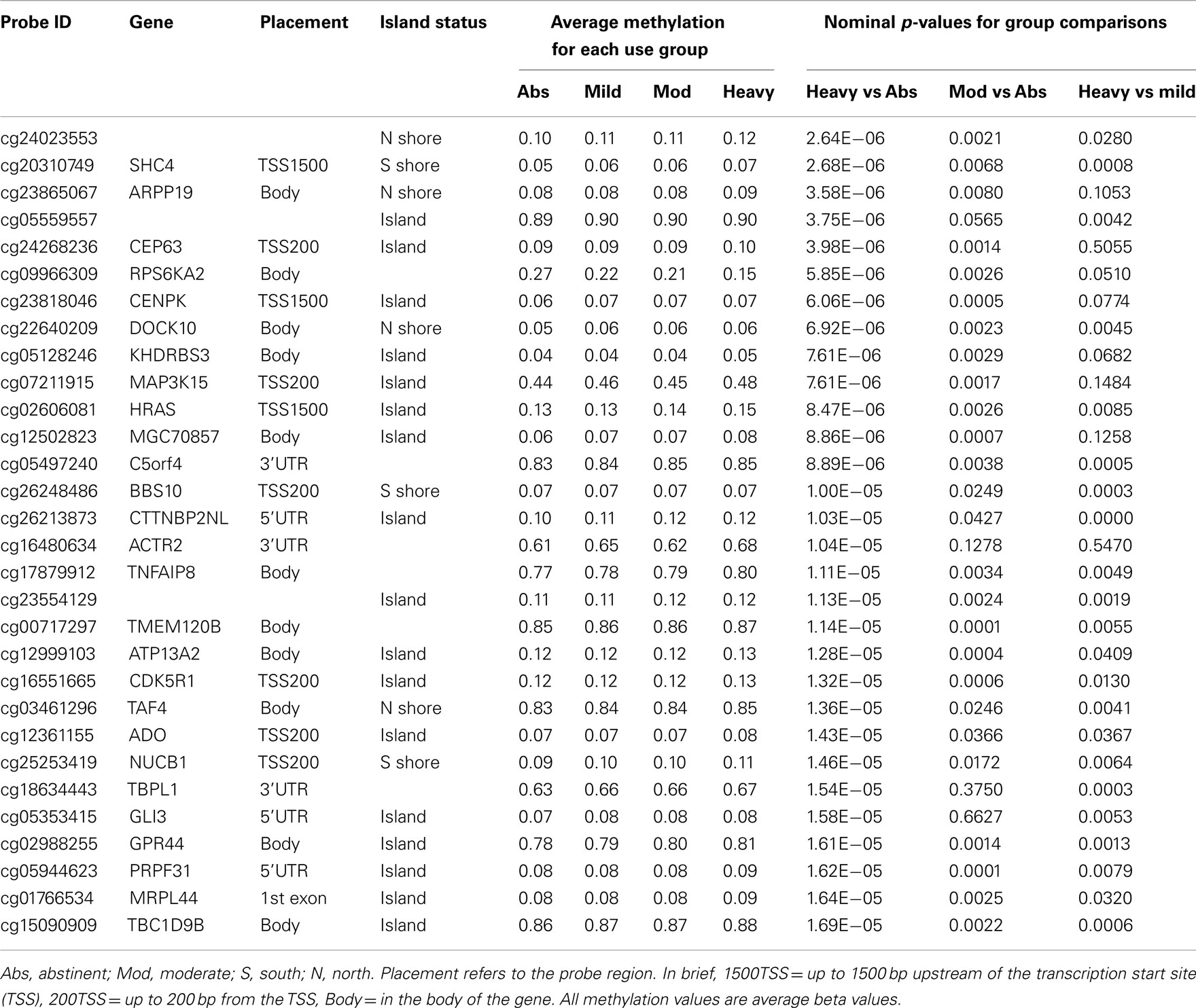
Table 2. The top 30 most significantly associated probes for individual alcohol group comparison.
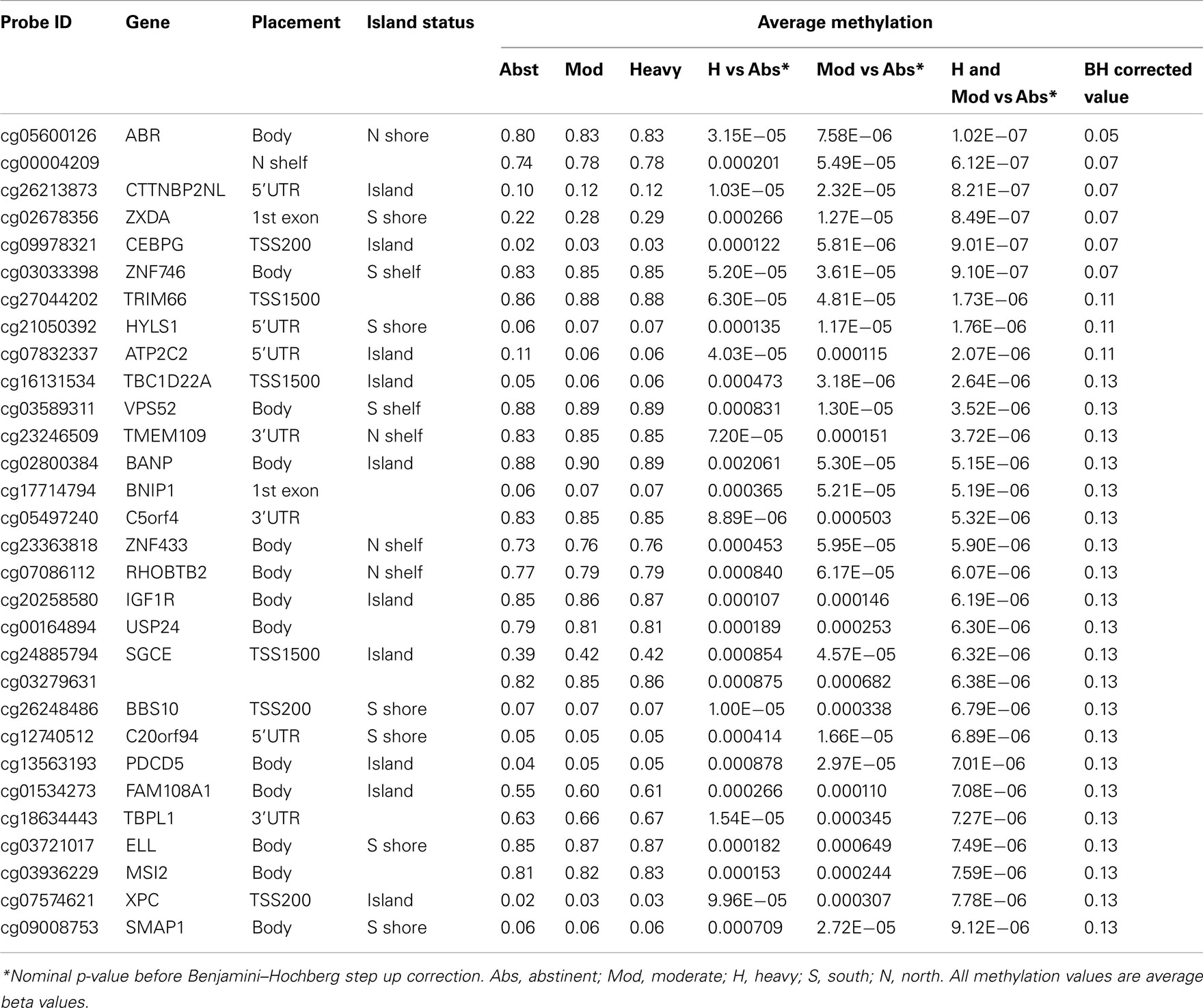
Table 3. The top 30 most significantly associated probes in the abstinent vs pooled moderate and heavy drinkers analysis.
There appears to be effect of the category of drinking frequency on both the strength of the overall comparisons (Table 4). Overall, 1711 of the 485,577 probes on the array were nominally significant at the p < 0.001 level in the heavy vs abstinent group comparison (expected value; 486 probes at p < 0.001). This number diminishes to 390 probes when comparing the moderate to the abstinent and 128 probes at the p < 0.001 level when comparing the mild drinkers to the abstinent drinkers despite the fact that the heavy drinker group was the smallest of the three groups.
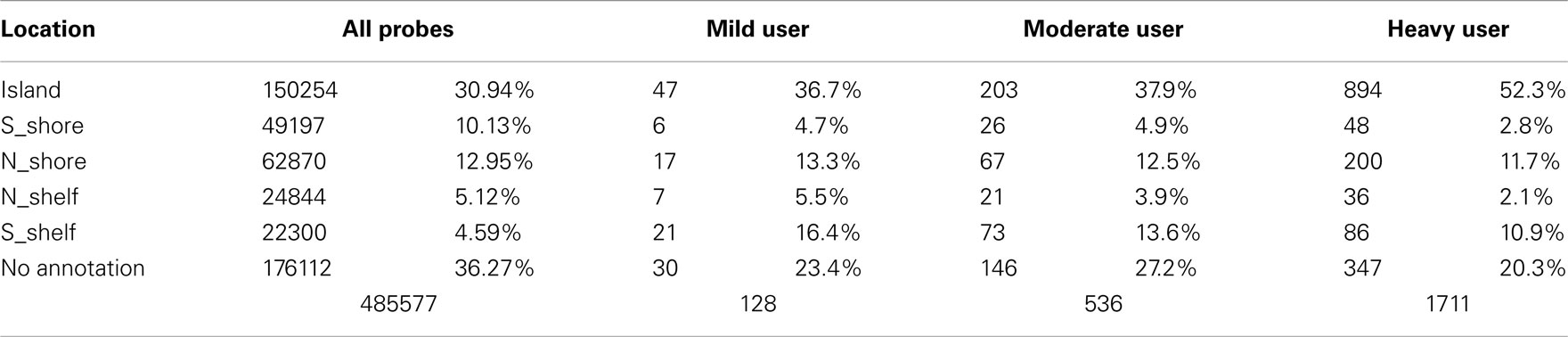
Table 4. Distribution of CpG values with respect to island status and extent of alcohol use.
This effect is also reflected in the distribution of the differentially methylated probes with respect to their island status. In previous work, Doi et al. (2009) showed that changes in cell fate preferentially affected CpG methylation based on the location of the residue with respect to their location in the CpG island. To examine whether this was happening with respect to alcohol use, we examined the location of the nominally significantly differentially methylated probes (at the p < 0.001 level) using the information contained in the Illumina file annotations. As Table 4 demonstrates, alcohol seems to preferentially affect the probes found in the center of the CpG islands with the proportion of all differentially methylated probes which localized to the center of the islands rising reaching to 52% (p < 0.0001) in the heavy drinking group as compared to the expected distribution of 30.9% localizing to the island.
Next, using a sliding 11-probe window, we examined whether using information from adjacent probes would strengthen the findings with respect with to alcohol use. The effect on significance was profound with values for 19 regional comparisons reaching genome wide significance at the p < 0.01 level (Table 5). Not surprisingly, many of the regions are overlapping with the top four region comparisons all being found in BLCAP, a chromosome 6 gene with 121 probes localizing to it. Please see Supplementary Table 1 for a complete list of the genomic location and context of all probes.
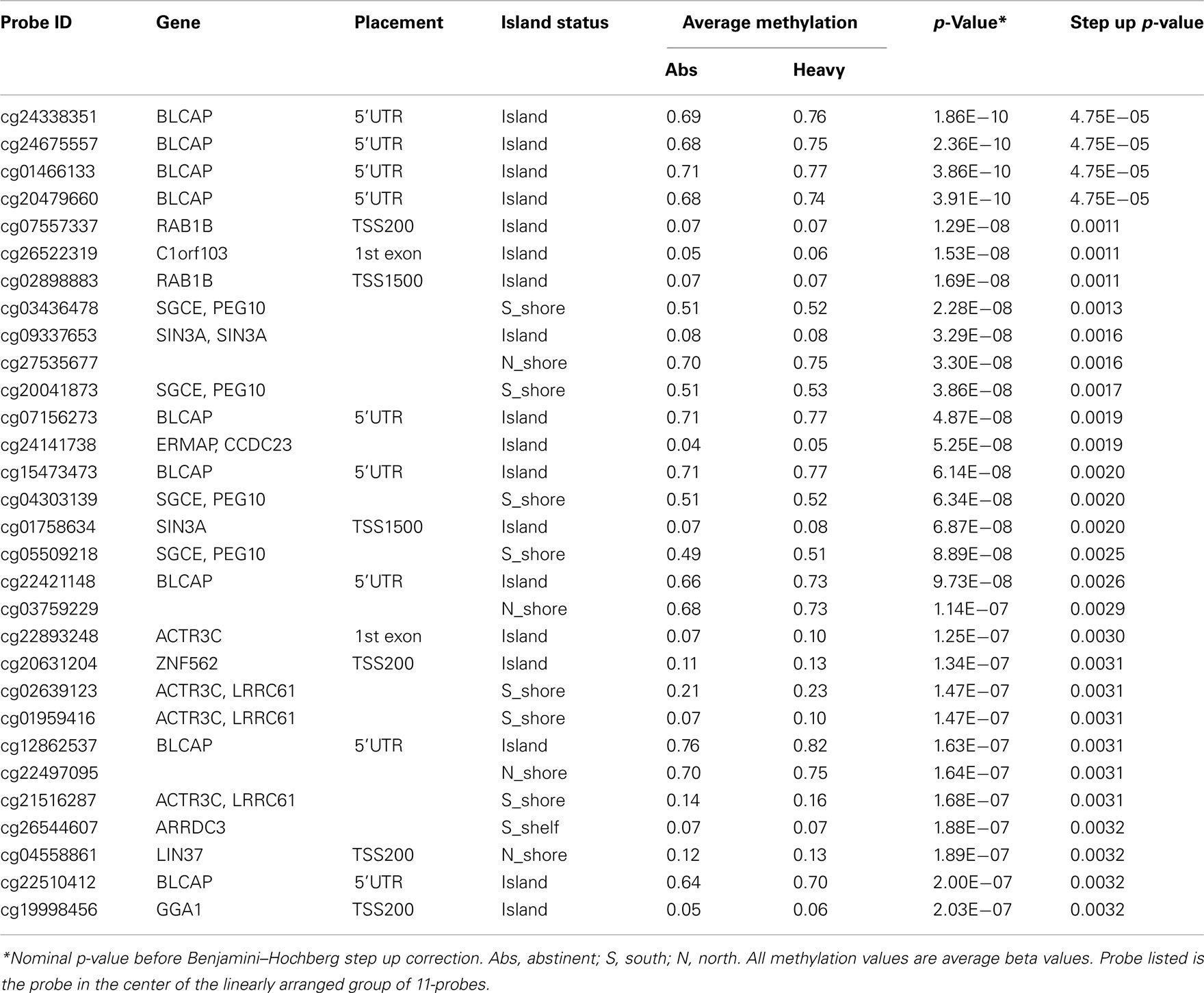
Table 5. The top 30 most significantly associated 11-probe regions.
Using the list of significantly differentiated unique genes (n = 24) from the sliding window comparison, we conducted pathway analyses using GoMiner. No significantly differentially methylated pathways were identified using this approach.
Finally, to determine whether our current array based measurements were valid, we compared the degree of methylation determined by the Illumina platform with the values determine previously for these 165 subjects at the serotonin transporter promoter associated CpG island (SLC6A4) using MALDI-TOF mass spectrophotometer. Overall, four CpG residues at this locus were surveyed by both approaches. At each CpG residue, the degree of methylation determined by each method was correlated with the average adjusted r2 equaling ∼0.34 which is similar to the findings by Bock et al. (2010) suggests that the current measurements are reliable.
Discussion
In summary, we present evidence that the pattern of alcohol use over the most recent 6 months is associated with widespread changes in the methylation of lymphoblast DNA derived from middle aged female subjects participating in the Iowa Adoption Studies. Limitations of the current findings include the fact that lymphoblasts are not primary human cells, the modest degree of differential methylation observed at any individual probe, the likelihood of tissue and cell dependent differences in methylation, and the likely confounding effect of prior alcohol use history on 6 month use history. Strengths of the manuscript include the high significant sliding window analyses, the internal consistency of the multiple comparisons, and the independent verification of methylation signatures at the SLC6A4 locus.
The dependency of differential DNA methylation on the frequency of drinking observed in the current study was to be expected. Depending on context, alcohol can be viewed as either as a drug or a potentially poisonous solvent. Under the latter viewpoint, the observed epigenetic changes described herein could then be explained as the gradated cellular responses to a exogenous toxin. However, before assuming that viewpoint it is important to acknowledge that the primary alcohol use variable employed in the current study was number of weeks in the past 6 months in which the subject drank which is more a measure of time of exposure not total amount ingested. To a certain extent, our choice to employ this measure is because our diagnostic instrument, the SSAGA, readily provides this as a recent use metric. It may well be that the choice of a different total consumption based metric, such as the number of alcoholic beverages consumed in the past 2 weeks, may have produced more robust findings. However, because of the manner in which the alcohol use questions are asked in our version of this instrument, information for all subjects is not always available or directly comparable. Hence, it may well be that other approaches to quantifying recent alcohol consumption may produce more robust findings. But we feel that the current classification system which captures the pattern of use over an extended period of time may be equally effective and that further analyses using replicate data sets using more complex (e.g., factor analyses) may be the best way to more adroitly define which recent alcohol use measures are most correlated with DNA methylation changes.
The effect of alcohol use severity on the distribution of the differentially methylated probes with respect to CpG island status is intriguing. In previous work, Doi et al. (2009) demonstrated that DNA methylation changes associated with assumption of cell fate preferentially affected the less dense outer area of the CpG islands referred to, poetically, as the “shore.” In this survey of the effects of alcohol intake, we observed that overall that greater consumption of alcohol is associated with increased levels of genome wide methylation and that the changes in the most chronically exposed subjects preferentially affects the centers of these islands. Because two-thirds of all CpG islands in the genome are promoter associated and hypermethylation of promoter associated CpG islands is thought to silence gene transcription (Suzuki and Bird, 2008), it is tempting to speculate that this increased methylation observed at these islands is associated with decreased gene expression at these loci. Indeed, in subset of these same cell lines, we have demonstrated that at the genome wide level, there are significant effects of methylation on gene expression (Plume et al., 2012). Unfortunately, because the magnitude of many of these changes in methylation are relatively small and others have observed that the relationship between DNA methylation and gene expression may be complex and weak (De Bustos et al., 2009; Fan and Zhang, 2009; Pai et al., 2011), directly demonstrating that these changes have biological relevance at any given locus may be difficult. However, the finding by Rodd et al. (2008) that the expression of BLCAP, a gene region that is significantly hypermethylated in our study, was significantly decreased in the nucleus accumbens of ethanol treated rats is encouraging.
The directionality of the overall changes observed herein is consistent with prior findings. In our single point and sliding window analyses, almost of all of the top 30 most significantly differentially methylated probes or regions were more methylated in heavy alcohol use group. This is very consistent with prior finding by ourselves and others (Bonsch et al., 2005; Philibert et al., 2008; Hillemacher et al., 2009a,b). However, there are a number of exceptions to this rule in this study and we expect that a fuller understanding of differentially regulated pathways in alcoholism to include a rich tapestry of both up-regulated and down-regulated genes.
It is also important to note that despite the fact that alcohol and nicotine use are frequently co-morbid, there were no differences in the frequency of smoking between the four alcohol use groups and controlling for smoking had no effect on the outcomes of the current study. Taken in conjunction with the previous finding by Breitling et al. (2011) as well as our own preliminary analyses, these findings suggest that cigarette smoke and alcohol ingestion present unique toxicological challenges to cells that have distinct effects on methylation. However, these effects do not have to be shared exactly. In our examinations that compared the smoking associated methylation signatures of whole blood DNA, lymphoblast DNA, and alveolar macrophages, we have found significant differences in individual CpG residues across loci whose signatures were otherwise quite well coordinated (Philibert et al., 2010; Monick et al., 2012). Because lymphoblasts are immortalized cell lines whose epigenetic signatures are partly affected by EBV transformation (Rollins et al., 2010) and to a certain extent have documented certain intercellular differences, integrated studies of different cell tissue preparations taken from the same individual would be beneficial in the generation of a more global understanding of the effects of alcohol in the entire body.
A critical question that is not addressed by the current study is the longevity of the methylation signatures associated with chronic alcohol use. In prior studies of the MAOA locus, we have demonstrated that cessation of smoking has dramatic effects on CpG methylation (Philibert et al., 2010). Unfortunately, the number of abstinent or nearly abstinent subjects contained within the current study is too small to conduct meaningful tests at the most significant loci for these purposes. Furthermore, not all abstinent individuals in this study were abstinent for the same reasons. Some are abstinent secondary to personal choice while others in our study are abstinent secondary to medical or legal necessity. Controlling for those and other potential confounders such as diet and lifestyle issues in small samples such as this may be difficult.
Assuming that the current findings are replicated, particularly in primary lymphocytes, the some of the most critical questions to be addressed concern the relationship of differential DNA methylation to the overall genomic tone of the cell. DNA methylation is assumed to be intimately involved in regulation of genomic tone. Hence, will reversal of the DNA methylation changes restore normal genomic tone? This is an important question because cells isolated from alcoholics also have more structural changes such as shorter telomeres and manifest other signs of cellular senescence such as abnormal post-translational modifications of proteins. Will these indicators of cellular dysfunction similarly revert if the methylation patterns can be reversed through dietary or pharmacological means? If so, defining the methods through which to accomplish this process could have substantial impact in the rehabilitation of those suffering from the mental and physical ravages of alcoholism.
In summary, we report that recent chronic alcohol intake is associated with significant changes in CpG methylation, and in particular, increased hypermethylation of CpG islands. We suggest further studies to confirm and extend these findings using primary cells and convergent epigenetic approaches are indicated.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in this study was supported by DA015789 and MH080898 to Dr. Philibert Additional support for these studies was derived from the Center for Contextual Genetics and Prevention Science (Grant Number P30 DA027827, Gene H. Brody) funded by the National Institute on Drug Abuse. On behalf of Dr. Philibert, the University of Iowa has filed intellectual property rights claims with respect to the content of this manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Behavioral_and_Psychiatric_Genetics/10.3389/fgene.2012.00054/abstract
References
Alter, M. D., and Hen, R. (2009). Is there a genomic tone? Implications for understanding development, adaptation and treatment. Dev. Neurosci. 31, 351–357.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Bleich, S., Lenz, B., Ziegenbein, M., Beutler, S., Frieling, H., Kornhuber, J., and Bonsch, D. (2006). Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol. Clin. Exp. Res. 30, 587–591.
Bock, C., Tomazou, E. M., Brinkman, A. B., Muller, F., Simmer, F., Gu, H., Jager, N., Gnirke, A., Stunnenberg, H. G., and Meissner, A. (2010). Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat. Biotechnol. 28, 1106–1114.
Bonsch, D., Lenz, B., Kornhuber, J., and Bleich, S. (2005). DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 16, 167–170.
Breitling, L. P., Yang, R., Korn, B., Burwinkel, B., and Brenner, H. (2011). Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 88, 450–457.
Bucholz, K. K., Cadoret, R., Cloninger, C. R., Dinwiddie, S. H., Hesselbrock, V. M., Nurnberger, J. I. Jr., Reich, T., Schmidt, I., and Schuckit, M. A. (1994). A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J. Stud. Alcohol 55, 149–158.
Burim, R. V., Canalle, R., Takahashi, C. S., Tavares, D. C., Martinelli, A. D. L. C., and Sakamoto-Hojo, E. T. (2004). Clastogenic effect of ethanol in chronic and abstinent alcoholics. Mutat. Res. 560, 187–198.
Caputo, J., Thompson, A., Mcclintock, P., Reid, Y., and Hay, R. (1991). An Effective method for establishing human B lymphoblastic cell lines using Epstein Barr virus. Methods Cell Sci. 13, 39–44.
De Bustos, C., Ramos, E., Young, J., Tran, R., Menzel, U., Langford, C., Eichler, E., Hsu, L., Henikoff, S., Dumanski, J., and Trask, B. (2009). Tissue-specific variation in DNA methylation levels along human chromosome 1. Epigenetics Chromatin 2, 7.
Dindot, S. V., Person, R., Strivens, M., Garcia, R., and Beaudet, A. L. (2009). Epigenetic profiling at mouse imprinted gene clusters reveals novel epigenetic and genetic features at differentially methylated regions. Genome Res. 19, 1374–1383.
Doi, A., Park, I.-H., Wen, B., Murakami, P., Aryee, M. J., Irizarry, R., Herb, B., Ladd-Acosta, C., Rho, J., Loewer, S., Miller, J., Schlaeger, T., Daley, G. Q., and Feinberg, A. P. (2009). Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 41, 1350–1353.
Esteban-Pretel, G., Marín, M. P., Romero, A. M., Ponsoda, X., Ballestin, R., Canales, J. J., and Renau-Piqueras, J. (2011). Protein traffic is an intracellular target in alcohol toxicity. Pharmaceuticals 4, 741–757.
Fan, S., and Zhang, X. (2009). CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem. Biophys. Res. Commun. 383, 421–425.
Farthing, C. R., Ficz, G., Ng, R. K., Chan, C.-F., Andrews, S., Dean, W., Hemberger, M., and Reik, W. (2008). Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 4, e1000116. doi:10.1371/journal.pgen.1000116
Fleiss, J. L. (1981). Statistical Methods for Rates and Proportions. New York, NY: John Wiley & Sons Inc.
Grant, B. F., Dawson, D. A., Stinson, F. S., Chou, S. P., Dufour, M. C., and Pickering, R. P. (2004). The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991-1992 and 2001-2002. Drug Alcohol Depend. 74, 223–234.
Harwood, H. (2000). Updating Estimates of the Economic Costs of Alcohol Abuse in the United States. Rockville, MD: Department of Health and Human Services.
Hillemacher, T., Frieling, H., Hartl, T., Wilhelm, J., Kornhuber, J., and Bleich, S. (2009a). Promoter specific methylation of the dopamine transporter gene is altered in alcohol dependence and associated with craving. J. Psychiatr. Res. 43, 388–392.
Hillemacher, T., Frieling, H., Luber, K., Yazici, A., Muschler, M. A., Lenz, B., Wilhelm, J., Kornhuber, J., and Bleich, S. (2009b). Epigenetic regulation and gene expression of vasopressin and atrial natriuretic peptide in alcohol withdrawal. Psychoneuroendocrinology 34, 555–560.
John, U., Meyer, C., Rumpf, H.-J., Schumann, A., Thyrian, J. R., and Hapke, U. (2003). Strength of the relationship between tobacco smoking, nicotine dependence and the severity of alcohol dependence syndrome criteria in a population based sample. Alcohol Alcohol. 38, 606–612.
Kaartinen, V., Nagy, A., Gonzalez-Gomez, I., Groffen, J., and Heisterkamp, N. (2002). Vestibular dysgenesis in mice lacking Abr and Bcr Cdc42/RacGAPs. Dev. Dyn. 223, 517–525.
Köhnke, M. D. (2008). Approach to the genetics of alcoholism: a review based on pathophysiology. Biochem. Pharmacol. 75, 160–177.
Lahiri, D. K., and Schnabel, B. (1993). DNA isolation by a rapid method from human blood samples: effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem. Genet. 31, 321–328.
Monick, M. M., Beach, S. R., Plume, J. T., Sears, R., Gerrard, M., Brody, G. H., and Philibert, R. (2012). Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159, 141–151.
Pace, A., De Weerth, A., Berna, M., Hillbricht, K., Tsokos, M., Bläker, M., Pueschel, K., and Lohse, A. W. (2009). Pancreas and liver injury are associated in individuals with increased alcohol consumption. Clin. Gastroenterol. Hepatol. 7, 1241–1246.
Pai, A. A., Bell, J. T., Marioni, J. C., Pritchard, J. K., and Gilad, Y. (2011). A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 7, e1001316. doi:10.1371/journal.pgen.1001316
Philibert, R. (2006). Merging genetic and environmental effects in the Iowa Adoption Studies: focus on depression. Ann. Clin. Psychiatry 18, 219–222.
Philibert, R. A., Beach, S. R., Gunter, T. D., Brody, G. H., Madan, A., and Gerrard, M. (2010). The effect of smoking on MAOA promoter methylation in DNA prepared from lymphoblasts and whole blood. Am. J. Med. Genet. 153B, 619–628.
Philibert, R. A., Gunter, T. D., Beach, S. R., Brody, G. H., and Madan, A. (2008). MAOA methylation is associated with nicotine and alcohol dependence in women. Am. J. Med. Genet. 147B, 565–570.
Plume, J. M., Beach, S. R. H., Brody, G. H., and Philibert, R. A. (2012). A cross platform genome wide comparison of the relationship of promoter DNA methylation to gene expression. Front. Genet. 3:12. doi:10.3389/fgene.2012.00012
Rodd, Z. A., Kimpel, M. W., Edenberg, H. J., Bell, R. L., Strother, W. N., Mcclintick, J. N., Carr, L. G., Liang, T., and Mcbride, W. J. (2008). Differential gene expression in the nucleus accumbens with ethanol self-administration in inbred alcohol-preferring rats. Pharmacol. Biochem. Behav. 89, 481–498.
Rollins, B., Martin, M. V., Morgan, L., and Vawter, M. P. (2010). Analysis of whole genome biomarker expression in blood and brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 919–936.
Sesso, H. D., Cook, N. R., Buring, J. E., Manson, J. E., and Gaziano, J. M. (2008). Alcohol consumption and the risk of hypertension in women and men. Hypertension 51, 1080–1087.
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476.
Welsh, J. P., Han, V. Z., Rossi, D. J., Mohr, C., Odagiri, M., Daunais, J. B., and Grant, K. A. (2011). Bidirectional plasticity in the primate inferior olive induced by chronic ethanol intoxication and sustained abstinence. Proc. Natl. Acad. Sci. U.S.A. 108, 10314–10319.
Yates, W., Cadoret, R., and Troughton, E. (1998). “The Iowa Adoption Studies methods and results,” in On the Way to Individuality: Methodological Issues in Behavioral Genetics, eds M. Labuda and E. Grigorenko (Hauppauge, NY: Nova Science Publishers), 95–125.
Keywords: ketogenic diet, neuroplasticity, epilepsy, neurological disorders
Citation: Philibert RA, Plume JM, Gibbons FX, Brody GH and Beach SRH (2012) The impact of recent alcohol use on genome wide DNA methylation signatures. Front. Gene. 3:54. doi: 10.3389/fgene.2012.00054
Received: 02 February 2012; Accepted: 21 March 2012;
Published online: 10 April 2012.
Edited by:
John McGeary, Brown University, USAReviewed by:
Lekki C. Wood, University of Alabama at Birmingham, USAJonathan Covault, University of Connecticut School of Medicine, USA
Alexandre Todorov, Washington University School of Medicine, USA
Copyright: © 2012 Philibert, Plume, Gibbons, Brody and Beach. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Robert A. Philibert, Department of Psychiatry, The University of Iowa, Room 2-126 MEB Psychiatry Research/MEB, Iowa City, IA 52242-1000, USA. e-mail:cm9iZXJ0LXBoaWxpYmVydEB1aW93YS5lZHU=