 Catharine E. Krebs1
Catharine E. Krebs1 Coro Paisán-Ruiz1,2,3,4*
Coro Paisán-Ruiz1,2,3,4*- 1 Department of Neurology, Mount Sinai School of Medicine, New York, NY, USA
- 2 Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY, USA
- 3 Department of Psychiatry, Mount Sinai School of Medicine, New York, NY, USA
- 4 The Friedman Brain Institute, Mount Sinai School of Medicine, New York, NY, USA
New advances in genomic technology are being introduced at a greater speed and are revolutionizing the field of genetics for both complex and Mendelian diseases. For instance, during the past few years, genome-wide association studies (GWAS) have identified a large number of significant associations between genomic loci and movement disorders such as Parkinson’s disease and progressive supranuclear palsy. GWAS are carried out through the use of high-throughput SNP genotyping arrays, which are also used to perform linkage analyses in families previously considered statistically underpowered for genetic analyses. In inherited movement disorders, using this latter technology, it has repeatedly been shown that mutations in a single gene can lead to different phenotypes, while the same clinical entity can be caused by mutations in different genes. This is being highlighted with the use of next-generation sequencing technologies and leads to the search for genes or genetic modifiers that contribute to the phenotypic expression of movement disorders. Establishing an accurate genome–epigenome–phenotype relationship is becoming a major challenge in the post-genomic research that should be facilitated through the implementation of both functional and cellular analyses. In this review, we summarize the latest genetic discoveries made by the use of NGS technologies and purpose future directions and challenges to truly understand the pathophysiology of MDs.
Introduction
Movement disorders (MDs) are a group of diseases and syndromes affecting the ability to produce and control movement and are often accompanied by secondary clinical presentations such as seizures, cognitive deficits, autoimmune deficiencies, and psychiatric symptoms, among others, causing major difficulties in diagnosis and treatment. MDs comprise the following clinical conditions: ataxia, dystonia, essential tremor (ET), Huntington’s disease (HD), multiple system atrophies (MSA), Parkinson’s disease (PD), progressive supranuclear palsy (PSP), restless legs syndrome (RLS), tics, Tourette’s syndrome (TS), and Wilson disease (WD). MDs are classified as monogenic (Mendelian) or multifactorial or polygenic (Complex). However, mutations in known MD genes account for only a small percentage of cases, indicating that more genes and higher-risk alleles await discovery (Kompoliti and Verhagen, 2010).
Although linkage analysis along with candidate gene screening was traditionally the preferred method for gene discovery in inherited MDs (Paisan-Ruiz et al., 2004, 2005), these are not appropriate for families with largely reduced penetrance or reduced number of affected individuals. And even though there has been considerable success in identifying genetic risk factors for PD and PSP through the application of genome-wide association studies (GWAS; Simon-Sanchez et al., 2009; Hamza et al., 2010; Hoglinger et al., 2011), a detailed mapping through targeted resequencing (TR) is still required to further identify both high-risk alleles and haplotypes (Tucci et al., 2010; Freedman et al., 2011). By contrast, next-generation sequencing (NGS) technologies, such as exome sequencing (ES) and whole-genome sequencing (WGS), enable respectively a complete scan resolution of the entire both exome and genome, allowing a rapid identification of disease-causing mutations and higher-risk alleles. This has already been evidenced in rare Mendelian diseases where large sample sizes are difficult to gather (Ng et al., 2009, 2010), and in families previously deemed statistically insufficient for positional cloning (Bilguvar et al., 2010; Marti-Masso et al., 2012). Likewise, de novo pathogenic mutations have recently been identified in autism spectrum disorders by performing trio-based ES in only 20 sporadic cases (O’Roak et al., 2011). In conclusion, NGS is an ideal approach to identify causal alleles for inherited MDs and its application will increase the candidate gene list associated with MDs, facilitating the subsequent diagnosis and understanding of the disease process (Singleton, 2011).
In this review, we first describe the phenotypic spectrum associated with MDs and second summarize the novel genetic findings achieved by the use of NGS in MDs. The genetic contribution of MDs is expanding by the discovery of novel genes and the association of novel neurological phenotypes to already known disease genes.
Movement Disorder Phenotypes in Which NGS has been Performed
Below the phenotypes and genetic backgrounds of some inherited MDs in which NGS technologies have been employed are described. Phenotypes are listed in alphabetical order.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that controls the voluntary muscle movement. Because it is sometimes accompanied by PD and related disorders and some MDs genes can also cause ALS (Bosco et al., 2011), we decided to include it here. ALS is characterized by upper and lower motor neuron dysfunction resulting in progressive paralysis generally followed by death due to respiratory failure (Johnston et al., 2006; Johnson et al., 2010). The majority of ALS is sporadic (∼90%), and the clinical features of familial ALS (FALS) and sporadic ALS (SALS) are virtually indistinguishable, aside from a slightly earlier age at onset of FALS. Even though all FALS genes have also been implicated in SALS, these only account for a small proportion of cases, leaving much of the genetics of this disorder to be discovered (Andersen and Al-Chalabi, 2011). Some ALS-associated genes may also cause other disorders, such as frontotemporal dementia (FTD), cerebellar ataxia, motor neuropathies, hereditary spastic paraplegia (HSP), and Parkinsonism.
Ataxia describes a lack of muscle coordination during voluntary movements usually due to pathology in the cerebellum and its connections, such as spinocerebellar and pontocerebellar pathways. Although it is a neurological symptom, there are several inherited disorders that are due to this unsteady movement of gait and limbs, including Friedreich’s ataxia (FA), spinocerebellar ataxia (SCA), episodic ataxia (EA), ataxia telangiectasia (AT), and Machado–Joseph disease (MJD). Ataxias can present as sporadic or hereditary and are divided into five main categories: mitochondrial, metabolic, defective DNA repair, abnormal protein folding and degradation, and channelopathies. Based on neurological examination, ataxias can be broadly classified into pure cerebellar ataxias and those in which additional neurological deficits are also present. Most of the ataxias are labeled with the term SCA followed by a number to denote the distinct locus. At present, there are over 30 SCA loci and at least 17 disease-related genes identified (Filla and De Michele, 2012; Subramony, 2012a,b).
Cerebral palsies (CPs) comprise a heterogeneous group of neurodevelopmental disorders characterized by motor and postural impairments (Pakula et al., 2009). CP is classified according to the movement disorder observed as spastic, ataxic, dystonic, or athetoid, and according to the limbs affected as monoplegic, hemiplegic, diplegic, or quadriplegic (Moreno-De-Luca et al., 2012). Furthermore, CP is often associated with other neurological complications such as intellectual disability (ID), psychosocial disorders, and seizure disorders, making diagnosis and management of symptoms difficult (Aisen et al., 2011).
Charcot–Marie–Tooth (CMT) disease is the most common inherited neurological disease characterized by distal muscle weakness and atrophy (Weedon et al., 2011), and sometimes it occurs with sensory loss, depressed tendon reflexes, and pes cavus (highly arched feet). There are two main forms of CMT: CMT1 or type 1 that causes demyelination and CMT2 or type 2 that affects nerve axons. CMT is quite genetically heterogeneous, with over 40 disease-associated loci identified, and presents with intra-familial phenotypic variability (Del Bo et al., 2006), making traditional screening techniques time consuming and genetic diagnosis difficult.
Dystonias comprise a clinically and genetically heterogeneous group of MDs characterized by involuntary sustained muscular contractions affecting one or more sites of the body that produce abnormal postures and repetitive movements. They are classified according their etiology as primary when dystonia is the sole manifestation or secondary when it is accompanied by other neurological conditions, and according to the affected muscle groups’ distribution as focal, segmental, multifocal, generalized, and hemidystonias. The most common type of primary dystonia is primary torsion dystonia, which is caused by mutations in either DYT1 or DYT6 genes (Ozelius et al., 2011). However, even though there are more than 20 loci associated with dystonia, most cases are idiopathic and all familial forms present with reduced penetrance, making traditional linkage analyses inaccurate (Fuchs and Ozelius, 2011).
Fatty acid hydroxylase-associated neurodegeneration (FAHN) is caused by a deficiency of fatty acid 2-hydroxylase (FA2H; Kruer et al., 2010). FA2H pathogenic mutations were first described in families with leukodystrophy, spastic paraplegia (SPG), and dystonia (Edvardson et al., 2008), but later in families with complex SPG (SPG35) and neurodegeneration with brain iron accumulation (NBIA; Dick et al., 2010; Kruer et al., 2010). NBIA is characterized by progressive extrapyramidal deterioration and high brain iron deposition, most consistently in the basal ganglia.
Leukoencephalopathies are white matter diseases, in which a movement dysfunction is often a symptom. Sensory, behavioral, and cognitive deficits are also common. For instance, hereditary diffuse leukoencephalopathy with spheroids (HDLS) is associated with variable behavioral, cognitive, and motor dysfunction (Rademakers et al., 2011). Clinically, it is difficult to diagnose because it often presents with intra-familial heterogeneity and a firm diagnosis often occurs post-mortem (Baba et al., 2006). Pelizaeus–Merzbacher disease (PMD) is an X-linked hypomyelinating leukodystrophy, manifested as impaired motor development followed by ataxia, dystonia, dysarthria, and progressive spasticity (Vaurs-Barriere et al., 2009).
Parkinson’s disease is the third most common neurodegenerative disorder, behind Alzheimer’s disease and ET, affecting approximately 1–2% of the population above the age of 65, and whose incidence increases steeply with age (Lang and Lozano, 1998). It is characterized by bradykinesia, resting tremor, muscular rigidity, and postural instability. Although approximately less than 20% of cases follow clear Mendelian inheritance and the majority of PD is sporadic (Lees et al., 2009), there are 16 PD loci reported and at least eight genes identified to date (Hardy et al., 2009).
Paroxysmal kinesigenic dyskinesias (PKD), also called paroxysmal kinesigenic choreoathetosis (PKC), is the most common paroxysmal movement disorder and is characterized by recurrent, brief attacks of dyskinesia that are induced by sudden voluntary movements. The attacks manifest in dystonia or choreoathetosis. There are two other types of paroxysmal dyskinesias: exercise-induced (PED) and non-kinesigenic (PNKD). PED is genetically heterogeneous and only a small percentage of familial and sporadic cases are explained by deficiency in the glucose transporter type 1 gene (GLUT1) whereas PNKD is mainly due to mutations in the myofibrillogenesis regulator 1 gene (MR-1; Bhatia, 2011). Most PKD cases follow an autosomal dominant (AD) inheritance pattern (65–72%). Benign familial infantile epilepsy (BFIE) clinically resembles PKD and thus both are often misdiagnosed. BFIE can be familial or sporadic and is characterized by brief, non-febrile convulsions starting in the first year of life (Rochette et al., 2008). The co-occurrence of BFIE and PKD within a single family gave rise to the distinct clinical entity called infantile convulsions and choreoathetosis (ICCA) syndrome.
Spastic paraplegias are a group of MDs characterized by progressive spasticity in the lower limbs. Clinically they are classified as pure when consisting mainly of spasticity, abnormal reflexes, and motor deficits, or complex when a wide range of neurological symptoms are also present (Salinas et al., 2008). Genetically, they can arise from mutations in at least 23 genes, with more still being discovered, as 48 SPG loci have already been reported. Spasticity is also a symptom often seen in many other neurological diseases, and mutations in some SPG genes are responsible for other neurodegenerative diseases such as atypical ALS and atypical Parkinsonism (Orlacchio et al., 2010; Paisan-Ruiz et al., 2010).
Tourette syndrome/chronic tic disorder (TS–CTD) is fairly common, affecting 1–10 per 1000 boys, with milder versions affecting up to 10% of children (Rampello et al., 2006). It is characterized by motor and vocal tics. Although multiple rare copy number variants within several genes have recently been found associated with TS (Sundaram et al., 2010), most of the genetic causes have yet to be determined, as candidate gene association and linkage studies have been inconsistent and irreproducible (Rampello et al., 2006).
As stated above and the literature illustrates, MDs are both genetically and phenotypically heterogeneous and thus NGS technologies are the preferred and more rapid method to elucidate their causal alleles.
Novel Movement Disorder Genes Identified through NGS
In the last 2 years, NGS technologies have seen great use in the field of movement disorder genetics in which eight novel genes have been elucidated and many other NGS-related manuscripts will probably come to light in the next couple of years. Novel genes have been identified for ALS, CMT type 2, PD, PKD, spinocerebellar ataxia, BFIE, ICCA syndrome, and TS–CTD (Table 1).
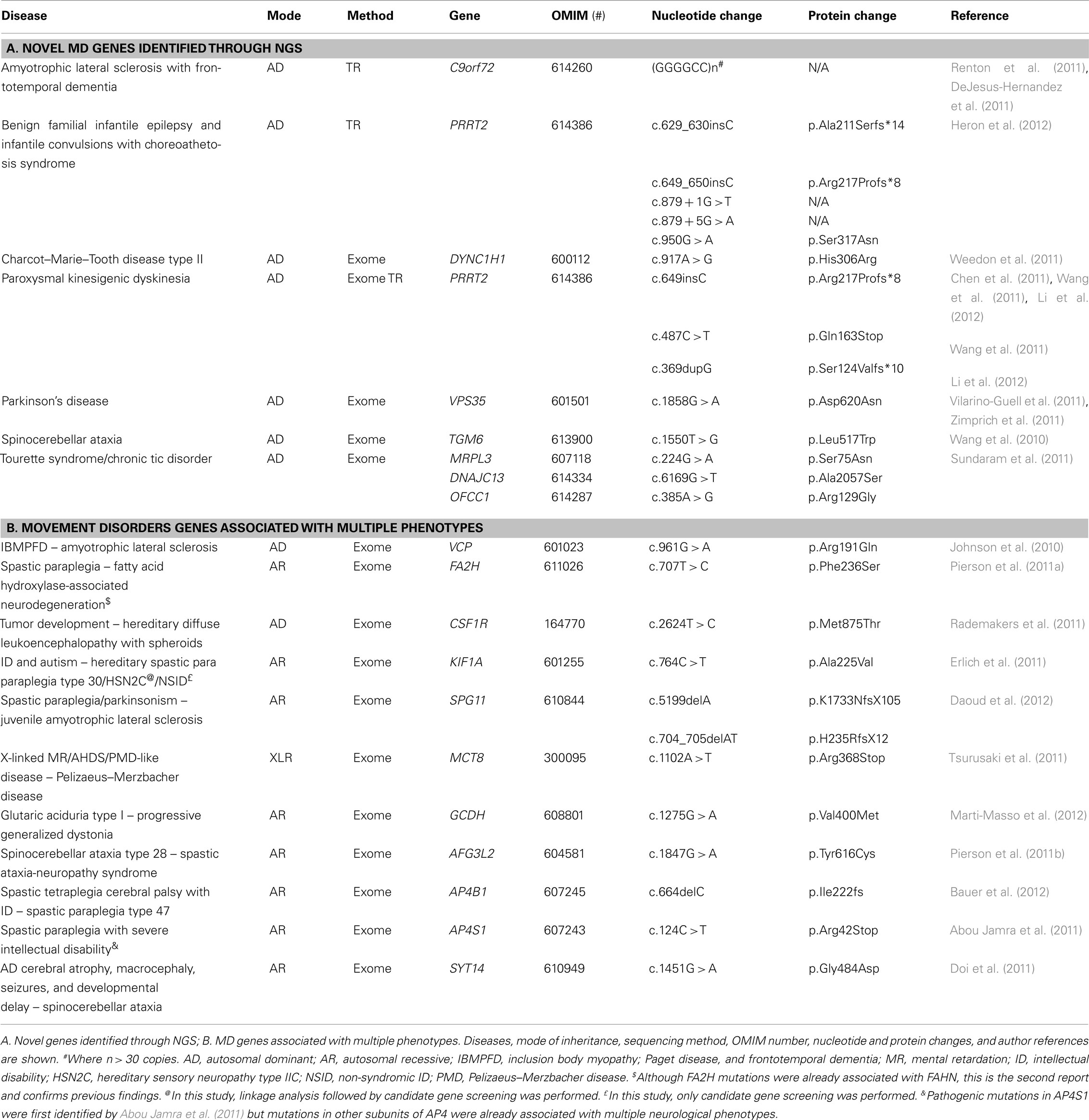
Table 1. Next-generation sequencing in movement disorders.
In some studies linkage analyses were carried out prior to NGS approaches. For instance, several studies identified the chromosome 9p21 as a causal locus for both ALS and FTD (Le Ber et al., 2009; Van Es et al., 2009; Shatunov et al., 2010). Although a large Swedish family was previously reported as presenting with mainly dementia in the first to third generation and motor neuron disease and mild dementia in the fourth generation (Gunnarsson et al., 1991), not until lately has the genetic cause for this link been identified: a massively expanded hexanucleotide repeat (GGGGCC). This hexanucleotide repeat was found in a non-coding region of C9orf72 located on chromosome 9p21 and is suggested to be the most common cause of both FALS and FTD identified to date (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The C9orf72-associated phenotype has recently been defined as an early-onset, AD neurodegenerative disease presenting with cognitive and behavioral impairment, specific neuroimaging changes, and reduced survival (Byrne et al., 2012). Similarly, PKD families were previously linked to chromosome 16p.11–q12.1 by different groups (Tomita et al., 1999), but not until now, taking advantage of ES technologies, has the causal gene, PRRT2, been identified (Chen et al., 2011; Wang et al., 2011; Li et al., 2012). PRRT2 mutations have additionally been reported in BFIE and ICCA families previously linked to chromosome 16p.11–q12.1 (Szepetowski et al., 1997; Caraballo et al., 2001; Heron et al., 2012), and in familial and sporadic cases with PKD, ICCA, PED, or PNKD-like disease (Liu et al., 2012), further confirming the PRRT2’s pathogenic role. TGM6 genetic variability has likewise identified in spinocerebellar ataxia-35 (SCA35), which was mapped to chromosome 20p13-12.2 and is characterized by a slowly progressive and relatively pure form of adult-onset cerebellar ataxia affecting both upper and lower limbs (Wang et al., 2010). TGM6, encoding transglutaminase 6, is expressed in Purkinje cells (Hadjivassiliou et al., 2008) and anti-transglutaminase 6 antibodies have been proved to cause ataxia in mice (Boscolo et al., 2010).
In conclusion, the pathogenesis of C9orf72, PRRT2, and TGM6 genes is fully disclosed by the linkage and disease-segregation data, the absence of pathogenic mutations in neurologically normal individuals, and their further replication in additional or animal studies.
But other studies performed ES directly or after exclusion of known genes. For instance, DYNC1H1 (cytoplasmic dynein heavy chain 1) genetic variability, which was previously shown to cause sensory neuropathy with motor neuron loss in mice (Chen et al., 2007), has recently found responsible for AD CMT2 in humans (Weedon et al., 2011). In addition, a mutation in the vacuolar protein sorting 35 (VPS35), p.Asp620Asn, identified by two independent groups in PD patients (Vilarino-Guell et al., 2011; Zimprich et al., 2011), has both been replicated and failed to be replicated in recent studies (Sheerin et al., 2011; Guella et al., 2012); and even though disease-segregating mutations in three genes, MRPL3, DNAJC13, and OFCC1, have recently identified in a three-generational TS–CTD pedigree (Sundaram et al., 2011), further studies are needed to corroborate this finding.
Movement Disorder Genes Associated with Multiple Phenotypes
With the advent of NGS technologies not only have novel MDs genes been identified, but also 10 already-disease-associated genes have been found mutated in other diseases, expanding the phenotypic spectrum associated with a single gene (Table 1). In most of these studies, SNPs-based arrays along with NGS were used to identify the genetic defects underlying disease. Examples are described below.
SPG11 mutation, although are known to be the most common causes of autosomal recessive (AR) HSP with thin corpus callosum (HSP-TCC; Hehr et al., 2007; Stevanin et al., 2007; Paisan-Ruiz et al., 2008a), has also been identified in AR both parkinsonism (Paisan-Ruiz et al., 2010) and juvenile ALS (ARJALS; Orlacchio et al., 2010). Through the use of ES, this latter association has recently been confirmed (Daoud et al., 2012) in a family consisting of two affected siblings, who one manifested with ALS and the other manifested with HSP-TCC, confirming the intra-familial phenotypic heterogeneity associated with SPG11 mutation, which is also seen in recessively inherited parkinsonism (Anheim et al., 2009). In conclusion, SPG11 genetic variability should be considered in SPG, ALS, and Parkinsonism, especially when they occur within the same family (Figure 1). Likewise, FA2H mutations were first known to cause complex forms of ARHSP (Edvardson et al., 2008; Dick et al., 2010) and later to cause NBIA, a syndrome that acquired the name of FAHN (Kruer et al., 2010). But recently, a compound heterozygous FA2H mutation has also been identified in a child with typical clinical features of NBIA and axonal sensory neuropathy, confirming previous findings and expanding the phenotypic spectrum associated with FA2H genetic variability, as axonal sensory neuropathy was not seen before in FAHN (Pierson et al., 2011b). KIF1A, already implicated in ID and autism (Galasso et al., 2008), has recently found mutated in hereditary sensory neuropathy type IIC (HSN2C; Riviere et al., 2011), non-syndromic ID (NSID; Hamdan et al., 2011), and HSPs (Erlich et al., 2011; Klebe et al., 2012). Due to their phenotypic heterogeneity, KIF1A mutations have been suggested to predict different phenotypes depending on their nature: nonsense mutations lead to a complete knockout of protein function and cause HSN2C while missense mutations in the kinesin motor domain lead to upper motor neuron dysfunction and cause SPG (Klebe et al., 2012).
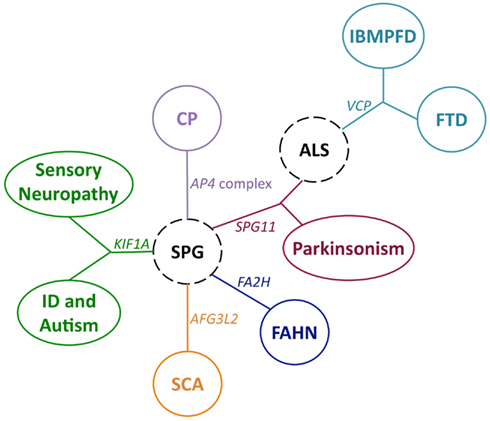
Figure 1. Movement disorder genes associated with multiple neurological phenotypes. Diseases and phenotypes are represented with circles and ovals with genes involved shown between implicated diseases. Dashed circles represent disorders in which more than one gene is involved. Disease abbreviations are described in the main text.
Previously, mutations in two subunits, AP4M1 and AP4E1, of the adaptor protein complex 4 (AP4) were found associated with AR spastic tetraplegia as well as cerebral palsy and microcephaly, respectively (Verkerk et al., 2009; Moreno-De-Luca et al., 2011). But recently, pathogenic mutations in three AP4 subunits, AP4S1, AP4B1, and AP4E1, have also been identified in consanguineous families presenting with early-onset complex SPG, severe ID, microcephaly, inability to walk, and epilepsy (Abou Jamra et al., 2011); AP4B1 pathogenic mutations are also responsible for SPG type 47 (Bauer et al., 2012). Since patients carrying AP4 mutations share many clinical features, the existence of a complex AP4-deficiency syndrome, characterized by severe ID, growth retardation, stereotypic laughter, progressive spasticity, cerebral palsy, and inability to walk, has been suggested, further supporting the key role of AP4-mediated trafficking in brain development and functioning (Abou Jamra et al., 2011).
Mutations in AFG3L2, encoding a subunit of a mitochondrial m-AAA protease, were previously associated with spinocerebellar ataxia type 28 (SCA28; Di Bella et al., 2010), but recently, a AFG3L2 mutation, p.Y616C, has also been identified in affected siblings presenting with early-onset SPG, progressive myoclonic epilepsy, and peripheral neuropathy (Pierson et al., 2011a), adding peripheral neuropathy to the AFG3L2-associated symptoms. Since AFG3L2 could form a homo-oligomeric isoenzyme with itself or a hetero-oligomeric complex with paraplegin, whose mutations are associated with an adult form of SPG (Casari et al., 1998), the AFG3L2 mutation, p.Y616C, has been suggested to specifically interrupt the formation of both the homo-oligomer and the hetero-oligomer with paraplegin, giving rise to a complex phenotype that combines symptoms of SPG7 and SCA28 along with mitochondrial symptoms like myoclonic epilepsy (Pierson et al., 2011a).
SYT14, previously found mutated in a 12-year-old girl with AD cerebral atrophy, macrocephaly, seizures, and developmental delay, in the form of a de novo balanced rearrangement [t(1;3)(q32.2;q25.2; Quintero-Rivera et al., 2007)], has lately been found mutated in AR forms of SCA (ARCA) and psychomotor retardation (Doi et al., 2011). Although both cases present with mental retardation and cerebellar atrophy, the ARCA phenotype differs in the mode of inheritance and presents with oculomotor apraxia, spasticity, peripheral neuropathy, retinal abnormality, and psychomotor retardation, largely increasing the phenotypic spectrum associated with SYT14 genetic variability.
Likewise, disease-segregating mutations in the valosin-containing protein, VCP, known to cause inclusion body myopathy, Paget disease, and FTD (IBMPFD), have recently been identified in FALS (Johnson et al., 2010), suggesting that the motor neuron degeneration seen in ALS patients should be included in the IBMPF-associated phenotype. Indeed, the presence of both Paget’s disease and FTD was subsequently reported in two ALS families carrying VCP mutations (Johnson et al., 2010).
Mutations in the colony-stimulating factor 1 receptor, CSF1R, previously implicated in tumor development, including myeloid and hematological malignancies (Ridge et al., 1990), have recently been identified in patients with HDLS, who reported having no bone-structure abnormalities (Rademakers et al., 2011); this suggests that CSF1R, which regulates the survival, proliferation, differentiation, and function of microglia in the brain (Stanley et al., 1997), may also be implicated in motor dysfunction. Likewise, SLC16A2 genetic variability, which was already implicated in X-linked mental retardation, Allan–Herndon–Dudley syndrome, and Pelizaeus–Merzbacher-like disease (Friesema et al., 2004; Dumitrescu et al., 2006; Frints et al., 2008; Vaurs-Barriere et al., 2009), has recently been associated with a X-linked form of leukoencephalopathy, clinically diagnosed as PMD (Tsurusaki et al., 2011). Although SLC16A2 encodes a thyroid hormone transporter known as monocarboxylate transporter 8, MCT8, none of PMD patients carrying SLC16A2 mutations reported thyroid dysfunction.
Lastly, mutations in GCDH, which encodes a glutaryl-CoA dehydrogenase, were known to cause glutaric aciduria type 1 (GA-1; Busquets et al., 2000), which is characterized by accumulation of glutaric acid (GA) and 3-hydroxyglutaric acid (3-OHGA) in brain and body fluids, leading to neurotoxicity, encephalopathy, macrocephaly, and eventually a dystonic–dyskinetic disorder (Jafari et al., 2011). However, a homozygous, disease-segregating GCDH mutation has recently been identified in two siblings who suffer from an AR progressive generalized dystonia (PGD; Marti-Masso et al., 2012) and whose urine samples showed excess of 3-OHGA. Since neither patient showed encephalopathy or macrocephaly, this finding associates GCDH deficiency with PGD, a phenotype not previously described in GA-1-associated GCDH deficiency.
Discussion
Next-generation sequencing technologies enable a comprehensive analysis of the entire genome and exome and as such have dramatically progressed the field of biological and biomedical research, especially in Mendelian diseases. Nonetheless, these novel methodologies also have several limitations; for instance, although the target coverage is continually improving, a complete coverage will probably never be reached since specific genomic regions such as GC-rich areas and repetitive elements are difficult to amplify and large copy number variations are hard to detect. Despite this, here we report on several movements disorder studies in which NGS technologies have successfully been employed for disease-causing mutations’ identification. Some of these findings have already been replicated. This is the case of C9orf72 causing ALS and PRRT2 causing PKD, ICCA syndrome, paroxysmal exercise-induced dyskinesias, and paroxysmal non-kinesigenic dyskinesias. In both cases, linkage analysis was performed years before the gene identification but the causal gene did not come to light until the use of NGS technologies. As for C9orf72, since the genetic defect is an expanded hexanucleotide repeat located in a non-coding region, it would have been difficult to find through traditional gene screening techniques, in which only coding variants are usually examined. Two other novel MD genes identified, TGM6 causing cerebellar ataxia and DYNC1H1 causing CMT type 2, were previously shown to cause similar symptoms in mice, further confirming their pathogenicity. Novel genes have also been identified for PD and TS–CTD; but in contrast, due to the lack of both linkage and functional data, further molecular studies are required to further prove their pathogenicity.
These studies have demonstrated that ES is a terrific tool to molecularly diagnose genetically and clinically heterogeneous MDs in which a large number of genes should be screened and a wide range of clinical features is usually present. In addition, recent NGS studies have shown that a single gene may be involved in multiple clinical entities. Accordingly, genes causing complex SPG may also be responsible for ALS, cerebral palsy, FAHN, Parkinsonism, sensory neuropathy, and spinocerebellar ataxia phenotypes; and VCP mutations already associated with IBMPFD, may also be present in amyotrophic lateral sclerosis (Figure 1). These phenotypic heterogeneities associated with a single gene clearly lead us to seek the molecular events by which mutations in a single gene can result in multiple phenotypes. This may lie in the nature of the mutation, as suggested for KIF1A mutations (Klebe et al., 2012), or the protein domain that is affected (De Angelis et al., 1999). However, the fact that the same mutation may sometimes cause multiple phenotypes requires the search of additional factors implicated in the phenotypic expression of a disease, such as interacting genes and other genetic and environmental modifiers. To face this ambitious challenge, the characterization of each gene function through cellular and animal studies is crucial. Today, novel molecular targets and subsequent cellular mechanisms may be determined by the employment of high-throughput techniques using either small animals such as Caenorhabditis elegans, Drosophila melanogaster, and Danio rerio, or automated cell culturing. In addition, these throughput techniques are perfect tools for drug discovery (Giacomotto and Segalat, 2010; Jain et al., 2011). Taken together, these analyses will definitely reveal novel insights into the disease-associated pathogenesis, which in turn are essential for developing more effective treatments.
We conclude that NGS, particularly in conjunction with other methods like SNPs-based arrays, has the robust capability to identify disease-causing genes in a very limited time and to some extent to expand the phenotypic spectrum associated with a single gene, improving diagnosis and leading to the development of more effective treatments. As NGS costs decrease, target coverage increases, and filtering databases grow, a trend toward sequencing the entire genome will come next, requiring advances in the understanding of non-coding genetic events and in the identification of both genetic and environmental modifiers. Finally, our understanding of the genetic contribution to MDs will surely increase in the coming years, but further functional and cellular analyses will be required for establishing the disease-associated mechanisms and building all molecular networks associated with MDs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Department of Neurology and the Friedman Brain Institute at the Mount Sinai School of Medicine for support.
Abbreviations
AD, autosomal dominant; AHDS, Allan–Herndon–Dudley syndrome; ALS, amyotrophic lateral sclerosis; AP4, adaptor protein complex 4; AR, autosomal recessive; AT, ataxia telangiectasia; BFIE, benign familial infantile epilepsy; ChAc, chorea-acanthocytosis; CMT, Charcot–Marie–Tooth; CMT1, Charcot–Marie–Tooth type 1; CMT2, Charcot–Marie–Tooth type 2; CP, cerebral palsy; DRD, dopa-responsive dystonia; EA, episodic ataxia; ES, exome sequencing; ET, essential tremor; FA, Friedreich’s ataxia; FAHN, fatty acid hydroxylase-associated neurodegeneration; FALS, familial ALS; FTD, frontotemporal dementia; GWAS, genome-wide association studies; HD, Huntington’s disease; HDL2, Huntington disease-like 2; HDLS, hereditary diffuse leukoencephalopathy with spheroids; HSN2C, hereditary sensory neuropathy type IIC; HSP, hereditary spastic paraplegia; IBMPFD, inclusion body myopathy, Paget disease, and frontotemporal dementia; ICCA, infantile convulsions and choreoathetosis syndrome; ID, intellectual disability; MDs, movement disorders; MJD, Machado–Joseph disease; MLS, X-linked McLeod syndrome; MSA, multiple system atrophies; MR, mental retardation; NA, neuroacanthocytosis; NGS, next-generation sequencing; NSID, non-syndromic ID; PD, Parkinson’s disease; PED, paroxysmal exercise-induced dyskinesias; PGD, progressive generalized dystonia; PKAN, pantothenate kinase-associated neurodegeneration; PKC, paroxysmal kinesigenic choreoathetosis; PKD, paroxysmal kinesigenic dyskinesias; PMD, Pelizaeus–Merzbacher disease; PMLD, Pelizaeus–Merzbacher-like disease; PNKD, Paroxysmal non-kinesigenic dyskinesias; PSP, progressive supranuclear palsy; RLS, restless legs syndrome; SALS, sporadic ALS; SCA, spinocerebellar ataxia; SPG, spastic paraplegia; TR, targeted resequencing; TS–CTD, Tourette syndrome/chronic tic disorder; TS, Tourette’s syndrome; WD, Wilson disease; WGS, whole-genome sequencing; XLD, X-linked dominant; XLR, X-linked recessive.
References
Abou Jamra, R., Philippe, O., Raas-Rothschild, A., Eck, S. H., Graf, E., Buchert, R., Borck, G., Ekici, A., Brockschmidt, F. F., Nothen, M. M., Munnich, A., Strom, T. M., Reis, A., and Colleaux, L. (2011). Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am. J. Hum. Genet. 88, 788–795.
Aisen, M. L., Kerkovich, D., Mast, J., Mulroy, S., Wren, T. A., Kay, R. M., and Rethlefsen, S. A. (2011). Cerebral palsy: clinical care and neurological rehabilitation. Lancet Neurol. 10, 844–852.
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615.
Anheim, M., Lagier-Tourenne, C., Stevanin, G., Fleury, M., Durr, A., Namer, I. J., Denora, P., Brice, A., Mandel, J. L., Koenig, M., and Tranchant, C. (2009). SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J. Neurol. 256, 104–108.
Baba, Y., Ghetti, B., Baker, M. C., Uitti, R. J., Hutton, M. L., Yamaguchi, K., Bird, T., Lin, W., Delucia, M. W., Dickson, D. W., and Wszolek, Z.K. (2006). Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 111, 300–311.
Bauer, P., Leshinsky-Silver, E., Blumkin, L., Schlipf, N., Schroder, C., Schicks, J., Lev, D., Riess, O., Lerman-Sagie, T., and Schols, L. (2012). Mutation in the AP4B1 gene cause hereditary spastic paraplegia type 47 (SPG47). Neurogenetics 13, 73–76.
Bilguvar, K., Ozturk, A. K., Louvi, A., Kwan, K. Y., Choi, M., Tatli, B., Yalnizoglu, D., Tuysuz, B., Caglayan, A. O., Gokben, S., Kaymakcalan, H., Barak, T., Bakircioglu, M., Yasuno, K., Ho, W., Sanders, S., Zhu, Y., Yilmaz, S., Dincer, A., Johnson, M. H., Bronen, R. A., Kocer, N., Per, H., Mane, S., Pamir, M. N., Yalcinkaya, C., Kumandas, S., Topcu, M., Ozmen, M., Sestan, N., Lifton, R. P., State, M. W., and Gunel, M. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210.
Bosco, D. A., Lavoie, M. J., Petsko, G. A., and Ringe, D. (2011). Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Biol. 3, a007500.
Boscolo, S., Lorenzon, A., Sblattero, D., Florian, F., Stebel, M., Marzari, R., Not, T., Aeschlimann, D., Ventura, A., Hadjivassiliou, M., and Tongiorgi, E. (2010). Anti transglutaminase antibodies cause ataxia in mice. PLoS ONE 5, e9698.
Busquets, C., Merinero, B., Christensen, E., Gelpi, J. L., Campistol, J., Pineda, M., Fernandez-Alvarez, E., Prats, J. M., Sans, A., Arteaga, R., Marti, M., Campos, J., Martinez-Pardo, M., Martinez-Bermejo, A., Ruiz-Falco, M. L., Vaquerizo, J., Orozco, M., Ugarte, M., Coll, M. J., and Ribes, A. (2000). Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically, and biochemically distinct. Pediatr. Res. 48, 315–322.
Byrne, S., Elamin, M., Bede, P., Shatunov, A., Walsh, C., Corr, B., Heverin, M., Jordan, N., Kenna, K., Lynch, C., Mclaughlin, R. L., Iyer, P. M., O’Brien, C., Phukan, J., Wynne, B., Bokde, A. L., Bradley, D. G., Pender, N., Al-Chalabi, A., and Hardiman, O. (2012). Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 11, 232–240.
Caraballo, R., Pavek, S., Lemainque, A., Gastaldi, M., Echenne, B., Motte, J., Genton, P., Cersosimo, R., Humbertclaude, V., Fejerman, N., Monaco, A. P., Lathrop, M. G., Rochette, J., and Szepetowski, P. (2001). Linkage of benign familial infantile convulsions to chromosome 16p12-q12 suggests allelism to the infantile convulsions and choreoathetosis syndrome. Am. J. Hum. Genet. 68, 788–794.
Casari, G., De Fusco, M., Ciarmatori, S., Zeviani, M., Mora, M., Fernandez, P., De Michele, G., Filla, A., Cocozza, S., Marconi, R., Durr, A., Fontaine, B., and Ballabio, A. (1998). Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93, 973–983.
Chen, W. J., Lin, Y., Xiong, Z. Q., Wei, W., Ni, W., Tan, G. H., Guo, S. L., He, J., Chen, Y. F., Zhang, Q. J., Li, H. F., Murong, S. X., Xu, J., Wang, N., and Wu, Z. Y. (2011). Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 43, 1252–1255.
Chen, X. J., Levedakou, E. N., Millen, K. J., Wollmann, R. L., Soliven, B., and Popko, B. (2007). Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J. Neurosci. 27, 14515–14524.
Daoud, H., Zhou, S., Noreau, A., Sabbagh, M., Belzil, V., Dionne-Laporte, A., Tranchant, C., Dion, P., and Rouleau, G. A. (2012). Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol. Aging 33, 839.e835–e839.
De Angelis, E., Macfarlane, J., Du, J. S., Yeo, G., Hicks, R., Rathjen, F. G., Kenwrick, S., and Brummendorf, T. (1999). Pathological missense mutations of neural cell adhesion molecule L1 affect homophilic and heterophilic binding activities. EMBO J. 18, 4744–4753.
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., Nicholson, A. M., Finch, N. A., Flynn, H., Adamson, J., Kouri, N., Wojtas, A., Sengdy, P., Hsiung, G. Y., Karydas, A., Seeley, W. W., Josephs, K. A., Coppola, G., Geschwind, D. H., Wszolek, Z. K., Feldman, H., Knopman, D. S., Petersen, R. C., Miller, B. L., Dickson, D. W., Boylan, K. B., Graff-Radford, N. R., and Rademakers, R. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256.
Del Bo, R., Locatelli, F., Corti, S., Scarlato, M., Ghezzi, S., Prelle, A., Fagiolari, G., Moggio, M., Carpo, M., Bresolin, N., and Comi, G. P. (2006). Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology 66, 752–754.
Di Bella, D., Lazzaro, F., Brusco, A., Plumari, M., Battaglia, G., Pastore, A., Finardi, A., Cagnoli, C., Tempia, F., Frontali, M., Veneziano, L., Sacco, T., Boda, E., Brussino, A., Bonn, F., Castellotti, B., Baratta, S., Mariotti, C., Gellera, C., Fracasso, V., Magri, S., Langer, T., Plevani, P., Di Donato, S., Muzi-Falconi, M., and Taroni, F. (2010). Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 42, 313–321.
Dick, K. J., Eckhardt, M., Paisan-Ruiz, C., Alshehhi, A. A., Proukakis, C., Sibtain, N. A., Maier, H., Sharifi, R., Patton, M. A., Bashir, W., Koul, R., Raeburn, S., Gieselmann, V., Houlden, H., and Crosby, A. H. (2010). Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum. Mutat. 31, E1251–E1260.
Doi, H., Yoshida, K., Yasuda, T., Fukuda, M., Fukuda, Y., Morita, H., Ikeda, S., Kato, R., Tsurusaki, Y., Miyake, N., Saitsu, H., Sakai, H., Miyatake, S., Shiina, M., Nukina, N., Koyano, S., Tsuji, S., Kuroiwa, Y., and Matsumoto, N. (2011). Exome sequencing reveals a homozygous SYT14 mutation in adult-onset, autosomal-recessive spinocerebellar ataxia with psychomotor retardation. Am. J. Hum. Genet. 89, 320–327.
Dumitrescu, A. M., Liao, X. H., Weiss, R. E., Millen, K., and Refetoff, S. (2006). Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147, 4036–4043.
Edvardson, S., Hama, H., Shaag, A., Gomori, J. M., Berger, I., Soffer, D., Korman, S. H., Taustein, I., Saada, A., and Elpeleg, O. (2008). Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am. J. Hum. Genet. 83, 643–648.
Erlich, Y., Edvardson, S., Hodges, E., Zenvirt, S., Thekkat, P., Shaag, A., Dor, T., Hannon, G. J., and Elpeleg, O. (2011). Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 21, 658–664.
Filla, A., and De Michele, G. (2012). Overview of autosomal recessive ataxias. Handb. Clin. Neurol. 103, 265–274.
Freedman, M. L., Monteiro, A. N., Gayther, S. A., Coetzee, G. A., Risch, A., Plass, C., Casey, G., De Biasi, M., Carlson, C., Duggan, D., James, M., Liu, P., Tichelaar, J. W., Vikis, H. G., You, M., and Mills, I. G. (2011). Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 43, 513–518.
Friesema, E. C., Grueters, A., Biebermann, H., Krude, H., Von Moers, A., Reeser, M., Barrett, T. G., Mancilla, E. E., Svensson, J., Kester, M. H., Kuiper, G. G., Balkassmi, S., Uitterlinden, A. G., Koehrle, J., Rodien, P., Halestrap, A. P., and Visser, T. J. (2004). Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364, 1435–1437.
Frints, S. G., Lenzner, S., Bauters, M., Jensen, L. R., Van Esch, H., Des Portes, V., Moog, U., Macville, M. V., Van Roozendaal, K., Schrander-Stumpel, C. T., Tzschach, A., Marynen, P., Fryns, J. P., Hamel, B., Van Bokhoven, H., Chelly, J., Beldjord, C., Turner, G., Gecz, J., Moraine, C., Raynaud, M., Ropers, H. H., Froyen, G., and Kuss, A. W. (2008). MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur. J. Hum. Genet. 16, 1029–1037.
Galasso, C., Lo-Castro, A., Lalli, C., Nardone, A. M., Gullotta, F., and Curatolo, P. (2008). Deletion 2q37: an identifiable clinical syndrome with mental retardation and autism. J. Child Neurol. 23, 802–806.
Giacomotto, J., and Segalat, L. (2010). High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 160, 204–216.
Guella, I., Solda, G., Cilia, R., Pezzoli, G., Asselta, R., Duga, S., and Goldwurm, S. (2012). The Asp620asn mutation in VPS35 is not a common cause of familial Parkinson’s disease. Mov. Disord.
Gunnarsson, L. G., Dahlbom, K., and Strandman, E. (1991). Motor neuron disease and dementia reported among 13 members of a single family. Acta Neurol. Scand. 84, 429–433.
Hadjivassiliou, M., Aeschlimann, P., Strigun, A., Sanders, D. S., Woodroofe, N., and Aeschlimann, D. (2008). Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann. Neurol. 64, 332–343.
Hamdan, F. F., Gauthier, J., Araki, Y., Lin, D. T., Yoshizawa, Y., Higashi, K., Park, A. R., Spiegelman, D., Dobrzeniecka, S., Piton, A., Tomitori, H., Daoud, H., Massicotte, C., Henrion, E., Diallo, O., Shekarabi, M., Marineau, C., Shevell, M., Maranda, B., Mitchell, G., Nadeau, A., D’Anjou, G., Vanasse, M., Srour, M., Lafreniere, R. G., Drapeau, P., Lacaille, J. C., Kim, E., Lee, J. R., Igarashi, K., Huganir, R. L., Rouleau, G. A., and Michaud, J. L. (2011). Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 88, 306–316.
Hamza, T. H., Zabetian, C. P., Tenesa, A., Laederach, A., Montimurro, J., Yearout, D., Kay, D. M., Doheny, K. F., Paschall, J., Pugh, E., Kusel, V. I., Collura, R., Roberts, J., Griffith, A., Samii, A., Scott, W. K., Nutt, J., Factor, S. A., and Payami, H. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 42, 781–785.
Hardy, J., Lewis, P., Revesz, T., Lees, A., and Paisan-Ruiz, C. (2009). The genetics of Parkinson’s syndromes: a critical review. Curr. Opin. Genet. Dev. 19, 254–265.
Hehr, U., Bauer, P., Winner, B., Schule, R., Olmez, A., Koehler, W., Uyanik, G., Engel, A., Lenz, D., Seibel, A., Hehr, A., Ploetz, S., Gamez, J., Rolfs, A., Weis, J., Ringer, T. M., Bonin, M., Schuierer, G., Marienhagen, J., Bogdahn, U., Weber, B. H., Topaloglu, H., Schols, L., Riess, O., and Winkler, J. (2007). Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann. Neurol. 62, 656–665.
Heron, S. E., Grinton, B. E., Kivity, S., Afawi, Z., Zuberi, S. M., Hughes, J. N., Pridmore, C., Hodgson, B. L., Iona, X., Sadleir, L. G., Pelekanos, J., Herlenius, E., Goldberg-Stern, H., Bassan, H., Haan, E., Korczyn, A. D., Gardner, A. E., Corbett, M. A., Gecz, J., Thomas, P. Q., Mulley, J. C., Berkovic, S. F., Scheffer, I. E., and Dibbens, L. M. (2012). PRRT2 Mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 90, 152–160.
Hoglinger, G. U., Melhem, N. M., Dickson, D. W., Sleiman, P. M., Wang, L. S., Klei, L., Rademakers, R., De Silva, R., Litvan, I., Riley, D. E., Van Swieten, J. C., Heutink, P., Wszolek, Z. K., Uitti, R. J., Vandrovcova, J., Hurtig, H. I., Gross, R. G., Maetzler, W., Goldwurm, S., Tolosa, E., Borroni, B., Pastor, P., Cantwell, L. B., Han, M. R., Dillman, A., Van Der Brug, M. P., Gibbs, J. R., Cookson, M. R., Hernandez, D. G., Singleton, A. B., Farrer, M. J., Yu, C. E., Golbe, L. I., Revesz, T., Hardy, J., Lees, A. J., Devlin, B., Hakonarson, H., Muller, U., and Schellenberg, G. D. (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705.
Jafari, P., Braissant, O., Bonafe, L., and Ballhausen, D. (2011). The unsolved puzzle of neuropathogenesis in glutaric aciduria type I. Mol. Genet. Metab. 104, 425–437.
Jain, S., Sondervan, D., Rizzu, P., Bochdanovits, Z., Caminada, D., and Heutink, P. (2011). The complete automation of cell culture: improvements for high-throughput and high-content screening. J. Biomol. Screen 16, 932–939.
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., Gibbs, J. R., Brunetti, M., Gronka, S., Wuu, J., Ding, J., Mccluskey, L., Martinez-Lage, M., Falcone, D., Hernandez, D. G., Arepalli, S., Chong, S., Schymick, J. C., Rothstein, J., Landi, F., Wang, Y. D., Calvo, A., Mora, G., Sabatelli, M., Monsurro, M. R., Battistini, S., Salvi, F., Spataro, R., Sola, P., Borghero, G., Galassi, G., Scholz, S. W., Taylor, J. P., Restagno, G., Chio, A., and Traynor, B. J. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864.
Johnston, C. A., Stanton, B. R., Turner, M. R., Gray, R., Blunt, A. H., Butt, D., Ampong, M. A., Shaw, C. E., Leigh, P. N., and Al-Chalabi, A. (2006). Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J. Neurol. 253, 1642–1643.
Klebe, S., Lossos, A., Azzedine, H., Mundwiller, E., Sheffer, R., Gaussen, M., Marelli, C., Nawara, M., Carpentier, W., Meyer, V., Rastetter, A., Martin, E., Bouteiller, D., Orlando, L., Gyapay, G., El-Hachimi, K. H., Zimmerman, B., Gamliel, M., Misk, A., Lerer, I., Brice, A., Durr, A., and Stevanin, G. (2012). KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur. J. Hum. Genet.
Kruer, M. C., Paisan-Ruiz, C., Boddaert, N., Yoon, M. Y., Hama, H., Gregory, A., Malandrini, A., Woltjer, R. L., Munnich, A., Gobin, S., Polster, B. J., Palmeri, S., Edvardson, S., Hardy, J., Houlden, H., and Hayflick, S. J. (2010). Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 68, 611–618.
Lang, A. E., and Lozano, A. M. (1998). Parkinson’s disease. First of two parts. N. Engl. J. Med. 339, 1044–1053.
Le Ber, I., Camuzat, A., Berger, E., Hannequin, D., Laquerriere, A., Golfier, V., Seilhean, D., Viennet, G., Couratier, P., Verpillat, P., Heath, S., Camu, W., Martinaud, O., Lacomblez, L., Vercelletto, M., Salachas, F., Sellal, F., Didic, M., Thomas-Anterion, C., Puel, M., Michel, B. F., Besse, C., Duyckaerts, C., Meininger, V., Campion, D., Dubois, B., and Brice, A. (2009). Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 72, 1669–1676.
Li, J., Zhu, X., Wang, X., Sun, W., Feng, B., Du, T., Sun, B., Niu, F., Wei, H., Wu, X., Dong, L., Li, L., Cai, X., Wang, Y., and Liu, Y. (2012). Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigenic choreoathetosis. J. Med. Genet. 49, 76–78.
Liu, Q., Qi, Z., Wan, X. H., Li, J. Y., Shi, L., Lu, Q., Zhou, X. Q., Qiao, L., Wu, L. W., Liu, X. Q., Yang, W., Liu, Y., Cui, L. Y., and Zhang, X. (2012). Mutations in PRRT2 result in paroxysmal dyskinesias with marked variability in clinical expression. J. Med. Genet. 49, 79–82.
Marti-Masso, J. F., Ruiz-Martinez, J., Makarov, V., De Munain, A. L., Gorostidi, A., Bergareche, A., Yoon, S., Buxbaum, J. D., and Paisan-Ruiz, C. (2012). Exome sequencing identifies GCDH (glutaryl-CoA dehydrogenase) mutations as a cause of a progressive form of early-onset generalized dystonia. Hum. Genet. 131, 435–442.
Moreno-De-Luca, A., Helmers, S. L., Mao, H., Burns, T. G., Melton, A. M., Schmidt, K. R., Fernhoff, P. M., Ledbetter, D. H., and Martin, C. L. (2011). Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J. Med. Genet. 48, 141–144.
Moreno-De-Luca, A., Ledbetter, D. H., and Martin, C. L. (2012). Genetic insights into the causes and classification of cerebral palsies. Lancet Neurol. 11, 283–292.
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M., Huff, C. D., Shannon, P. T., Jabs, E. W., Nickerson, D. A., Shendure, J., and Bamshad, M. J. (2010). Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet. 42, 30–35.
Ng, S. B., Turner, E. H., Robertson, P. D., Flygare, S. D., Bigham, A. W., Lee, C., Shaffer, T., Wong, M., Bhattacharjee, A., Eichler, E. E., Bamshad, M., Nickerson, D. A., and Shendure, J. (2009). Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461, 272–276.
Orlacchio, A., Babalini, C., Borreca, A., Patrono, C., Massa, R., Basaran, S., Munhoz, R. P., Rogaeva, E. A., St George-Hyslop, P. H., Bernardi, G., and Kawarai, T. (2010). SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133, 591–598.
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., Karakoc, E., Mackenzie, A. P., Ng, S. B., Baker, C., Rieder, M. J., Nickerson, D. A., Bernier, R., Fisher, S. E., Shendure, J., and Eichler, E. E. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589.
Ozelius, L. J., Lubarr, N., and Bressman, S. B. (2011). Milestones in dystonia. Mov. Disord. 26, 1106–1126.
Paisan-Ruiz, C., Dogu, O., Yilmaz, A., Houlden, H., and Singleton, A. (2008a). SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70, 1384–1389.
Paisan-Ruiz, C., Guevara, R., Federoff, M., Hanagasi, H., Sina, F., Elahi, E., Schneider, S. A., Schwingenschuh, P., Bajaj, N., Emre, M., Singleton, A. B., Hardy, J., Bhatia, K. P., Brandner, S., Lees, A. J., and Houlden, H. (2010). Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov. Disord. 25, 1791–1800.
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., Van Der Brug, M., Lopez De Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., De Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W., and Singleton, A. B. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600.
Paisan-Ruiz, C., Saenz, A., Lopez De Munain, A., Marti, I., Martinez Gil, A., Marti-Masso, J. F., and Perez-Tur, J. (2005). Familial Parkinson’s disease: clinical and genetic analysis of four basque families. Ann. Neurol. 57, 365–372.
Pakula, A. T., Van Naarden Braun, K., and Yeargin-Allsopp, M. (2009). Cerebral palsy: classification and epidemiology. Phys. Med. Rehabil. Clin. N. Am. 20, 425–452.
Pierson, T. M., Adams, D., Bonn, F., Martinelli, P., Cherukuri, P. F., Teer, J. K., Hansen, N. F., Cruz, P., Mullikin for the Nisc Comparative Sequencing Program, J. C., Blakesley, R. W., Golas, G., Kwan, J., Sandler, A., Fuentes Fajardo, K., Markello, T., Tifft, C., Blackstone, C., Rugarli, E. I., Langer, T., Gahl, W. A., and Toro, C. (2011a). Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 7, e1002325.
Pierson, T. M., Simeonov, D. R., Sincan, M., Adams, D. A., Markello, T., Golas, G., Fuentes-Fajardo, K., Hansen, N. F., Cherukuri, P. F., Cruz, P., Blackstone, C., Tifft, C., Boerkoel, C. F., and Gahl, W. A. (2011b). Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration. Eur. J. Hum. Genet. 20, 476–479.
Quintero-Rivera, F., Chan, A., Donovan, D. J., Gusella, J. F., and Ligon, A. H. (2007). Disruption of a synaptotagmin (SYT14) associated with neurodevelopmental abnormalities. Am. J. Med. Genet. A 143, 558–563.
Rademakers, R., Baker, M., Nicholson, A. M., Rutherford, N. J., Finch, N., Soto-Ortolaza, A., Lash, J., Wider, C., Wojtas, A., Dejesus-Hernandez, M., Adamson, J., Kouri, N., Sundal, C., Shuster, E. A., Aasly, J., Mackenzie, J., Roeber, S., Kretzschmar, H. A., Boeve, B. F., Knopman, D. S., Petersen, R. C., Cairns, N. J., Ghetti, B., Spina, S., Garbern, J., Tselis, A. C., Uitti, R., Das, P., Van Gerpen, J. A., Meschia, J. F., Levy, S., Broderick, D. F., Graff-Radford, N., Ross, O. A., Miller, B. B., Swerdlow, R. H., Dickson, D. W., and Wszolek, Z. K. (2011). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44, 200–205.
Rampello, L., Alvano, A., Battaglia, G., Bruno, V., Raffaele, R., and Nicoletti, F. (2006). Tic disorders: from pathophysiology to treatment. J. Neurol. 253, 1–15.
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., Schymick, J. C., Laaksovirta, H., Van Swieten, J. C., Myllykangas, L., Kalimo, H., Paetau, A., Abramzon, Y., Remes, A. M., Kaganovich, A., Scholz, S. W., Duckworth, J., Ding, J., Harmer, D. W., Hernandez, D. G., Johnson, J. O., Mok, K., Ryten, M., Trabzuni, D., Guerreiro, R. J., Orrell, R. W., Neal, J., Murray, A., Pearson, J., Jansen, I. E., Sondervan, D., Seelaar, H., Blake, D., Young, K., Halliwell, N., Callister, J. B., Toulson, G., Richardson, A., Gerhard, A., Snowden, J., Mann, D., Neary, D., Nalls, M. A., Peuralinna, T., Jansson, L., Isoviita, V. M., Kaivorinne, A. L., Holtta-Vuori, M., Ikonen, E., Sulkava, R., Benatar, M., Wuu, J., Chio, A., Restagno, G., Borghero, G., Sabatelli, M., Heckerman, D., Rogaeva, E., Zinman, L., Rothstein, J. D., Sendtner, M., Drepper, C., Eichler, E. E., Alkan, C., Abdullaev, Z., Pack, S. D., Dutra, A., Pak, E., Hardy, J., Singleton, A., Williams, N. M., Heutink, P., Pickering-Brown, S., Morris, H. R., Tienari, P. J., and Traynor, B. J. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268.
Ridge, S. A., Worwood, M., Oscier, D., Jacobs, A., and Padua, R. A. (1990). FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc. Natl. Acad. Sci. U.S.A. 87, 1377–1380.
Riviere, J. B., Ramalingam, S., Lavastre, V., Shekarabi, M., Holbert, S., Lafontaine, J., Srour, M., Merner, N., Rochefort, D., Hince, P., Gaudet, R., Mes-Masson, A. M., Baets, J., Houlden, H., Brais, B., Nicholson, G. A., Van Esch, H., Nafissi, S., De Jonghe, P., Reilly, M. M., Timmerman, V., Dion, P. A., and Rouleau, G. A. (2011). KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 89, 219–230.
Rochette, J., Roll, P., and Szepetowski, P. (2008). Genetics of infantile seizures with paroxysmal dyskinesia: the infantile convulsions and choreoathetosis (ICCA) and ICCA-related syndromes. J. Med. Genet. 45, 773–779.
Salinas, S., Proukakis, C., Crosby, A., and Warner, T. T. (2008). Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127–1138.
Shatunov, A., Mok, K., Newhouse, S., Weale, M. E., Smith, B., Vance, C., Johnson, L., Veldink, J. H., Van Es, M. A., Van Den Berg, L. H., Robberecht, W., Van Damme, P., Hardiman, O., Farmer, A. E., Lewis, C. M., Butler, A. W., Abel, O., Andersen, P. M., Fogh, I., Silani, V., Chio, A., Traynor, B. J., Melki, J., Meininger, V., Landers, J. E., Mcguffin, P., Glass, J. D., Pall, H., Leigh, P. N., Hardy, J., Brown, R. H. Jr., Powell, J. F., Orrell, R. W., Morrison, K. E., Shaw, P. J., Shaw, C. E., and Al-Chalabi, A. (2010). Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 9, 986–994.
Sheerin, U. M., Charlesworth, G., Bras, J., Guerreiro, R., Bhatia, K., Foltynie, T., Limousin, P., Silveira-Moriyama, L., Lees, A., and Wood, N. (2011). Screening for VPS35 mutations in Parkinson’s disease. Neurobiol. Aging 33, 838.e831–e835.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S. W., Hernandez, D. G., Kruger, R., Federoff, M., Klein, C., Goate, A., Perlmutter, J., Bonin, M., Nalls, M. A., Illig, T., Gieger, C., Houlden, H., Steffens, M., Okun, M. S., Racette, B. A., Cookson, M. R., Foote, K. D., Fernandez, H. H., Traynor, B. J., Schreiber, S., Arepalli, S., Zonozi, R., Gwinn, K., Van Der Brug, M., Lopez, G., Chanock, S. J., Schatzkin, A., Park, Y., Hollenbeck, A., Gao, J., Huang, X., Wood, N. W., Lorenz, D., Deuschl, G., Chen, H., Riess, O., Hardy, J. A., Singleton, A. B., and Gasser, T. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312.
Stanley, E. R., Berg, K. L., Einstein, D. B., Lee, P. S., Pixley, F. J., Wang, Y., and Yeung, Y. G. (1997). Biology and action of colony – stimulating factor-1. Mol. Reprod. Dev. 46, 4–10.
Stevanin, G., Santorelli, F. M., Azzedine, H., Coutinho, P., Chomilier, J., Denora, P. S., Martin, E., Ouvrard-Hernandez, A. M., Tessa, A., Bouslam, N., Lossos, A., Charles, P., Loureiro, J. L., Elleuch, N., Confavreux, C., Cruz, V. T., Ruberg, M., Leguern, E., Grid, D., Tazir, M., Fontaine, B., Filla, A., Bertini, E., Durr, A., and Brice, A. (2007). Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 39, 366–372.
Subramony, S. H. (2012b). Overview of autosomal dominant ataxias. Handb. Clin. Neurol. 103, 389–398.
Sundaram, S. K., Huq, A. M., Sun, Z., Yu, W., Bennett, L., Wilson, B. J., Behen, M. E., and Chugani, H. T. (2011). Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann. Neurol. 69, 901–904.
Sundaram, S. K., Huq, A. M., Wilson, B. J., and Chugani, H. T. (2010). Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 74, 1583–1590.
Szepetowski, P., Rochette, J., Berquin, P., Piussan, C., Lathrop, G. M., and Monaco, A. P. (1997). Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am. J. Hum. Genet. 61, 889–898.
Tomita, H., Nagamitsu, S., Wakui, K., Fukushima, Y., Yamada, K., Sadamatsu, M., Masui, A., Konishi, T., Matsuishi, T., Aihara, M., Shimizu, K., Hashimoto, K., Mineta, M., Matsushima, M., Tsujita, T., Saito, M., Tanaka, H., Tsuji, S., Takagi, T., Nakamura, Y., Nanko, S., Kato, N., Nakane, Y., and Niikawa, N. (1999). Paroxysmal kinesigenic choreoathetosis locus maps to chromosome 16p11.2-q12.1. Am. J. Hum. Genet. 65, 1688–1697.
Tsurusaki, Y., Okamoto, N., Suzuki, Y., Doi, H., Saitsu, H., Miyake, N., and Matsumoto, N. (2011). Exome sequencing of two patients in a family with atypical X-linked leukodystrophy. Clin. Genet. 80, 161–166.
Tucci, A., Nalls, M. A., Houlden, H., Revesz, T., Singleton, A. B., Wood, N. W., Hardy, J., and Paisan-Ruiz, C. (2010). Genetic variability at the PARK16 locus. Eur. J. Hum. Genet. 18, 1356–1359.
Van Es, M. A., Veldink, J. H., Saris, C. G., Blauw, H. M., Van Vught, P. W., Birve, A., Lemmens, R., Schelhaas, H. J., Groen, E. J., Huisman, M. H., Van Der Kooi, A. J., De Visser, M., Dahlberg, C., Estrada, K., Rivadeneira, F., Hofman, A., Zwarts, M. J., Van Doormaal, P. T., Rujescu, D., Strengman, E., Giegling, I., Muglia, P., Tomik, B., Slowik, A., Uitterlinden, A. G., Hendrich, C., Waibel, S., Meyer, T., Ludolph, A. C., Glass, J. D., Purcell, S., Cichon, S., Nothen, M. M., Wichmann, H. E., Schreiber, S., Vermeulen, S. H., Kiemeney, L. A., Wokke, J. H., Cronin, S., Mclaughlin, R. L., Hardiman, O., Fumoto, K., Pasterkamp, R. J., Meininger, V., Melki, J., Leigh, P. N., Shaw, C. E., Landers, J. E., Al-Chalabi, A., Brown, R. H. Jr., Robberecht, W., Andersen, P. M., Ophoff, R. A., and Van Den Berg, L. H. (2009). Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087.
Vaurs-Barriere, C., Deville, M., Sarret, C., Giraud, G., Des Portes, V., Prats-Vinas, J. M., De Michele, G., Dan, B., Brady, A. F., Boespflug-Tanguy, O., and Touraine, R. (2009). Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann. Neurol. 65, 114–118.
Verkerk, A. J., Schot, R., Dumee, B., Schellekens, K., Swagemakers, S., Bertoli-Avella, A. M., Lequin, M. H., Dudink, J., Govaert, P., Van Zwol, A. L., Hirst, J., Wessels, M. W., Catsman-Berrevoets, C., Verheijen, F. W., De Graaff, E., De Coo, I. F., Kros, J. M., Willemsen, R., Willems, P. J., Van Der Spek, P. J., and Mancini, G. M. (2009). Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am. J. Hum. Genet. 85, 40–52.
Vilarino-Guell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., Soto-Ortolaza, A. I., Cobb, S. A., Wilhoite, G. J., Bacon, J. A., Behrouz, B., Melrose, H. L., Hentati, E., Puschmann, A., Evans, D. M., Conibear, E., Wasserman, W. W., Aasly, J. O., Burkhard, P. R., Djaldetti, R., Ghika, J., Hentati, F., Krygowska-Wajs, A., Lynch, T., Melamed, E., Rajput, A., Rajput, A. H., Solida, A., Wu, R. M., Uitti, R. J., Wszolek, Z. K., Vingerhoets, F., and Farrer, M. J. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167.
Wang, J. L., Cao, L., Li, X. H., Hu, Z. M., Li, J. D., Zhang, J. G., Liang, Y., San, A., Li, N., Chen, S. Q., Guo, J. F., Jiang, H., Shen, L., Zheng, L., Mao, X., Yan, W. Q., Zhou, Y., Shi, Y. T., Ai, S. X., Dai, M. Z., Zhang, P., Xia, K., Chen, S. D., and Tang, B. S. (2011). Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 134, 3493–3501.
Wang, J. L., Yang, X., Xia, K., Hu, Z. M., Weng, L., Jin, X., Jiang, H., Zhang, P., Shen, L., Guo, J. F., Li, N., Li, Y. R., Lei, L. F., Zhou, J., Du, J., Zhou, Y. F., Pan, Q., Wang, J., Li, R. Q., and Tang, B. S. (2010). TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 133, 3510–3518.
Weedon, M. N., Hastings, R., Caswell, R., Xie, W., Paszkiewicz, K., Antoniadi, T., Williams, M., King, C., Greenhalgh, L., Newbury-Ecob, R., and Ellard, S. (2011). Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 89, 308–312.
Zimprich, A., Benet-Pages, A., Struhal, W., Graf, E., Eck, S. H., Offman, M. N., Haubenberger, D., Spielberger, S., Schulte, E. C., Lichtner, P., Rossle, S. C., Klopp, N., Wolf, E., Seppi, K., Pirker, W., Presslauer, S., Mollenhauer, B., Katzenschlager, R., Foki, T., Hotzy, C., Reinthaler, E., Harutyunyan, A., Kralovics, R., Peters, A., Zimprich, F., Brucke, T., Poewe, W., Auff, E., Trenkwalder, C., Rost, B., Ransmayr, G., Winkelmann, J., Meitinger, T., and Strom, T. M. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175.
Keywords: next-generation sequencing, movement disorders, gene discovery, novel neurological phenotypes
Citation: Krebs CE and Paisán-Ruiz C (2012) The use of next-generation sequencing in movement disorders. Front. Gene. 3:75. doi: 10.3389/fgene.2012.00075
Received: 15 February 2012; Paper pending published: 27 February 2012;
Accepted: 21 April 2012; Published online: 14 May 2012.
Edited by:
Berit Kerner, University of California Los Angeles, USAReviewed by:
Berit Kerner, University of California Los Angeles, USAYijin Yan, The University of Chicago Medical Center, USA
Copyright: © 2012 Krebs and Paisán-Ruiz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Coro Paisán-Ruiz, Department of Neurology, Mount Sinai School of Medicine, Box 1137, 1468 Madison Avenue, Room A22-24C, New York, NY 10029, USA. e-mail:Y29yby5wYWlzYW4tcnVpekBtc3NtLmVkdQ==