

- 1 Department of Genetics and Developmental Biology, University of Connecticut Health Center, Farmington, CT, USA
- 2 Department of Neuroscience, University of Connecticut Health Center, Farmington, CT, USA
- 3 Department of Molecular, Microbial and Structural Biology, University of Connecticut Health Center, Farmington, CT, USA
Neurons modulate gene expression with subcellular precision through excitation-coupled local protein synthesis, a process that is regulated in part through the involvement of microRNAs (miRNAs), a class of small non-coding RNAs. The biosynthesis of miRNAs is reviewed, with special emphasis on miRNA families, the subcellular localization of specific miRNAs in neurons, and their potential roles in the response to drugs of abuse. For over a decade, DNA microarrays have dominated genome-wide gene expression studies, revealing widespread effects of drug exposure on neuronal gene expression. We review a number of recent studies that explore the emerging role of miRNAs in the biochemical and behavioral responses to cocaine. The more powerful next-generation sequencing technology offers certain advantages and is supplanting microarrays for the analysis of complex transcriptomes. Next-generation sequencing is unparalleled in its ability to identify and quantify low-abundance transcripts without prior sequence knowledge, facilitating the accurate detection and quantification of miRNAs expressed in total tissue and miRNAs localized to postsynaptic densities (PSDs). We previously identified cocaine-responsive miRNAs, synaptically enriched and depleted miRNA families, and confirmed cocaine-induced changes in protein expression for several bioinformatically predicted target genes. The miR-8 family was found to be highly enriched and cocaine-regulated at the PSD, where its members may modulate expression of cell adhesion molecules. An integrative approach that combines mRNA, miRNA, and protein expression profiling in combination with focused single gene studies and innovative behavioral paradigms should facilitate the development of more effective therapeutic approaches to treat addiction.
miRNAs – Essential Background
microRNAs (miRNAs) are a class of endogenous 21–25 nucleotide small RNAs that modulate gene expression through binding to complementary sequences in the 3′-untranslated regions (3′-UTRs) of target mRNAs (Kim et al., 2009). Based on their sequences, the 672 annotated mouse miRNAs (http://www.mirbase.org) can be grouped into 253 families; there are over 1000 miRNAs in humans, and several human miRNAs not yet officially recognized in the mouse genome have been identified (Henry et al., 2011). Family members share a common seven to eight nucleotide (nt) seed sequence at their 5′-end and are thought to interact with the same target genes; the sequences of the five members of the miR-8 family are shown in Figure 1. Although miRNAs have only been intensively studied for the last decade, their role in regulating gene expression is now widely accepted. One to 3% of the genome is devoted to miRNAs, which are typically found in clusters in intergenic regions (Mongroo and Rustgi, 2010; Henry et al., 2011; Law and Wong, 2011). About a quarter of the known miRNAs occur within the introns of genes encoding proteins (hence “mirtrons”), in which case expression of the miRNAs is controlled by expression of the “host” gene (Mongroo and Rustgi, 2010; Law and Wong, 2011). Hundreds of miRNAs are expressed in the mature mammalian brain (Lagos-Quintana et al., 2002; Krichevsky et al., 2003; Miska et al., 2004; Landgraf et al., 2007), where they are involved in the control of synapse development and neuronal plasticity (Banerjee et al., 2009; Schratt, 2009b; Siegel et al., 2011). The binding of multiple miRNAs to sites in the 3′-UTR of a target gene can repress translation or cause mRNA degradation (Figure 1), with only a partial match required to alter translation. MicroRNAs are thought to target more than half of mRNAs encoded in the genome, with any one miRNA binding up to several thousand target mRNAs. Since any given target mRNA may have binding sites for over a 100 miRNAs, a consortium of miRNAs work together to regulate the translational rate and stability of that target mRNA (Mongroo and Rustgi, 2010; Henry et al., 2011; Law and Wong, 2011; Li and van der Vaart, 2011; Siegel et al., 2011). MicroRNAs mediate their effects on protein expression when packaged into the RNA-induced silencing complex? (RISC) along with several important proteins (Figure 1).
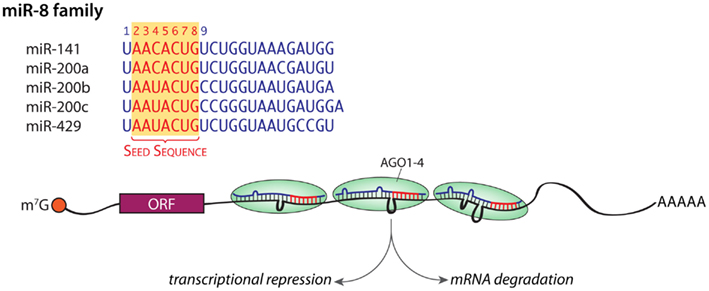
Figure 1. miRNA families. The sequences of the five members of the miR-8 family are shown; the shared seed sequences are shown in red. An mRNA with RISC bound to three different loci in its 3′-UTR is shown; the net effect of these interactions can be diminished translation or mRNA degradation. m7G, 7-methylguanosine in the 5′-cap; orange ball, cap-binding complex; ORF, open reading frame; AAAAA, poly A tail. Mismatches are shown as loops.
In the biogenesis of mature miRNAs, long primary miRNA (pri-miRNA) transcripts encoding a single miRNA or a cluster of miRNAs are first cleaved into shorter preliminary miRNA (pre-miRNA) hairpins by a complex of Drosha, an RNase III, and the cofactor DGCR8 (DiGeorge syndrome critical region gene 8; Figure 2A). About half of mammalian miRNA loci encode multiple miRNAs and are transcribed as a polycistronic transcript (Kim et al., 2009); 65 miRNA clusters encompass 45% (300) of all miRNA genes scattered throughout the mouse genome. Once cleaved from the pri-miRNA, the ∼70 nucleotide pre-miRNA is exported from the nucleus by Exportin-5. The pre-miRNA is then cleaved by Dicer, another RNase III enzyme, in complex with TRBP (HIV transactivating response RNA-binding protein or TARBP2) to make an approximately 22 nt duplex RNA. One strand of the double stranded intermediate (miRNA/miRNA* duplex) is then loaded into an Argonaute protein to form the RISC complex (Kim et al., 2009; Ender and Meister, 2010; Esteller, 2011; Figure 2A). The red sequence represents the mature or “guide” miRNA while the blue sequence is the “star” or “passenger” strand, which is often degraded when not loaded into a RISC. The mature miRNA/RISC targets the 3′-UTR of target mRNAs and regulates protein translation from or stability of the target mRNAs (Guo et al., 2010; Mongroo and Rustgi, 2010; Law and Wong, 2011).
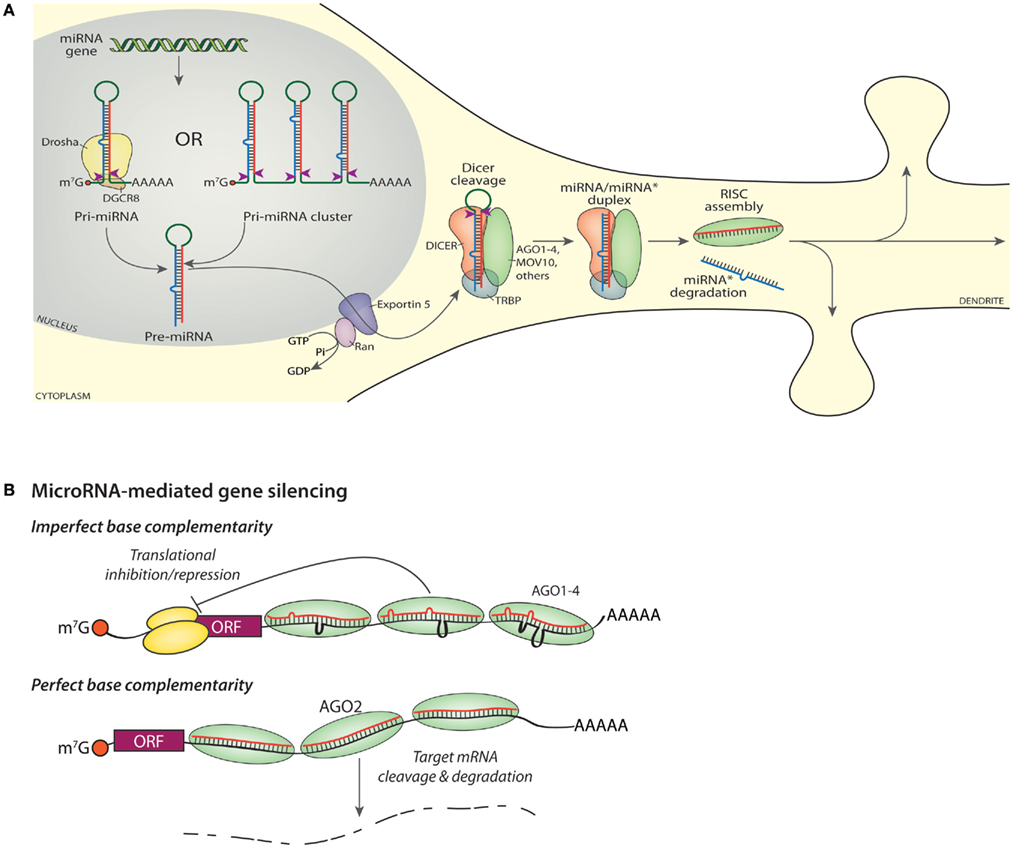
Figure 2. miRNA biogenesis and gene silencing. (A) Transcription of a miRNA gene yields a capped, polyadenylated Pri-miRNA shown undergoing cleavage (red arrowheads) by Drosha (top, left). Transcription of a miRNA cluster yields a transcript encoding multiple miRNAs (top, right), each of which must be cleaved into a Pre-miRNA by Drosha. The mature or “guide” miRNA is shown in red, with the “star” or “passenger” strand shown in blue. Nuclear export of the Pre-miRNA requires Exportin-5. Cleavage of the Pre-miRNA by DICER (red arrowheads) generates a mature guide and star strand duplex; cleavage occurs in a protein complex that includes DICER, an Argonaute protein (AGO1-4), MOV10, TRBP, and additional proteins such as FMR1, DDX9, PUM2 (Schratt, 2009b; Czech and Hannon, 2011; De and MacRae, 2011). The star strand is lost from the final RISC and ultimately degraded, while the assembled RISC is available to bind to cognate RNAs and modulate protein expression. (B) Imperfect base complementarity. When the sequence of the miRNA guide strand does not perfectly match the target mRNA, translation of that mRNA is inhibited. Bound RISCs may contain any one of the four Argonaute proteins. The orange ball is the cap-binding complex; the large and small ribosomal subunits are shown in yellow. Perfect base complementarity. When the sequence of the miRNA guide strand matches the target mRNA perfectly, the target mRNA is sliced by AGO2.
Each fully assembled RISC contains an Argonaute protein (AGO1-4 in human and mouse), an enzyme similar to RNase H (Song et al., 2004), which cleaves the RNA strand of RNA-DNA hybrids. RISC also includes TRBP, the RNA helicase MOV10, a single mature miRNA, and several additional proteins (Höck and Meister, 2008; Banerjee et al., 2009; Vo et al., 2010; De and MacRae, 2011). The RISC interacts with target mRNAs via complementary base pairing interactions specified by the miRNA sequence (Figure 2B). AGO2 is the only catalytically active AGO family member in humans and mice; it has endonuclease (“slicer”) activity and the ability to cleave target mRNAs (Kim et al., 2009). Mutant, catalytically inactive AGO2 silences translation as well as active AGO2 (Broderick et al., 2011).
miRNAs and their Emerging Role in Alcohol and Drug Abuse
miRNAs likely participate in long-lasting forms of synaptic plasticity through the modulation of regulatory pathways that involve controlled dendritic mRNA trafficking, excitation-coupled modulation of synaptic mRNA translation, alterations to the actin cytoskeleton, neurotransmitter metabolism, and peptide hormone processing (Schratt, 2009a). They play an important role in the local control of dendritic morphology through fine-tuning translation of synaptically localized mRNAs (Schratt, 2009b). miRNAs are implicated in the pathophysiology of a variety of neuropsychiatric disorders and mental retardation syndromes, including Alzheimer’s disease, Parkinson’s disease (Conn et al., 2005), Huntington’s disease (Martí et al., 2010), schizophrenia (Beveridge et al., 2010; Santarelli et al., 2011), bipolar disorder (Moreau et al., 2011), alcoholism (Lewohl et al., 2011), Fragile X mental retardation (Li and Jin, 2009), and Rett syndrome (Urdinguio et al., 2010; Wu et al., 2010).
A particularly intriguing example of the coordinated manner in which miRNAs alter function comes from an analysis of the effects of alcohol on expression of miR-9, the most prevalent miRNA in the nucleus accumbens (NAc; Pietrzykowski et al., 2008; Treistman and Martin, 2009; Eipper-Mains et al., 2011). Expression of the BK channel, a large conductance Ca2+ and voltage-activated K+ channel that plays a major role in neuronal excitability, responds to alcohol in a region-specific manner and is known to play a key role in alcohol tolerance. Exposure of striatal cultures to alcohol results in a rapid (15 min) decrease in BK mRNA levels, with loss of specific splice variants. This is due to an alcohol-induced increase in miR-9 expression, which results in degradation of BK splice variants that contain a miR-9 binding site in their 3′-UTR (Pietrzykowski et al., 2008; Treistman and Martin, 2009). The remaining BK variants are those least affected by the presence of alcohol.
Several recent studies have investigated the role of miRNAs and AGO2 in biochemical and behavioral responses to cocaine (Chandrasekar and Dreyer, 2009, 2011; Hollander et al., 2010; Im et al., 2010; Schaefer et al., 2010; Saba et al., 2012). Using qPCR and in situ analysis of bioinformatically determined miRNAs, let-7d, miR-124, and miR-181a were identified as cocaine-regulated in rats (Chandrasekar and Dreyer, 2009, 2011). Brain derived neurotrophic factor (BDNF) expression declined in response to expression of miR-124 while expression of let-7d diminished expression of a dopamine receptor (Drd3). Using lentiviral vectors to over-express or silence these same miRNAs in the NAc, their role in the ability of rats to exhibit conditioned place preference for cocaine was demonstrated (Chandrasekar and Dreyer, 2009, 2011). Expression of the GluA2 subunit of the AMPA receptor is diminished by the binding of miR-181a to a site in its 3′-UTR (Saba et al., 2012).
Two studies by the Kenny group demonstrated induction of miR-132 and miR-212 in dorsal striatum after 7 days of cocaine self-administration in rats and implicate miR-212 in the behavioral and motivational response to cocaine through CREB, MeCP2, and BDNF signaling (Hollander et al., 2010; Im et al., 2010). In addition, Schaefer et al. (2010) identified an overlapping subset of cocaine-induced and AGO2-knockdown-depleted miRNAs in the NAc neurons that express theDrd2 dopamine receptor. Depletion of AGO2 from Drd2-expressing neurons resulted in reduced cocaine self-administration. In a different study, array screens were used to identify 32 miRNAs whose expression increased in a similar manner in multiple regions of the mouse brain in response to repeated injections of nicotine, cocaine, or amphetamine (Lippi et al., 2011); when these miRNAs were compared to miRNAs up-regulated at the time of synaptogenesis, the miR-29a/b and miR-182/183 clusters were singled out. The ability of miR-29a/b to reduce mushroom spine formation in primary hippocampal neurons was then associated with its ability to diminish the expression of Arpc3, a component of the ARP2/3 actin nucleation complex (Lippi et al., 2011).
In addition to miRNA array analyses of postmortem brains from human alcoholics and controls (Lewohl et al., 2011), arrays have been used to identify subsets of miRNAs affected by alcohol treatment of primary mouse neuronal cultures and human neuroblastoma cell lines (Yadav et al., 2011; Guo et al., 2012). Similarly, studies targeted at specific miRNAs have been used to study the actions of morphine and other opiates in model systems such as primary neuronal cultures, neuronal cell lines, and developing zebrafish embryos (Wu et al., 2009; He et al., 2010; Sanchez-Simon et al., 2010; Zheng et al., 2010).
Together, these studies demonstrate that miRNA-mediated gene regulation plays an important role in the complex effects that chronic exposure to cocaine or other drugs of abuse have on the nervous system. However, each of these studies was targeted to a small number of candidate miRNAs or used microarrays, which are limited by cross-hybridization, high background signal, low dynamic range, and the inability to identify unknown miRNAs (Metzker, 2010). Newly developed next-generation sequencing approaches provide a more complete picture of drug-induced changes in both mRNA and miRNA expression.
System-Wide Analysis of Gene Expression: The Advantages of Sequencing
DNA microarrays have dominated genome-wide studies of gene expression for more than a decade, but next-generation sequencing methods are rapidly becoming the method of choice for the analysis of complex transcriptomes (Graveley, 2008). Microarrays have been instrumental in the interrogation and profiling of DNA-protein interactions, identification of single-nucleotide polymorphisms, and comparative analyses of mRNA expression (Shendure, 2008). However, microarrays are limited in a number of ways that next-generation sequencing is not. Since microarrays rely on base complementarity between the probe and the mRNA or cDNA, the technology is prone to artifacts of cross-hybridization of highly homologous genes or isoforms (Metzker, 2010). Microarrays contain a pre-defined set of oligonucleotide probes designed using existing gene annotations for organisms with known genome sequences (Graveley, 2008), whereas sequencing facilitates the identification and absolute quantification of low-abundance transcripts without prior knowledge of the sequence (Cloonan et al., 2008; Mortazavi et al., 2008; Wang et al., 2009b; Metzker, 2010). The sequencing approach has a low background because of the unambiguous nature of sequence mapping and has increased sensitivity for low-abundance transcripts (Graveley, 2008; Wang et al., 2009b). Additionally, sequencing enables the detection and investigation of alternative splicing (Cloonan et al., 2008; Mortazavi et al., 2008; Wang et al., 2008a), imprinting and allele specific expression (Wang et al., 2008b), sequence variation (Wang et al., 2009b), and RNA editing (Wahlstedt et al., 2009) much more easily and robustly than microarrays. Finally, microarrays provide far less data than current next-generation sequencing technologies, which can produce gigabases (109 nucleotides) of sequence from a single experiment, enabling detection of very minor transcripts (Graveley, 2008).
DNA sequencing began in earnest in 1975 (Maxam and Gilbert, 1977; Sanger et al., 1977), but the first truly high-throughput sequencing device was not introduced until the 1990s by Lynx Therapeutics. Massively parallel signature sequencing of mRNAs employed microbead arrays and involved in vitro cloning of cDNA templates on a monolayer of slide-fixed microscopic beads, producing “signature” sequences ranging from 16 to 20 nucleotides in length (Brenner et al., 2000). In 2005, two new methods of next-generation sequencing were introduced: pyrosequencing, or “sequencing-by-synthesis,” which detects pyrophosphate release on nucleotide incorporation (Margulies et al., 2005), and multiplex “sequencing-by-ligation” of mate-paired polymerase colonies (“polonies”; Shendure et al., 2005). In 2006, Solexa introduced the Genome Analyzer, which provided up to 1 gigabase (Gb) of sequence in a single sequencing run, and in 2007, Illumina acquired Solexa, and its sequencers can generate up to 600 Gb of sequence per run (Illumina, 2011). Rapid sequencing research has exploded, as evidenced by the exponential increase in publications in the past few years, increasing from 20 papers in 2005 to nearly 1600 papers in 2011.
Next-generation sequencing of RNA requires the preparation of libraries (Figure 3); 1 mg of total RNA is sufficient to purify the small RNA population and prepare a library for miRNA sequencing. For sequencing of miRNAs, total RNA from tissue (such as NAc) or a subcellular fraction (such as postsynaptic densities, PSD) is subjected to size selection; gel purification can be used to isolate 18–35 nt RNAs. Adaptors are ligated to the 5′ and then 3′ ends, and then the RNAs are reverse transcribed and amplified by polymerase chain reaction (PCR). PCR products are then gel purified and sequenced for 40–50 cycles. The adaptor sequences are trimmed from the reads, and data are then aligned with annotated miRNAs (for example, from miRBase).
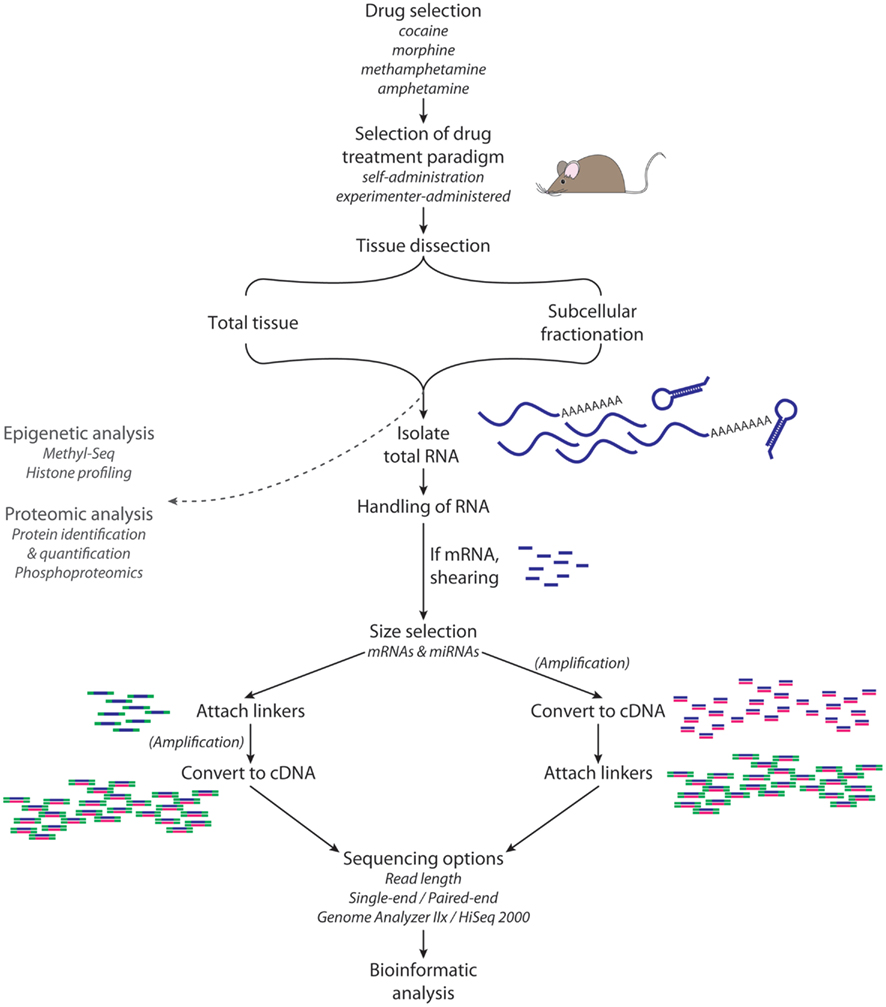
Figure 3. RNA-Seq. High-throughput sequence analysis requires isolation of high quality RNA from the tissue, cell type, or subcellular organelle of interest. For the analysis of miRNAs, a size selection step yields RNA of the appropriate size (e.g., 18–35 nt); for the analysis of mRNAs, transcripts of the desired size are isolated and then sheared. A linker is attached to the 5′-end of each RNA fragment; a different linker is then attached to the 3′-end. Reverse transcription followed by PCR yields sufficient material for analysis. Adaptor sequences are computationally removed from the sequencing data, and then sequences are aligned with annotated miRNAs.
High-throughput sequencing of RNA is an extremely powerful tool, but it is not without limitations. The density of sequence reads varies along the length of a transcript, indicating the existence of sequence bias at some point during the library preparation or sequencing process (Fu et al., 2009; Oshlack and Wakefield, 2009). There are data showing an under-representation of AT-rich and GC-rich regions in Illumina RNA-Seq data, which is likely attributable to sequence bias during the reverse transcription and amplification steps of library preparation (Hansen et al., 2010; Levin et al., 2010; Metzker, 2010). Strand-specific sequencing, in which the sequencing adapters are ligated to the RNA fragment prior to amplification, may show a more even distribution of aligning reads along the length of the gene; this method, however, requires additional manipulation of highly labile RNA prior to its conversion to more stable cDNA (Ozsolak and Milos, 2011). Other difficulties of traditional RNA-Seq include issues of quantifying very low-abundance transcripts, problems with profiling repetitive regions of the genome, and trouble in quantifying small RNAs and RNA with a short half-life (Ozsolak and Milos, 2011). Next–next-generation (third generation) sequencing, in which single molecules are sequenced without prior amplification, is considered the future of the sequencing field (Ozsolak and Milos, 2011; Hayden, 2012).
Challenges accompany the rapidly improving technology and accumulation of datasets containing vast amounts of information. Many biologists struggle to make scientific sense of the data at hand, while many computer scientists are not equipped with the biology training to tackle the relevant questions on their own. A growing number of tools are available to assist in the analysis of high-throughput datasets, including packages for sequence alignment (Langmead et al., 2009), de novo genome and transcriptome assembly (Trapnell et al., 2010), analysis of alternative splicing (Trapnell et al., 2009), functional category assessment, and identification of signaling pathway enrichment (Kanehisa and Goto, 2000; Dennis et al., 2003; Thomas et al., 2003; Li et al., 2008). At present, few “user friendly” and fully customizable packages exist for the comprehensive analysis of sequencing data.
Picking the System to Study
Abundant evidence, from both animals and humans, identifies the limbic system as the convergence point for the actions of all drugs of abuse (Koob and Le Moal, 2001; Nestler, 2001, 2005b; Di Chiara, 2002; Volkow et al., 2004; Wise, 2004). The mesocorticolimbic dopamine pathway, which involves a subset of all limbic structures, is activated by normal physiologic rewards such as food, drink, social interaction, and sex (Wise, 1996, 2002; Nestler, 2005a). This system includes dopaminergic projections from the ventral tegmental area (VTA) to the NAc and prefrontal cortex (PFC) as well as glutamatergic projections from the PFC to the NAc (Kauer and Malenka, 2007; Figure 4A). Although drugs of abuse originate from a diverse array of chemical classes and have a variety of different primary targets, their effects all ultimately converge on the dopamine neurons of the VTA (Wolf, 2006). Other neurotransmitters, including γ-aminobutyric acid (GABA), opioid peptides, serotonin, acetylcholine, and endogenous cannabinoids, are also involved in the signaling of addiction (Spangler et al., 1996; Hyman and Malenka, 2001; Ross and Peselow, 2009). The convergence of these myriad signaling systems has led many to hypothesize that the NAc is the key integration point in the rewarding effects seen with drugs of abuse (Nestler and Aghajanian, 1997; Berke and Hyman, 2000; Kalivas et al., 2005; Nestler, 2005b; Hyman et al., 2006).
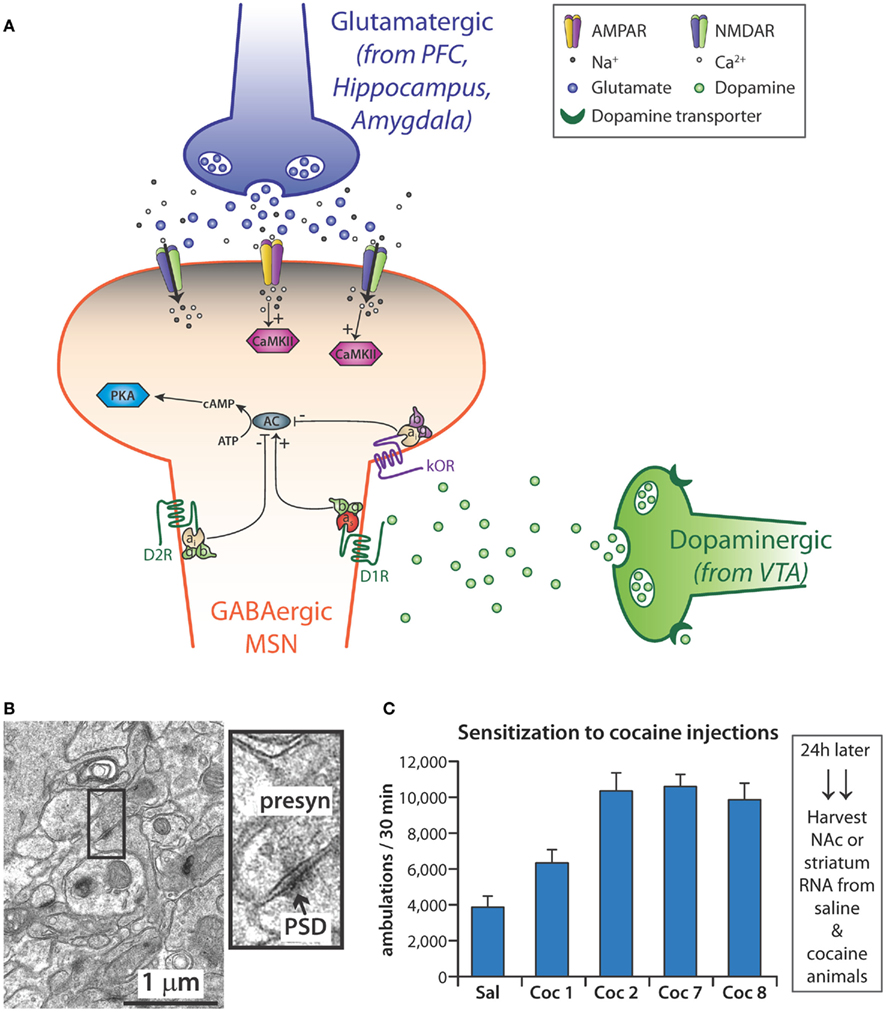
Figure 4. (A) Medium spiny neurons (MSN), which use γ-NH2-butyric acid (GABA) as their major transmitter, are the primary cell type in the nucleus accumbens (NAc). Some MSNs express mostly the D1 dopamine receptor while others express primarily the D2 dopamine receptor. Dopamine released from the endings of dopaminergic neurons in the ventral tegmental area (VTA) signals through both types of receptor, affecting the activation of adenylate cyclase (AC); reuptake of dopamine by the dopamine transporter (DAT) is blocked by cocaine. MSNs receive excitatory inputs from glutamatergic neurons in the prefrontal cortex (PFC), hippocampus, and amygdala. Activation of AMPA and NMDA receptors localized to the spine-studded dendrites of MSNs leads to the influx of Ca2+ and activation of CaMKII. In addition, MSNs are responsive to various neuropeptides, which bind to G-protein coupled receptors such as the κ-opioid receptor (κOR). (B) Standard procedures have been developed to purify postsynaptic densities (PSDs) from tissue homogenates; miRNAs enriched at the PSD are thought to allow local regulation of mRNA translation and stability. The electron micrograph (7 week mouse hippocampus) illustrates the complex system from which PSDs must be purified: presynaptic terminals can be recognized by their content of synaptic vesicles; PSDs are closely apposed to the presynaptic endings. (C) Sensitization to the locomotor stimulating effects of cocaine requires repeated exposure to the drug. Adult mice typically are typically given 10–20 mg cocaine/kg; locomotor activity (ambulations) is monitored for the next 30 min in an activity chamber with infrared light sensors. “Sal” is the average locomotor data for all mice given saline initially and the mice given saline throughout; Coc1 are data for mice give 10 mg/kg cocaine on day 1; Coc2 and Coc7 are data from days 2 and 7 for mice given 20 mg/kg cocaine daily for days 2–7; Coc8 are the data for the cocaine treated mice given 10 mg/kg on day 8; substantial locomotor sensitization is apparent compared to Coc1. Tissue harvested soon after drug administration allows analysis of the immediate response, while tissue harvested 24 h after final drug administration allows evaluation of the steady state response. N = 10 each for saline and cocaine.
Neurons modulate gene expression with subcellular precision; polyribosomes and mRNAs are found at the base of dendritic spines and are involved in localized protein synthesis (Rao and Steward, 1991; Kiebler and DesGroseillers, 2000). Individual synapses may act independently in the local control of excitation-coupled protein translation (Schratt, 2009b; Zukin et al., 2009). The morphological changes that occur in dendritic spines as a result of synaptic activity cannot occur without de novo protein synthesis, and a blockade of protein synthesis blunts formation of long-term memory (Schratt, 2009b). Modulating synthesis of selected synaptic proteins gives neurons the ability to fine-tune and integrate extracellular cues at single dendrites or at individual, specified synapses (Wang et al., 2009a). Subcellular fractionation is an important technique that enables the differentiation of events occurring at the synapse from those occurring in the cell soma. Despite the complex interactions between presynaptic endings and their target receptors on dendritic spines (Figure 4B), PSD-enriched fractions can be prepared and allow identification of postsynaptically enriched components.
miRNAs are known to play a role in modulating localized translation of mRNAs in dendrites (Kim et al., 2004). Localization of the miRNA processing enzyme DICER and the RISC component AGO2 to the PSD in mouse cortical and hippocampal slices by electron microscopy supports the possibility of local regulation (Lugli et al., 2005). Further studies identified the RISC component and RNA helicase MOV10 (homologous to Drosophila Armitage) at the synapse and established a role for RNA-induced silencing in the control of synaptic gene expression, learning, and memory (Ashraf et al., 2006; Banerjee et al., 2009; Vo et al., 2010). Through its effects on LIMK1, miR-134, which is synapto-dendritically localized, plays a role in the activity-dependent regulation of spine size (Schratt et al., 2006; Siegel et al., 2011). Similarly, miR-132 and miR-138 play a role in dendritic remodeling and synaptogenesis (Wayman et al., 2008; Siegel et al., 2009).
Sensitization is the progressive and persistent amplification of behavioral and motivational responses to a fixed dose of drug (Berke and Hyman, 2000; Hyman et al., 2006). It persists for weeks, months, and years after cessation of drug taking and is thought to play an important role in the risk of a reformed addict for relapse to drug taking behavior (Paulson et al., 1991; Castner and Goldman-Rakic, 1999; Robinson and Berridge, 2001). We therefore exposed adult male mice to a treatment paradigm (eight daily intraperitoneal injections of cocaine or saline) that reliably yielded locomotor sensitization before harvesting tissue for library preparation (Figure 4C). We harvested NAc 24 h after the final injection of saline or cocaine and constructed small miRNA-Seq libraries for miRNA analysis (Eipper-Mains et al., 2011).
In order to identify miRNAs localized to synapses, we prepared another cohort of saline-injected and cocaine sensitized mice; 24 h after the final injection, we purified PSD from their striata and isolated the total RNA associated with this fraction (Figure 4B; Eipper-Mains et al., 2011). While NAc was used to analyze the total miRNA population, striatum was used to ensure availability of sufficient PSD RNA. Sequencing libraries were prepared and analyzed as outlined in Figure 3 (Eipper-Mains et al., 2011). As methods for library preparation improve, it will be possible to work with RNA purified from even smaller brain regions.
Sequence analysis identifies all of the RNAs present in a given sample (Figure 5). While miRNAs accounted for over 70% of the reads in the NAc libraries, they accounted for slightly under half of the reads in the PSD libraries. Sequences mapping to rRNAs and tRNAs were about five-times more prevalent in the PSD libraries than in the NAc libraries. Sequences that mapped to the transcriptome accounted for about 15% of the reads in both types of sample and most likely represent products of mRNA degradation isolated during size selection. Although minor components, snoRNAs, snRNAs, and SINE (short interspersed element) are present and their sequences can also be analyzed.
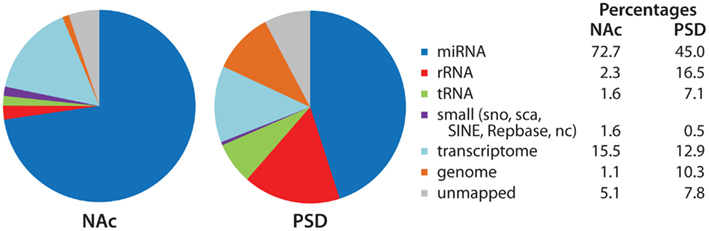
Figure 5. Identity of mapped miRNA sequences in NAc and PSD libraries. Adaptor sequences were trimmed from the extracted sequence data. Bowtie (version 0.12.7; Langmead et al., 2009) was used to align sequences 18–30 nt in length to the miRBase annotated mouse microRNA database (version 16; Griffiths-Jones et al., 2006, 2008); perfect alignment was required. The Rfam, Repbase, and NON-CODE databases were used to identify sequences that aligned with annotated non-coding and repetitive elements. Sequences were then aligned to the mouse transcriptome (exons, introns, exon–exon junctions) and mouse genome. For non-miRNA alignments, 1-mismatch was allowed. Data (% total reads mapped to each category) for the NAc and PSD libraries from saline-injected animals are shown.
miRNA-Seq analysis identifies the exact miRNA species present (Figure 6). As shown in Figure 2, the opposite ends of each miRNA are generated by Drosha and DICER. While the major miR-375 sequence identified corresponded to that predicted, multiple miR-182 and miR-212-3p species (isomirs) were found in all four libraries; of note, levels of both miR-182 and miR-212-3p had previously been identified as cocaine-responsive (Hollander et al., 2010; Im et al., 2010; Lippi et al., 2011). This heterogeneity has important consequences when comparing miRNA-Seq data with microarray and qPCR data, since miRNA-Seq can unambiguously identify many sequences as belonging to one miRNA (e.g., miR-212-3p), while qPCR and microarrays will miss most of the variants. For miRNAs with a single major species which conforms to the accepted annotation (e.g., miR-375), the agreement across methods should be much better, enabling firm comparisons of RNA-Seq with qPCR data (Eipper-Mains et al., 2011). Tools for analysis include websites such as http://www.microrna.org and http://www.targetscan.org.
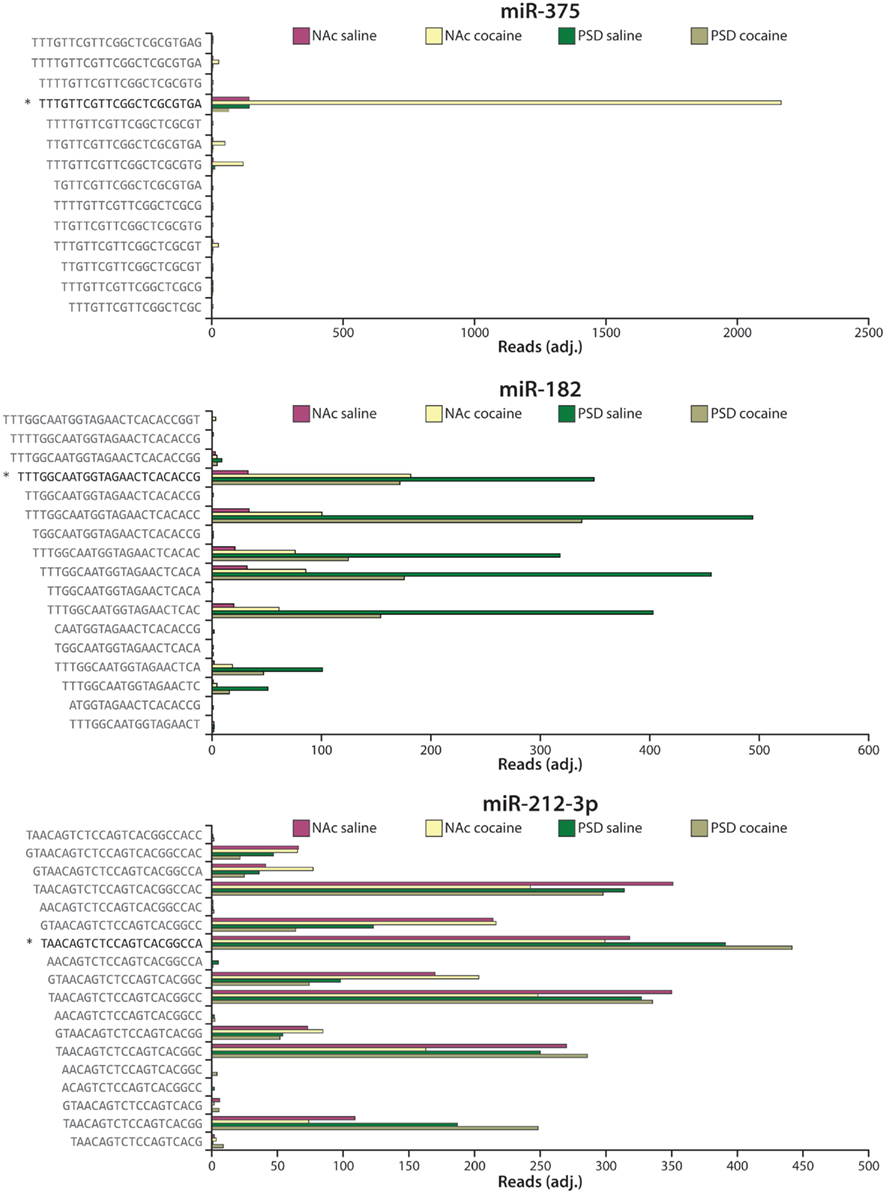
Figure 6. Heterogeneity in miRNA sequence. Expression data for the four different libraries (NAc saline, NAc cocaine, PSD saline, PSD cocaine) are shown as reads (adjusted for equal total reads per sample) for sequences mapping to miR-375 (top), miR-182 (middle), and miR-212-3p (bottom). The arrowhead ( ) marks the miRBase (version 16) annotated mature miRNA sequence (Griffiths-Jones et al., 2008). Commercially available qPCR primers will only amplify the sequence marked by the arrowhead.
) marks the miRBase (version 16) annotated mature miRNA sequence (Griffiths-Jones et al., 2008). Commercially available qPCR primers will only amplify the sequence marked by the arrowhead.
Global Analysis Reveals Role for miRNA Clusters and Families
In a tour de force, Lagos-Quintana et al. (2002) cloned hundreds of miRNAs from the cortex, cerebellum, and midbrain of 18.5-week-old mice. One year later, microarray technology was used to study regulation of miRNAs during neuronal development (Krichevsky et al., 2003). We chose to use ultra-high-throughput sequencing of miRNAs with the overarching goal of attaining a more complete picture of the response of the brain to chronic cocaine exposure (Eipper-Mains et al., 2011). Previous genome-wide characterizations of drug-induced gene expression focused on mRNA transcripts rather than miRNAs, were carried out using microarray technology, and often looked at other regions of the brain using different experimental paradigms (McClung and Nestler, 2008). The data reviewed here represent the first look at striatal miRNA synaptic enrichment and cocaine-regulation by deep sequencing.
Eighteen of the synaptically enriched miRNAs identified in our analysis are members of genomic miRNA clusters. For both the total lysate and PSD samples, the expression patterns of the clustered miRNAs are quite similar across the entire cluster. Especially instructive examples of clusters include the miR-8 family clusters on chromosomes 4 and 6 (miR-429/200a/b and miR-141/200c), thelet-7 family cluster on the X chromosome (let-7f/miR-98) and the clusters for miR-1/133a (chromosome 2), miR-182/96/183(chromosome 6), and miR-216a/217(chromosome 11; Figure 7). In contrast, little concordance is seen across the large miR-379-410 cluster, in which only miR-485 is synaptically enriched (Figure 7).
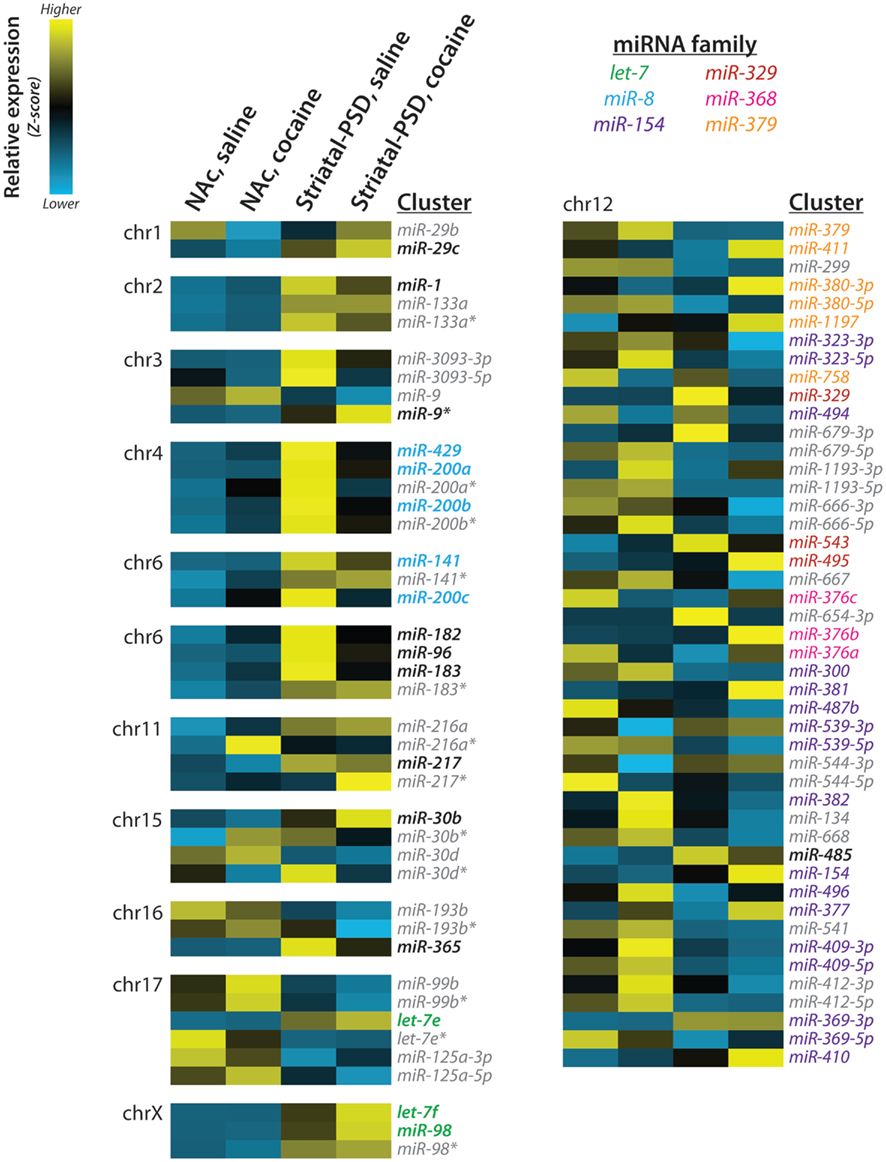
Figure 7. miRNA clusters and families. Expression of striatal PSD-enriched miRNAs and other members of each genomic cluster is shown as a heat map; the chromosomal (chr) localization of each cluster is indicated. Z-scores were computed using normalized miRNA frequency across all samples: blue, low expression; yellow, high expression. miRNA families are indicated by text color; PSD-enriched miRNAs are indicated by bold; miRNAs not enriched at striatal PSDs and/or not belonging to the listed miR families are shown in gray italic. Reproduced from Eipper-Mains et al. (2011) with permission.
Lugli and colleagues used microarrays to identify the subcellular localization of mature and precursor miRNAs in adult (2-month-old) mouse cortex and hippocampus. They found that synaptically enriched miRNAs were primarily expressed in evolutionarily newer species (Lugli et al., 2008). We compared our synaptic enrichment ratios to this earlier data set, revealing significant overlap between the most synaptically enriched and synaptically depleted miRNAs in our striatal samples and the previous hippocampal and cortical samples (Eipper-Mains et al., 2011). Importantly, most of the miR-8 family (miR-200a/b/c/429) are markedly enriched in both datasets. In addition, miR-182 and miR-183 are dramatically enriched in PSDs in both datasets. By contrast, miRs-126, -143, -145, -150, and -451 are depleted in both synaptic datasets. A separate study used microarrays to identify synaptosomally enriched and depleted miRNAs from rat P15 total forebrain samples (Siegel et al., 2009). In a direct comparison, miRs-219-5p, -21, -377, -98, -376b, -218, -7a/b, and -29a are synaptically enriched in both datasets, and in both datasets miRs-143, -145, and -150 are depleted. The remarkable extent of agreement between these three datasets, which were obtained using different experimental platforms to analyze synaptically enriched samples prepared in different ways from multiple brain regions taken from animals of different ages, may be emblematic of the preponderance of glutamatergic synapses and additional similar properties in all of these brain regions. Whether the identities and expression levels of miRNAs differ substantially from one synapse to the next, dictated by the local microenvironment, remains to be determined.
Particularly striking was the synaptic enrichment of members of several miRNA clusters (Figure 7). The miR-8 family has been studied most extensively in relation to cancer pathophysiology and has been shown to inhibit the first step of cancer metastasis, the epithelial-mesenchymal transition (Inui et al., 2010). An expression atlas of miRNAs indicates enrichment of all members of the miR-8 family, except miR-429, in endocrine glands and tissues, kidney, and the reproductive system (Landgraf et al., 2007). Expression of this family of miRNAs is thought to maintain the epithelial phenotype through direct targeting of ZEB1 and ZEB2, transcriptional repressors of the cell adhesion molecule ECAD (Inui et al., 2010). Members of the cadherin family of cell surface glycoproteins mediate calcium-dependent cell–cell adhesion and have critical roles in early brain development, axonal outgrowth, and synaptogenesis (Martinek and Gaul, 1997). ECAD is present at the synapse, and antibodies which block homophilic interactions between pre- and postsynaptic ECAD attenuate induction of long-term potentiation (Tang et al., 1998).
Members of the highly conserved let-7 family of miRNAs, with almost a dozen members in the mammalian genome and key roles in development and cancer suppression (Roush and Slack, 2008; McCarty, 2012), are also highly enriched at the PSD. The let-7 family is highly expressed in pituitary, hypothalamus, and pancreatic islets (Landgraf et al., 2007), and its members play a major role in controlling insulin secretion in response to a glucose load and tissue responsiveness to insulin (Frost and Olson, 2011). The let-7 family members localized at the PSD may participate in the control of vesicle trafficking and secretion, both of which play essential roles at the synapse. The miR-183 family has not been studied as extensively, but clearly plays a crucial role in the development and function of sensory cells such as photoreceptors and hair cells (Li et al., 2010; Weston et al., 2011; Zhu et al., 2011).
Response to Cocaine
Our finding that PSD levels of AGO2 protein rose in the NAc of mice sensitized to cocaine led to our analysis of the effects of cocaine on miRNA expression and localization (Eipper-Mains et al., 2011). One particularly intriguing pattern emerged from our data analysis: many of the miRNAs that were up-regulated in NAc tissue lysate after chronic cocaine were down-regulated at the synapse. Specifically, members of the miR-8 family, which were very highly enriched in striatal PSDs, exhibited opposite changes in tissue lysate versus PSDs after cocaine.
As summarized above, several recent studies investigated miRNA regulation in the mesolimbic dopamine system after cocaine administration. The first used qPCR and in situ hybridization of specific bioinformatically identified miRNAs to identify three cocaine-regulated miRNAs in rats receiving cocaine for 15 days, let-7d (decreased in NAc), miR-124 (decreased in NAc and dorsal striatum), and miR-181a (increased in NAc and dorsal striatum; Chandrasekar and Dreyer, 2009). Our data, from mice receiving cocaine for 7 days, indicate no cocaine-regulation of let-7d or miR-124 and a modest increase in miR-181a in NAc lysates (Eipper-Mains et al., 2011). Two related studies identified induction of miR-132 and miR-212 in dorsal striatum after 7 days of cocaine self-administration in rats and implicate miR-212 in the behavioral and motivational response to cocaine through CREB, MeCP2, and BDNF signaling (Hollander et al., 2010; Im et al., 2010). Our data support induction of miR-132 in the NAc and a decrease in miR-132 in striatal PSDs. miR-212, with 10 major isomirs identified using miRNA-Seq (Figure 6), exhibits a complex response to cocaine: tissue levels of three isomirs increase while tissue levels of five isomirs decrease and two are unchanged (Eipper-Mains et al., 2011). In the fourth study, miRNA microarrays were used to identify the overlapping subset of cocaine-induced and AGO2-knockdown-depleted miRNAs in Drd2-expressing neurons of the NAc (Schaefer et al., 2010). Direct comparison of our data to this study is not possible because the experimental paradigms differ so significantly; it is, however, interesting to note that ablation of AGO2 caused a marked reduction in the level of miR-182 (Schaefer et al., 2010), identified in our data as cocaine-regulated and synaptically enriched. Future studies will need to address specific cell-types and subcellular localization of the entire complement of miRNAs, to determine more precisely how miRNAs are involved in the response of the brain to cocaine.
Bioinformatically Predicted Targets
We used an established list (Suzuki et al., 2007) of PSD-enriched mRNAs to identify potential target genes for the 16 most cocaine-regulated synaptically localized miRNAs (Eipper-Mains et al., 2011). Bioinformatic analysis using miRanda yielded a mirSVR score for each gene. A striking feature of this list was the number of miRNA binding sites predicted for any given transcript. Transcripts encoding metadherin (Mtdh), which is also known as AEG-1 or Lyric, had the most negative mirSVR score, with five sites for PSD-localized miRNAs down-regulated by cocaine and four sites for PSD-localized miRNAs up-regulated by cocaine; sites predicted for each of the PSD-enriched cocaine-regulated miRNAs are shown (Figure 8A).
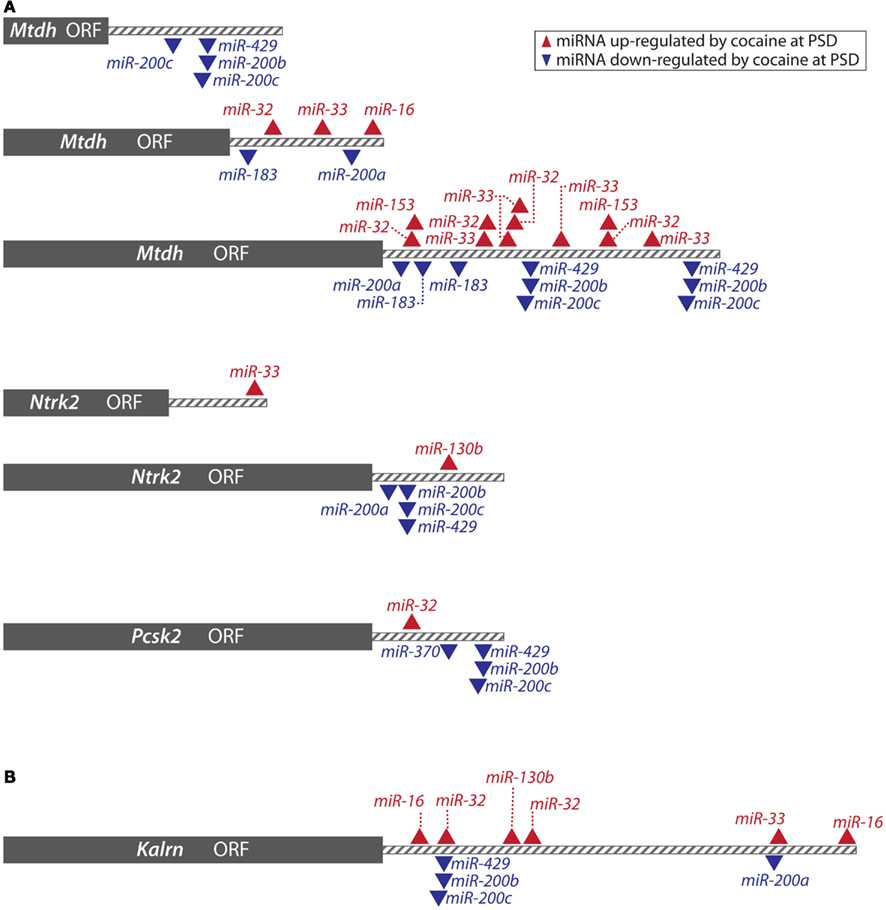
Figure 8. miRanda predictions. (A) The transcripts shown are all PSD-enriched; based on miRanda analysis using the 16 most cocaine-regulated striatal PSD miRNAs, it was predicted that cocaine exposure might affect expression of these genes via regulation of miRNAs. Cocaine-regulation of metadherin, Ntrk2 (BDNF receptor), and PC2 (PCSK2) protein expression at the PSD was verified by Western blot analysis. Gray box indicates the open reading frame (ORF) for each transcript; the 3′-UTR is shown by gray stripes; arrowheads indicate miRNA binding sites predicted for miRNAs up ( ) and down (
) and down ( ) regulated by cocaine. Reproduced from Eipper-Mains et al. (2011) with permission. (B) Kalrn was included in the list of synaptically enriched transcripts targeted by cocaine-regulated PSD-enriched miRNAs. Shown are the miRanda predicted binding sites for the 16 most cocaine-regulated striatal PSD miRNAs in the 3′-UTR of mouse KAL7.
) regulated by cocaine. Reproduced from Eipper-Mains et al. (2011) with permission. (B) Kalrn was included in the list of synaptically enriched transcripts targeted by cocaine-regulated PSD-enriched miRNAs. Shown are the miRanda predicted binding sites for the 16 most cocaine-regulated striatal PSD miRNAs in the 3′-UTR of mouse KAL7.
Primarily studied for its role in cancer progression, Mtdh is expressed in most tissue types and has been localized to epithelial tight junctions (Hu et al., 2009). Using Western blot analysis, we first documented the presence of metadherin at the PSD, and observed a 42% increase in PSD-localized metadherin protein following chronic cocaine treatment (Eipper-Mains et al., 2011). Ectopic expression of Mtdh in primary human fetal astrocytes inhibits expression of the glial high-affinity glutamate transporter (SLC1A2 or EAAT2; Kang et al., 2005). An expanding body of evidence implicates glutamatergic signaling in the response of the brain to cocaine (Kalivas, 2009), and more rapid removal of glutamate from the glutamatergic synapses formed by neurons of the PFC onto the medium spiny neurons of the NAc would be of functional significance.
In addition to Mtdh, Pcdh8 (protocadherin 8; also known as arcadlin for activity-regulated cadherin-like protein), and Pcdh12 (protocadherin 12) were identified as targets for synaptically localized miRNAs (Eipper-Mains et al., 2011). Expression of Pcdh8, which is known to be localized to synapses, is rapidly and transiently increased in response to seizures, and plays a role in long-term potentiation (Yamagata et al., 1999). A comparison of the gene expression profiles in the hippocampus of cocaine addicts versus drug-free age-matched controls identified PCDH8 in a group of up-regulated genes, many of which are involved in regulating the extracellular matrix (Mash et al., 2007). The role of Pcdh12 in the brain has not yet been addressed.
Ntrk2, the BDNF receptor or TrkB, was also identified as a potential target for the cocaine-regulated PSD-localized miRNAs; predicted binding sites are shown in Figure 8A. Prior studies have shown that the level of TrkB increases in the NAc of rats after chronic cocaine self-administration (Graham et al., 2009) and TrkB-BDNF signaling in the NAc is thought to modulate behavioral responses to cocaine (Lobo et al., 2010). Using an antibody to the BDNF receptor, we found increased expression in striatal PSDs following cocaine treatment (Eipper-Mains et al., 2011).
Two of the predicted miRNA targets, Pcsk2 (prohormone convertase 2, PC2) and Penk (proenkephalin) are part of the peptidergic system, which is known to play multiple roles in addiction; predicted binding sites for cocaine-regulated PSD-enriched miRNAs are shown in Figure 8A. PC2, a Ca2+-dependent subtilisin-like endoprotease, is the major prohormone convertase in the brain (Winsky-Sommerer et al., 2000). Expressed at relatively high levels in the striatum, PC2 is a key enzyme in the cleavage of all three opioid precursors, proenkephalin (Penk), prodynorphin (Pdyn), and proopiomelanocortin (Pomc), as well as CART (Cartpt, cocaine, and amphetamine-regulated transcript; Pan et al., 2006). Based primarily on studies of neuroendocrine cells, PC2 has been localized to the trans-Golgi network and secretory granules. Peptide-containing secretory granules are found in presynaptic axon terminals and in dendrites, at the base of spines (Winsky-Sommerer et al., 2000). Using Western blots, we confirmed the presence of PC2 at the synapse and demonstrated an increase of 67% in levels of mature PC2 at the synapse after chronic cocaine treatment (Eipper-Mains et al., 2011). PC2 is synthesized with a signal sequence that guides the nascent chain into the lumen of the secretory pathway. The signal sequence is removed co-translationally; proPC2, which is inactive, is activated by an endoproteolytic cleavage that releases an autoinhibitory N-terminal peptide (Helwig et al., 2011). Our data suggest that this entire process can occur in the vicinity of the synapse and may respond in part to synaptic activity.
Penk, another bioinformatically predicted miRNA target, is expressed primarily in the medium spiny neurons that express Drd2 (Curran and Watson, 1995). Penk is involved in the behavioral response to cocaine and its mRNA and peptide levels were known to be cocaine-regulated (Przewlocka and Lason, 1995; Daunais et al., 1997; Crespo et al., 2001). Striatal expression of CART peptides increases after cocaine exposure and is hypothesized to modulate the effects of the enhanced dopaminergic signaling in response to cocaine (Hubert et al., 2008). The increased levels of PC2 could play an essential role in their appearance.
Based on the same bioinformatic analysis, miRNA-regulated expression of Kalirin (Kalrn), a Rho guanine nucleotide exchange factor that regulates dendritic spine morphology and function (Ma et al., 2008), was predicted (Figure 8B). Cocaine-regulated expression of Kalrn is of special interest because of the well-documented morphological effects of chronic cocaine exposure on the mesolimbic dopaminergic pathway (Wolf, 2006). Increased dendritic spine size and density are observed in the MSNs of mice following both experimenter-administered (Robinson and Kolb, 1997, 1999; Kiraly et al., 2010a,b) and self-administered cocaine (Robinson et al., 2001). Chronic cocaine exposure is known to result in an increase in expression of Kal7, the predominant adult isoform of Kalrn, in the NAc (Kiraly et al., 2010a; Mains et al., 2011), and an increase in Kal7 protein at the PSD (Kiraly et al., 2010a; Mains et al., 2011). Mice lacking Kal7 fail to form additional spines in response to chronic cocaine treatment (Kiraly et al., 2010a; Mains et al., 2011). The 3′-UTR of mouse Kal7 contains multiple predicted binding sites for cocaine-regulated PSD miRNAs (Figure 8B).
Next – Conclusion; Future Studies
The number of studies examining the effects of cocaine on one or a small number of genes still dwarf the number of genome-wide expression studies. These focused studies have revealed widespread changes in multiple neurotransmitter systems (dopamine, glutamate, peptidergic), in proteins controlling cytoskeletal organization and in multiple transcription factors. Examples include several dopamine receptors and the dopamine transporter, a number of ionotropic and metabotropic glutamate receptors, opiate receptors and opiate peptides, other neuropeptides, the synaptic vesicle associated proteins α-synuclein and synaptotagmin, clathrin, signaling pathway protein such as the mitogen-activated protein kinases (MAPKs), calmodulin, and Ca2+/calmodulin-dependent protein kinases along with components of the machinery regulating the actin cytoskeleton (Self and Nestler, 1995; Lehrmann et al., 2003; Mash et al., 2003; McClung and Nestler, 2003; Norrholm et al., 2003; Morabito et al., 2004; Rossman et al., 2005; Benavides et al., 2007; Anderson et al., 2008; Li et al., 2008; Liu et al., 2009; Kiraly et al., 2010a,b; Mains et al., 2011).
A number of comprehensive analyses of cocaine-induced gene expression in animal models and human postmortem brain, primarily using microarray, have been published over the years. Like our global analysis of cocaine-regulated miRNA expression, these studies have further expanded the scope of targets affected by cocaine. In particular, these global approaches have consistently highlighted the effects of cocaine on components of the extracellular matrix, including cadherins, integrins, and matrix metalloproteinases (Lehrmann et al., 2003; Albertson et al., 2004; Bannon et al., 2005; Mash et al., 2007). Particularly appealing is the idea that some of the long-lived effects of cocaine may reflect changes in the extracellular matrix. Drugs designed to target multiple participants in these chronic effects of cocaine may prove to be more effective than drugs targeted to individual intracellular signaling pathways in the treatment of drug abusers.
Knockout and transgenic mouse models have been invaluable for investigations into the behavioral, biochemical, and molecular effects mediated by single genes. It is becoming increasingly clear, however, that the changes that occur as a result of drug abuse are incredibly complex and widespread, affecting multiple brain regions and cell-types in a system-wide manner. These global studies emphasize the broad effects of drug exposure on neuronal gene expression, underscoring the need for an integrative approach that combines mRNA, miRNA, and protein expression profiling in combination with focused single gene studies and innovative behavioral paradigms in the quest to uncover what drives addiction and develop integrated therapeutic approaches. With improved methodologies it will be possible to study the role of miRNAs in specific cell-types in animals self-administering drugs or undergoing withdrawal.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health Grants DA-15464, DA-23082, DK-32948, and GM062516. We thank Dr. Brenton R. Graveley for thoughtful comments on the manuscript.
References
Albertson, D. N., Pruetz, B., Schmidt, C. J., Kuhn, D. M., Kapatos, G., and Bannon, M. J. (2004). Gene expression profile of the nucleus accumbens of human cocaine abusers: evidence for dysregulation of myelin. J. Neurochem. 88, 1211–1219.
Anderson, S. M., Famous, K. R., Sadri-Vakili, G., Kumaresan, V., Schmidt, H. D., Bass, C. E., Terwilliger, E. F., Cha, J.-H. J., and Pierce, R. C. (2008). CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci. 11, 344–353.
Ashraf, S. I., McLoon, A. L., Sclarsic, S. M., and Kunes, S. (2006). Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell 124, 191–205.
Banerjee, S., Neveu, P., and Kosik, K. S. (2009). A coordinated local translational control point at the synapse involving relief from silencing and MOV10 degradation. Neuron 64, 871–884.
Bannon, M., Kapatos, G., and Albertson, D. (2005). Gene expression profiling in the brains of human cocaine abusers. Addict. Biol. 10, 119–126.
Benavides, D. R., Quinn, J. J., Zhong, P., Hawasli, A. H., DiLeone, R. J., Kansy, J. W., Olausson, P., Yan, Z., Taylor, J. R., and Bibb, J. A. (2007). Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J. Neurosci. 27, 12967–12976.
Berke, J. D., and Hyman, S. E. (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25, 515–532.
Beveridge, N. J., Gardiner, E., Carroll, A. P., Tooney, P. A., and Cairns, M. J. (2010). Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol. Psychiatry 15, 1176–1189.
Brenner, S., Johnson, M., Bridgham, J., Golda, G., Lloyd, D. H., Johnson, D., Luo, S., McCurdy, S., Foy, M., Ewan, M., Roth, R., George, D., Eletr, S., Albrecht, G., Vermaas, E., Williams, S. R., Moon, K., Burcham, T., Pallas, M., DuBridge, R. B., Kirchner, J., Fearon, K., Mao, J., and Corcoran, K. (2000). Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 18, 630–634.
Broderick, J. A., Salomon, W. E., Ryder, S. P., Aronin, N., and Zamore, P. D. (2011). Argonaute protein identity and pairing geometry determine cooperativity in mammalian RNA silencing. RNA 17, 1858–1869.
Castner, S. A., and Goldman-Rakic, P. S. (1999). Long-lasting psychotomimetic consequences of repeated low-dose amphetamine exposure in rhesus monkeys. Neuropsychopharmacology 20, 10–28.
Chandrasekar, V., and Dreyer, J.-L. (2009). microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol. Cell. Neurosci. 42, 350–362.
Chandrasekar, V., and Dreyer, J.-L. (2011). Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology 36, 1149–1164.
Cloonan, N., Forrest, A. R. R., Kolle, G., Gardiner, B. B. A., Faulkner, G. J., Brown, M. K., Taylor, D. F., Steptoe, A. L., Wani, S., Bethel, G., Robertson, A. J., Perkins, A. C., Bruce, S. J., Lee, C. C., Ranade, S. S., Peckham, H. E., Manning, J. M., McKernan, K. J., and Grimmond, S. M. (2008). Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5, 613–619.
Conn, P. J., Battaglia, G., Marino, M. J., and Nicoletti, F. (2005). Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat. Rev. Neurosci. 6, 787–798.
Crespo, J. A., Manzanares, J., Oliva, J. M., Corchero, J., Palomo, T., and Ambrosio, E. (2001). Extinction of cocaine self-administration produces a differential time-related regulation of proenkephalin gene expression in rat brain. Neuropsychopharmacology 25, 185–194.
Curran, E. J., and Watson, S. J. (1995). Dopamine receptor mRNA expression patterns by opioid peptide cells in the nucleus accumbens of the rat: a double in situ hybridization study. J. Comp. Neurol. 361, 57–76.
Czech, B., and Hannon, G. J. (2011). Small RNA sorting: matchmaking for Argonautes. Nat. Rev. Genet. 12, 19–31.
Daunais, J. B., Nader, M. A., and Porrino, L. J. (1997). Long-term cocaine self-administration decreases striatal preproenkephalin mRNA in rhesus monkeys. Pharmacol. Biochem. Behav. 57, 471–475.
De, N., and MacRae, I. J. (2011). Purification and assembly of human Argonaute, Dicer, and TRBP complexes. Methods Mol. Biol. 725, 107–119.
Dennis, G., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C., and Lempicki, R. A. (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3.
Di Chiara, G. (2002). Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav. Brain Res. 137, 75–114.
Eipper-Mains, J. E., Kiraly, D. D., Palakodeti, D., Mains, R. E., Eipper, B. A., and Graveley, B. R. (2011). microRNA-Seq reveals cocaine-regulated expression of striatal microRNAs. RNA 17, 1529–1543.
Frost, R. J. A., and Olson, E. N. (2011). Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc. Natl. Acad. Sci. U.S.A. 108, 21075–21080.
Fu, X., Fu, N., Guo, S., Yan, Z., Xu, Y., Hu, H., Menzel, C., Chen, W., Li, Y., Zeng, R., and Khaitovich, P. (2009). Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 10, 161. doi:10.1186/1471-2164-10-161
Graham, D. L., Krishnan, V., Larson, E. B., Graham, A., Edwards, S., Bachtell, R. K., Simmons, D., Gent, L. M., Berton, O., Bolanos, C. A., DiLeone, R. J., Parada, L. F., Nestler, E. J., and Self, D. W. (2009). Tropomyosin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biol. Psychiatry 65, 696–701.
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A., and Enright, A. J. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144.
Griffiths-Jones, S., Saini, H. K., van Dongen, S., and Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158.
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840.
Guo, Y., Chen, Y., Carreon, S., and Qiang, M. (2012). Chronic intermittent ethanol exposure and its removal induce a different miRNA expression pattern in primary cortical neuronal cultures. Alcohol Clin. Exp. Res. 36, 1058–1066.
Hansen, K. D., Brenner, S. E., and Dudoit, S. (2010). Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 38, e131.
Hayden, E. C. (2012). Nanopore genome sequencer makes its debut. Nature doi:10.1038/nature.2012.10051
He, Y., Yang, C., Kirkmire, C. M., and Wang, Z. J. (2010). Regulation of opioid tolerance by let-7 family microRNA targeting the mu opioid receptor. J. Neurosci. 30, 10251–10258.
Helwig, M., Lee, S.-N., Hwang, J. R., Ozawa, A., Medrano, J. F., and Lindberg, I. (2011). Dynamic modulation of prohormone convertase 2 (PC2)-mediated precursor processing by 7B2 protein: preferential effect on glucagon synthesis. J. Biol. Chem. 286, 42504–42513.
Henry, J. C., Azevedo-Pouly, A. C. P., and Schmittgen, T. D. (2011). MicroRNA replacement therapy for cancer. Pharm. Res. 28, 3030–3042.
Hollander, J. A., Im, H.-I., Amelio, A. L., Kocerha, J., Bali, P., Lu, Q., Willoughby, D., Wahlestedt, C., Conkright, M. D., and Kenny, P. J. (2010). Striatal microRNA controls cocaine intake through CREB signalling. Nature 466, 197–202.
Hu, G., Wei, Y., and Kang, Y. (2009). The multifaceted role of MTDH/AEG-1 in cancer progression. Clin. Cancer Res. 15, 5615–5620.
Hubert, G. W., Jones, D. C., Moffett, M. C., Rogge, G., and Kuhar, M. J. (2008). CART peptides as modulators of dopamine and psychostimulants and interactions with the mesolimbic dopaminergic system. Biochem. Pharmacol. 75, 57–62.
Hyman, S. E., and Malenka, R. C. (2001). Addiction and the brain: the neurobiology of compulsion and its persistence. Nat. Rev. Neurosci. 2, 695–703.
Hyman, S. E., Malenka, R. C., and Nestler, E. J. (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 29, 565–598.
Illumina. (2011). Technology: History of Solexa Sequencing. Available at: http://www.illumina.com [accessed May 13, 2011].
Im, H.-I., Hollander, J. A., Bali, P., and Kenny, P. J. (2010). MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat. Neurosci. 13, 1120–1127.
Inui, M., Martello, G., and Piccolo, S. (2010). MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 11, 252–263.
Kalivas, P. W. (2009). The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 10, 561–572.
Kalivas, P. W., Volkow, N., and Seamans, J. (2005). Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45, 647–650.
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30.
Kang, D.-C., Su, Z.-Z., Sarkar, D., Emdad, L., Volsky, D. J., and Fisher, P. B. (2005). Cloning and characterization of HIV-1-inducible astrocyte elevated gene-1, AEG-1. Gene 353, 8–15.
Kauer, J. A., and Malenka, R. C. (2007). Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–858.
Kiebler, M. A., and DesGroseillers, L. (2000). Molecular insights into mRNA transport and local translation in the mammalian nervous system. Neuron 25, 19–28.
Kim, J., Krichevsky, A., Grad, Y., Hayes, G. D., Kosik, K. S., Church, G. M., and Ruvkun, G. (2004). Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc. Natl. Acad. Sci. U.S.A. 101, 360–365.
Kim, V. N., Han, J., and Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139.
Kiraly, D. D., Ma, X.-M., Mazzone, C. M., Xin, X., Mains, R. E., and Eipper, B. A. (2010a). Behavioral and morphological responses to cocaine require kalirin7. Biol. Psychiatry 68, 249–255.
Kiraly, D. D., Eipper-Mains, J. E., Mains, R. E., and Eipper, B. A. (2010b). Synaptic plasticity, a symphony in GEF. ACS Chem. Neurosci. 1, 348–365.
Koob, G. F., and Le Moal, M. (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129.
Krichevsky, A. M., King, K. S., Donahue, C. P., Khrapko, K., and Kosik, K. S. (2003). A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 9, 1274–1281.
Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739.
Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N., Aravin, A., Pfeffer, S., Rice, A., Kamphorst, A. O., Landthaler, M., Lin, C., Socci, N. D., Hermida, L., Fulci, V., Chiaretti, S., Foà, R., Schliwka, J., Fuchs, U., Novosel, A., Müller, R.-U., Schermer, B., Bissels, U., Inman, J., Phan, Q., Chien, M., Weir, D. B., Choksi, R., de Vita, G., Frezzetti, D., Trompeter, H.-I., Hornung, V., Teng, G., Hartmann, G., Palkovits, M., di Lauro, R., Wernet, P., Macino, G., Rogler, C. E., Nagle, J. W., Ju, J., Papavasiliou, F. N., Benzing, T., Lichter, P., Tam, W., Brownstein, M. J., Bosio, A., Borkhardt, A., Russo, J. J., Sander, C., Zavolan, M., and Tuschl, T. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414.
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25.
Law, P. T.-Y., and Wong, N. (2011). Emerging roles of microRNA in the intracellular signaling networks of hepatocellular carcinoma. J. Gastroenterol. Hepatol. 26, 437–449.
Lehrmann, E., Oyler, J., Vawter, M. P., Hyde, T. M., Kolachana, B., Kleinman, J. E., Huestis, M. A., Becker, K. G., and Freed, W. J. (2003). Transcriptional profiling in the human prefrontal cortex: evidence for two activational states associated with cocaine abuse. Pharmacogenomics J. 3, 27–40.
Levin, J. Z., Yassour, M., Adiconis, X., Nusbaum, C., Thompson, D. A., Friedman, N., Gnirke, A., and Regev, A. (2010). Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715.
Lewohl, J. M., Nunez, Y. O., Dodd, P. R., Tiwari, G. R., Harris, R. A., and Mayfield, R. D. (2011). Up-regulation of MicroRNAs in brain of human alcoholics. Alcohol. Clin. Exp. Res. 35, 1928–1937.
Li, C.-Y., Mao, X., and Wei, L. (2008). Genes and (common) pathways underlying drug addiction. PLoS Comput. Biol. 4, e2. doi:10.1371/journal.pcbi.0040002
Li, H., Kloosterman, W., and Fekete, D. M. (2010). MicroRNA-183 family members regulate sensorineural fates in the inner ear. J. Neurosci. 30, 3254–3263.
Li, M. D., and van der Vaart, A. D. (2011). MicroRNAs in addiction: adaptation’s middlemen? Mol. Psychiatry 16, 1159–1168.
Li, X., and Jin, P. (2009). Macro role(s) of microRNAs in fragile X syndrome? Neuromolecular Med. 11, 200–207.
Lippi, G., Steinert, J. R., Marczylo, E. L., D’Oro, S., Fiore, R., Forsythe, I. D., Schratt, G., Zoli, M., Nicotera, P., and Young, K. W. (2011). Targeting of the Arpc3 actin nucleation factor by miR-29a/b regulates dendritic spine morphology. J. Cell Biol. 194, 889–904.
Liu, X.-Y., Mao, L.-M., Zhang, G.-C., Papasian, C. J., Fibuch, E. E., Lan, H.-X., Zhou, H.-F., Xu, M., and Wang, J. Q. (2009). Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron 61, 425–438.
Lobo, M. K., Covington, H. E., Chaudhury, D., Friedman, A. K., Sun, H., Damez-Werno, D., Dietz, D. M., Zaman, S., Koo, J. W., Kennedy, P. J., Mouzon, E., Mogri, M., Neve, R. L., Deisseroth, K., Han, M.-H., and Nestler, E. J. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390.
Lugli, G., Larson, J., Martone, M. E., Jones, Y., and Smalheiser, N. R. (2005). Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J. Neurochem. 94, 896–905.
Lugli, G., Torvik, V. I., Larson, J., and Smalheiser, N. R. (2008). Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J. Neurochem. 106, 650–661.
Ma, X.-M., Kiraly, D. D., Gaier, E. D., Wang, Y., Kim, E.-J., Levine, E. S., Eipper, B. A., and Mains, R. E. (2008). Kalirin-7 is required for synaptic structure and function. J. Neurosci. 28, 12368–12382.
Mains, R. E., Kiraly, D. D., Eipper-Mains, J. E., Ma, X., and Eipper, B. A. (2011). Kalrn promoter usage and isoform expression respond to chronic cocaine exposure. BMC Neurosci. 12, 20. doi:10.1186/1471-2202-12-20
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., Berka, J., Braverman, M. S., Chen, Y.-J., Chen, Z., Dewell, S. B., Du, L., Fierro, J. M., Gomes, X. V., Godwin, B. C., He, W., Helgesen, S., Ho, C. H., Ho, C. H., Irzyk, G. P., Jando, S. C., Alenquer, M. L. I., Jarvie, T. P., Jirage, K. B., Kim, J.-B., Knight, J. R., Lanza, J. R., Leamon, J. H., Lefkowitz, S. M., Lei, M., Li, J., Lohman, K. L., Lu, H., Makhijani, V. B., McDade, K. E., McKenna, M. P., Myers, E. W., Nickerson, E., Nobile, J. R., Plant, R., Puc, B. P., Ronan, M. T., Roth, G. T., Sarkis, G. J., Simons, J. F., Simpson, J. W., Srinivasan, M., Tartaro, K. R., Tomasz, A., Vogt, K. A., Volkmer, G. A., Wang, S. H., Wang, Y., Weiner, M. P., Yu, P., Begley, R. F., and Rothberg, J. M. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380.
Martí, E., Pantano, L., Bañez-Coronel, M., Llorens, F., Miñones-Moyano, E., Porta, S., Sumoy, L., Ferrer, I., and Estivill, X. (2010). A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 38, 7219–7235.
Martinek, S., and Gaul, U. (1997). Neural development: how cadherins zipper up neural circuits. Curr. Biol. 7, R712–R715.
Mash, D. C., Ffrench-Mullen, J., Adi, N., Qin, Y., Buck, A., and Pablo, J. (2007). Gene expression in human hippocampus from cocaine abusers identifies genes which regulate extracellular matrix remodeling. PLoS ONE 2, e1187. doi:10.1371/journal.pone.0001187
Mash, D. C., Ouyang, Q., Pablo, J., Basile, M., Izenwasser, S., Lieberman, A., and Perrin, R. J. (2003). Cocaine abusers have an overexpression of alpha-synuclein in dopamine neurons. J. Neurosci. 23, 2564–2571.
Maxam, A. M., and Gilbert, W. (1977). A new method for sequencing DNA. Proc. Natl. Acad. Sci. U.S.A. 74, 560–564.
McCarty, M. F. (2012). Metformin may antagonize Lin28 and/or Lin28B activity, thereby boosting let-7 levels and antagonizing cancer progression. Med. Hypotheses 78, 262–269.
McClung, C. A., and Nestler, E. J. (2003). Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat. Neurosci. 6, 1208–1215.
McClung, C. A., and Nestler, E. J. (2008). Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology 33, 3–17.
Miska, E. A., Alvarez-Saavedra, E., Townsend, M., Yoshii, A., Sestan, N., Rakic, P., Constantine-Paton, M., and Horvitz, H. R. (2004). Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5, R68.
Mongroo, P. S., and Rustgi, A. K. (2010). The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 10, 219–222.
Morabito, M. A., Sheng, M., and Tsai, L.-H. (2004). Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J. Neurosci. 24, 865–876.
Moreau, M. P., Bruse, S. E., David-Rus, R., Buyske, S., and Brzustowicz, L. M. (2011). Altered MicroRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol. Psychiatry 69, 188–193.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628.
Nestler, E. J. (2001). Molecular basis of long-term plasticity underlying addiction. Nat. Rev. Neurosci. 2, 119–128.
Nestler, E. J. (2005b). Is there a common molecular pathway for addiction? Nat. Neurosci. 8, 1445–1449.
Nestler, E. J., and Aghajanian, G. K. (1997). Molecular and cellular basis of addiction. Science 278, 58–63.
Norrholm, S. D., Bibb, J. A., Nestler, E. J., Ouimet, C. C., Taylor, J. R., and Greengard, P. (2003). Cocaine-induced proliferation of dendritic spines in nucleus accumbens is dependent on the activity of cyclin-dependent kinase-5. Neuroscience 116, 19–22.
Oshlack, A., and Wakefield, M. J. (2009). Transcript length bias in RNA-seq data confounds systems biology. Biol. Direct 4, 14.
Ozsolak, F., and Milos, P. M. (2011). RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98.
Pan, H., Che, F.-Y., Peng, B., Steiner, D. F., Pintar, J. E., and Fricker, L. D. (2006). The role of prohormone convertase-2 in hypothalamic neuropeptide processing: a quantitative neuropeptidomic study. J. Neurochem. 98, 1763–1777.
Paulson, P. E., Camp, D. M., and Robinson, T. E. (1991). Time course of transient behavioral depression and persistent behavioral sensitization in relation to regional brain monoamine concentrations during amphetamine withdrawal in rats. Psychopharmacology (Berl.) 103, 480–492.
Pietrzykowski, A. Z., Friesen, R. M., Martin, G. E., Puig, S. I., Nowak, C. L., Wynne, P. M., Siegelmann, H. T., and Treistman, S. N. (2008). Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron 59, 274–287.
Przewlocka, B., and Lason, W. (1995). Adaptive changes in the proenkephalin and D2 dopamine receptor mRNA expression after chronic cocaine in the nucleus accumbens and striatum of the rat. Eur. Neuropsychopharmacol. 5, 465–469.
Rao, A., and Steward, O. (1991). Evidence that protein constituents of postsynaptic membrane specializations are locally synthesized: analysis of proteins synthesized within synaptosomes. J. Neurosci. 11, 2881–2895.
Robinson, T. E., and Berridge, K. C. (2001). Incentive-sensitization and addiction. Addiction 96, 103–114.
Robinson, T. E., Gorny, G., Mitton, E., and Kolb, B. (2001). Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse 39, 257–266.
Robinson, T. E., and Kolb, B. (1997). Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J. Neurosci. 17, 8491–8497.
Robinson, T. E., and Kolb, B. (1999). Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur. J. Neurosci. 11, 1598–1604.
Ross, S., and Peselow, E. (2009). The neurobiology of addictive disorders. Clin. Neuropharmacol. 32, 269–276.
Rossman, K. L., Der, C. J., and Sondek, J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180.
Saba, R., Störchel, P. H., Aksoy-Aksel, A., Kepura, F., Lippi, G., Plant, T. D., and Schratt, G. M. (2012). Dopamine-regulated microRNA MiR-181a controls GluA2 surface expression in hippocampal neurons. Mol. Cell. Biol. 32, 619–632.
Sanchez-Simon, F. M., Zhang, X. X., Loh, H. H., Law, P.-Y., and Rodriguez, R. E. (2010). Morphine regulates dopaminergic neuron differentiation via miR-133b. Mol. Pharmacol. 78, 935–942.
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74, 5463–5467.
Santarelli, D. M., Beveridge, N. J., Tooney, P. A., and Cairns, M. J. (2011). Upregulation of dicer and microRNA expression in the dorsolateral prefrontal cortex Brodmann area 46 in schizophrenia. Biol. Psychiatry 69, 180–187.
Schaefer, A., Im, H.-I., Venø, M. T., Fowler, C. D., Min, A., Intrator, A., Kjems, J., Kenny, P. J., O’Carroll, D., and Greengard, P. (2010). Argonaute 2 in dopamine 2 receptor-expressing neurons regulates cocaine addiction. J. Exp. Med. 207, 1843–1851.
Schratt, G. (2009a). Fine-tuning neural gene expression with microRNAs. Curr. Opin. Neurobiol. 19, 213–219.
Schratt, G. M., Tuebing, F., Nigh, E. A., Kane, C. G., Sabatini, M. E., Kiebler, M., and Greenberg, M. E. (2006). A brain-specific microRNA regulates dendritic spine development. Nature 439, 283–289.
Self, D. W., and Nestler, E. J. (1995). Molecular mechanisms of drug reinforcement and addiction. Annu. Rev. Neurosci. 18, 463–495.
Shendure, J., Porreca, G. J., Reppas, N. B., Lin, X., McCutcheon, J. P., Rosenbaum, A. M., Wang, M. D., Zhang, K., Mitra, R. D., and Church, G. M. (2005). Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732.
Siegel, G., Obernosterer, G., Fiore, R., Oehmen, M., Bicker, S., Christensen, M., Khudayberdiev, S., Leuschner, P. F., Busch, C. J. L., Kane, C., Hübel, K., Dekker, F., Hedberg, C., Rengarajan, B., Drepper, C., Waldmann, H., Kauppinen, S., Greenberg, M. E., Draguhn, A., Rehmsmeier, M., Martinez, J., and Schratt, G. M. (2009). A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol. 11, 705–716.
Siegel, G., Saba, R., and Schratt, G. (2011). microRNAs in neurons: manifold regulatory roles at the synapse. Curr. Opin. Genet. Dev. 21, 491–497.
Song, J.-J., Smith, S. K., Hannon, G. J., and Joshua-Tor, L. (2004). Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305, 1434–1437.
Spangler, R., Ho, A., Zhou, Y., Maggos, C. E., Yuferov, V., and Kreek, M. J. (1996). Regulation of kappa opioid receptor mRNA in the rat brain by “binge” pattern cocaine administration and correlation with preprodynorphin mRNA. Brain Res. Mol. Brain Res. 38, 71–76.
Suzuki, T., Tian, Q. B., Kuromitsu, J., Kawai, T., and Endo, S. (2007). Characterization of mRNA species that are associated with postsynaptic density fraction by gene chip microarray analysis. Neurosci. Res. 57, 61–85.
Tang, L., Hung, C. P., and Schuman, E. M. (1998). A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron 20, 1165–1175.
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., Diemer, K., Muruganujan, A., and Narechania, A. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141.
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., and Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515.
Treistman, S. N., and Martin, G. E. (2009). BK channels: mediators and models for alcohol tolerance. Trends Neurosci. 32, 629–637.
Urdinguio, R. G., Fernandez, A. F., Lopez-Nieva, P., Rossi, S., Huertas, D., Kulis, M., Liu, C.-G., Croce, C., Calin, G. A., and Esteller, M. (2010). Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 5, 656–663.
Vo, N. K., Cambronne, X. A., and Goodman, R. H. (2010). MicroRNA pathways in neural development and plasticity. Curr. Opin. Neurobiol. 20, 457–465.
Volkow, N. D., Fowler, J. S., Wang, G.-J., and Swanson, J. M. (2004). Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol. Psychiatry 9, 557–569.
Wahlstedt, H., Daniel, C., Ensterö, M., and Ohman, M. (2009). Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 19, 978–986.
Wang, D. O., Kim, S. M., Zhao, Y., Hwang, H., Miura, S. K., Sossin, W. S., and Martin, K. C. (2009a). Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science 324, 1536–1540.
Wang, Z., Gerstein, M., and Snyder, M. (2009b). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Wang, E., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S., Schroth, G., and Burge, C. (2008a). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Wang, X., Sun, Q., McGrath, S. D., Mardis, E. R., Soloway, P. D., and Clark, A. G. (2008b). Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS ONE 3, e3839. doi:10.1371/journal.pone.0003839
Wayman, G. A., Davare, M., Ando, H., Fortin, D., Varlamova, O., Cheng, H.-Y. M., Marks, D., Obrietan, K., Soderling, T. R., Goodman, R. H., and Impey, S. (2008). An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. U.S.A. 105, 9093–9098.
Weston, M. D., Pierce, M. L., Jensen-Smith, H. C., Fritzsch, B., Rocha-Sanchez, S., Beisel, K. W., and Soukup, G. A. (2011). MicroRNA-183 family expression in hair cell development and requirement of microRNAs for hair cell maintenance and survival. Dev. Dyn. 240, 808–819.
Winsky-Sommerer, R., Benjannet, S., Rovère, C., Barbero, P., Seidah, N. G., Epelbaum, J., and Dournaud, P. (2000). Regional and cellular localization of the neuroendocrine prohormone convertases PC1 and PC2 in the rat central nervous system. J. Comp. Neurol. 424, 439–460.
Wolf, M. E. (2006). “Addiction,” in Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, Vol. 7, eds G. J. Siegel, B. W. Agranoff, R. W. Albers, S. K. Fisher and M. D. Uhler (New York: American Society for Neurochemistry), 911–926.
Wu, H., Tao, J., Chen, P. J., Shahab, A., Ge, W., Hart, R. P., Ruan, X., Ruan, Y., and Sun, Y. E. (2010). Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 18161–18166.
Wu, Q., Zhang, L., Law, P.-Y., Wei, L.-N., and Loh, H. H. (2009). Long-term morphine treatment decreases the association of mu-opioid receptor (MOR1) mRNA with polysomes through miRNA23b. Mol. Pharmacol. 75, 744–750.
Yadav, S., Pandey, A., Shukla, A., Talwelkar, S. S., Kumar, A., Pant, A. B., and Parmar, D. (2011). miR-497 and miR-302b regulate ethanol-induced neuronal cell death through BCL2 protein and cyclin D2. J. Biol. Chem. 286, 37347–37357.
Yamagata, K., Andreasson, K. I., Sugiura, H., Maru, E., Dominique, M., Irie, Y., Miki, N., Hayashi, Y., Yoshioka, M., Kaneko, K., Kato, H., and Worley, P. F. (1999). Arcadlin is a neural activity-regulated cadherin involved in long term potentiation. J. Biol. Chem. 274, 19473–11979.
Zheng, H., Chu, J., Zeng, Y., Loh, H. H., and Law, P.-Y. (2010). Yin Yang 1 phosphorylation contributes to the differential effects of mu-opioid receptor agonists on microRNA-190 expression. J. Biol. Chem. 285, 21994–22002.
Zhu, Q., Sun, W., Okano, K., Chen, Y., Zhang, N., Maeda, T., and Palczewski, K. (2011). Sponge transgenic mouse model reveals important roles for the microRNA-183 (miR-183)/96/182 cluster in postmitotic photoreceptors of the retina. J. Biol. Chem. 286, 31749–31760.
Keywords: cocaine, RNA-Seq, postsynaptic density, cell adhesion, miR-8, microRNAs, synaptic plasticity
Citation: Eipper-Mains JE, Eipper BA and Mains RE (2012) Global approaches to the role of miRNAs in drug-induced changes in gene expression. Front. Gene. 3:109. doi: 10.3389/fgene.2012.00109
Received: 01 March 2012; Accepted: 29 May 2012;
Published online: 13 June 2012.
Edited by:
Ashok Sharma, Georgia Health Sciences University, USAReviewed by:
Timothy Bowen, Cardiff University School of Medicine, UKAshok Sharma, Georgia Health Sciences University, USA
Navjotsingh Pabla, California Institute of Technology, USA
Melanie Carless, Texas Biomedical Research Institute, USA
Copyright: © 2012 Eipper-Mains, Eipper and Mains. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Richard E. Mains, Department of Neuroscience, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030-3401, USA. e-mail:bWFpbnNAbnNvLnVjaGMuZWR1