


- 1 Canadian Centre for Behavioural Neuroscience, University of Lethbridge, Lethbridge, AB, Canada
- 2 Department of Biological Sciences, University of Lethbridge, Lethbridge, AB, Canada
Epigenetic regulation modulates gene expression without altering the DNA sequence to facilitate rapid adjustments to dynamically changing environmental conditions. The formation of an epigenetic memory allows passing on this information to subsequent generations. Here we propose that epigenetic memories formed by adverse environmental conditions and stress represent a critical determinant of health and disease in the F3 generation and beyond. Transgenerational programming of epigenetic regulation may represent a key to understand adult-onset complex disease pathogenesis and cumulative effects of life span and familial disease etiology. Ultimately, the mechanisms of generating an epigenetic memory may become of potentially promising diagnostic and therapeutic relevance due to their reversible nature. Exploring the role of environmental factors, such as stress, in causing variations in epigenetic profiles may lead to new avenues of personalized, preventive medicine based on epigenetic signatures and interventions.
Introduction and Scope
It is well established that the risk of many human complex diseases is determined by a heritable component. Recent evidence suggests that non-genomic and epigenetic processes participate in the heritable origins of disease with effects that extend beyond a single generation. The evolutionary conservation of these mechanisms is central to produce viable and apt offspring. Since the dynamic formation of a heritable memory may persist through multiple generations it may also poorly prepare the offspring to a dynamically changing environment, thus contributing to disease and compromising longevity. Therefore, programming by transgenerational inheritance represents a central mechanism by which environmental conditions may influence disease risk across multiple generations.
The concept of transgenerational inheritance refers to three main mechanisms. Aside from altered maternal endocrine responses and postnatal parental behavior, transmission of modifications in the epigenome appears to be a major component of transgenerational programming of disease (Jablonka and Lamb, 1989; Gluckman et al., 2007). Epigenetic transgenerational inheritance refers to the passage of epigenetic marks to the next generation without being altered or erased (Reik and Walter, 2001). Epigenetic processes readily respond to environmental conditions and so allow rapid modifications to an adverse environment, such as stress. Here we argue that human complex diseases, including psychiatric and metabolic disorders, are related to transgenerational epigenetic inheritance of maladaptive responses to environmental stress (Figure 1).
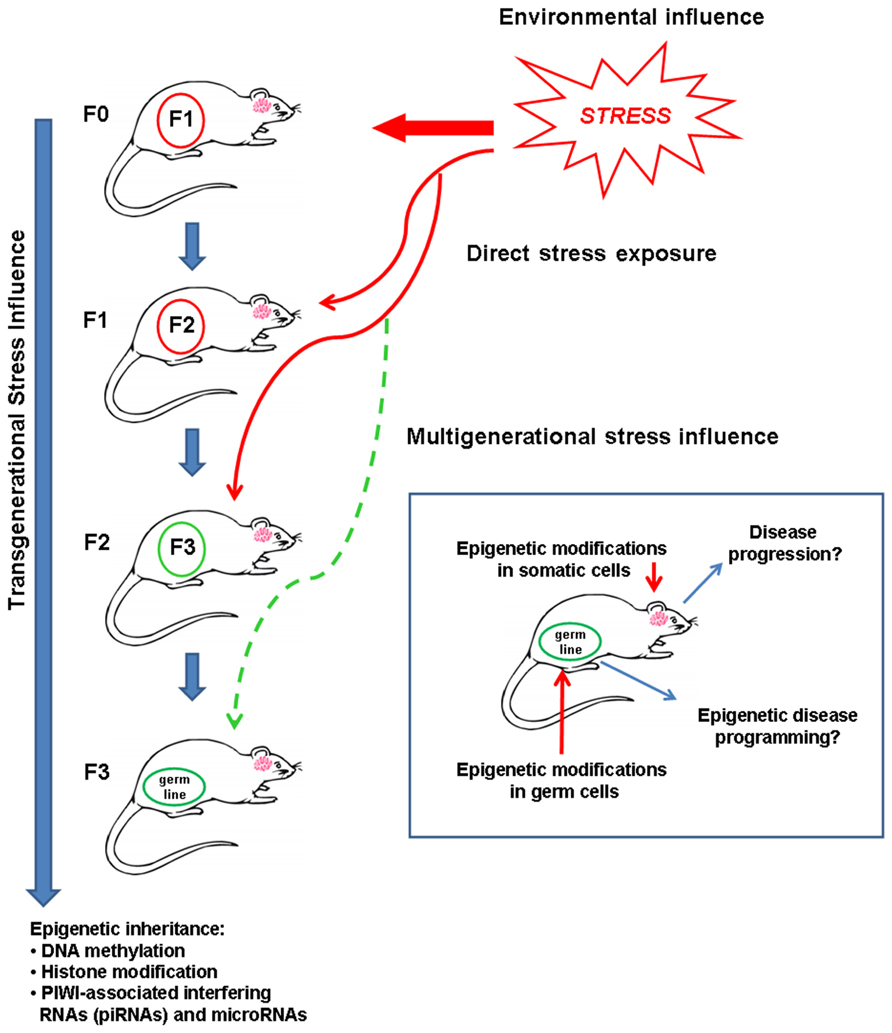
FIGURE 1. Inheritance of an epigenetic memory caused by an adverse environment, such as stressful experiences, produce phenotypes in next generations that may influence disease risk. Solid red arrow indicates direct exposure to stress in an animal; thin red arrows indicate direct stress exposure leading to intergenerational stress influence in F1 and F2 generations; dashed thin green arrow indicates possible transgenerational inheritance of a multigenerational stress phenotype in the F3 generation. To prove genuine epigenetic programming, it is necessary to determine phenotype transmission to the third generation (F3).
The main hypothesis of this review is that an adverse environment affects disease predisposition and outcome beyond a single life span to create a heritable or familial contribution to disease. According to Skinner (2008), we propose that epimutations need to propagate to at least three generations in order to represent genuine epigenetic inheritance. Very few studies have in fact investigated transgenerational inheritance of parental experience through three subsequent generations. We will first review the basic concepts of epigenetic regulation of gene expression and transgenerational programming. We will then discuss the role of environmental adversity, such as stress, in shaping the predisposition to future disease within a single lifespan and across generations. We will provide evidence supporting the conclusion that health and disease are programmed across multiple generations via epigenetic mechanisms.
Unraveling the Secret Language of Destiny: Basic Concepts of Epigenetic Programming
Epigenetic regulation can “program” the genetic information and cell fate and thus largely determine the functionality of an organ including the brain. These mechanisms may facilitate an adaptive response to a changing environment to optimize the chances of survival and reproductive success. Epigenetic programming, or re-programming by changing experience, permits reversible and heritable modulation of gene expression without altering the DNA sequence to rapidly adjust cellular processes to constantly changing environmental conditions (Champagne and Meaney, 2006, 2007; Champagne et al., 2008; Migicovsky and Kovalchuk, 2011; Skinner, 2011; Skinner et al., 2011; Babenko et al., 2012a,b).
Epigenetic changes are mitotically stable mechanisms responsible for gene expression modulation and/or their inheritance by molecular adjustments. The major epigenetic events include DNA cytosine methylation, histone modifications, transcriptional, and posttranscriptional control of gene expression through PIWI-interacting RNA (piRNA), microRNA (miRNA), and heterochromatin gene silencing (Figure 2; Ilnytskyy et al., 2008; Yauk et al., 2008; Filkowski et al., 2010; Migicovsky and Kovalchuk, 2011). Recent studies have recognized the importance of differential expression of miRNA-regulated pathways in complex epigenetic selfregulation (Bartel, 2004). For instance, miR-184, a brain-specific miRNA suspected in the etiology of autism spectrum disorder, is upregulated by the release of the methyl-CpG-binding protein 2 (MeCP2) from paternal allele-specific expression (Nomura et al., 2008). While MeCP2 is integral to the function of mature neurons, the presence of a methyl-CpG-binding domain and interaction with miR-184 provides a link between epigenetic pathways involving small regulatory RNAs and DNA methylation. These pathways may synergistically affect transgenerational programming of disease via genomic imprinting.
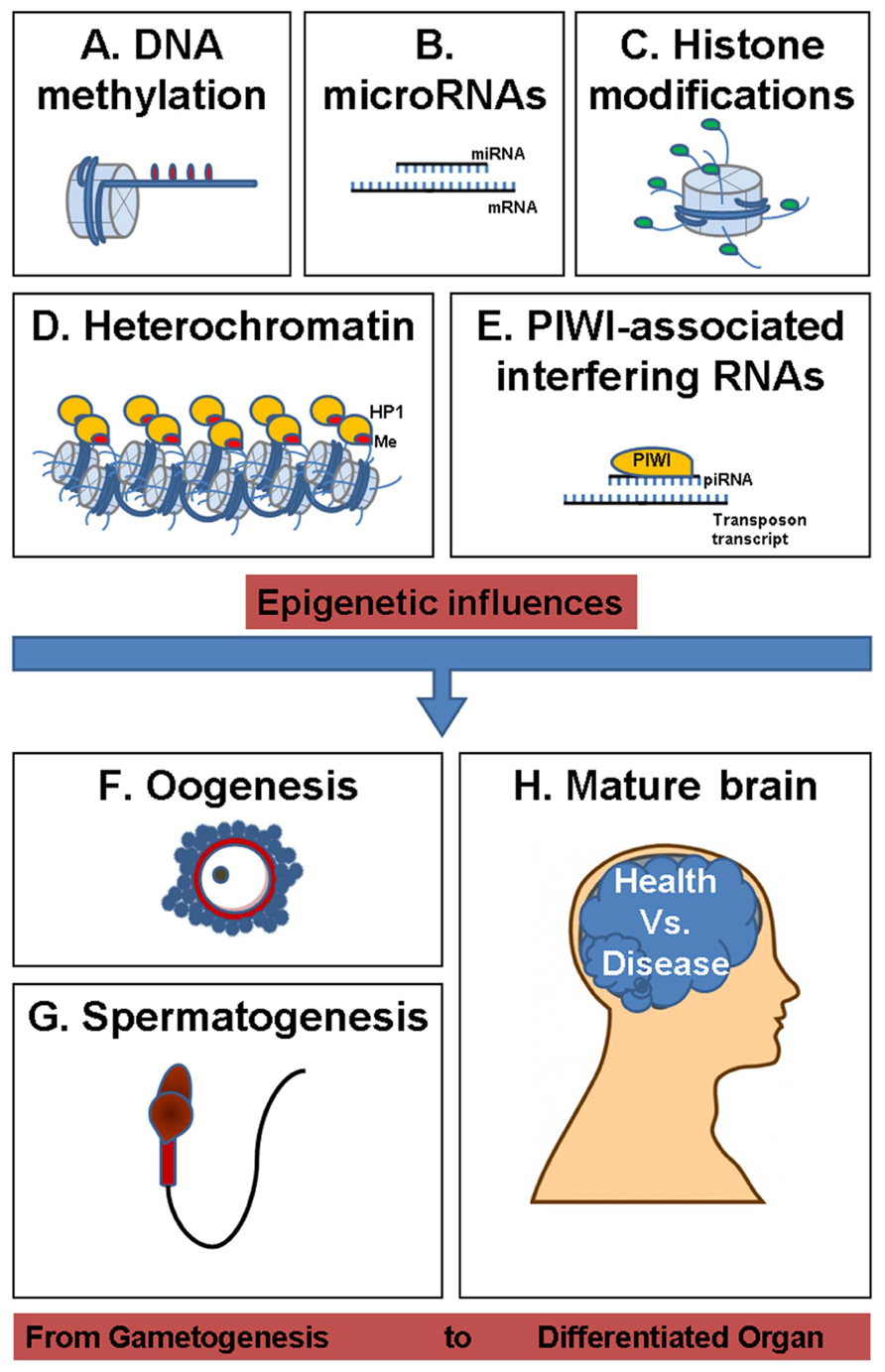
FIGURE 2. Overview of epigenetic influences shaping health and disease from gametogenesis to differentiated organ. (A) DNA methylation (DNA containing methyl groups, which is packed in a nucleosome core histone complex). (B) microRNAs (microRNA–mRNA complex). (C) Histone modifications (histone tail acetylation, methylation, ubiquitination, sumoylation, or phosphorylation). (D) Heterochromatin (containing hypoacetylated, methylated H3K9 histone tail). (E), PIWI-associated interfering RNAs (PIWI protein and piRNA guide forming a complex with transposon transcript). (F) Oogenesis (ovulum). (G) Spermatogenesis (spermatozoid). (H) Mature brain.
Genomic imprinting refers to a process that involves chromatin modifications to achieve monoallelic gene expression (Reik and Walter, 2001). One of the first evidences of genomic imprinting was derived from an observation linking Angelman syndrome, a brain developmental disorder including speech and motor deficits (Angelman, 1965), to Prader–Willi syndrome, a congenital condition that is characterized by insatiable appetite and delayed motor maturation (Curfs and Fryns, 1992). These two very distinct human phenotypes are both linked to loss of function of the ubiquitin UBE3A pathway, encoded by a nucleotide sequence on chromosome 15 (Magenis et al., 1987; Adams, 2008). Usually the sequence derived from the mother encodes function of this pathway while the paternal contribution is silenced. Thus, the paternal gene is unable to compensate for the loss of the maternal gene in Angelman syndrome, leading to this characteristic phenotype (Jiang et al., 1998; Rougeulle et al., 1998). In Prader–Willi syndrome, however, the neighboring region SNRPN on chromosome 15, encoding a pathway for mRNA splicing, shows the opposite pattern of gene imprinting. Here, the paternal contribution of the gene is normally being transcribed and the maternal contribution is silenced. If function of the paternal gene becomes lost, the methylated maternal gene is unable to correct for this loss and the phenotype of Prader–Willi syndrome may occur (Cassidy et al., 2012). While this kind of inherent genomic imprinting is a potent influence on future health and disease, the epigenome, in particular methylation patterns, readily respond to changes in life style.
Methylation has been the most widely investigated mechanism of genomic imprinting in health and disease. Based on the observation that alleles may be expressed from only one of the two parental chromosomes (Reik and Walter, 2001; Wilkins and Haig, 2003), recent studies suggested that DNA methylation may represent a central mechanism to facilitate phenotypic variation of complex traits (Itier et al., 1998; Wilkinson et al., 2007; Wang et al., 2010). Methylation patterns are highly responsive to parental life style and environment. For instance, prenatal exposure to maternal tobacco smoking may elevate DNA methylation of the gene encoding brain-derived neurotrophic factor (BDNF; Toledo-Rodriguez et al., 2010), a vital growth factor in brain development, plasticity, and psychiatric disease (Balaratnasingam and Janca, 2012). Methylation of the BDNF-6 exon causes reduction in BDNF expression and possibly facilitates variations in brain development in the fetus exposed to maternal smoking (Toledo-Rodriguez et al., 2010). Thus, one may argue that transgenerational variation in BDNF expression may contribute to the programming of psychiatric diseases. Aside from substance use, the maternal psychological state may also determine the psychiatric health of the offspring through altered DNA methylation status. For example, maternal depressed mood during pregnancy was found to be related to methylation of the SLC6A promoter, which encodes a transmembrane serotonin transporter, in both mother and infant (Devlin et al., 2010). The authors of this study showed that maternal depressed mood in the second trimester was associated with reduced maternal and newborn SLC6A4 methylation status (Devlin et al., 2010), which represents a risk factor for greater vulnerability to post-traumatic stress disorder (Koenen et al., 2011). These findings suggest that maternal mood and wellbeing can have profound consequences on offspring health.
Epigenetic Mechanisms as an Interface Between Stress and Disease Across All Ages
Epigenetic mechanisms represent a critical interface between a stressful environment and the genome. The term stress refers to perturbation of homeostasis by an adverse experience or environmental threats that activate the hypothalamic–pituitary–adrenal (HPA) axis and associated neuroendocrine changes (Seckl, 2008). The onset of the stress response results in the release of primary stress hormones, such as cortisol in primates, which bind to glucocorticoid receptors in the cytoplasm. As a hormone–receptor complex, cortisol then translocates to the nucleus to regulate gene expression and protein synthesis (Zucchi et al., 2010). The genomic response to stress mediates a set of complex neural, metabolic, and endocrine adaptations that may have long-term consequences (Drake et al., 2005). Above and beyond these regulatory activities, stress may also leave an epigenomic imprint to affect health and disease (Babenko et al., 2012a,b). A particularly sensitive time window to programming of long-term physiology and health through epigenetic marks is the perinatal period.
Perinatal programming refers to the priming of long-term physical and psychological health outcomes during critical periods in early development. Excessive glucocorticoid levels caused by severe maternal stress during pregnancy may saturate the protective placental enzyme barrier, 11-beta-hydroxysteroid dehydrogenase (11beta-HSD), to reach the fetus (Miller and Chernoff, 1995; Welberg et al., 2000; Brummelte et al., 2010) and permanently modulate fetal HPA axis activity (Drake et al., 2005) and behavior. For example, perinatal stress influences anxiety- and depression-like behaviors and cognitive abilities (Champagne et al., 2003, 2008; Richards and Sacker, 2003; Gatt et al., 2009; Kinsella and Monk, 2009; Rokyta et al., 2008) as well as central dopaminergic system function to modulate rewarding behavior in later life (McArthur et al., 2007; Mueller and Bale, 2008). Many of the behavioral effects of stress likely include an epigenetic component (Colvis et al., 2005; Champagne et al., 2006) that facilitates robust perinatal programming. A striking example of this link was proposed by studies of maternal care. Variations in maternal care may program individual differences in stress reactivity across generations by epigenetic mechanisms, including DNA methylation (Francis et al., 1999; Champagne et al., 2003; Weaver et al., 2004). This early life experience has long-lasting effects on the progeny and is transmitted to the next generation (Francis et al., 1999; Caldji et al., 2003).
Prenatal glucocorticoid exposure and postnatal care by a stressed mother not only program the response to recurrent stress in later life, but may also affect the predisposition to disease in adulthood. The concept that vulnerability to disease is acquired in the perinatal period was developed more than 30 years ago (e.g., Dörner et al., 1984). Studies by David Barker’s group in the 1990s have first associated adverse perinatal environment with higher incidence of obesity and cardiovascular disease in the aged offspring (Law et al., 1992; Barker, 2007). Experience during gestation and/or early postnatal life may prime the susceptibility for disease in later life, including metabolic (e.g., type 2 diabetes mellitus, obesity), neurological (Mabandla et al., 2009; Mabandla and Russell, 2010; Babenko et al., 2012b), and cardiovascular disease (Newnham, 2001; Zambrano et al., 2005; Cottrell and Seckl, 2009; Zambrano, 2009; Singhal, 2010; Thompson and Einstein, 2010). Thus, while the information given to the developing fetus may support immediate adaptation to the postnatal environment, it may not support successful aging because these endocrine adjustments to an adverse environment come at a high metabolic expense (Figure 3).
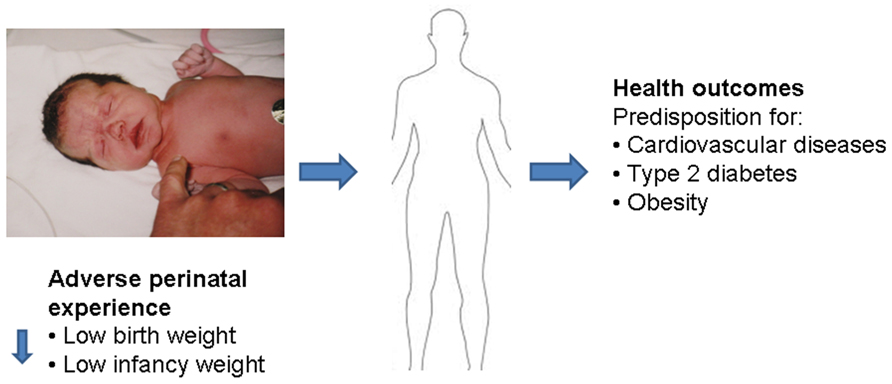
FIGURE 3. Adverse perinatal experiences may influence the risk of cardiovascular and metabolic diseases in adulthood. Low birth weight and low infancy weight (first year) has been correlated with a predisposition of adult onset diseases, such as high blood pressure and risk of cardiovascular disease, insulin resistance and risk of diabetes, and adulthood obesity.
Later in life, stress-induced changes in epigenetic regulation are suspected to contribute to the enhanced susceptibility to neurological and psychiatric disease through regulation of components of the HPA axis activation, such as corticotrophin-releasing factor (Sterrenburg et al., 2011). Moreover, prenatal stress causes region-specific changes in miRNA expression resulting in dysmasculinization of the male brain (Morgan and Bale, 2011). During critical periods of sexual differentiation, methylation patterns change regardless to neonatal hormone exposure and are dynamically regulated throughout the life span (Schwarz et al., 2010). Accordingly, evidence suggests that risk factors for cardiovascular disease, including stress and an unbalanced diet, are associated with modified epigenetic markers (Ordovás and Smith, 2010). Possible epigenetic correlates of elevated cardiovascular risk were reported to include global DNA hypermethylation (Stenvinkel et al., 2007) and miRNA-29b (miR-29b)-induced regulation of genes that influence atherosclerosis risk (Chen et al., 2011). The latter study showed that miR-29b upregulation results in DNMT3b down-regulation in human aortic smooth muscle cells and altered MMP-2/MMP-9 gene expression involved in cell migration (Chen et al., 2011). Moreover, central genomic and epigenomic responses to psychological stress or associated endocrine changes involve DNA methylation (Mychasiuk et al., 2011; Sterrenburg et al., 2011) and miRNA regulation (Babenko et al., 2012a). Interestingly, age-related epigenetic re-programming may also be responsible for a positive relationship between advanced maternal or grandmaternal age at the time of birth and an increased risk of psychiatric conditions, such as autism spectrum disorder (Golding et al., 2010) or memory impairments (Burns and Mery, 2010) in the offspring. These studies concur that epigenetic activity closely influences brain development and neurological functions (Figure 2).
Aside from perinatal programming, the cumulative effects of life-long adverse stimuli may also modify the risk of functional decline and disease in adulthood and old age (Mora et al., 2007; Miller and O’Callaghan, 2008; Merrett et al., 2010; Schreier et al., 2011). Epigenetic signatures are central to adult-onset complex diseases including psychiatric and neurological (Glaser et al., 2010; Ronald et al., 2010; Babenko et al., 2012b), cardiovascular (Stenvinkel et al., 2007; Kaneda et al., 2009; Chen et al., 2011; Stein et al., 2011), and metabolic conditions (Ling et al., 2008; Begum et al., 2012; Volkmar et al., 2012). It may be the cumulative experience to stress throughout life that, through epigenetic regulation, precipitates or accelerates pathological processes that promote neurodegenerative events (Babenko et al., 2012b). Accordingly, methylation patterns in the brain are correlated with chronological age (Hernandez et al., 2011) and with life style (Kaliman et al., 2011). Notably, the aging of the brain and age-related decline in motor and cognitive functions corresponds to a global reduction in gene expression (Lu et al., 2004) and a diminished capacity for epigenetic memory in chromatin of oligodendrocytes (Shen et al., 2008). The age-associated loss of the epigenetic memory may significantly alter cellular function over time.
Central targets of epigenetic programming in the brain at any age are neurotrophic molecules, which are highly sensitive to the effects of stress and other environmental factors (Kobilo et al., 2011; Taliaz et al., 2011; Johansen et al., 2012). Neurotrophic and growth factors that are modulated by epigenetic regulation include glial cell line-derived neurotrophic factor (GDNF; Uchida et al., 2011), BDNF (Fuchikami et al., 2010; Zeng et al., 2011), and nerve growth factor (NGF; Sen and Snyder, 2011). Epigenetic regulation of histones mediates neurotrophin actions with histone acetylation enhancing cAMP response element-binding (CREB)-associated transcription elicited by BDNF and NGF (Sen and Snyder, 2011). BDNF activates neuronal nitric oxide synthase with the nitrosylated GAPDH/seven in absentia homolog (Siah) complex translocating to the nucleus. Degradation of the histone–lysine N-methyltransferase SUV39H1 by Siah facilitates histone H3K9 acetylation, CREB binding to DNA, and enhances expression of CREB-regulated genes and neurite outgrowth (Sen and Snyder, 2011). Histone deacetylases are the first identified intranuclear targets of nitric oxide, but, due to its highly diffusible nature, it is likely that many other nuclear factors are directly regulated by nitric oxide (Nott and Riccio, 2009).
Dysregulation of neurotrophic molecules is a central component in the pathogenesis of major psychiatric and neurologic disorders, such as schizophrenia, obsessive-compulsive disorder, Alzheimer’s and Huntington diseases (Mattson, 2008). The cumulative adverse effects of stressful experiences throughout life may accelerate age-related decline through loss of neurotrophic factors and neuroplasticity (Pham et al., 2002; Gatt et al., 2009), reduced gene expression (Bishop et al., 2010), and altered epigenetic status (Siegmund et al., 2007). Interestingly, susceptibility and adaptation to adverse environments during a lifetime may be closely linked to the epigenetic status of the GDNF gene (Uchida et al., 2011). Importantly, epigenetic signatures of neurotrophic factors may be passed on to the fetus (Toledo-Rodriguez et al., 2010) and propagate to subsequent generations.
Forming an Epigenetic Memory Across Generations
Epigenetic memory refers to transgenerationally stable, yet dynamic re-programming of the germline epigenome that will transfer epigenetic information from one generation to the next in the absence of DNA sequence mutations (Zambrano, 2009; Migicovsky and Kovalchuk, 2011). A pioneering example of an epigenetic memory was generated by studies focusing on the agouti locus in mice that controls coat color (Morgan et al., 1999). In mice expressing the viable yellow (Avy) allele the coat color is determined by the epigenetic status of intra-cisternal A particle (IAP) retrotransposons. Without methylation, the transcription of an IAP retrotransposon inserted upstream of the agouti gene will initiate the expression of agouti protein, resulting in yellow fur along with abnormal metabolic responses and predisposition to tumors (Morgan et al., 1999). In the hypermethylated state, however, silencing of the promoter in the IAP I will result in a black coat and mice become indistinguishable from wild-type (A/A) mice (Morgan et al., 1999). Such profiles may persist to subsequent generations to form an epigenetic memory. Epigenetic modifications may propagate to the offspring by means of two different processes and at different stages in a mammal’s lifecycle: programming of somatic cells and the germline, as illustrated in Figure 1.
Programming of somatic cells, including the brain, may mediate transgenerational inheritance. Environmental conditions may impact the organization of brain organogenesis and phenotype through epigenetic reprogramming rendering the individual susceptible to adult-onset neurological disease. These acquired memories may lead to organ- or tissue-specific metabolic imbalance or disease predisposition. The mechanisms also include behavioral and physiological modifications in response to the environment that shape intergenerational transmission (Champagne and Meaney, 2006). Thus, a behavioral phenotype may perpetuate across generations via changes in chromatin. For example, a behavioral trait such as a certain type of maternal care may be passed on to the subsequent generation. Rats raised by a mother displaying poor maternal care, such as low levels of licking and grooming toward her pups, also are more likely to exhibit poor maternal care toward their own offspring (Weaver et al., 2004, 2007; Cameron, 2011). This behavioral phenotype is linked to hypermethylation of the BDNF gene and consequently low expression of BDNF in the prefrontal cortex of offspring (Roth et al., 2009). Transgenerational behavioral traits of maternal care may also determine survival in other ways. In the biparental burying beetle Nicrophorus vespilloides, grandparents and parents of both sexes cumulatively influence offspring development (Lock, 2012). In the biparental condition, females spend most of their time caring for the larvae, while males spend most time with indirect care by maintaining the carcass in which the larvae lives. The existence of an intergenerational influence from F0 to F2 generations provide a selection pressure for the division of parental investment in biparental species across different taxa, which seems to enhance survival of this species (Lock, 2012).
The second mechanism of transgenerational programming by stress may be mediated through the germline. These modifications may be passed on to future generations without direct exposure (Chong and Whitelaw, 2004). The germline-mediated epigenetic transgenerational inheritance is based on marks in the chromatin that influence the gonadal development and reproductive success of the affected individual (Whitelaw and Whitelaw, 2008). Usually, DNA methylation sites are selectively erased during gonadal sex determination in embryogenesis. Some DNA marks will survive this critical time window, however, and persist throughout development and maturation (Migicovsky and Kovalchuk, 2011). The methylated DNA then may integrate in the gametes and be passed on to the embryo of the next generation. These genes “imprinted” with a pattern of DNA methylation may propagate stress-induced modifications to the subsequent generations (Skinner et al., 2008). An example of germline transmission is represented by the effects of environmental toxins, such as the endocrine disruptor bisphenol-A (BPA), which resembles the actions of reproductive hormones and thus influences the reproductive success of females in the next generation (Dolinoy et al., 2007). The hypothesis that diseases might be programmed by transgenerational influences, such as stress through epigenetic mechanisms seems reasonable in this framework, considering that the environment shapes current (somatic cells) and future (germ cells) generations.
Transgenerational epigenetic inheritance may be defined as a phenotype that is transmitted to more than one generation by means other than Mendelian genetics (Whitelaw and Whitelaw, 2008; Migicovsky and Kovalchuk, 2011; Daxinger and Whitelaw, 2012). Thus, physiological and/or environmental factors affecting the parent (F0 generation) can have consequences for the next, unexposed generation (F1 generation) and possibly for further subsequent generations (F2, F3) as well. A recent concept argues that only epigenetic alterations persisting to the F3 generation may be considered truly heritable when pregnant females are concerned (Skinner, 2008). Skinner (2008) defined transgenerational epigenetic inheritance as changes in unexposed generations (F3 and beyond). This concept excludes F0 generation exposure to a given environmental factor during pregnancy that directly also affects the phenotype of the F1 embryo and F2 primordial germ cells. Therefore, as illustrated in Figure 1, the interpretation of genuine transgenerational effects requires analysis of the F3 phenotype. In this context, transgenerational phenotypes are defined as traits acquired from an initial exposure, persistently transmitted to the next generation in the absence of continued exposure in any of the successive generations (Skinner, 2011). An intergenerational phenotype refers to transmission to the F1 and F2 generations that are not (yet) known to persistently alter the F3 phenotype. By contrast, multigenerational phenotypes are characterized by direct exposure of multiple generations to an environmental factor or toxin (Skinner, 2011). For instance, endocrine disrupting agents and pesticides induce gene imprinting (F1–F4; Anway et al., 2005; Jirtle and Skinner, 2007; Manikkam et al., 2012), anxiety-like behavior (Skinner et al., 2008), and adult-onset disease (Anway et al., 2006) in a genuine transgenerational pattern. According to these studies, the present review will consider the definition of transgenerational inheritance to the F3 generation, and our hypothesis is that disease is programmed by epigenetic inheritance across three successive generations.
An epigenetic transgenerational phenotype will ultimately affect gene expression, molecular adjustments, cellular function, physiological parameters, and behavioral profiles. These processes may perturb the integrity of somatic and/or germ cells in the offspring later in life, leading to compromised health. The short-term adaptation that results from epigenetic modifications may facilitate rapid adaptation to adverse environmental conditions and immediate survival of an individual. In the long-term, however, acquired physiological and epigenetic profiles, such as those in response to stress, may produce a mismatch to later-life challenges and enhance the vulnerability to disease.
Life-Time Versus Transgenerational Epigenetic Programming
Epigenetic programming represents a major mechanism to allow passing on the effects of experience to subsequent generations. This process generates an epigenetic memory that is now recognized as a central component in the transmission of information about early experience, environmental conditions, and life style factors to the progeny (Boyko et al., 2010; Boyko and Kovalchuk, 2010; Migicovsky and Kovalchuk, 2011). For example, in the womb epigenetic re-programming seems to occur rapidly to facilitate adaptation of the fetus to current environmental conditions (Breton et al., 2009; Guerrero-Preston et al., 2010; Toledo-Rodriguez et al., 2010). While initially a beneficial response to prime developmental plasticity (Mousseau and Fox, 1998; Cropley et al., 2012), transgenerational transmission of epigenetic alterations in the long-term may have maladaptive consequences that compromise overall health and successful aging. Thus, epigenetic programming may represent a key to understand diseases processes, propagation through generations, and cumulative effects of life span and familial disease etiology.
Epigenetic programming might direct evolution through inheritance of certain epimutations across generations. Experience throughout life, however, can modify the imprints of perinatal programming and disease risk. Accordingly, positive experiences such as education, physical activity, and social environment can ameliorate maladaptive programming and disease vulnerability (Wirdefeldt et al., 2005; Carlson et al., 2008; James et al., 2011). Moreover, positive experience in rats, including exposure to an enriched environment, can diminish consequences of neurodegenerative influences and age-related functional decline (Escorihuela et al., 1994; Mattson et al., 2001; Laviola et al., 2004; Jadavji et al., 2006; Paban et al., 2009; Segovia et al., 2009). Accordingly, the sum of cumulative favorable and adverse experiences creates age-associated epigenetic drift, which was shown in animals (Bennett-Baker et al., 2003) and in monozygotic twins (Fraga et al., 2005; Martin, 2005; Poulsen et al., 2007). Fraga et al. (2005) demonstrated that advancing age leads to greater global and locus-specific differences in DNA methylation and histone H3 and H4 acetylation in peripheral blood lymphocytes, buccal mucosal epithelial cells, skeletal muscle biopsies, and adipose tissue of identical twins. Different epigenetic patterns were found in almost all telomeres and certain gene-rich regions of chromosomes (Fraga et al., 2005). These findings illustrate that epigenetic modifications in somatic cells may perpetuate throughout the life span (Siegmund et al., 2007; Hernandez et al., 2011; Uchida et al., 2011) likely by stable mitosis (in cell lines, tissues, or organs) to facilitate adaptation to ongoing environmental conditions (Skinner, 2011).
The effects of parental exposure to environmental factors may persist to the F2 generation and enhance vulnerability to adult-onset disease across multiple generations (Gluckman et al., 2007; Zambrano, 2009; Nolan et al., 2011; Feil and Fraga, 2012). For example, a recent collaboration between Swedish and British scientists provided evidence of sex-specific programming of longevity in humans. A transgenerational correlation was found between the nutritional status during early life of the paternal grandparents and the grandchild’s chances of longevity, including associations with cardiovascular and diabetic disease (Pembrey, 2010). A prominent case of intergenerational programming of disease stems from a systematic study of the Dutch Famine Birth Cohort. Exposure to poor nutrition during pregnancy in the Dutch winter 1944/45 led to smaller babies (Stein and Lumey, 2000), who developed an increased risk of insulin resistance in adulthood (Painter et al., 2005). Furthermore, granddaughters of women pregnant during the famine were smaller, more prone to neonatal adipositas and poor health in later life (Painter et al., 2008). These findings confirm that consequences of prenatal adverse experience may reach down across several generations.
Another link to intergenerational HPA axis programming is illustrated by lower cortisol levels in offspring of Holocaust survivors with post-traumatic stress disorder (Yehuda et al., 2007). HPA axis changes may also be involved in the observation that grandchildren of Holocaust survivors present with higher rates of psychiatric illness (Sigal et al., 1988), elevated levels of fear, neurotic behavior, and aggression (Scharf, 2007) and depression (Felsen and Erlich, 1990). On the other hand, adaptive changes may reflect in improved coping skills (Sigal and Weinfeld, 2001). Thus, ancestral programming may result in both beneficial and detrimental physiological and behavioral outcomes.
In rat and guinea pig models, intergenerational stress-induced changes lasting to the F2 generation have been shown for caloric restriction (Benyshek et al., 2006; Bertram et al., 2008; Pinheiro et al., 2008) or overfeeding (Pentinat et al., 2010). A protein-restricted diet in the parental generation led to reduced birth and brain weight and altered glucose metabolism in the unexposed F2 generation (Zamenhof et al., 1971; Benyshek et al., 2006). Altered glucose metabolism in these studies is usually characterized by high blood sugar, increased blood insulin, and insulin resistance, which may be indicative of diabetes (Figure 3; Pinheiro et al., 2008). Various mechanisms may participate in the perinatal programming of these phenotypes, including altered gestational endocrine milieu and maternal behavior, as well as gene imprinting by epigenetic factors (Migicovsky and Kovalchuk, 2011; Matthews and Phillips, 2012). An epigenetic component to intergenerational programming was demonstrated for depressive-like behaviors induced by maternal separation stress in mice (Franklin et al., 2010). These behavioral changes were accompanied by altered DNA methylation profiles in the germline and brains of the offspring of stressed males (Franklin et al., 2010).
Support for the notion that stress-induced intergenerational long-term adaptations are epigenetically programmed originates from a recent longitudinal study in children. Essex et al. (2011) showed that maternal stress in infancy and paternal stress in preschool years are predictive of permanent differences in DNA methylation patterns in adolescent school children. The association between adversity in early childhood and the DNA methylation profiles in adolescents suggests that early experience creates a lasting imprint on the epigenomic status. The neonatal environment may be a particularly important determinant of the onset of age-related complications. Although life-long experience may change these patterns, the stability of DNA methylation patterns through development and adolescence suggests that parentally programmed or inherited epigenetic profiles create an epigenetic blueprint that persists into adulthood and potentially into old age.
Although the importance of transgenerational programming of disease has been recognized, a limited number of reports have investigated the F3 generation and beyond. Transgenerational inheritance refers to the ability of environmental factors to promote an intergenerational phenotype or disease state beyond the exposed mother, her female offspring and grandoffspring (Skinner et al., 2008). Truly transgenerational effects were shown for factors such as nutrition (Benyshek et al., 2006; Dunn and Bale, 2011), physiological disturbance (Aerts and van Assche, 1979), and endocrine disruptors (Blatt et al., 2003; Anway et al., 2005; Brouwers et al., 2006; Nilsson et al., 2008; Skinner et al., 2008; Guerrero-Bosagna et al., 2010). By contrast, if a phenotype was caused by a direct effect of experience on the germline and somatic cells without lasting heritable genomic imprints, these effects are expected to disappear by the F3 generation. For example, Drake et al. (2005) observed that prenatal treatment with the synthetic steroid dexamethasone in rats resulted in low birth weight and metabolic markers for elevated diabetes risk, such as hyperglycemia, hyperinsulinemia, and increased activity of gluconeogenic enzyme phosphoenolpyruvate carboxykinase, in the progeny. These changes, however, were resolved by the third generation (Drake et al., 2005). This observation indicates that perinatal programming by steroid hormones may be caused by direct effects on the primordial germ line rather than stable epigenetic imprinting. Similarly, Avy hypermethylation induced by maternal methyl supplementation to induce the agouti phenotype in mice is also not inherited transgenerationally from F0 to F3 generations (Waterland et al., 2007). However, transgenerational inheritance of stress responses and associated epigenetic correlates are well established in plant models (Boyko et al., 2010; Boyko and Kovalchuk, 2011; Bilichak et al., 2012). Information about the causative mechanisms of transgenerational inheritance in mammals is still extremely limited, however, selective breeding experiments may offer a welcome venue for such studies.
Studies of selective breeding during domestication support the notion of lasting transgenerational traits, although a possible epigenetic component still remains to be determined. For instance, long-term multigenerational selection for traits of docility and tolerance toward human company of the silver fox (Vulpes vulpes) resulted in complex feature changes including attenuated HPA axis activity (Trut et al., 2009). This selection was accompanied by a change in 3% of the transcriptome in the fox prefrontal cortex (Kukekova et al., 2011). Aside from selection for genetic variants epigenetic regulation may have largely contributed to the changes in gene expression associated with the domestication of this species. Another interesting insight provides a multigenerational study of breeding 11 generations of mice selected for anxiety traits (Filiou et al., 2011). Anxiety-like behaviors were linked to altered proteomic status and metabolic pathway function, along with substantial differences in the transcriptome (Czibere et al., 2011; Filiou et al., 2011).
Although the above criteria of transgenerational programming do not apply to invertebrates, a recent study showed intriguing evidence of multigenerational programming of longevity. An example of epigenetic transmission of an adaptive parental trait to future generations was recently described in the nematode Caenorhabditis elegans (Greer et al., 2011). In this study, deficiency in the chromatin modifying histone H3 lysine 4 trimethylation complex, which regulates C. elegans lifespan, can be passed on to three subsequent generations to extend their lifespan (Greer et al., 2011). Through methylation-induced gene silencing, RNA or protein inheritance a histone modification involving H3K4me3, a regulatory complex associated with longevity, was shown to persist to the F4 generation of a C. elegans population (Greer et al., 2011). In this study, however, epigenetic inheritance of longevity disappeared in the F5 generation. The mechanisms involved in developing transgenerational behavioral, physiological, and metabolic traits remain to be elucidated.
The findings discussed above suggest widespread and drastic phenotypic changes after transgenerational pre- or postnatal adverse environment with an emphasis on epigenetic regulation. A growing body of evidence provides examples of transgenerational stability of epigenetic changes, such as DNA methylation patterns and miRNA expression (Zambrano, 2009; Boyko et al., 2010; Matthews and Phillips, 2010, 2012) and alterations associated with aging processes (Fraga et al., 2005; Rodríguez-Rodero et al., 2010). Transgenerational differences and the effects of age on miRNA expression are robust (Jukic et al., 2010). The specific mechanisms of transgenerational transmission are not entirely known, but DNA methylation and miRNA expression changes are likely involved (Boyko et al., 2007, 2010; Skinner and Guerrero-Bosagna, 2009) and these may offer an exciting potential to identify epigenetic signatures of prognostic significance for disease. Interestingly, recent findings support the potential to obtain predictive epigenetic markers of disease through blood samples or placenta (Tsui et al., 2010; Du et al., 2011; Hahn et al., 2011), which bears important therapeutic potential.
Implications and Conclusion
While life-long experience constantly modulates epigenomic patterns, ancestral programming sets the stage for responses to stress, disease, and aging across the lifespan. In this review, we proposed that epigenetic imprints of environmental information may persist through multiple generations. Such epigenetic memories may represent a unifying component to phenotypic plasticity to influence development, performance, and disease (Petronis, 2010). Most mechanistic studies of epigenetic inheritance have focused on F0–F1 transmission. Fewer reports of transgenerational phenotypes and epigenotypes including and beyond the F3 generation exist, however, transgenerational programming of brain and developmental plasticity, metabolic, neurological and psychiatric functions is a critical and rewarding field of investigation.
Transgenerational programming of disease risk may explain the general difficulty to identify the causes of complex adult-onset diseases. Understanding the possibly substantial contribution of ancestors to disease risk in an individual may significantly advance prognostic medicine. The impact of transgenerational programming versus life-long experience on health and disease is a central issue for personalized medicine and disease prevention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Alberta Innovates – Health Solutions Interdisciplinary Preterm Birth and Healthy Outcomes Team award and the Canadian Institutes of Health Research. Gerlinde A. Metz is a senior scholar of Alberta Innovates-Health Solutions.
References
Adams, J. (2008). Imprinting and genetic disease: Angelman, Prader–Willi and Beckwith–Wiedemann syndromes. Nat. Educ. 1.
Aerts, L., and van Assche, F. A. (1979). Is gestational diabetes an acquired condition? J. Dev. Physiol. 1, 219–225.
Angelman, H. (1965). “Puppet” children: a report of three cases. Dev. Med. Child Neurol. 7, 681–688.
Anway, M. D., Cupp, A. S., Uzumcu, M., and Skinner, M. K. (2005). Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469.
Anway, M. D., Leathers, C., and Skinner, M. K. (2006). Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 147, 5515–5523.
Babenko, O., Golubov, A., Ilnytskyy, Y., Kovalchuk, I., and Metz, G. A. (2012a). Genomic and epigenomic responses to chronic stress involve miRNA-mediated programming. PLoS ONE 7, e29441. doi: 10.1371/journal.pone.0029441
Babenko, O., Kovalchuk, I., and Metz, G. A. (2012b). Epigenetic programming of neurodegenerative diseases by an adverse environment. Brain Res. 1444, 96–111.
Balaratnasingam, S., and Janca, A. (2012). Brain derived neurotrophic factor: a novel neurotrophin involved in psychiatric and neurological disorders. Pharmacol. Ther. 134, 116–124.
Begum, G., Stevens, A., Smith, E. B., Connor, K., Challis, J. R., Bloomfield, F., and White, A. (2012). Epigenetic changes in fet al hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 26, 1694–1703.
Bennett-Baker, P. E., Wilkowski, J., and Burke, D. T. (2003). Age-associated activation of epigenetically repressed genes in the mouse. Genetics 165, 2055–2062.
Benyshek, D. C., Johnston, C. S., and Martin, J. F. (2006). Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49, 1117–1119.
Bertram, C., Khan, O., Ohri, S., Phillips, D. I., Matthews, S. G., and Hanson, M. A. (2008). Transgenerational effects of prenatal nutrient restriction on cardiovascular and hypothalamic–pituitary–adrenal function. J. Physiol. 586, 2217–2229.
Bilichak, A., Ilnystkyy, Y., Hollunder, J., and Kovalchuk, I. (2012). The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 7, e30515. doi: 10.1371/journal.pone.0030515
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535.
Blatt, J., Van Le, L., Weiner, T., and Sailer, S. (2003). Ovarian carcinoma in an adolescent with transgenerational exposure to diethylstilbestrol. J. Pediatr. Hematol. Oncol. 25, 635–636.
Boyko, A., Blevins, T., Yao, Y., Golubov, A., Bilichak, A., Ilnytskyy, Y., Hollunder, J., Meins, F. Jr., and Kovalchuk, I. (2010). Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 5, e9514. doi: 10.1371/journal.pone.0009514
Boyko, A., Kathiria, P., Zemp, F. J., Yao, Y., Pogribny, I., and Kovalchuk, I. (2007). Transgenerational changes in the genome stability and methylation in pathogen-infected plants (virus-induced plant genome instability). Nucleic Acids Res. 35, 1714–1725.
Boyko, A., and Kovalchuk, I. (2010). Transgenerational response to stress in Arabidopsis thaliana. Plant Signal. Behav. 5, 995–998.
Boyko, A., and Kovalchuk, I. (2011). Genome instability and epigenetic modification – heritable responses to environmental stress? Curr. Opin. Plant Biol. 14, 260–266.
Breton, C. V., Byun, H. M., Wenten, M., Pan, F., Yang, A., and Gilliland, F. D. (2009). Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 180, 462–467.
Brouwers, M. M., Feitz, W. F., Roelofs, L. A., Kiemeney, L. A., de Gier, R. P., and Roeleveld, N. (2006). Hypospadias: a transgenerational effect of diethylstilbestrol? Hum. Reprod. 21, 666–669.
Brummelte, S., Schmidt, K. L., Taves, M. D., Soma, K. K., and Galea, L. A. (2010). Elevated corticosterone levels in stomach milk, serum, and brain of male and female offspring after maternal corticosterone treatment in the rat. Dev. Neurobiol. 70, 714–725.
Burns, J. G., and Mery, F. (2010). Transgenerational memory effect of ageing in Drosophila. J. Evol. Biol. 23, 678–686.
Caldji, C., Diorio, J., and Meaney, M. J. (2003). Variations in maternal care alter GABA(A) receptor subunit expression in brain regions associated with fear. Neuropsychopharmacology 28, 1950–1959.
Cameron, N. M. (2011). Maternal programming of reproductive function and behavior in the female rat. Front. Evol. Neurosci. 3:10. doi: 10.3389/fnevo.2011.00010
Carlson, M. C., Helms, M. J., Steffens, D. C., Burke, J. R., Potter, G. G., and Plassman, B. L. (2008). Midlife activity predicts risk of dementia in older male twin pairs. Alzheimers Dement. 4, 324–331.
Cassidy, S. B., Schwartz, S., Miller, J. L., and Driscoll, D. J. (2012). Prader–Willi syndrome. Genet. Med. 14, 10–26.
Champagne, D. L., Bagot, R. C., van Hasselt, F., Ramakers, G., Meaney, M. J., de Kloet, E. R., Joëls, M., and Krugers, H. (2008). Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 28, 6037–6045.
Champagne, F. A., Francis, D. D., Mar, A., and Meaney, M. J. (2003). Variations in maternal care in the rat as a mediating influence for the effects of environment on development. Physiol. Behav. 79, 359–371.
Champagne, F. A., and Meaney, M. J. (2006). Stress during gestation alters postpartum maternal care and the development of the offspring in a rodent model. Biol. Psychiatry 59, 1227–1235.
Champagne, F. A., and Meaney, M. J. (2007). Transgenerational effects of social environment on variations in maternal care and behavioral response to novelty. Behav. Neurosci. 121, 1353–1363.
Champagne, F. A., Weaver, I. C., Diorio, J., Dymov, S., Szyf, M., and Meaney, M. J. (2006). Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology 147, 2909–2915.
Chen, K. C., Liao, Y. C., Hsieh, I. C., Wang, Y. S., Hu, C. Y., and Juo, S. H. (2011). OxLDL causes both epigenetic modification and signaling regulation on the microRNA-29b gene: novel mechanisms for cardiovascular diseases. J. Mol. Cell. Cardiol. 52, 587–595.
Chong, S., and Whitelaw, E. (2004). Epigenetic germline inheritance. Curr. Opin. Genet. Dev. 14, 692–696.
Colvis, C. M., Pollock, J. D., Goodman, R. H., Impey, S., Dunn, J., Mandel, G., Champagne, F. A., Mayford, M., Korzus, E., Kumar, A., Renthal, W., Theobald, D. E., and Nestler, E. J. (2005). Epigenetic mechanisms and gene networks in the nervous system. J. Neurosci. 25, 10379–10389.
Cottrell, E. C., and Seckl, J. R. (2009). Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 3:19. doi: 10.3389/neuro.08.019.2009
Cropley, J. E., Dang, T. H., Martin, D. I., and Suter, C. M. (2012). The penetrance of an epigenetic trait in mice is progressively yet reversibly increased by selection and environment. Proc. Biol. Sci. 279, 2347–2353.
Curfs, L. M., and Fryns, J. P. (1992). Prader–Willi syndrome: a review with special attention to the cognitive and behavioral profile. Birth Defects Orig. 28, 99–104.
Czibere, L., Baur, L. A., Wittmann, A., Gemmeke, K., Steiner, A., Weber, P., Pütz, B., Ahmad, N., Bunck, M., Graf, C., Widner, R., Kühne, C., Panhuysen, M., Hambsch, B., Rieder, G., Reinheckel, T., Peters, C., Holsboer, F., Landgraf, R., and Deussing, J. M. (2011). Profiling trait anxiety: transcriptome analysis reveals cathepsin B (Ctsb) as a novel candidate gene for emotionality in mice. PLoS ONE 6, e23604. doi: 10.1371/journal.pone.0023604
Daxinger, L., and Whitelaw, E. (2012). Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 13, 153–162.
Devlin, A. M., Brain, U., Austin, J., and Oberlander, T. F. (2010). Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS ONE 5, e12201. doi: 10.1371/journal.pone.0012201
Dolinoy, D. C., Huang, D., and Jirtle, R. L. (2007). Maternal nutrient supplementation counteracts bisphenol A induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. U.S.A. 104, 13056–13061.
Dörner, G., Steindel, E., Thoelke, H., and Schliack, V. (1984). Evidence for decreasing prevalence of diabetes mellitus in childhood apparently produced by prevention of hyperinsulinism in the foetus and newborn. Exp. Clin. Endocrinol. 84, 134–142.
Drake, A. J., Walker, B. R., and Seckl, J. R. (2005). Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R34–R38.
Du, Y., Zhang, J., Wang, H., Yan, X., Yang, Y., Yang, L., Luo, X., Chen, Y., Duan, T., and Ma, D. (2011). Hypomethylated DSCR4 is a placenta-derived epigenetic marker for trisomy 21. Prenat. Diagn. 31, 207–214.
Dunn, G. A., and Bale, T. L. (2011). Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 152, 2228–2236.
Escorihuela, R. M., Tobena, A., and Fernandez-Teruel, A. (1994). Environmental enrichment reverses the detrimental action of early inconsistent stimulation and increases the beneficial effects of postnatal handling on shuttlebox learning in adult rats. Behav. Brain Res. 61, 169–173.
Essex, M. J., Thomas Boyce, W., Hertzman, C., Lam, L. L., Armstrong, J. M., Neumann, S. M., and Kobor, M. S. (2011). Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. doi: 10.1111/j.1467-8624.2011.01641.x [Epub ahead of print].
Feil, R., and Fraga, M. F. (2012). Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109.
Felsen, I., and Erlich, H. S. (1990). Identification patterns of offspring of Holocaust survivors with their parents. Am. J. Orthopsychiatry 60, 506–520.
Filiou, M. D., Zhang, Y., Teplytska, L., Reckow, S., Gormanns, P., Maccarrone, G., Frank, E., Kessler, M. S., Hambsch, B., Nussbaumer, M., Bunck, M., Ludwig, T., Yassouridis, A., Holsboer, F., Landgraf, R., and Turck, C. W. (2011). Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol. Psychiatry 70, 1074–1082.
Filkowski, J. N., Ilnytskyy, Y., Tamminga, J., Koturbash, I., Golubov, A., Bagnyukova, T., Pogribny, I. P., and Kovalchuk, O. (2010). Hypomethylation and genome instability in the germline of exposed parents and their progeny is associated with altered miRNA expression. Carcinogenesis 31, 1110–1115.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., Heine-Suñer, D., Cigudosa, J. C., Urioste, M., Benitez, J., Boix-Chornet, M., Sanchez-Aguilera, A., Ling, C., Carlsson, E., Poulsen, P., Vaag, A., Stephan, Z., Spector, T. D., Wu, Y. Z., Plass, C., and Esteller, M. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609.
Francis, D. D., Caldji, C., Champagne, F., Plotsky, P. M., and Meaney, M. J. (1999). The role of corticotropin-releasing factor – norepinephrine systems in mediating the effects of early experience on the development of behavioral and endocrine responses to stress. Biol. Psychiatry 46, 1153–1166.
Franklin, T. B., Russig, H., Weiss, I. C., Gräff, J., Linder, N., Michalon, A., Vizi, S., and Mansuy, I. M. (2010). Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 68, 408–415.
Fuchikami, M., Yamamoto, S., Morinobu, S., Takei, S., and Yamawaki, S. (2010). Epigenetic regulation of BDNF gene in response to stress. Psychiatry Investig. 7, 251–256.
Gatt, J. M., Nemeroff, C. B., Dobson-Stone, C., Paul, R. H., Bryant, R. A., Schofield, P. R., Gordon, E., Kemp, A. H., and Williams, L. M. (2009). Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol. Psychiatry 14, 681–695.
Glaser, B., Ades, A. E., Lewis, S., Emmet, P., Lewis, G., Smith, G. D., and Zammit, S. (2010). Perinatal folate-related exposures and risk of psychotic symptoms in the ALSPAC birth cohort. Schizophr. Res. 120, 177–183.
Gluckman, P. D., Hanson, M. A., and Beedle, A. S. (2007). Non-genomic transgenerational inheritance of disease risk. Bioessays 29, 145–154.
Golding, J., Steer, C., and Pembrey, M. (2010). Parental and grandparental ages in the autistic spectrum disorders: a birth cohort study. PLoS ONE 5, e9939. doi: 10.1371/journal.pone.0009939
Greer, E. L., Maures, T. J., Ucar, D., Hauswirth, A. G., Mancini, E., Lim, J. P., Benayoun, B. A., Shi, Y., and Brunet, A. (2011). Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479, 365–371.
Guerrero-Bosagna, C., Settles, M., Lucker, B., and Skinner, M. K. (2010). Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE 5, e13100. doi: 10.1371/journal.pone.0013100
Guerrero-Preston, R., Goldman, L. R., Brebi-Mieville, P., Ili-Ganga, C., Lebron, C., Witter, F. R., Apelberg, B. J., Hernández-Roystacher, M., Jaffe, A., Halden, R. U., and Sidransky, D. (2010). Global hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics 5, 539–546.
Hahn, S., Rusterholz, C., Hösli, I., and Lapaire, O. (2011). Cell-free nucleic acids as potential markers for preeclampsia. Placenta 32, S17–S20.
Hernandez, D. G., Nalls, M. A., Gibbs, J. R., Arepalli, S., van der Brug, M., Chong, S., Moore, M., Longo, D. L., Cookson, M. R., Traynor, B. J., and Singleton, A. B. (2011). Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 20, 1164–1172.
Ilnytskyy, Y., Zemp, F. J., Koturbash, I., and Kovalchuk, O. (2008). Altered microRNA expression patterns in irradiated hematopoietic tissues suggest a sex-specific protective mechanism. Biochem. Biophys. Res. Commun. 377, 41–45.
Itier, J. M., Tremp, G. L., Léonard, J. F., Multon, M. C., Ret, G., Schweighoffer, F., Tocqué, B., Bluet-Pajot, M. T., Cormier, V., and Dautry, F. (1998). Imprinted gene in postnatal growth role. Nature 393, 125–126.
Jablonka, E., and Lamb, M. J. (1989). The inheritance of acquired epigenetic variations. J. Theor. Biol. 139, 69–83.
Jadavji, N. M., Kolb, B., and Metz, G. A. (2006). Enriched environment improves motor function in intact and unilateral dopamine-depleted rats. Neuroscience 140, 1127–1138.
James, B. D., Boyle, P. A., Buchman, A. S., Barnes, L. L., and Bennett, D. A. (2011). Life space and risk of Alzheimer disease, mild cognitive impairment, and cognitive decline in old age. Am. J. Geriatr. Psychiatry 19, 961–969.
Jiang, Y., Tsai, T. F., Bressler, J., and Beaudet, A. L. (1998). Imprinting in Angelman and Prader–Willi syndromes. Curr. Opin. Genet. Dev. 8, 334–342.
Jirtle, R. L., and Skinner, M. K. (2007). Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 8, 253–262.
Johansen, I. B., Sørensen, C., Sandvik, G. K., Nilsson, G. E., Höglund, E., Bakken, M., and Overli, O. (2012). Neural plasticity is affected by stress and heritable variation in stress coping style. Comp. Biochem. Physiol. Part D Genomics Proteomics 7, 161–171.
Jukic, D. M., Rao, U. N., Kelly, L., Skaf, J. S., Drogowski, L. M., Kirkwood, J. M., and Panelli, M. C. (2010). MicroRNA profiling analysis of differences between the melanoma of young adults and older adults. J. Transl. Med. 8, 27.
Kaliman, P., Párrizas, M., Lalanza, J. F., Camins, A., Escorihuela, R. M., and Pallàs, M. (2011). Neurophysiological and epigenetic effects of physical exercise on the aging process. Ageing Res. Rev. 10, 475–486.
Kaneda, R., Takada, S., Yamashita, Y., Choi, Y. L., Nonaka-Sarukawa, M., Soda, M., Misawa, Y., Isomura, T., Shimada, K., and Mano, H. (2009). Genome-wide histone methylation profile for heart failure. Genes Cells 14, 69–77.
Kinsella, M. T., and Monk, C. (2009). Impact of maternal stress, depression and anxiety on fetal neurobehavioral development. Clin. Obstet. Gynecol. 52, 425–440.
Kobilo, T., Liu, Q. R., Gandhi, K., Mughal, M., Shaham, Y., and van Praag, H. (2011). Running is the neurogenic and neurotrophic stimulus in environmental enrichment. Learn. Mem. 18, 605–609.
Koenen, K. C., Uddin, M., Chang, S. C., Aiello, A. E., Wildman, D. E., Goldmann, E., and Galea, S. (2011). SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depress. Anxiety 28, 639–647.
Kukekova, A. V., Johnson, J. L., Teiling, C., Li, L., Oskina, I. N., Kharlamova, A. V., Gulevich, R. G., Padte, R., Dubreuil, M. M., Vladimirova, A. V., Shepeleva, D. V., Shikhevich, S. G., Sun, Q., Ponnala, L., Temnykh, S. V., Trut, L. N., and Acland, G. M. (2011). Sequence comparison of prefrontal cortical brain transcriptome from a tame and an aggressive silver fox (Vulpes vulpes). BMC Genomics 12, 482. doi: 10.1186/1471-2164-12-482
Laviola, G., Rea, M., Morley-Fletcher, S., Di Carlo, S., Bacosi, A., De Simone, R., Bertini, M., and Pacifici, R. (2004). Beneficial effects of enriched environment on adolescent rats from stressed pregnancies. Eur. J. Neurosci. 20, 1655–1664.
Law, C. M., Barker, D. J. P., Osmond, C., Fall, C. H. D., and Simmonds, S. J. (1992). Early growth and abdominal fatness in adult life. J. Epidemiol. Comm. Health 46, 184–186.
Ling, C., Del Guerra, S., Lupi, R., Rönn, T., Granhall, C., Luthman, H., Masiello, P., Marchetti, P., Groop, L., and Del Prato, S. (2008). Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51, 615–622.
Lock, J. E. (2012). Transgenerational effects of parent and grandparent gender on offspring development in a biparental beetle species. Biol. Lett. 8, 408–411.
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., and Yankner, B. A. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891.
Mabandla, M. V., Kellaway, L. A., Daniels, W. M., and Russell, V. A. (2009). Effect of exercise on dopamine neuron survival in prenatally stressed rats. Metab. Brain Dis. 24, 525–539.
Mabandla, M. V., and Russell, V. A. (2010). Voluntary exercise reduces the neurotoxic effects of 6-hydroxydopamine in maternally separated rats. Behav. Brain Res. 211, 16–22.
Magenis, R. E., Brown, M. G., Lacy, D. A., Budden, S., and LaFranchi, S. (1987). Is Angelman syndrome an alternate result of del(15)(q11q13)? Am. J. Med. Genet. 28, 829–838.
Manikkam, M., Guerrero-Bosagna, C., Tracey, R., Haque M. M., and Skinner, M. K. (2012). Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS ONE 7, e31901. doi: 10.1371/journal.pone.0031901
Martin, G. M. (2005). Epigenetic drift in aging identical twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10413–10414.
Matthews, S. G., and Phillips, D. I. (2010). Minireview: transgenerational inheritance of the stress response: a new frontier in stress research. Endocrinology 151, 7–13.
Matthews, S. G., and Phillips, D. I. (2012). Transgenerational inheritance of stress pathology. Exp. Neurol. 233, 95–101.
Mattson, M. P. (2008). Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N.Y. Acad. Sci. 1144, 97–112.
Mattson, M. P., Duan, W., Lee, J., and Guo, Z. (2001). Suppression of brain aging and neurodegenerative disorders by dietary restriction and environmental enrichment: molecular mechanisms. Mech. Ageing Dev. 122, 757–778.
McArthur, S., McHale, E., and Gillies, G. E. (2007). The size and distribution of midbrain dopaminergic populations are permanently altered by perinatal glucocorticoid exposure in a sex- region- and time-specific manner. Neuropsychopharmacology 32, 1462–1476.
Merrett, D. L., Kirkland, S. W., and Metz, G. A. (2010). Synergistic effects of age and stress in a rodent model of stroke. Behav. Brain Res. 214, 55–59.
Migicovsky, Z., and Kovalchuk, I. (2011). Epigenetic memory in mammals. Front. Genet. 2:28. doi: 10.3389/fgene.2011.00028
Miller, D. B., and Chernoff, N. (1995). Restraint-induced stress in pregnant mice – degree of immobilization affects maternal indices of stress and developmental outcome in offspring. Toxicology 98, 177–186.
Miller, D. B., and O’Callaghan, J. P. (2008). Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism 57, S44–S49.
Mora, F., Segovia, G., and del Arco, A. (2007). Aging, plasticity and environmental enrichment: structural changes and neurotransmitter dynamics in several areas of the brain. Brain Res. Rev. 55, 78–88.
Morgan, C. P., and Bale, T. L. (2011). Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J. Neurosci. 31, 11748–11755.
Morgan, H. D., Sutherland, H. G., Martin, D. I., and Whitelaw, E. (1999). Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 23, 314–318.
Mousseau, T. A., and Fox, C. W. (1998). The adaptive significance of maternal effects. Trends Ecol. Evol. 13, 403–407.
Mueller, B. R., and Bale, T. L. (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28, 9055–9065.
Mychasiuk, R., Ilnytskyy, S., Kovalchuk, O., Kolb, B., and Gibb, R. (2011). Intensity matters: brain, behaviour and the epigenome of prenatally stressed rats. Neuroscience 180, 105–110.
Newnham, J. P. (2001). Is prenatal glucocorticoid administration another origin of adult disease? Clin. Exp. Pharmacol. Physiol. 28, 957–961.
Nilsson, E. E., Anway, M. D., Stanfield, J., and Skinner, M. K. (2008). Transgenerational epigenetic effects of the endocrine disruptor vinclozolin on pregnancies and female adult onset disease. Reproduction 135, 713–721.
Nolan, C. J., Damm, P., and Prentki, M. (2011). Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet 378, 169–181.
Nomura, T., Kimura, M., Horii, T., Morita, S., Soejima, H., Kudo, S., and Hatada, I. (2008). MeCP2-dependent repression of an imprinted miR-184 released by depolarization. Hum. Mol. Genet. 17, 1192–1199.
Nott, A., and Riccio, A. (2009). Nitric oxide-mediated epigenetic mechanisms in developing neurons. Cell Cycle 1, 725–730.
Ordovás, J. M., and Smith, C. E. (2010). Epigenetics and cardiovascular disease. Nat. Rev. Cardiol. 7, 510–519.
Paban, V., Chambon, C., Manrique, C., Touzet, C., and Alescio-Lautier, B. (2009). Neurotrophic signaling molecules associated with cholinergic damage in young and aged rats: environmental enrichment as potential therapeutic agent. Neurobiol. Aging 32, 470–485.
Painter, R. C., Osmond, C., Gluckman, P., Hanson, M., Phillips, D. I., and Roseboom, T. J. (2008). Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 115, 1243–1249.
Painter, R. C., Roseboom, T. J., and Bleker, O. P. (2005). Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod. Toxicol. 20, 345–352.
Pembrey, M. E. (2010). Male-line transgenerational responses in humans. Hum. Fertil. (Camb.) 13, 268–271.
Pentinat, T., Ramon-Krauel, M., Cebria, J., Diaz, R., and Jimenez-Chillaron, J. C. (2010). Transgenerational inheritance of glucose intolerance in a mouse model of neonatal overnutrition. Endocrinology 151, 5617–5623.
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465, 721–727.
Pham, T. M., Winblad, B., Granholm, A. C., and Mohammed, A. H. (2002). Environmental influences on brain neurotrophins in rats. Pharmacol. Biochem. Behav. 73, 167–175.
Pinheiro, A. R., Salvucci, I. D., Aguila, M. B., and Mandarim-de-Lacerda, C. A. (2008). Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin. Sci. (Lond.) 114, 381–392.
Poulsen, P., Esteller, M., Vaag, A., and Fraga, M. F. (2007). The epigenetic basis of twin discordance in age-related diseases. Pediatr. Res. 61, 38R–42R.
Reik, W., and Walter, J. (2001). Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2, 21–32.
Richards, M., and Sacker, A. (2003). Lifetime antecedents of cognitive reserve. J. Clin. Exp. Neuropsychol. 25, 614–624.
Rodríguez-Rodero, S., Fernández-Morera, J. L., Fernandez, A. F., Menéndez-Torre, E., and Fraga, M. F. (2010). Epigenetic regulation of aging. Discov. Med. 10, 225–233.
Rokyta, R., Yamamotová, A., Slamberová, R., Franek, M., Vaculín, S., Hrubá, L., Schutová, B., and Pometlová, M. (2008). Prenatal and perinatal factors influencing nociception, addiction and behavior during ontogenetic development. Physiol. Res. 57, S79–S88.
Ronald, A., Pennell, C. E., and Whitehouse, A. J. (2010). Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front. Psychol. 1:223. doi: 10.3389/fpsyg.2010.00223
Roth, T. L., Lubin, F. D., Funk, A. J., and Sweatt, J. D. (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 65, 760–769.
Rougeulle, C., Cardoso, C., Fontés, M., Colleaux, L., and Lalande, M. (1998). An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet. 19, 15–16.
Scharf, M. (2007). Long-term effects of trauma: psychosocial functioning of the second and third generation of Holocaust survivors. Dev. Psychopathol. 19, 603–622.
Schreier, N. K., Moltchanova, E. V., Blomstedt, P. A., Kajantie, E., and Eriksson, J. G. (2011). Prenatal exposure to wartime stress – long-term effect on coronary heart disease in later life. Ann. Med. 43, 555–561.
Schwarz, J. M., Nugent, B. M., and McCarthy, M. M. (2010). Developmental and hormone-induced epigenetic changes to estrogen and progesterone receptor genes in brain are dynamic across the life span. Endocrinology 151, 4871–4881.
Seckl, J. R. (2008). Glucocorticoids, developmental “programming” and the risk of affective dysfunction. Prog. Brain Res. 167, 17–34.
Segovia, G., del Arco, A., and Mora, F. (2009). Environmental enrichment, prefrontal cortex, stress, and aging of the brain. J. Neural Transm. 116, 1007–1016.
Sen, N., and Snyder, S. H. (2011). Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 108, 20178–20183.
Shen, S., Liu, A., Li, J., Wolubah, C., and Casaccia-Bonnefil, P. (2008). Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol. Aging 29, 452–463.
Siegmund, K. D., Connor, C. M., Campan, M., Long, T. I., Weisenberger, D. J., Biniszkiewicz, D., Jaenisch, R., Laird, P. W., and Akbarian, S. (2007). DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE 2, e895. doi: 10.1371/journal.pone.0000895
Sigal, J. J., DiNicola, V. F., and Buonvino, M. (1988). Grandchildren of survivors: can negative effects of prolonged exposure to excessive stress be observed two generations later? Can. J. Psychiatry 33, 207–212.
Sigal, J. J., and Weinfeld, M. (2001). Do children cope better than adults with potentially traumatic stress? A 40-year follow-up of Holocaust survivors. Psychiatry 64, 69–80.
Singhal, A. (2010). Does early growth affect long-term risk factors for cardiovascular disease? Nestle Nutr. Workshop Ser. Pediatr. Program 65, 55–64.
Skinner, M. K. (2008). What is an epigenetic transgenerational phenotype? F3 or F2. Reprod. Toxicol. 25, 2–6.
Skinner, M. K. (2011). Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 6, 838–842.
Skinner, M. K., Anway, M. D., Savenkova, M. I., Gore, A. C., and Crews, D. (2008). Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS ONE 3, e3745. doi: 10.1371/journal.pone.0003745
Skinner, M. K., and Guerrero-Bosagna, C. (2009). Environmental signals and transgenerational epigenetics. Epigenomics 1, 111–117.
Skinner, M. K., Manikkam, M., and Guerrero-Bosagna, C. (2011). Epigenetic transgenerational actions of endocrine disruptors. Reprod. Toxicol. 31, 337–343.
Stein, A. B., Jones, T. A., Herron, T. J., Patel, S. R., Day, S. M., Noujaim, S. F., Milstein, M. L., Klos, M., Furspan, P. B., Jalife, J., and Dressler, G. R. (2011). Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J. Clin. Invest. 121, 2641–2650.
Stein, A. D., and Lumey, L. H. (2000). The relationship between maternal and offspring birth weights after maternal prenatal famine exposure: the Dutch Famine Birth Cohort Study. Hum. Biol. 72, 641–654.
Stenvinkel, P., Karimi, M., Johansson, S., Axelsson, J., Suliman, M., Lindholm, B., Heimbürger, O., Barany, P., Alvestrand, A., Nordfors, L., Qureshi, A. R., Ekström, T. J., and Schalling, M. (2007). Impact of inflammation on epigenetic DNA methylation – a novel risk factor for cardiovascular disease? J. Intern. Med. 261, 488–499.
Sterrenburg, L., Gaszner, B., Boerrigter, J., Santbergen, L., Bramini, M., Elliott, E., Chen, A., Peeters, B. W., Roubos, E. W., and Kozicz, T. (2011). Chronic stress induces sex-specific alterations in methylation and expression of corticotropin-releasing factor gene in the rat. PLoS ONE 6, e28128. doi: 10.1371/ journal.pone.0028128
Taliaz, D., Loya, A., Gersner, R., Haramati, S., Chen, A., and Zangen, A. (2011). Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J. Neurosci. 31, 4475–4483.
Thompson, R. F., and Einstein, F. H. (2010). Epigenetic basis for fetal origins of age-related disease. J. Womens Health (Larchmt) 19, 581–587.
Toledo-Rodriguez, M., Lotfipour, S., Leonard, G., Perron, M., Richer, L., Veillette, S., Pausova, Z., and Paus, T. (2010). Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 1350–1354.
Trut, L., Oskina, I., and Kharlamova, A. (2009). Animal evolution during domestication: the domesticated fox as a model. Bioessays 31, 349–360.
Tsui, D. W., Lam, Y. M., Lee, W. S., Leung, T. Y., Lau, T. K., Lau, E. T., Tang, M. H., Akolekar, R., Nicolaides, K. H., Chiu, R. W., Lo, Y. M., and Chim, S. S. (2010). Systematic identification of placental epigenetic signatures for the noninvasive prenatal detection of Edwards syndrome. PLoS ONE 5, e15069. doi: 10.1371/journal.pone.0015069
Uchida, S., Hara, K., Kobayashi, A., Otsuki, K., Yamagata, H., Hobara, T., Suzuki, T., Miyata, N., and Watanabe, Y. (2011). Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69, 359–372.
Volkmar, M., Dedeurwaerder, S., Cunha, D. A., Ndlovu, M. N., Defrance, M., Deplus, R., Calonne, E., Volkmar, U., Igoillo-Esteve, M., Naamane, N., Del Guerra, S., Masini, M., Bugliani, M., Marchetti, P., Cnop, M., Eizirik, D. L., and Fuks, F. (2012). DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 31, 1405–1426.
Wang, C., Wang, Z., Luo, J., Li, Q., Li, Y., Ahn, K., Prows, D. R., and Wu, R. (2010). A model for transgenerational imprinting variation in complex traits. PLoS ONE 5, e11396. doi: 10.1371/journal.pone.0011396
Waterland, R. A., Travisano, M., and Tahiliani, K. G. (2007). Diet-induced hypermethylation at agouti viable yellow is not inherited transgenerationally through the female. FASEB J. 21, 3380–3385.
Weaver, I. C., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., Dymov, S., Szyf, M., and Meaney, M. J. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854.
Weaver, I. C., D’Alessio, A. C., Brown, S. E., Hellstrom, I. C., Dymov, S., Sharma, S., Szyf, M., and Meaney, M. J. (2007). The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J. Neurosci. 27, 1756–1768.
Welberg, L. A., Seckl, J. R., and Holmes, M. C. (2000). Inhibition of 11beta-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur. J. Neurosci. 12, 1047–1054.
Whitelaw, N. C., and Whitelaw, E. (2008). Transgenerational epigenetic inheritance in health and disease. Curr. Opin. Genet. Dev. 18, 273–279.
Wilkins, J., and Haig, D. (2003). What good is genomic imprinting: the function of parent-specific gene expression. Nat. Rev. Genet. 4, 359–368.
Wilkinson, L., Davies, W., and Isles, A. (2007). Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci. 8, 832–843.
Wirdefeldt, K., Gatz, M., Pawitan, Y., and Pedersen, N. L. (2005). Risk and protective factors for Parkinson’s disease: a study in Swedish twins. Ann. Neurol. 57, 27–33.
Yauk, C., Polyzos, A., Rowan-Carroll, A., Somers, C. M., Godschalk, R. W., Van Schooten, F. J., Berndt, M. L., Pogribny, I. P., Koturbash, I., Williams, A., Douglas, G. R., and Kovalchuk, O. (2008). Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc. Natl. Acad. Sci. U.S.A. 105, 605–610.
Yehuda, R., Teicher, M. H., Seckl, J. R., Grossman, R. A., Morris, A., and Bierer, L. M. (2007). Parental posttraumatic stress disorder as a vulnerability factor for low cortisol trait in offspring of holocaust survivors. Arch. Gen. Psychiatry 64, 1040–1048.
Zambrano, E. (2009). The transgenerational mechanisms in developmental programming of metabolic diseases. Rev. Invest. Clin. 61, 41–52.
Zambrano, E., Bautista, C. J., Deás, M., Martínez-Samayoa, P. M., González-Zamorano, M., Ledesma, H., Morales, J., Larrea, F., and Nathanielsz, P. W. (2005). A low maternal protein diet during pregnancy and lactation has sex- and window of exposure-specific effects on offspring growth and food intake, glucose metabolism and serum leptin in the rat. J. Physiol. 571, 221–230.
Zamenhof, S., van Marthens, E., and Grauel, L. (1971). DNA (cell number) in neonatal brain: second generation (F2) alteration by maternal (F0) dietary protein restriction. Science 172, 850–851.
Zeng, Y., Tan, M., Kohyama, J., Sneddon, M., Watson, J. B., Sun, Y. E., and Xie, C. W. (2011). Epigenetic enhancement of BDNF signaling rescues synaptic plasticity in aging. J. Neurosci. 31, 17800–17810.
Keywords: epigenetics, microRNA, methylation, histone, programming by perinatal stress, adverse experience, neurological and psychiatric diseases, metabolic diseases
Citation: Zucchi FCR, Yao Y and Metz GA (2012) The secret language of destiny: stress imprinting and transgenerational origins of disease. Front. Gene. 3:96. doi: 10.3389/fgene.2012.00096
Received: 01 March 2012; Accepted: 10 May 2012;
Published online: 04 June 2012.
Edited by:
Igor Kovalchuk, University of Lethbridge, CanadaReviewed by:
Sheridan Gentili, University of South Australia, AustraliaFeng Zhou, Indiana University School Medicine, USA
Copyright: © 2012 Zucchi, Yao and Metz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gerlinde A. Metz, Canadian Centre for Behavioural Neuroscience, University of Lethbridge, 4401 University Drive, Lethbridge, AB, Canada T1K 3M4. e-mail:Z2VybGluZGUubWV0ekB1bGV0aC5jYQ==