 D. Zhi1†
D. Zhi1† M. R. Irvin2† C. C. Gu3
M. R. Irvin2† C. C. Gu3 A. J. Stoddard4 R. Lorier4 A. Matter4 D. C. Rao3 V. Srinivasasainagendra1 H. K. Tiwari1
A. J. Stoddard4 R. Lorier4 A. Matter4 D. C. Rao3 V. Srinivasasainagendra1 H. K. Tiwari1 A. Turner4 U. Broeckel4†
A. Turner4 U. Broeckel4† D. K. Arnett2*†
D. K. Arnett2*†- 1 Department of Biostatistics, University of Alabama at Birmingham, Birmingham, AL, USA
- 2 Department of Epidemiology, University of Alabama at Birmingham, Birmingham, AL, USA
- 3 Division of Biostatistics, Washington University in St. Louis, St. Louis, MO, USA
- 4 Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, USA
Rationale: Left ventricular hypertrophy (LVH) is a heritable predictor of cardiovascular disease, particularly in blacks. Objective: Determine the feasibility of combining evidence from two distinct but complementary experimental approaches to identify novel genetic predictors of increased LV mass. Methods: Whole-exome sequencing (WES) was conducted in seven African-American sibling trios ascertained on high average familial LV mass indexed to height (LVMHT) using Illumina HiSeq technology. Identified missense or nonsense (MS/NS) mutations were examined for association with LVMHT using linear mixed models adjusted for age, sex, body weight, and familial relationship. To functionally assess WES findings, human induced pluripotent stem cell-derived cardiomyocytes (induced pluripotent stem cell-CM) were stimulated to induce hypertrophy; mRNA sequencing (RNA-seq) was used to determine gene expression differences associated with hypertrophy onset. Statistically significant findings under both experimental approaches identified LVH candidate genes. Candidate genes were further prioritized by seven supportive criteria that included additional association tests (two criteria), regional linkage evidence in the larger HyperGEN cohort (one criterion), and publically available gene and variant based annotations (four criteria). Results: WES reads covered 91% of the target capture region (of size 37.2 MB) with an average coverage of 65×. WES identified 31,426 MS/NS mutations among the 21 individuals. A total of 295 MS/NS variants in 265 genes were associated with LVMHT with q-value <0.25. Of the 265 WES genes, 44 were differentially expressed (P < 0.05) in hypertrophied cells. Among the 44 candidate genes identified, 5, including HLA-B, HTT, MTSS1, SLC5A12, and THBS1, met 3 of 7 supporting criteria. THBS1 encodes an adhesive glycoprotein that promotes matrix preservation in pressure-overload LVH. THBS1 gene expression was 34% higher in hypertrophied cells (P = 0.0003) and a predicted conserved and damaging NS variant in exon 13 (A2099G) was significantly associated with LVHMT (P = 4 × 10−6). Conclusion: Combining evidence from cutting-edge genetic and cellular experiments can enable identification of novel LVH risk loci.
Introduction
Echocardiographic measurement of increased left ventricular (LV) mass predicts cardiovascular morbidity and mortality across demographic groups (Benjamin and Levy, 1999). Beyond the established risk factors for LV hypertrophy (LVH; race, age, hypertension, obesity), research convincingly suggests there is a genetic basis of disease (Harshfield et al., 1990). African-American populations may be enriched for risk variants as LVH burden is about twofold greater in African Americans compared to Caucasians and LV mass is strongly correlated in hypertensive African-American siblings (Arnett et al., 2001). Previous linkage (Arnett et al., 2009a; Tang et al., 2009), candidate gene (Rasmussen-Torvik et al., 2005), and genome-wide association studies (GWAS; Arnett et al., 2009b, 2011; Wineinger et al., 2011) in carefully ascertained and well characterized African-American cohorts have uncovered significant LVH risk loci; however, these loci explain only a fraction of the phenotypic variation attributable to familial inheritance.
Advances in targeted capture and high throughput sequencing have recently made whole-exome sequencing (WES) affordable for clinical studies and offer advantages over array-based genotyping for the identification of novel disease-associated loci. The approach has proved powerful for the study of Mendelian disease where the identification of rare, mostly coding mutations clustering in patients is achieved through strict, discrete bioinformatics filters (Robinson et al., 2011). Indeed, this approach has been applied to identify genes underlying familial dilated cardiomyopathy (Norton et al., 2011) and others (Ng et al., 2010a,b; Majewski et al., 2011). Recently, this approach has been extended to non-Mendelian disease (O’Roak et al., 2011; Ramagopalan et al., 2011; Kim et al., 2012). It has been suggested that rare variation within pedigrees can contribute substantially to the heritability of common traits, offering advantages to family based designs (Manolio et al., 2009; Shi and Rao, 2011) such as the non-random ascertainment of extreme phenotype families (Shi and Rao, 2011). The application of WES to uncover genes associated with cardiovascular disease-related quantitative traits such as those that measure LVH has not yet been reported.
A major challenge of genomic studies is the functional assessment of a statistically significant finding in the relevant tissue, which, in the case of LVH, is cardiomyocytes. Until the recent development of stem cell technology, human adult cardiomyocytes were generally not available for functional analyses. Although human adult ventricular myocytes can be cultured, they are not optimal for biochemical and molecular biologic investigations (Berry et al., 2007). Recently, the use of human induced pluripotent stem cell (iPSC)-derived cardiomyocytes allows direct interrogation of the molecular mechanisms occurring in these cells under differing experimental conditions. Further, this model system aims to provide the ability to identify disease pathways and therapeutic targets, which could ultimately lead to more specific, tailored LVH treatment. Therefore, an approach using WES in combination with functional studies in iPSC-derived cardiomyocytes can provide a powerful platform for disease gene discovery.
In the current study we conducted WES to isolate novel loci for LVH in seven hypertensive African-American sibling trios from the Hypertension Genetic Epidemiology Network (HyperGEN) that were enriched for increased LV mass. Concurrently, we implemented an experimental protocol using a novel LVH model system based on human iPSC-derived cardiomyocytes. These cardiomyocytes were subjected to conditions to produce hypertrophy, and mRNA expression was measured in comparison to control cardiomyocytes generated from the same iPSC line. We show combining results from WES and differential gene expression in the LVH model system, may identify novel candidate genes supported by statistical and annotation-based criteria.
Materials and Methods
Study Population
The HyperGEN study has been previously described (Williams et al., 2000). Briefly, HyperGEN is part of the Family Blood Pressure Program funded by the National Heart Lung and Blood Institute and was designed to study the genetics of hypertension and related conditions. Families were drawn from population-based cohorts or the community-at-large if sibships had ≥2 siblings who had been diagnosed with hypertension before age 60. The study was later extended to include siblings and offspring of the original sibpairs. Hypertension was defined as current antihypertensive medication use or having an average systolic blood pressure ≥140 mm Hg and/or diastolic blood pressure ≥90 mm Hg measured at two clinic visits. Two of four centers (AL, NC) recruited 1,264 African Americans making up 470 families. This study was approved by all local institutional review boards (University of Minnesota’s Human Research Protection Program Institutional Review Board, University of Alabama at Birmingham’s Institutional Review Board for Human Use, University of Utah’s Institutional Review Board, University of North Carolina at Chapel Hill’s Office of Human Research Ethics Biomedical Institutional Review Board, Boston University’s Medical Campus Institutional Review Board, Medical College of Wisconsin’s Institutional Review Board); all subjects gave informed consent. In the current study, seven African-American sibling trios with history of hypertension and average age <55 years ascertained on the highest average familial LVMHT were selected for exome sequencing.
Blood Pressure Measurement and Antihypertensive Medications
Systolic and diastolic blood pressure is reported as the average of the second and third measures of a series of six sitting blood pressure measurements. Antihypertensive medication treatment was defined as use of drug(s) belonging to one of the following six classes at the time of the study including diuretics, ace inhibitors, beta blockers, alpha blockers, calcium channel blockers, and angiotensin 2 receptor antagonists.
Echocardiography
Doppler, two-dimensional (2D), and M-mode (2D-guided) echocardiograms were performed following a standardized protocol previously described (Devereux and Roman, 1995). Certified sonographers from each center were trained at the echocardiography reading center (New York Hospital-Cornell Medical Center). Measurements were made at the echocardiography reading center using a computerized review station equipped with a digitizing tablet and monitor overlay used for calibration and quantification (Digisonics, Inc., Houston, TX, USA). LVM was calculated using end-diastolic dimensions by an anatomically validated formula and indexed to height (m2.7; Devereux et al., 1984).
Whole-Exome Sequencing
Exome capture
Using Agilent SureSelect All Exome Capture 21, index-tagged, paired-end libraries were prepared. As recommended, 3 μg of genomic DNA diluted in 1× Low TE was sheared using a Covaris instrument with a subsequent end repair step. Prior to end repair, 1 μL of DNA was analyzed on an Agilent 2100 Bioanalyzer DNA 1000 chip. All samples recovered at least 2 μg of DNA post-shearing and had an electropherogram distribution peak between 150 and 200 nucleotides on the DNA 1000 chip.
After end repair, the samples were purified using Agencourt AMPure XP beads, and the purified DNA then had “A” Bases added to the 3′ end of the DNA fragments. After “A” base addition, the DNA was again purified with AMPure XP beads. Ligation of the indexing-specific, paired-end adapter was done, and the product was AMPure XP bead purified, then PCR amplified using the Illumina InPE1.0 (forward) PCR primer and the SureSelect Indexing Pre-Capture PCR primer. For PCR, five cycles of amplification were used.
After PCR amplification, amplified product was AMPure XP bead purified and 1 μL was analyzed on an Agilent 2100 Bioanalyzer DNA 1000 chip. All samples showed an electropherogram distribution peak between 250 and 275 nucleotides on the DNA 1000 chip and had a concentration of at least 147 ng/μL.
For hybridization, all samples needed to be at a concentration of 147 ng/μL. To achieve this, an Eppendorf Vacufuge Plus concentrator was used to completely lyophilize the samples. Using the concentrations calculated from results obtained with the DNA 1000 chip, an appropriate amount of nuclease-free water was added to each sample post-complete lyophilization to bring the concentration to 147 ng/μL. Hybridization using the Agilent Hybridization protocol and the Agilent SureSelect All Exome Capture Library was performed. Hybridization was stopped at 24 h. All samples contained at least 20 μL, indicating an optimal capture, and samples were then bead selected using the SureSelect selection protocol utilizing Dynal MyOne Streptavidin T1 (Invitrogen) magnetic beads.
Once selected, the captured libraries were then AMPure XP bead purified and Index barcode tags were added. As recommended for pooling two samples of our capture size, Agilent Index’s #6 (GCCAAT) and #12 (CTTGTA) were used. Each sample received one tag, with half of the samples receiving #6 and the other half #12, one bar-coded with #6 and one with #12, and pooled together on one sequencing flow cell lane. During addition of the index tag, the libraries were amplified with SureSelect Indexing Post-Capture PCR (Forward) Primer and Index PCR (Reverse) Primer, with the Index PCR Primer being the sample-specific index barcode tag. For PCR, 16 cycles of amplification were used.
The prepared libraries were AMPure XP bead purified and 1 μL of library was run out on an Agilent 2100 Bioanalyzer DNA High Sensitivity chip. All samples had an electropherogram distribution peak between 300 and 325 nucleotides on the DNA High Sensitivity chip. To more accurately quantify the libraries for pooling, samples were quantified using the Agilent QPCR NGS Library Quantification Kit.
After quantification samples were pooled to a volume of 20 μL with an equimolar amount of 10 nM, following Agilent’s multiplexing pooling protocol. Each pool was spiked with 1% phiX control to improve base calling while sequencing, as was recommended by Illumina for pooling of two libraries.
Illumina sequencing
Following Illumina HiSeq sequencing and cBot protocols, each of the pooled, multiplexed, index-tagged, paired-end libraries was denatured, underwent cluster generation onto a HiSeq v1.5 flow cell and was sequenced.
Each of the 10 nM libraries was denatured using the 4–8 pM procedure to generate a final concentration of 5 or 7 pM to load per lane for cluster generation. Once cluster generation onto flow cells was complete, samples were sequenced using the Illumina HiSeq Sequencing Kit (200 cycles) and multiplexing sequencing chemistry.
Exome read mapping and variant calling method
Basecalling, demultiplexing, read mapping, and initial SNP calling were done using Illumina’s CASAVA v1.7 software. Read mapping was to the whole autosomal sequence from UCSC hg19 (also known as GRCh37), using the default parameters to the ELAND2 program.
Autosomal SNPs were called using the CASAVA v1.7 variant caller with default parameters, except that the chromosomal coverage variation filter was turned off to account for exome capture. SNPs were called where the CASAVA v1.7 score was greater than the default threshold of 10. Genotypes calling at the SNP sites (variant calling) were done by the SAMTools program v0.1.7-6 (r530; Li et al., 2009; Li, 2011). Multi-sampling joint variant calling for all SNP sites discovered by CASAVA was carried out by the mpileup command line with the default parameters. Genotype calls in the target capture region with PHRED-like genotype quality score GQ ≥ 30, totaling 102,089 variants sites, were annotated by the ANNOVAR program (version 2010-12-02; Wang et al., 2010) against the UCSC hg19 refGene annotation.
Statistical and Bioinformatics Methods for Whole-Exome Sequencing
In order to identify missense or nonsense (MS/NS) single-nucleotide variants (SNVs) associated with LVMHT we conducted a mixed model regression with LVMHT as the dependent variable controlling for kinship structures as well as age, sex, and weight as covariates using the kinship R package program LMEKIN (Lourenco et al., 2011). We used a false discovery rate (FDR) criterion of q-value <0.25 (P-value <0.00258) for significance; this is more flexible than the usual Bonferroni criterion given our small sample size (N = 21).
Induced Pluripotent Stem Cell-Derived Cardiomyocyte Experiments
Cell culture
For the cardiomyocyte studies, we utilized human iCell™ Cardiomyocytes derived from iPSCs (Cellular Dynamics International, Madison, WI, USA). iCells were recovered from frozen culture as recommended by the manufacturer and allowed to recover at least 12 days in iCell Maintenance Medium (iCMM) before experimentation. For experiments, cells were plated at 1.5 × 105 cells/well in iCMM, as recommended by the manufacturer for cell-based assays, and grown in pre-coated (0.1% gelatin solution), 12-well dishes at 37°C in a 95% air: 7% CO2 humidified atmosphere. After 14 days of recovery, iCells were washed twice with warmed PBS and starved in iCell serum-free Maintenance Medium (iCSM) for 48 h, then stimulated in iCMM, as described below.
Cellular model of LVH
Human iCell Cardiomyocytes were plated to confluence, allowed to recover before starving for 48 h, and stimulated with an established stimulant for the beta-adrenergic system, isopreterenol (ISO), for up to 72 h. As a correlate of developing LV hypertrophy, cell culture changes in cell surface area were measured. At least 200 randomly selected stimulated cells were measured as well as unstimulated controls. As shown in Figure 1, our data demonstrate that iCell Cardiomyocytes respond to hypertrophic stimuli by a clear and significant increase in relative cell size (ISO treatment versus control P = 0.0043). We also observed an up-regulation of hypertrophy markers such as intermediate-early genes C-FOS and C-JUN measured by immunochemistry (Lijnen and Petrov, 1999). Together, these data demonstrate successful establishment of a myocyte hypertrophy model in iPS cell-derived cardiomyocytes representative of the hypertrophy phenotype.
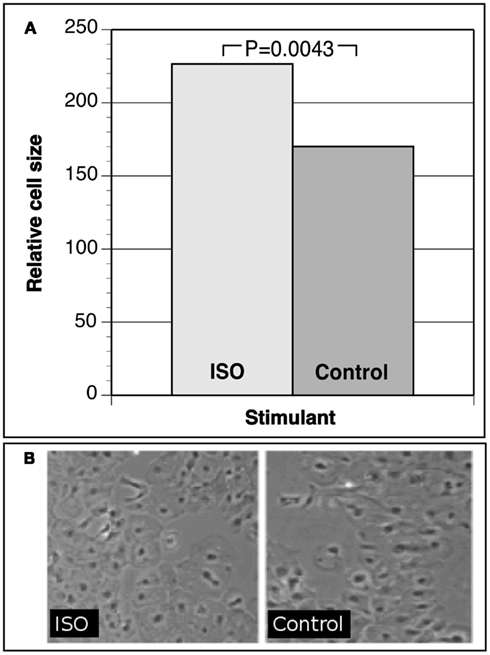
Figure 1. (A) Relative cell size of iCell cardiomyocytes under isoproterenol (ISO) hypertrophic stimulation versus control cells. (B) Photographs of iCell cardiomyocytes after hypertrophic stimulation versus controls.
Stimulation, cell harvest, and RNA extraction
iCells were stimulated for 72 h with ISO (10−5 M; Sigma Aldrich), replenishing every 24 h. After stimulation, cells were harvested with Trizol reagent and the Purlink™ RNA Mini Kit (Invitrogen). Total RNA was extracted per manufacturer’s recommendations, resuspended in nuclease-free water, and quantified/checked for integrity by UV spectrophotometry (NanoDrop™ 2000, Thermo Scientific).
RNA sequencing
Six paired-end cDNA libraries (three biological replicates of control iPSC cardiomyocytes, three biological replicates of isoproterenol stimulated cardiomyocytes) were prepared and sequenced using Illumina TruSeq RNA Sample Preparation Kit.
Total RNA was extracted and quantified. Following the TruSeq RNA sample preparation low-throughput protocol, 500 ng of total RNA was used to generate index-tagged paired-end cDNA libraries. During library preparation samples were each tagged with Agilent Index #2 (CGATGT), #4 (TGACCA), #5 (ACAGTG), or #6 (GCCAAT). After cDNA libraries were generated using the sample preparation kit, quality of the libraries was checked using 1 μL of sample run on an Agilent 2100 Bioanalyzer DNA 1000 chip. All samples showed an electropherogram peak at ∼260 base pair. Samples were then quantified using the Agilent QPCR NGS Library Quantification Kit.
After quantification, samples were pooled for multiplexing to a volume of 20 μL with an equimolar amount of 10 nM, following Agilent’s multiplexing pooling protocol. Each pool was spiked with 1% phiX control to improve base calling while sequencing, as was recommended by Illumina for pooling of two libraries.
Illumina sequencing
Following Illumina HiSeq sequencing and cBot protocols, each of the pooled, multiplexed, index-tagged, paired-end libraries was denatured, underwent cluster generation onto a HiSeq v1.5 flow cell and was sequenced.
Each of the 10 nM libraries was denatured using the 4–8 pM procedure to generate a final concentration of 6 pM to load per lane for cluster generation. Once cluster generation onto flow cells was complete, samples were sequenced using the Illumina HiSeq Sequencing Kit (200 cycles) and multiplexing sequencing chemistry.
RNA-seq assembly
Six paired-end cDNA libraries were sequenced (three biological replicates of control iPSC cardiomyocytes, three biological replicates of isoproterenol stimulated cardiomyocytes). Basecalling and demultiplexing were performed using CASAVA v1.8 from Ilumina. Paired-end fastq sequence reads from each sample were assembled against hg19 using the splicing aligner Tophat v1.3.1 (Trapnell et al., 2009) with the Illumina-supplied hg19 gene-model annotation file (gtf annotation).
RNA-seq differential expression
Splice-aligned reads were assigned to gene models using the software package HTSeq v0.5.3p3 (http://www-huber.embl.de/users/anders/HTSeq) and the hg19 gtf annotation. Fold-change and differential expression significance values were calculated from gene-level read counts using the DESeq Bioconductor R package v1.2.1 (Anders and Huber, 2010).
Bioinformatic and Statistical Methods for Prioritization of Candidate Genes
Variants with an association with LVMHT of q-value <0.25 are designated as candidate variants. For each candidate variant, its minor allele frequency (MAF) among African Americans in the NHLBI Exome Sequencing Project (ESP) was obtained from the ESP exome variant server v.0.0.6 http://snp.gs.washington.edu/EVS/ (released September 9, 2011); its GERP score and PolyPhen score were obtained from the SeattleSeqAnnotation131.
Candidate gene prioritization strategy
Information from different statistical analyses and bioinformatic functional annotations were used to generate a composite measure similar to that described by Gu et al. (2007) about the relevance of candidate genes for their potential roles in LVH. We prioritized candidate genes (identified through the intersection of WES findings and differential RNA expression in the cellular model of LVH) by the following seven criteria.
Variant association after exclusion of outlying case (P20). Many candidate variants significantly associated with LVMHT after correction for multiple testing described above are influenced by an extreme case with LVMHT of 114 g/m2.7 whom we considered may harbor important LVH variants. However, this individual will also harbor false positive findings so we additionally considered the association of candidate variants after exclusion of the extreme case using the regression models described above (N = 20). Add 1 to the composite prioritization score if any candidate variant P < 0.05, otherwise 0. Rationale: More weight is given to variants that are associated with LVMHT both with and without the most extreme case considered.
Gene-based association (GB). We calculated gene-based P-values for the association of a genetic score with LVMHT for 21 individuals. Gene-based genetic score was calculated as the weighted sum of variants, similar to Madsen and Browning (2009). Genetic score for gene j was defined as where K is the number MS/NS variants in the gene, Iij is the minor allele counts at variant i, ni is the number of individuals among 21 having a high quality genotype calling, and pi is the MAF of African Americans in ESP. For alleles with frequency 0 in ESP, we used an allele frequency of 0.0002, corresponding to the lowest allele frequency in the ESP exome collection, 1 heterozygous among ∼2,500 individuals. If only one missense or non-synonymous variant was present in a gene, the gene-based P-value was set to 1. Association of the gene-based score with LVMHT was tested using mixed linear regression similarly to the single-variant analysis described. Add 1 to the composite prioritization score if P < 0.05, otherwise 0. Rationale: Allelic heterogeneity may be a probable scenario for common disease where multiple rare variation considered together may explain a larger portion of the genetic basis of disease (Madsen and Browning, 2009).
Conservation (GERP). We considered the highest Genomic Evolutionary Rate Profiling (GERP) score (Davydov et al., 2010) among all candidate variants. We added 1 to the composite prioritization score if GERP score ≥5, otherwise 0. Rationale: GERP can be used to characterize genomic regions that have been subjected to purifying selection and are enriched for functional elements that may be predisposing to human disease (Cooper et al., 2005).
Functional annotation (PH). We used the Polymorphism Phenotyping (PolyPhen) annotation of candidate variants. We added 1 to the composite prioritization score if any of candidate gene variant was annotated as “probably damaging,” otherwise 0. Rationale: PolyPhen is predictive of the possible impact of an amino acid substitution on the structure and function of a human protein using physical and comparative considerations (Ramensky et al., 2002).
Minor allele frequency. We considered the minimal MAF of all candidate variants among African Americans from the NHLBI exome sequencing project (ESP). Add 1 to the composite prioritization score if MAF <0.01, otherwise 0. Rationale: Rare frequency of an SNV in a general reference population suggests enrichment of the SNV in an extreme phenotype population may be related to disease (Madsen and Browning, 2009).
Gene expression in cardiomyocyte (GNF). GNF GeneAtlas2 (Su et al., 2004) gene expression was obtained from the UCSC genome browser database. Add 1 to the composite prioritization score if expression score >1, otherwise 0. Rationale: This confirms expression in the relevant disease tissue.
Information from existing linkage studies (Linkage). Existing linkage analysis results for LV mass of the HyperGEN cohort was obtained from the results of a previous work in HyperGEN (Arnett et al., 2009a). Linkage peaks of LOD >1.75 were considered a hit (Rao and Province, 2000). We added 1 to the composite prioritization score if gene was within 50 kb region centered at a linkage hit, otherwise 0. Rationale: Linkage constitutes an independent statistical genetic approach for identifying rare and functional variants within multiply affected families.
Results
Table 1 presents demographic and phenotypic data measured for each of the seven sibling trios. Hypertension was well controlled in this population by medication. The average number of antihypertensive medications reported at the time of blood pressure measurement was 1.7 ± 1. In comparison to the entire HyperGEN African-American stratum (N = 1264, average LVMHT 42 ± 12 g/m2.7 and 25% LVH) this subset is enriched for LVH (LVMHT 59 ± 16 g/m2.7 and 66% LVH; Arnett et al., 2011). Additionally, intra-family LVH case counts ranging from 1 to 3 provided phenotypic variability necessary for gene-to-trait association analyses.
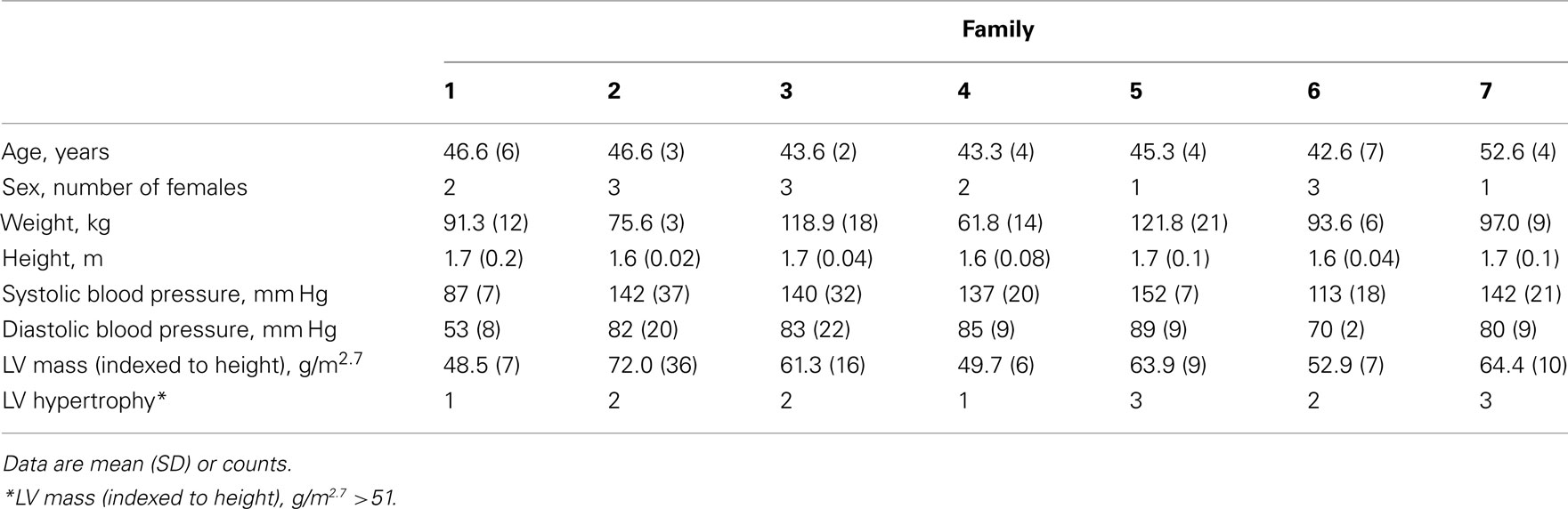
Table 1. Phenotypic values for seven hypertensive African-American sibling trios.
Whole-exome sequencing reads covered 91% of the 37.2 MB target capture region with an average coverage of 65× (Table 2). After applying variant calling and quality control filters described we identified 102,089 SNVs among the 21 individuals (Table 3). Of those variants, 31,426 are MS/NS mutations (Table 4) which were examined for association with LVMHT. For tallies of variant type and total per individual, see Table 3.
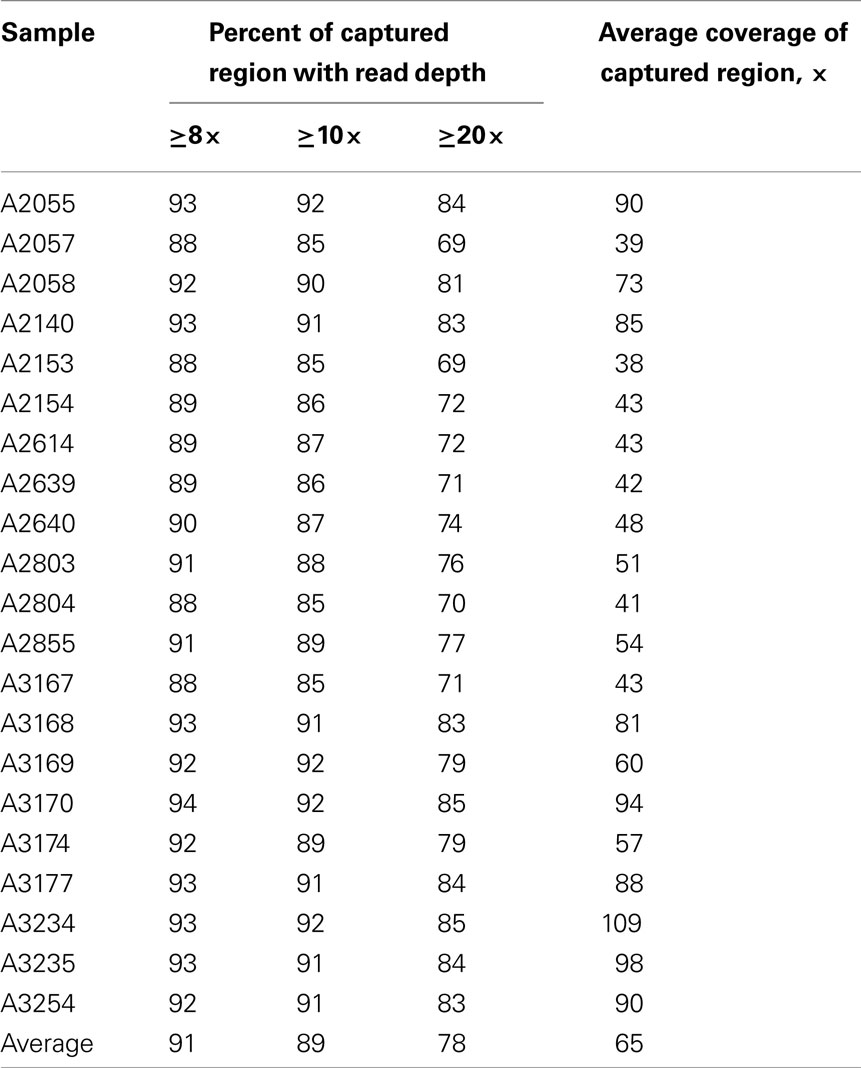
Table 2. Basic statistics of exome sequencing per sample.
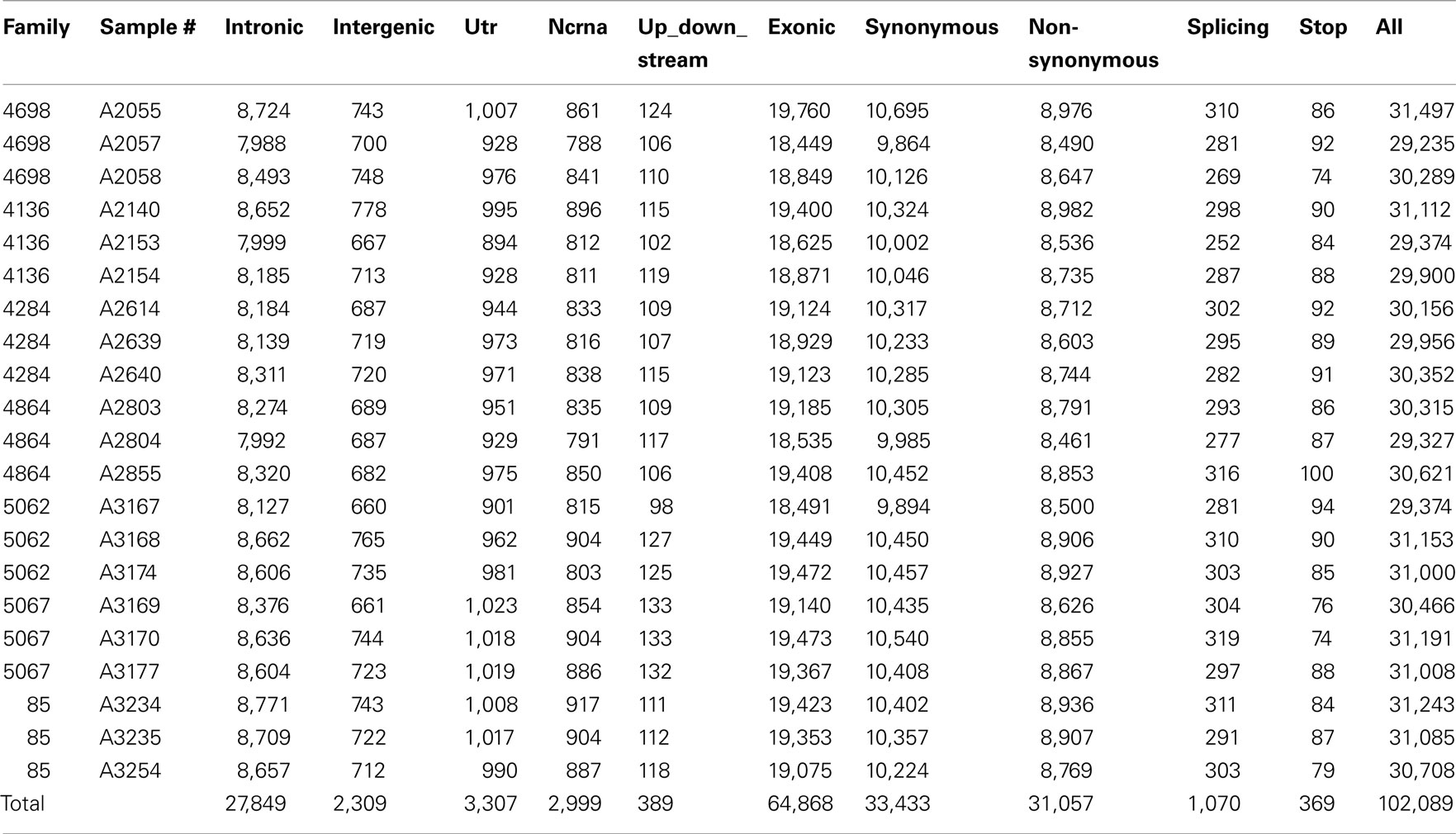
Table 3. Number of variants within individual by category.
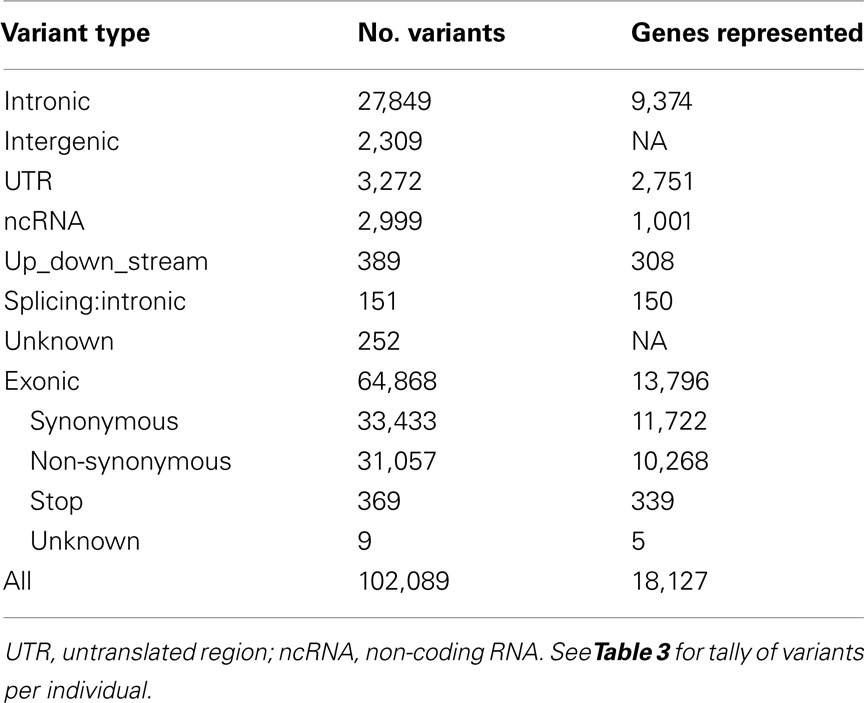
Table 4. Genetic variants found by WES and annotated by ANNOVAR.
Regression analyses yielded 295 MS/NS candidate variants in 265 genes (“WES genes”) that passed significance criteria for multiple testing (see Supplementary Material). DEseq RNA differential expression results revealed a total of 44 of the 265 WES genes were also differentially expressed with P < 0.05 in the iPSC model of LVH (see Supplementary Material). Those 44 “candidate genes” were further prioritized based on 7 supportive criteria (Table 5). We focus here on five genes that satisfy at least three of the seven criteria in Table 5. Among these five genes are major histocompatibility complex, class I, B (HLA-B), huntingtin (HTT), metastasis suppressor 1 (MTSS1), solute carrier family 5 (sodium/glucose cotransporter), member 12 (SLC5A12), and thrombospondin 1 (THBS1). Adjustment of DEseq P-values (Padj) for the genome-wide list of tested genes (N = 11,746 genes expressed in at least one experimental condition) yielded 11 of 44 of candidate genes significantly differently expressed (Padj < 0.05) including THBS1 (Padj = 0.009), but not any of the remaining prioritized candidates.
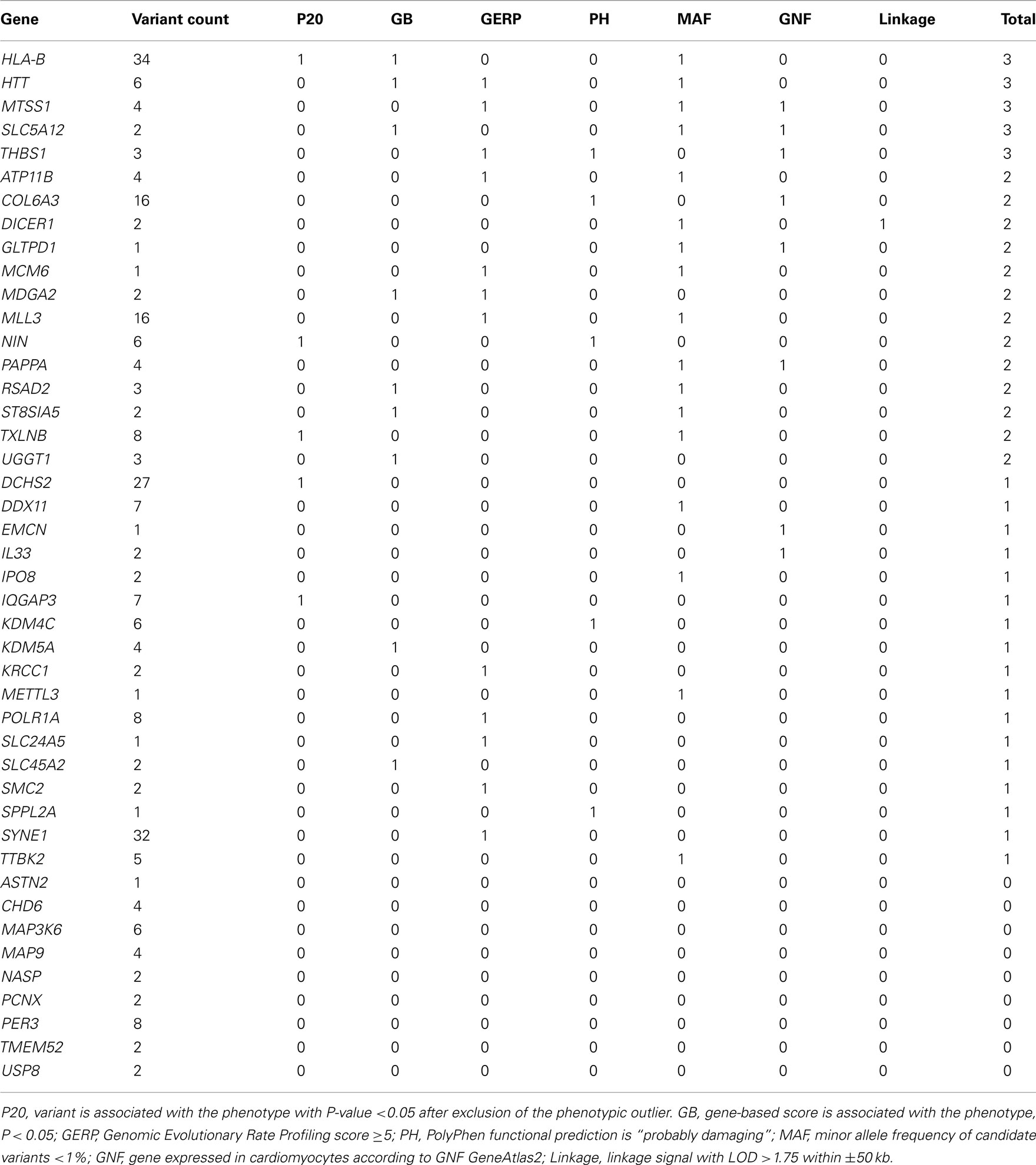
Table 5. Candidate gene (N = 44) prioritization given a composite score of seven supportive functional and/or statistical categories.
Discussion
Whole-exome sequencing provides new genetic information by identifying rare and potentially novel protein-coding variants not available on existing genotyping microarrays. Like previous genomic studies, the functional assessment of novel gene variants associated with LVH pathology identified through WES poses significant challenges. Here we present the first WES analysis of any common, quantitative trait in an African-American sample. We identified 295 variants in 265 genes associated with LV mass indexed to height through WES in 7 hypertensive sibling trios. To functionally assess our findings, we combined evidence obtained using RNA sequencing in a molecular model of LVH using human iPSC-derived cardiomyocytes. Using this approach we discovered 44 genes with evidence of a role in disease pathology and statistical association with LVMHT. We refined the list to five genes applying a prioritization strategy incorporating statistical and annotation-based bioinformatic filters. Among the five genes, THBS1 has previously been shown to promote matrix preservation and prevent chamber dilation in an animal model of LVH (Vanhoutte and Heymans, 2011; Xia et al., 2011) while the other genes are novel LVH candidates. Due to several limitations the findings presented in this manuscript are suggestive. Still we provide proof of concept that a novel cellular model of LVH is a promising platform for the functional assessment of genes highlighted via genomic discovery efforts.
THBS1 is an adhesive glycoprotein that mediates cell-to-cell and cell-to-matrix interactions. It inhibits angiogenesis and activates latent transforming growth factor beta, a protein related to cellular differentiation in many tissues. In a recent report, THBS1-null mice had accentuated cardiac hypertrophy (Xia et al., 2011). In the RNA-seq experiment we observed a 1.34-fold increase in THBS1 expression (P = 0.003) after ISO stimulation (see Supplementary Material) consistent with the up-regulation of the protein in disease. These points make genetic disruption of the protein in humans an interesting topic for follow-up research.
Among the remaining genes we report on, MTSS1 functions in cell proliferation. It is a suspected scaffold protein that interacts with multiple partners to regulate actin dynamics; its down-regulation has been observed in multiple cancer types (Xie et al., 2011). HLA-B is part of a family of genes making up the immune system’s HLA complex which aids in the body’s reaction to a wide range of pathogens. A SNP near HLA-B (rs2523586) was recently shown to be associated (P = 1 × 10−6) with diastolic blood pressure (DBP) in African Americans as part of the Candidate Gene Association Resource (CARe) consortium (N = 8,592) although this effect did not replicate in an independent African-American population (Fox et al., 2011). Trinucleotide repeats in HTT are known to cause Huntington’s disease, although a biological link to LVH is unlikely. Finally, SLC5A12 is a sodium-coupled monocarboxylate transporter indicated in the renal handling of lactate and urate (Thangaraju et al., 2006; Ganapathy et al., 2008). Uric acid has a strong link to CVD and hyperuricemia has been linked to ventricular remodeling in an animal model (Chen et al., 2011; Isik et al., 2012).
We note several limitations to our study. Specifically, our sample size was small and not sufficiently powered to identify single genes and variants associated with LV mass solely through statistical modeling approaches. Additionally, we did not directly test the functional effect of the identified variants using the human iPSC-derived cardiomyocyte model of LVH, rather we relied on differential RNA expression of the corresponding gene to suggest variant functional association to the observed pathology. Many questions remain whether the expression pattern in a cell culture model fully resembles the molecular changes of cardiomyocytes in a complex organ such as the heart (Kong et al., 2010). However, we and others (Carvajal-Vergara et al., 2010) have observed well established changes previously described as characteristic for LVH. Plus, iPSC cardiomyocytes have been used extensively for the study of other cardiovascular disease phenotypes, for example human cardiac cellular electrophysiology (Yokoo et al., 2009; Moretti et al., 2010; Germanguz et al., 2011; Itzhaki et al., 2011). Finally, we employed several cutoff criteria throughout our procedures which, if altered, could influence our findings. This includes FDR criteria for variant association with LVMHT and an un-weighted candidate gene prioritization strategy. Therefore, some false positive and alternatively, false negative findings, may have resulted and further replication is required. Still, we present a procedure designed to limit such false findings by combining evidence from genetic and cellular experiments and further prioritizing our results based on rich evidence from existing studies and publically available databases.
In conclusion, we employed an innovative, iterative approach to identify protein-coding variants associated with LVH in African-American hypertensives. The identified genes with significant variants are linked to cell proliferation, cell adhesion, solute handing, and injury repair. One candidate, THBS1, is involved in injury response in multiple tissues, has been linked to cardiac hypertrophy in an animal model, and is upregulated in our novel cellular model of disease. Results necessitate replication and questions remain about the mechanistic relevance of the specific variants in the detected genes, however the results presented support the expansion of this research. Ultimately, we describe how progress in the discovery of genetic risk factors for LVH may benefit from a tiered approach that integrates evidence from new and existing data including a novel cellular model of LVH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
HyperGEN: Genetics of Left Ventricular Hypertrophy, ancillary to the Family Blood Pressure Program, http://clinicaltrials.gov/ct/show/NCT00005267. Funding sources included National Heart, Lung, and Blood Institute grant R01 HL55673 and cooperative agreements (U01) with the National Heart, Lung, and Blood Institute: HL54471, HL54515 (UT); HL54472, HL54496 (MN); HL54473 (DCC); HL54495 (AL); HL54509 (NC), and HL107437.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Applied_Genetic_Epidemiology/10.3389/fgene.2012.00092/abstract
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106.
Arnett, D. K., Devereux, R. B., Rao, D. C., Li, N., Tang, W., Kraemer, R., Claas, S. A., Leon, J. M., and Broeckel, U. (2009a). Novel genetic variants contributing to left ventricular hypertrophy: the HyperGEN study. J. Hypertens. 27, 1585–1593.
Arnett, D. K., Li, N., Tang, W., Rao, D. C., Devereux, R. B., Claas, S. A., Kraemer, R., and Broeckel, U. (2009b). Genome-wide association study identifies single-nucleotide polymorphism in KCNB1 associated with left ventricular mass in humans: the HyperGEN Study. BMC Med. Genet. 10, 43. doi:10.1186/1471-2350-10-43
Arnett, D. K., Hong, Y., Bella, J. N., Oberman, A., Kitzman, D. W., Hopkins, P. N., Rao, D. C., and Devereux, R. B. (2001). Sibling correlation of left ventricular mass and geometry in hypertensive African Americans and whites: the HyperGEN study. Hypertension genetic epidemiology network. Am. J. Hypertens. 14, 1226–1230.
Arnett, D. K., Meyers, K. J., Devereux, R. B., Tiwari, H. K., Gu, C. C., Vaughan, L. K., Perry, R. T., Patki, A., Claas, S. A., Sun, Y. V., Broeckel, U., and Kardia, S. L. (2011). Genetic variation in NCAM1 contributes to left ventricular wall thickness in hypertensive families. Circ. Res. 108, 279–283.
Benjamin, E. J., and Levy, D. (1999). Why is left ventricular hypertrophy so predictive of morbidity and mortality? Am. J. Med. Sci. 317, 168–175.
Berry, J. M., Haris Naseem, R., Rothermel, B. A., and Hill, J. A. (2007). Models of cardiac hypertrophy and transition to heart failure. Drug Discov. Today Dis. Models 4, 197–206.
Carvajal-Vergara, X., Sevilla, A., D’Souza, S. L., Ang, Y. S., Schaniel, C., Lee, D. F., Yang, L., Kaplan, A. D., Adler, E. D., Rozov, R., Ge, Y., Cohen, N., Edelmann, L. J., Chang, B., Waghray, A., Su, J., Pardo, S., Lichtenbelt, K. D., Tartaglia, M., Gelb, B. D., and Lemischka, I. R. (2010). Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465, 808–812.
Chen, C. C., Hsu, Y. J., and Lee, T. M. (2011). Impact of elevated uric acid on ventricular remodeling in infarcted rats with experimental hyperuricemia. Am. J. Physiol. Heart Circ. Physiol. 301, H1107–H1117.
Cooper, G. M., Stone, E. A., Asimenos, G., Green, E. D., Batzoglou, S., and Sidow, A. (2005). Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913.
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025. doi:10.1371/journal.pcbi.1001025
Devereux, R. B., Lutas, E. M., Casale, P. N., Kligfield, P., Eisenberg, R. R., Hammond, I. W., Miller, D. H., Reis, G., Alderman, M. H., and Laragh, J. H. (1984). Standardization of M-mode echocardiographic left ventricular anatomic measurements. J. Am. Coll. Cardiol. 4, 1222–1230.
Devereux, R. B., and Roman, M. J. (1995). “Evaluation of cardiac function and vascular structure and function by echocardiography and other noninvasive techniques,” in Hypertension: Pathophysiology, Diagnosis, and Management, eds J. H. Laragh and B. M. Brenner (New York: Raven P), 1969–1985.
Fox, E. R., Young, J. H., Li, Y., Dreisbach, A. W., Keating, B. J., Musani, S. K., Liu, K., Morrison, A. C., Ganesh, S., Kutlar, A., Ramachandran, V. S., Polak, J. F., Fabsitz, R. R., Dries, D. L., Farlow, D. N., Redline, S., Adeyemo, A., Hirschorn, J. N., Sun, Y. V., Wyatt, S. B., Penman, A. D., Palmas, W., Rotter, J. I., Townsend, R. R., Doumatey, A. P., Tayo, B. O., Mosley, T. H. Jr., Lyon, H. N., Kang, S. J., Rotimi, C. N., Cooper, R. S., Franceschini, N., Curb, J. D., Martin, L. W., Eaton, C. B., Kardia, S. L., Taylor, H. A., Caulfield, M. J., Ehret, G. B., Johnson, T., Chakravarti, A., Zhu, X., and Levy, D. (2011). Association of genetic variation with systolic and diastolic blood pressure among African Americans: the Candidate Gene Association Resource study. Hum. Mol. Genet. 20, 2273–2284.
Ganapathy, V., Thangaraju, M., Gopal, E., Martin, P. M., Itagaki, S., Miyauchi, S., and Prasad, P. D. (2008). Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 10, 193–199.
Germanguz, I., Sedan, O., Zeevi-Levin, N., Shtrichman, R., Barak, E., Ziskind, A., Eliyahu, S., Meiry, G., Amit, M., Itskovitz-Eldor, J., and Binah, O. (2011). Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J. Cell. Mol. Med. 15, 38–51.
Gu, C. C., Hunt, S. C., Kardia, S., Turner, S. T., Chakravarti, A., Schork, N., Olshen, R., Curb, D., Jaquish, C., Boerwinkle, E., and Rao, D. C. (2007). An investigation of genome-wide associations of hypertension with microsatellite markers in the family blood pressure program (FBPP). Hum. Genet. 121, 577–590.
Harshfield, G. A., Grim, C. E., Hwang, C., Savage, D. D., and Anderson, S. J. (1990). Genetic and environmental influences on echocardiographically determined left ventricular mass in black twins. Am. J. Hypertens. 3, 538–543.
Isik, T., Ayhan, E., Ergelen, M., and Uyarel, H. (2012). Uric acid: a novel prognostic marker for cardiovascular disease. Int. J. Cardiol. 156, 328–329.
Itzhaki, I., Maizels, L., Huber, I., Zwi-Dantsis, L., Caspi, O., Winterstern, A., Feldman, O., Gepstein, A., Arbel, G., Hammerman, H., Boulos, M., and Gepstein, L. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229.
Kim, J. J., Park, Y. M., Baik, K. H., Choi, H. Y., Yang, G. S., Koh, I., Hwang, J. A., Lee, J., Lee, Y. S., Rhee, H., Kwon, T. S., Han, B. G., Heath, K. E., Inoue, H., Yoo, H. W., Park, K., and Lee, J. K. (2012). Exome sequencing and subsequent association studies identify five amino acid-altering variants influencing human height. Hum. Genet. 131, 471–478.
Kong, C. W., Akar, F. G., and Li, R. A. (2010). Translational potential of human embryonic and induced pluripotent stem cells for myocardial repair: insights from experimental models. Thromb. Haemost. 104, 30–38.
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., and Durbin, R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079.
Lijnen, P., and Petrov, V. (1999). Renin-angiotensin system, hypertrophy and gene expression in cardiac myocytes. J. Mol. Cell. Cardiol. 31, 949–970.
Lourenco, V. M., Pires, A. M., and Kirst, M. (2011). Robust linear regression methods in association studies. Bioinformatics 27, 815–821.
Madsen, B. E., and Browning, S. R. (2009). A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 5, e1000384. doi:10.1371/journal.pgen.1000384
Majewski, J., Schwartzentruber, J., Lalonde, E., Montpetit, A., and Jabado, N. (2011). What can exome sequencing do for you? J. Med. Genet. 48, 580–589.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., McCarthy, M. I., Ramos, E. M., Cardon, L. R., Chakravarti, A., Cho, J. H., Guttmacher, A. E., Kong, A., Kruglyak, L., Mardis, E., Rotimi, C. N., Slatkin, M., Valle, D., Whittemore, A. S., Boehnke, M., Clark, A. G., Eichler, E. E., Gibson, G., Haines, J. L., MacKay, T. F., McCarroll, S. A., and Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753.
Moretti, A., Bellin, M., Welling, A., Jung, C. B., Lam, J. T., Bott-Flugel, L., Dorn, T., Goedel, A., Hohnke, C., Hofmann, F., Seyfarth, M., Sinnecker, D., Schomig, A., and Laugwitz, K. L. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409.
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., McMillin, M. J., Gildersleeve, H. I., Beck, A. E., Tabor, H. K., Cooper, G. M., Mefford, H. C., Lee, C., Turner, E. H., Smith, J. D., Rieder, M. J., Yoshiura, K., Matsumoto, N., Ohta, T., Niikawa, N., Nickerson, D. A., Bamshad, M. J., and Shendure, J. (2010a). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793.
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M., Huff, C. D., Shannon, P. T., Jabs, E. W., Nickerson, D. A., Shendure, J., and Bamshad, M. J. (2010b). Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet. 42, 30–35.
Norton, N., Li, D., Rieder, M. J., Siegfried, J. D., Rampersaud, E., Zuchner, S., Mangos, S., Gonzalez-Quintana, J., Wang, L., McGee, S., Reiser, J., Martin, E., Nickerson, D. A., and Hershberger, R. E. (2011). Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 88, 273–282.
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., Karakoc, E., MacKenzie, A. P., Ng, S. B., Baker, C., Rieder, M. J., Nickerson, D. A., Bernier, R., Fisher, S. E., Shendure, J., and Eichler, E. E. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589.
Ramagopalan, S. V., Dyment, D. A., Cader, M. Z., Morrison, K. M., Disanto, G., Morahan, J. M., Berlanga-Taylor, A. J., Handel, A., De Luca, G. C., Sadovnick, A. D., Lepage, P., Montpetit, A., and Ebers, G. C. (2011). Rare variants in the CYP27B1 gene are associated with multiple sclerosis. Ann. Neurol. 70, 881–886.
Ramensky, V., Bork, P., and Sunyaev, S. (2002). Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900.
Rao, D. C., and Province, M. A. (2000). The future of path analysis, segregation analysis, and combined models for genetic dissection of complex traits. Hum. Hered. 50, 34–42.
Rasmussen-Torvik, L. J., North, K. E., Gu, C. C., Lewis, C. E., Wilk, J. B., Chakravarti, A., Chang, Y. P., Miller, M. B., Li, N., Devereux, R. B., and Arnett, D. K. (2005). A population association study of angiotensinogen polymorphisms and haplotypes with left ventricular phenotypes. Hypertension 46, 1294–1299.
Robinson, P. N., Krawitz, P., and Mundlos, S. (2011). Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin. Genet. 80, 127–132.
Shi, G., and Rao, D. C. (2011). Optimum designs for next-generation sequencing to discover rare variants for common complex disease. Genet. Epidemiol. 35, 572–579.
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., Zhang, J., Soden, R., Hayakawa, M., Kreiman, G., Cooke, M. P., Walker, J. R., and Hogenesch, J. B. (2004). A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067.
Tang, W., Devereux, R. B., Li, N., Oberman, A., Kitzman, D. W., Rao, D. C., Hopkins, P. N., Claas, S. A., and Arnett, D. K. (2009). Identification of a pleiotropic locus on chromosome 7q for a composite left ventricular wall thickness factor and body mass index: the HyperGEN Study. BMC Med. Genet. 10, 40. doi:10.1186/1471-2350-10-40
Thangaraju, M., Ananth, S., Martin, P. M., Roon, P., Smith, S. B., Sterneck, E., Prasad, P. D., and Ganapathy, V. (2006). c/ebpdelta null mouse as a model for the double knock-out of slc5a8 and slc5a12 in kidney. J. Biol. Chem. 281, 26769–26773.
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111.
Vanhoutte, D., and Heymans, S. (2011). Thrombospondin 1: a protective “matri-cellular” signal in the stressed heart. Hypertension 58, 770–771.
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164.
Williams, R. R., Rao, D. C., Ellison, R. C., Arnett, D. K., Heiss, G., Oberman, A., Eckfeldt, J. H., Leppert, M. F., Province, M. A., Mockrin, S. C., and Hunt, S. C. (2000). NHLBI family blood pressure program: methodology and recruitment in the HyperGEN network. Hypertension genetic epidemiology network. Ann. Epidemiol. 10, 389–400.
Wineinger, N. E., Patki, A., Meyers, K. J., Broeckel, U., Gu, C. C., Rao, D. C., Devereux, R. B., Arnett, D. K., and Tiwari, H. K. (2011). Genome-wide joint SNP and CNV analysis of aortic root diameter in African Americans: the HyperGEN study. BMC Med. Genomics 4, 4. doi:10.1186/1755-8794-4-4
Xia, Y., Dobaczewski, M., Gonzalez-Quesada, C., Chen, W., Biernacka, A., Li, N., Lee, D. W., and Frangogiannis, N. G. (2011). Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 58, 902–911.
Xie, F., Ye, L., Ta, M., Zhang, L., and Jiang, W. G. (2011). MTSS1: a multifunctional protein and its role in cancer invasion and metastasis. Front. Biosci. 3, 621–631.
Keywords: hypertrophy, left ventricular mass, cardiomyocyte, exome, genomics
Citation: Zhi D, Irvin MR, Gu CC, Stoddard AJ, Lorier R, Matter A, Rao DC, Srinivasasainagendra V, Tiwari HK, Turner A, Broeckel U and Arnett DK (2012) Whole-exome sequencing and an iPSC-derived cardiomyocyte model provides a powerful platform for gene discovery in left ventricular hypertrophy. Front. Gene. 3:92. doi: 10.3389/fgene.2012.00092
Received: 22 March 2012; Accepted: 08 May 2012;
Published online: 28 May 2012.
Edited by:
Karen T. Cuenco, University of PIttsburgh, USAReviewed by:
Kai Wang, University of Southern California, USAJesus Sainz, Consejo Superior de Investigaciones Cientificas, Spain
Copyright: © 2012 Zhi, Irvin, Gu, Stoddard, Lorier, Matter, Rao, Srinivasasainagendra, Tiwari, Turner, Broeckel and Arnett. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: D. K. Arnett, Department of Epidemiology, University of Alabama at Birmingham, 1530 3rd Avenue South, Birmingham, AL 35294-0022, USA. e-mail:YXJuZXR0QHVhYi5lZHU=
†D. Zhi and M. R. Irvin contributed equally as co-first authors, U. Broeckel and D. K. Arnett as co-senior authors in the execution of this study.