



- 1Center for Innovative Cancer Research, Ottawa Hospital Research Institute, Ottawa, ON, Canada
- 2Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
- 3Ontario Cancer Institute, Princess Margaret Cancer Center/University Health Network, Toronto, ON, Canada
- 4Ontario Institute for Cancer Research, Toronto, ON, Canada
- 5Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada
Oncolytic viruses (OVs) comprise a versatile and multi-mechanistic therapeutic platform in the growing arsenal of anticancer biologics. These replicating therapeutics find favorable conditions in the tumor niche, characterized among others by increased metabolism, reduced anti-tumor/antiviral immunity, and disorganized vasculature. Through a self-amplification that is dependent on multiple cancer-specific defects, these agents exhibit remarkable tumor selectivity. With several OVs completing or entering Phase III clinical evaluation, their therapeutic potential as well as the challenges ahead are increasingly clear. One key hurdle is tumor heterogeneity, which results in variations in the ability of tumors to support productive infection by OVs and to induce adaptive anti-tumor immunity. To this end, mounting evidence suggests tumor epigenetics may play a key role. This review will focus on the epigenetic landscape of tumors and how it relates to OV infection. Therapeutic strategies aiming to exploit the epigenetic identity of tumors in order to improve OV therapy are also discussed.
Introduction
While genetic information establishes the primary blueprint for cellular identity, multiple regulatory layers responsive to extra and intra-cellular signals ultimately control the manifestation of this blueprint. Changes in cellular state, including initiation of DNA synthesis, activation of apoptotic programs, or triggering of antiviral defense mechanisms, result from an integrated response to stimuli received by the cell. These are controlled in large part by gene/protein expression profiles unique to each cell. It is now well understood that activation of transcription factors that bind in a DNA sequence-specific manner at promoter and enhancer elements is responsible for many of the changes in gene expression that occur in response to environmental or developmental cues. However transcription factors and their associated gene targets are themselves further regulated by the accessibility of DNA sequences. Since the genome resides in the finite space provided by the nucleus, it interacts with proteins known as histones to form chromatin and facilitate its compaction. The configuration of chromatin compaction is modulated by epigenetic modification and is a key determinant for transcription factor-mediated activation of gene transcription (Magnani et al., 2011).
Epigenetic modifications create a reversible imprint that may be inherited through cell division. For example, DNA methylated at promoter CpG islands is associated with gene silencing and can be reversed by treatment with DNA methyltransferase inhibitors such as 5-AZA (5-aza-2′-deoxycytidine) leading to the reactivation of silenced genes (Baylin and Jones, 2011; Krecmerova and Otmar, 2012). Similarly, chromatin structure can alter accessibility to the DNA template and can be readily remodeled by histone post-translational modifications (PTMs). PTMs including acetylation, methylation, phosphorylation, ubiquitination, and many others can be added to numerous residues of histone proteins (Bannister and Kouzarides, 2011). Different PTMs will favor chromatin compaction while others will increase its accessibility to DNA binding proteins. Histone modifications and DNA methylation are highly interdependent processes and define the epigenetic code (Cedar and Bergman, 2009). The epigenetic code is regulated by a complex interplay of enzymatic erasers, readers, and writers that exhibit specificities toward different histones and residues (Rice and Allis, 2001). For example, the level of histone acetylation is regulated by the relative activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs), proteins with opposing enzymatic activities that are often found in the same protein complexes (Johnsson et al., 2009; Peserico and Simone, 2010). This also applies to histone lysine methyltransferases (KMTs) and lysine demethylases (KDMs). Consequently, modulating the activity of histone-modifying enzymes can profoundly alter the epigenetic profile of a cell (Egger et al., 2004; Yoo and Jones, 2006).
Given their critical role in the regulation of normal cellular physiology, it is not surprising that aberrations in epigenetic modifications can contribute to the manifestations of human disease. For example, a cell’s epigenetic profile can impact the progression of acute microbial diseases (discussed in more detail below) as well as the development and treatment of chronic diseases such as cancer. DNA hypermethylation is often observed in cancer cells (Patel et al., 2012). The genome-wide distribution of histone modifications can also be altered in the course of cancer development (Akhtar-Zaidi et al., 2012; Magnani et al., 2013). As well, the activity of various histone-modifying enzymes can be altered through mutations (Taylor et al., 2011), aberrant expression (Schildhaus et al., 2011; Bennani-Baiti et al., 2012) and/or recruitment to target histone residues via oncogenic fusion proteins (Lubieniecka et al., 2008). Consequently, many cancers are sensitive to epigenetic modulators such as 5-AZA, HDAC, or KDM inhibitors (Hurtubise et al., 2008; Taylor et al., 2011; Schenk et al., 2012) and epigenetic modifications have been shown to influence the response to chemotherapy (Glasspool et al., 2006; Magnani et al., 2013).
Oncolytic Virotherapy
While epigenetic modulators hold promise as anticancer agents, it is clear that like for many other cancer therapies, tumor-specificity is of paramount importance. Tremendous efforts have been made over the past decades to tackle the difficult task of developing more selective cancer therapies, aiming to exploit the sometimes-subtle differences between normal tissues and tumors. One promising new class of therapeutics comes to us from the field of virology. Since the early 1900s it has been observed that cancers can be uniquely susceptible to virus infection (Dock, 1904). While the first clinical trials using replication-competent viruses to treat cancer began in the seventies (Asada, 1974; Kelly and Russell, 2007; Pol et al., 2013), approval of the first oncolytic virus (OV) is only now in the foreseeable future in North America (Carroll, 2011; Galanis et al., 2012; Heo et al., 2013). The more recent clinical success of OVs is in large part due to our more complete understanding of the molecular biology of both cancer cells and viruses that allowed us to create virus strains with improved selectivity and anti-tumor activity, and clinical safety profile (Breitbach et al., 2011). Rapid proliferation and deregulated metabolism (Fritz and Fajas, 2010), disorganized vasculature (Jain, 2005), and defective antiviral innate immune responses (Dunn et al., 2006) in malignant tumors are hallmarks that not only define cancer, but also favor viral growth. Building on these observations, several OVs have been engineered or selected to take advantage of one or more of these features (Russell et al., 2012). A variety of OV platforms are currently under clinical evaluation including those based on herpes simplex virus (HSV), Reovirus, vaccinia virus (VV), Adenovirus, Measles virus, and vesicular stomatitis virus (VSV; U.S. National Library of Medicine, 2013).
Oncolytic Virotherapy and the Cellular Innate Antiviral Response
It is now well established that cancer cells that evolve to frank malignancies often acquire defects in their ability to mount a successful antiviral response and this attribute/deficit contributes to the selectivity of many if not all OVs (Norman and Lee, 2000; Stojdl et al., 2000, 2003). This is often a consequence of the observation that approximately 65–70% of tumors are unable to produce or respond to type I interferon (IFN), a key mediator of the cellular antiviral response (Stojdl et al., 2003; Dunn et al., 2006). IFNs are antiviral cytokines induced following recognition of viral proteins and nucleic acids by cellular pattern recognition receptors such as Toll-like receptors (TLRs) that signal through to transcription factors such as interferon regulatory factors (IRFs). There are many isoforms of IFN, which can be functionally sub-divided in at least three types (types I/II/III). While type I/III IFNs (e.g., IFN-α, IFN-β/IFN-λ) stimulate cellular antimicrobial immunity; type II IFNs (e.g., IFN-γ) coordinate the host immune response. IFNs elicit their transcriptional effects through autocrine and paracrine activation of IFN receptors and signaling through the Jak/STAT signaling pathway (Borden et al., 2007). This induces the transcriptional up-regulation of interferon-stimulated genes (ISGs), many of which have direct antiviral/pro-apoptotic activities (e.g., RNAseL, TNF-α, TRAIL) and/or immune-stimulatory properties (e.g., components of major histocompatibility complex).
Oncolytic Viruses and the Generation of an Anti-Tumor Immune Response
In addition to taking advantage of a niche provided by aberrations unique to cancer and the tumor microenvironment, OVs have been used as platforms to express a range of therapeutic transgenes, from suicide genes to immune-stimulatory cytokines (Merrick et al., 2009; Maldonado et al., 2010; Chai et al., 2012; Stephenson et al., 2012; Lange et al., 2013). In this regard, it is now well recognized that beyond simply lysing infected tumor cells, OVs effectively “de-cloak” tumors by stimulating immune cells to recognize cancer antigens, ultimately leading to tumor destruction and in some cases, long-term cures (Sobol et al., 2011; Huang et al., 2012). Many tumors evade immune recognition due to a dysfunctional antigen presentation pathway, which is under tight multilayered transcriptional control ultimately dictated by type I/II IFNs and the class II transactivator (CIITA). This transcription factor controls the expression of numerous genes involved in antigen presentation, including class I and II MHC molecules, which display tumor or pathogen derived peptides to killer T cells (CD4+/CD8+; LeibundGut-Landmann et al., 2004).
The antigen presentation pathway is influenced by both tumorigenesis and OV therapy. Many tumor cells including leukemias, lymphomas, and carcinomas, avoid immune recognition due to a dysfunctional antigen presentation pathway, largely caused by epigenetic silencing (e.g., histone deacetylation or DNA methylation) of MHC2TA, the gene encoding CIITA (LeibundGut-Landmann et al., 2004). OV therapies can enhance tumor-associated antigen presentation through various mechanisms. In response to OV infection, type I and II IFN secretion by infected cells within the tumor environment (which also includes normal tumor infiltrating cells) leads to the up-regulation of hundreds of ISGs including IRF-1, which up-regulates CIITA expression (Muhlethaler-Mottet et al., 1998). Notably, this response is dependent upon the ability to respond to IFN, which can be limited in many cancer cells (Stojdl et al., 2003; Dunn et al., 2006).
Oncolytic virotherapy can have a positive influence on antigen presentation and the anti-tumor response. Some OVs including HSV, reovirus, and measles virus, induce syncytia formation in infected and neighboring cells. These large multinucleated tumor cells secrete an abundance of “syncytiosomes,” which are exosome-like vesicles that present tumor-associated antigens via MHC molecules (Bateman et al., 2000, 2002). Finally, destruction of cancer cells following infection by OVs provides an additional source of tumor antigens available for capture by antigen-presenting immune cells. The immunostimulatory nature of the virus itself, through activation of TLRs and subsequent cellular production of pro-inflammatory cytokines stimulates the recruitment of antigen-presenting cells that sample tumor-derived and virus-expressed antigens. Presentation of tumor antigens to killer T cells (CD4+/CD8+) through MHC molecules in the presence of inflammatory cytokines can thus lead to generation of a robust and long-lasting immune responses directed against the tumor.
To capitalize on these beneficial immunological effects, some groups have developed OV/vaccine hybrid strategies. These strategies are designed specifically to re-educate the adaptive immune system to recognize and respond to tumor antigens. Thus, OVs can be engineered to express not only immune-stimulatory cytokines but also tumor-specific antigens to further stimulate an anti-tumor immune response following OV infection of cancer cells (Diaz et al., 2007; Pulido et al., 2012). Indeed, several studies have shown that this “tumor antigen vaccination” effect can be further amplified using a prime-boost strategy, by priming with an antigen then boosting the response using an OV expressing the same antigen (Bridle et al., 2010, 2013). As discussed below, it is possible to use epigenetic modifiers to further fine-tune this oncolytic vaccine approach. It is also possible to take advantage of this vaccine effect by infecting cancer cells ex vivo and re-injecting the inactivated “oncolysate” to generate prophylactic and even therapeutic anticancer immune responses. The resulting up-regulation of MHCs and co-regulatory factors and presentation of tumor antigens at the surface of OV infected cells as well as the presence of immune-stimulating virus is thought to be at the root of this effect (Lemay et al., 2012). Overall, these studies emphasize the important role of antigen expression/presentation in OV-stimulated anti-tumoral responses.
Tumor Heterogeneity: Inherent Barrier to OV Therapy
Despite promising clinical data, it is clear that there is considerable inter- (and likely intra-) tumor heterogeneity in the responsiveness to OV therapy in vitro as well as in vivo in both pre-clinical and clinical settings (Breitbach et al., 2011; Sobol et al., 2011). Because overcoming the innate cellular antiviral response and generating a robust anti-tumor response are critical to observe meaningful therapeutic benefits from oncolytic virotherapy, it is important to understand what tumorigenic processes influence these closely linked pathways in order to manipulate them to improve therapeutic outcomes.
Given the profound epigenetic divergence that prevails in tumor cells (Akhtar-Zaidi et al., 2012; De Carvalho et al., 2012), it is foreseeable that tumor-specific gene expression response profiles induced by virus infection may be altered by epigenetic modifications and that this could contribute to the heterogeneity of tumor responsiveness to OVs. As discussed previously, epigenetic reprogramming is well known to play an important role in oncogenic transformation and numerous reviews extensively cover the role of epigenetics in cancer (Muntean and Hess, 2009; Baylin and Jones, 2011; Hatziapostolou and Iliopoulos, 2011; Suva et al., 2013). Thus, the remainder of this review aims to highlight current knowledge of genes epigenetically regulated in cancer that are also involved in pathways critical for OV therapy, namely the IFN-mediated antiviral response and antigen presentation (Table 1), and how this contributes to tumor heterogeneity (Figure 1).
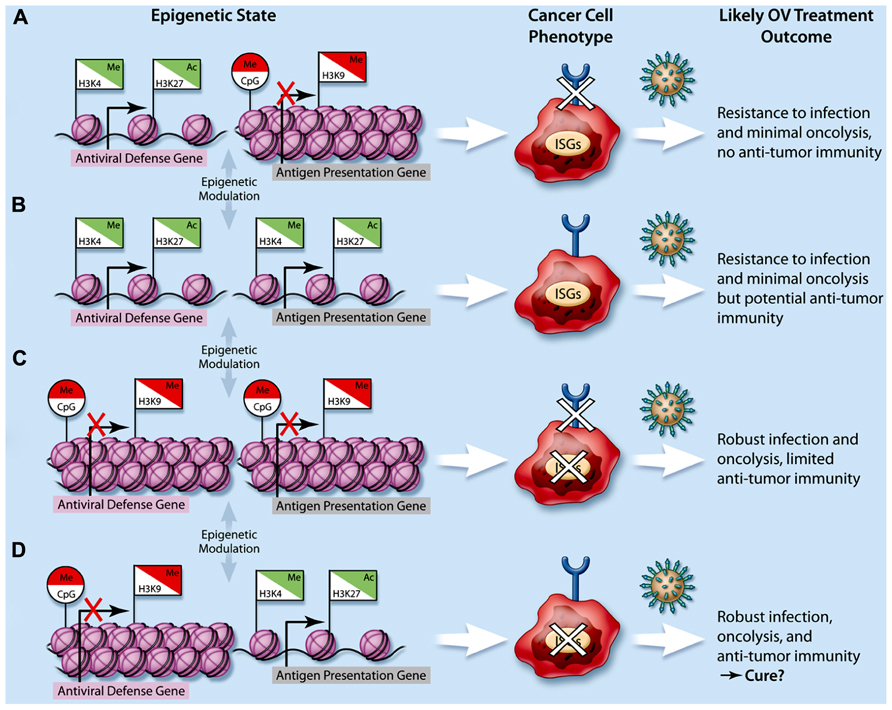
FIGURE 1. Impact of cancer epigenetics on oncolytic virotherapy. The integration of repressive epigenetic marks such as DNA CpG methylation (Me, circle flags) and histone H3K9 methylation (Me, square flags), and activating epigenetic marks such as histone H3K4 methylation and histone H3K27 acetylation (Ac, square flags) lead to higher-order nucleosome packaging and repression (red flags) or open chromatin and gene expression (green flags). In cancer cells, dysregulation of epigenetic processes leads to various possible epigenetic states with respect to genes involved in the antiviral response (e.g., type I IFN, interferon stimulated genes or ISGs) as well as those involved in antigen presentation (e.g., MHC I/II expression, represented by a semi-circle at the end of a stick). This ultimately leads to a variety of cancer cell phenotypes (A–D) and subsequently, a variety of potential therapeutic responses to oncolytic viruses (OVs, represented by spiked green circles).
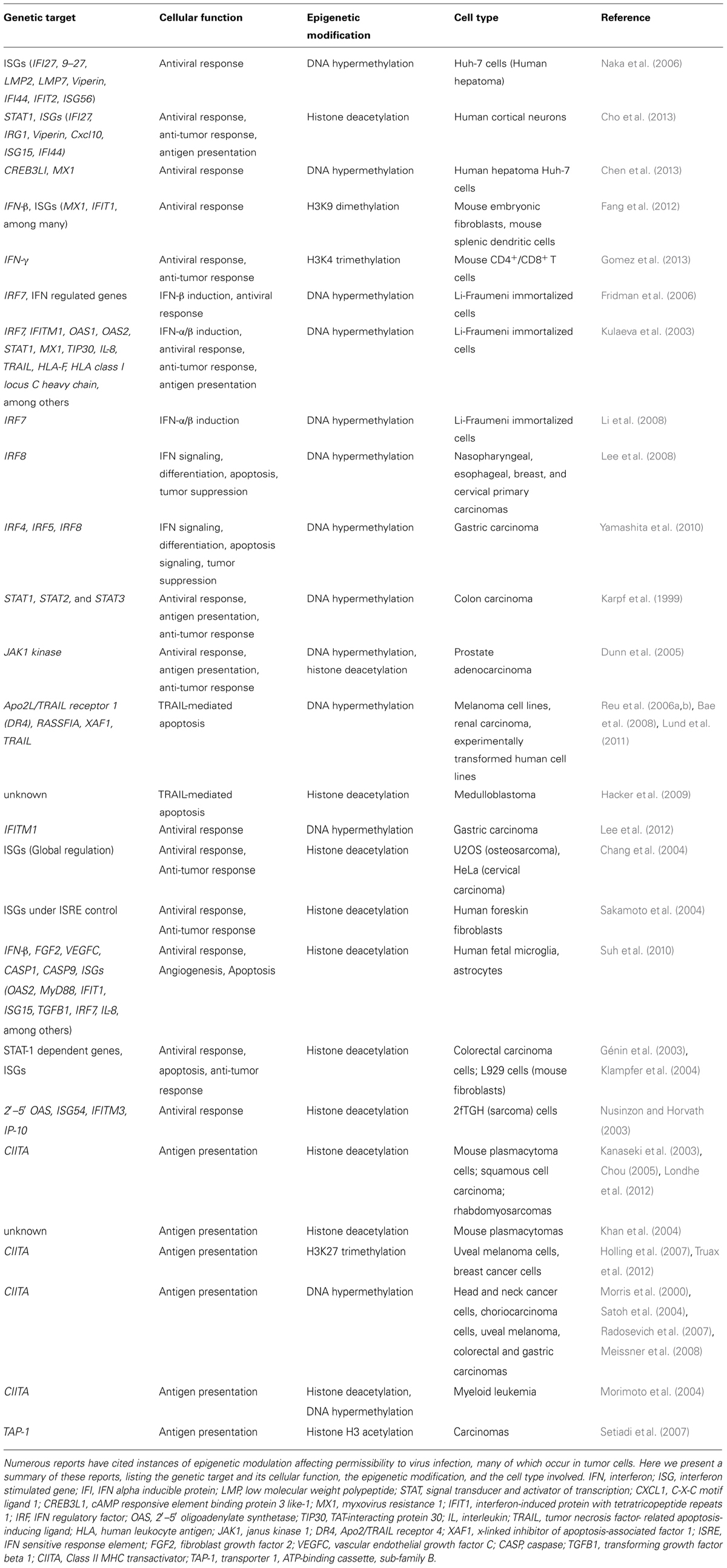
TABLE 1. Epigenetic control: implications in cancer and OV therapy.
The Role of Epigenetics in Host Susceptibility to Viral Infection
Epigenetic regulation of innate and adaptive immune processes is emerging as a key determinant of susceptibility to viral infection. Several reports suggest that cell type-specific epigenetic regulation of antiviral ISGs leads to differences in permissibility to virus infections in both normal and tumor cells (Naka et al., 2006; Nguyen et al., 2008; Fang et al., 2012; Chen et al., 2013; Cho et al., 2013). Recently, histone H3K9 di-methylation, a repressive heterochromatin mark, was found to be elevated within IFN genes and ISGs in non-professional IFN-producing cells (e.g., fibroblasts) as compared to professional IFN-producing plasmacytoid dendritic cells (pDCs). Interestingly, inhibiting the KMT G9a by both genetic and pharmacological means led to increased IFN production and responsiveness in fibroblasts. In line with this, G9a-ablated fibroblasts were also rendered more resistant to infection by viruses (Fang et al., 2012; Figure 1).
Another recent study in mice harboring the murine viral susceptibility locus Tmevp3 revealed the intriguing role of NeST, a long non-coding RNA (lncRNA) adjacent to the IFN-γ locus in both mice and humans (Vigneau et al., 2001). NeST was found to function as an epigenetically driven enhancer element (Gomez et al., 2013) leading to increased IFN-γ production in mouse CD8+ T cells by directly interacting with the H3K4 histone methyltransferase complex and increasing H3K4 trimethylation, an activating mark. This novel epigenetic modification culminated in heightened susceptibility to persistent viral infection in mice (Gomez et al., 2013; Figure 1). Although the role of NeST in human epigenetic regulation is currently unknown, it is likely lncRNAs contribute to epigenetic regulation and manifestation of cell phenotypes including permissiveness to virus infection and cancer.
Cancer Epigenetics Impact the Regulation of Antiviral Response Genes
As previously discussed, the majority (but not all) of cancer cells are dysfunctional in their ability to produce and/or respond to IFN (Dunn et al., 2006). While crosstalk between oncogenic signals and the antiviral response pathways have been shown to play a role (Farassati et al., 2001; Shmulevitz et al., 2005); epigenetic events are also likely contributors to this phenotype. One indication of this comes from a series of studies on cells derived from cancer-prone Li-Fraumeni syndrome patients. Cells from these patients spontaneously immortalize when serially passaged in tissue culture due to mutations in the tumor suppressor p53, however transformation is inhibited upon treatment with 5-AZA (Kulaeva et al., 2003; Fridman et al., 2006). DNA methylation profiling of these immortalized cells revealed hypermethylation at the promoters of numerous genes involved in the type I IFN pathway, including IRF7 (Kulaeva et al., 2003; Fridman et al., 2006; Li et al., 2008). Interestingly, these immortalized Li-Fraumeni patient-derived cells were inherently more sensitive to VSV infection (Fridman et al., 2006; Figure 1).
Indeed, epigenetic repression of IFN and associated genes correlates with IFN insensitivity in many cancers. IRFs 4, 5, 7, and 8 are the target of DNA methylation, leading to dysfunctional responsiveness to type I and II IFNs in gastric cancer (Yamashita et al., 2010), while IRF8 is silenced by the same mechanism in several carcinomas (Lee et al., 2008). Similarly, IFN responsiveness was found to be suppressed in colon carcinoma cells due to DNA methylation at STAT1, STAT2, and STAT3, which can be restored following 5-AZA treatment (Karpf et al., 1999; Figure 1). Along the same signaling axis, epigenetic silencing of JAK1 in prostate adenocarcinoma cells was associated with unresponsiveness to both type I and type II IFNs (Dunn et al., 2005).
IFN-induced apoptosis is mediated by ISGs including Apo2L/TRAIL, which are also often dysfunctional in cancers (Reu et al., 2006b; Borden, 2007; Bae et al., 2008; Burton et al., 2013). Genes involved in Apo2L/TRAIL signaling, including TRAIL, the TRAIL receptor DR4, RASSF1A, and XAF1 are epigenetically silenced in melanomas (Reu et al., 2006a, b; Bae et al., 2008), leukemia (Soncini et al., 2013), renal carcinoma (Reu et al., 2006a) and experimentally transformed cells (Lund et al., 2011). Interestingly, 5-AZA treatment can restore TRAIL-mediated apoptosis induced by type I and II IFN (Reu et al., 2006a,b; Bae et al., 2008; Lund et al., 2011; Soncini et al., 2013; Figure 1). However, this cell death pathway is likely also epigenetically silenced through histone PTMs given that in medulloblastoma, IFN-γ could induce apoptosis via TRAIL only following treatment with the HDAC inhibitor valproic acid (Hacker et al., 2009).
Overall, these studies highlight multiple epigenetic mechanisms that transcriptionally repress IFN-associated genes, culminating in dysfunctional and non-responsive IFN signaling across various cancer subtypes. However, in some instances alterations to epigenetic modifications in cancer lead to the up-regulation of antiviral factors. In both gastric tumors and gliomas, overexpression of the ISG IFITM1 promotes cancer cell migration and invasion, and its elevated expression is linked to reduced CpG methylation levels (Yu et al., 2011; Lee et al., 2012). Alongside its oncogenic properties, IFITM1 has antiviral properties, through its ability to inhibit viral membrane fusion (Li et al., 2013; Figure 1).
It is also notable that while most cancers display IFN pathway defects, approximately a third of cancer cells are fully functional in their ability to produce and respond to IFN (Stojdl et al., 2003; Norman and Lee, 2000). Importantly, several studies have shown that HDAC inhibition using a variety of chemical inhibitors modulate IFN-induced expression of ISGs, type I IFN, and TLR3/4 (Génin et al., 2003; Nusinzon and Horvath, 2003; Chang et al., 2004; Klampfer et al., 2004; Sakamoto et al., 2004; Suh et al., 2010), which leads to increased OV activity in resistant cells (Nguyen et al., 2008). This further highlights the key role of epigenetic regulation in the generation of an antiviral response and suggests that it may be possible to improve OV efficacy in resistant tumors by manipulating the cancer epigenome as will be discussed shortly.
Cancer Cells Epigenetically Regulate Genes Involved in Antigen Presentation
In addition to inactivating the antiviral response to escape anti-proliferative/pro-death signals, tumors must also evade immune recognition and clearance. To this end, many tumor types epigenetically suppress CIITA expression by mechanisms including histone deacetylation/methylation and DNA promoter methylation, resulting in suppressed IFN-γ mediated MHC-I and MHC-II gene expression and dysfunctional antigen presentation (Morris et al., 2000; Kanaseki et al., 2003; Morimoto et al., 2004; Satoh et al., 2004; Chou, 2005; Holling et al., 2007; Radosevich et al., 2007; Meissner et al., 2008; Londhe et al., 2012; Truax et al., 2012; Figure 1). Interestingly, treatment of cancer cells with HDAC inhibitors can promote antigen presentation and ultimately help to induce anti-tumor immunity (Khan et al., 2004; Chou, 2005). For example, trichostatin A (TSA)-treated irradiated B16 melanoma cells administered prophylactically as a cancer vaccine are significantly more effective then control irradiated B16 cells at protecting from a subsequent challenge with live B16 tumor cells (Khan et al., 2007). Cancer immune evasion can also be mediated by dampened expression of the transporter associated with antigen processing 1 (TAP1), a key factor for antigen presentation by MHC molecules (Johnsen et al., 1999). In carcinoma cells, decreased TAP1 expression was attributed to reduced levels of histone H3 acetylation at the TAP-1 promoter (Setiadi et al., 2007; Figure 1).
In addition to these direct epigenetic effects on components of the antigenic response within cancer cells, the tumor microenvironment has also been shown to epigenetically drive tumor infiltrating CD4+ T cells to tolerance. In colon cancer, infiltrating CD4+ lymphocytes displayed high levels of DNA methylation at the IFN-γ promoter, and consequently required treatment with 5-AZA to enable tumor antigen-stimulated IFN-γ production (Janson et al., 2008; Figure 1). Overall, these studies highlight the role of epigenetic control in conferring “stealth” status to tumor cells such that they may evade the immune surveillance.
HDAC Inhibitors Can Alter Susceptibility to Oncolytic Viruses
As alluded to earlier, defects in the IFN pathway are common in many malignancies but a significant proportion of tumors retain an active antiviral response (Stojdl et al., 2003; Dunn et al., 2006). Overcoming this antiviral response has been identified as a key barrier to the success of OV therapy and is the focus of many research groups including our own (Parato et al., 2005; Chiocca, 2008; Diallo et al., 2010; Liikanen et al., 2011; Russell et al., 2012). To overcome this barrier, many groups have looked at the possibility of using HDAC inhibitors in combination with OV therapy due to their repressive effects on the IFN-mediated antiviral response.
In one of the earliest reports, the anti-tumor effect of oncolytic adenovirus (OBP-301) in human lung cancer cells was found to synergize with FR901228 (Romidepsin), a class I HDAC inhibitor (Watanabe et al., 2006). However, in this report, increased activity was attributed to the upregulation of coxsackie adenovirus receptor (CAR) expression in cancer cells as opposed to direct effects on the antiviral response. Intriguingly, valproic acid, a class I/II HDAC inhibitor was found by another group in parallel to inhibit oncolytic adenovirus through the up-regulation of p21 (WAF1/CIP1; Hoti et al., 2006). Subsequently, TSA and valproic acid, two pan-HDAC inhibitors were found to enhance HSV oncolysis in squamous cell carcinoma and glioma cells (Otsuki et al., 2008; Katsura et al., 2009). Around the same time, Nguyen et al. (2008) showed that several HDIs could synergize with the oncolytic VSV-Δ51, an attenuated oncolytic VSV-mutant that is incapable of blocking IFN production (Stojdl et al., 2003). Combination treatment with HDIs resulted in synergistic cell killing, due to both enhanced induction of cell death and increased viral output (typically over 100-fold). Enhanced viral spreading of VV and semliki forest virus (SFV) was also observed in this study. Subsequent to this, TSA was shown to be particularly effective for improving VV-based OVs in several resistant cancer cell lines in vitro and in subcutaneous xenograft and syngeneic lung metastasis mouse models (MacTavish et al., 2011). Importantly, the impacts of HDAC inhibitors on OV spread and efficacy remain restricted to tumors and not normal cells, presumably because cancer cells exhibit a number of additional aberrations, such as increased metabolism, that promote viral growth independent of the status of the antiviral response.
HDAC Inhibitors as Modulators of Oncolytic Virus-Associated Anti-Tumor Immunity
While initial experiences with HDAC inhibitors in combination with OVs exploited mainly the ability of these epigenetic modifiers to improve the infectivity of resistant tumors, at least in part by dampening the innate cellular antiviral response, more recent studies have further exploited the broader immunological effects of HDAC inhibitors. For example, one report showed that valproic acid suppresses NK cell activity by blocking STAT5/T-BET signaling leading to enhanced oncolytic HSV activity (Alvarez-Breckenridge et al., 2012). Also of note, a recent report by Bridle et al. (2013) demonstrated significant improvements in the generation of an anti-tumor immune response elicited against aggressive melanoma following a heterologous prime-boost vaccination strategy. After the establishment of intracranial melanomas, immune-competent mice were primed with a non-replicating adenovirus expressing the dopachrome tautomerase (hDCT) melanoma antigen, and then boosted with oncolytic VSV expressing hDCT. While this prolonged survival, mice were fully cured (64%) only when VSV-hDCT was administered in combination with the class I HDAC inhibitor MS-275. Remarkably, MS-275 reduced VSV-specific neutralizing antibodies and memory CD8+ T cells while maintaining prime-induced levels of humoral and cellular immunity against the tumor antigen. Interestingly, MS-275 also ablated autoimmune vitiligo typically observed following immunization against the melanocyte-expressed antigen (Bridle et al., 2013).
Use of other Epigenetic Modulators to Improve Oncolytic Virotherapy?
Given the epigenetic regulation of the antiviral response and antigen presentation pathways, it is tempting to speculate that other epigenetic modulators, in addition to HDAC inhibitors, may also be used to amplify therapeutic responses in combination with OVs. To this end, a recent study by Okemoto et al. (2012) showed that 5-AZA treatment could enhance HSV replication when co-administered with IL-6 (Figure 1). However, given numerous reports of cancers epigenetically silencing antiviral genes by DNA methylation (Table 1), we would expect that in general 5-AZA and other DNA methyltransferase inhibitors should be ineffective at overcoming the cellular antiviral response. On the other hand, the advent of new pharmacological inhibitors of KMTs and KDMs brings forth new possibilities for improving OV efficacy. For example, given the finding that histone H3K9 dimethylation observed at ISGs correlates with repression and reduced IFN response/expression, investigating the potential utility of H3K9-demetylase inhibitors for enhancing OV spread in resistant tumors seems warranted. However, it is of critical importance that, as is observed for HDAC inhibitors, OV-enhancing effects remain tumor-selective.
Conclusion
While genetic mutations are believed to be essential initiators of carcinogenesis, it is clear that epigenetic deregulation plays a key role in augmenting and/or maintaining the tumor phenotype. OVs are promising biotherapeutics that among others take advantage of the epigenetic silencing of cellular antiviral response genes and in many ways unmask cancer antigens as they destroy cancer cells and promote an inflammatory response. While additional studies on the impact of epigenetic regulation on the antiviral and immunological responses are needed, it is already recognized from studies using HDAC inhibitors that epigenetic modulators can positively impact OV efficacy. Additional in vitro and in vivo studies evaluating the effect of other epigenetic modulators are needed to determine whether these could be used in combination with promising OV platforms anticipated to reach the clinic in the near future, to further improve their therapeutic impact.
Conflict of Interest Statement
John C. Bell is CSO and co-founder of Jennerex Biotherapeutics.
Acknowledgment
This work was supported by grants from the Terry Fox Research Institute (Jean-Simon Diallo and John C. Bell).
References
Akhtar-Zaidi, B., Cowper-Sal-Lari, R., Corradin, O., Saiakhova, A., Bartels, C. F., Balasubramanian, D., et al. (2012). Epigenomic enhancer profiling defines a signature of colon cancer. Science 336, 736–739. doi: 10.1126/science.1217277
Alvarez-Breckenridge, C. A., Yu, J., Price, R., Wei, M., Wang, Y., Nowicki, M. O., et al. (2012). The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 86, 4566–4577. doi: 10.1128/JVI.05545-11
Asada, T. (1974). Treatment of human cancer with mumps virus. Cancer 34, 1907–1928. doi:10.1002/1097-0142(197412)34:6<1907::AID-CNCR2820340609>3.0.CO;2-4
Bae, S. I., Cheriyath, V., Jacobs, B. S., Reu, F. J., and Borden, E. C. (2008). Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene 27, 490–498.
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Bateman, A., Bullough, F., Murphy, S., Emiliusen, L., Lavillette, D., Cosset, F. L., et al. (2000). Fusogenic membrane glycoproteins as a novel class of genes for the local and immune-mediated control of tumor growth. Cancer Res. 60, 1492–1497.
Bateman, A. R., Harrington, K. J., Kottke, T., Ahmed, A., Melcher, A. A., Gough, M. J., et al. (2002). Viral fusogenic membrane glycoproteins kill solid tumor cells by non-apoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res. 62, 6566–6578.
Baylin, S. B., and Jones, P. A. (2011). A decade of exploring the cancer epigenome – biological and translational implications. Nat. Rev. Cancer 11, 726–734.
Bennani-Baiti, I. M., Machado, I., Llombart-Bosch, A., and Kovar, H. (2012). Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum. Pathol. 43, 1300#x02013;1307.
Borden, E. C. (2007). Augmentation of effects of interferon-stimulated genes by reversal of epigenetic silencing: potential application to melanoma. Cytokine Growth Factor Rev. 18, 491–501. doi: 10.1016/j.cytogfr.2007.06.022
Borden, E. C., Sen, G. C., Uze, G., Silverman, R. H., Ransohoff, R. M., Foster, G. R., et al. (2007). Interferons at age 50: past, current, and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990. doi: 10.1038/nrd2422
Breitbach, C. J., Burke, J., Jonker, D., Stephenson, J., Haas, A. R., Chow, L. Q., et al. (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477, 99–102. doi: 10.1038/nature10358
Bridle, B. W., Chen, L., Lemay, C. G., Diallo, J.-S., Pol, J., Nguyen, A., et al. (2013). HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol. Ther. 21, 887–894. doi: 10.1038/mt.2012.265
Bridle, B. W., Stephenson, K. B., Boudreau, J. E., Koshy, S., Kazdhan, N., Pullenayegum, E., et al. (2010). Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 18, 1430–1439. doi: 10.1038/mt.2010.98
Burton, T. R., Henson, E. S., Azad, M. B., Brown, M., Eisenstat, D. D., and Gibson, S. B. (2013). BNIP3 acts as transcriptional repressor of death receptor-5 expression and prevents TRAIL-induced cell death in gliomas. Cell Death Dis. 4, e587. doi: 10.1038/cddis.2013.100
Carroll, J. (2011). Amgen bets $1B on BioVex’s dual-mechanism cancer therapy. Fierce Biotech. Available at: http://www.fiercebiotech.com/story/amgen-bets-1b-biovexs-dual-mechanism-cancer-therapy/2011-01-25 (accessed April 3, 2013).
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. doi: 10.1038/nrg2540
Chai, L., Liu, S., Mao, Q., Wang, D., Li, X., Zheng, X., et al. (2012). A novel conditionally replicating adenoviral vector with dual expression of IL-24 and arresten inserted in E1 and the region between E4 and fiber for improved melanoma therapy. Cancer Gene Ther. 19, 247–254. doi: 10.1038/cgt.2011.84
Chang, H. M., Paulson, M., Holko, M., Rice, C. M., Williams, B. R., Marie, I., et al. (2004). Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc. Natl. Acad. Sci. U.S.A. 101, 9578–9583. doi: 10.1073/pnas.0400567101
Chen, Q., Denard, B., Huang, H., and Ye, J. (2013). Epigenetic silencing of antiviral genes renders clones of Huh-7 cells permissive for hepatitis C virus replication. J. Virol. 87, 659–665. doi: 10.1128/jvi.01984-12
Cho, H., Proll, S. C., Szretter, K. J., Katze, M. G., Gale, M., and Diamond, M. S. (2013). Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med.
Chou, S. D. (2005). Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int. Immunol. 17, 1483–1494. doi: 10.1093/intimm/dxh326
De Carvalho, D. D., Sharma, S., You, J. S., Su, S. F., Taberlay, P. C., Kelly, T. K., et al. (2012). DNA methylation screening identifies driver epigenetic events of cancer cell. Cancer Cell 21, 655–667. doi: 10.1016/j.ccr.2012.03.045
Diallo, J. S., Le Boeuf, F., Lai, F., Cox, J., Vaha-Koskela, M., Abdelbary, H., et al. (2010). A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol. Ther. 18, 1123–1129. doi: 10.1038/mt.2010.67
Diaz, R. M., Galivo, F., Kottke, T., Wongthida, P., Qiao, J., Thompson, J., et al. (2007). Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 67, 2840–2848. doi: 10.1158/0008-5472.can-06-3974
Dock, G. (1904). The influence of complicating diseases upon leukemia. Am. J. Med. Sci. 127, 563–592.
Dunn, G. P., Koebel, C. M., and Schreiber, R. D. (2006). Interferons, immunity, and cancer immunoediting. Nat. Rev. Immunol. 6, 836–848. doi: 10.1038/nri1961
Dunn, G. P., Sheehan, K. C., Old, L. J., and Schreiber, R. D. (2005). IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 65, 3447–3453. doi: 10.1158/0008-5472.can-04-4316
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463. doi: 10.1038/nature02625
Fang, T. C., Schaefer, U., Mecklenbrauker, I., Stienen, A., Dewell, S., Chen, M. S., et al. (2012). Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 209, 661–669. doi: 10.1084/jem.20112343
Farassati, F., Yang, A. D., and Lee, P. W. (2001). Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 3, 745–750. doi: 10.1038/35087061
Fridman, A. L., Tang, L., Kulaeva, O. I., Ye, B., Li, Q., Nahhas, F., et al. (2006). Expression profiling identifies three pathways altered in cellular immortalization: interferon, cell cycle, and cytoskeleton. J. Gerontol. A Biol. Sci. Med. Sci. 61, 879–889. doi: 10.1093/gerona/61.9.879
Fritz, V., and Fajas, L. (2010). Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 29, 4369–4377. doi: 10.1038/onc.2010.182
Galanis, E., Markovic, S. N., Suman, V. J., Nuovo, G. J., Vile, R. G., Kottke, T. J., et al. (2012). Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol. Ther. 20, 1998–2003. doi: 10.1038/mt.2012.146
Génin, P., Morin, P., and Civas, A. (2003). Impairment of interferon-induced IRF-7 gene expression due to inhibition of ISGF3 formation by trichostatin A. J. Virol. 77, 7113–7119. doi: 10.1128/JVI.77.12.7113-7119.2003
Glasspool, R. M., Teodoridis, J. M., and Brown, R. (2006). Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 94, 1087–1092. doi: 10.1038/sj.bjc.6603024
Gomez, J. A., Wapinski, O. L., Yang, Y. W., Bureau, J.-F., Gopinath, S., Monack, D. M., et al. (2013). The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 152, 743–754. doi: 10.1016/j.cell.2013.01.015
Hacker, S., Dittrich, A., Mohr, A., Schweitzer, T., Rutkowski, S., Krauss, J., et al. (2009). Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene 28, 3097–3110. doi: 10.1038/onc.2009.161
Hatziapostolou, M., and Iliopoulos, D. (2011). Epigenetic aberrations during oncogenesis. Cell. Mol. Life. Sci. 68, 1681–1702. doi: 10.1007/s00018-010-0624-z
Heo, J., Reid, T., Ruo, L., Breitbach, C. J., Rose, S., Bloomston, M., et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 19, 329–336. doi: 10.1038/nm.3089
Holling, T. M., Bergevoet, M. W., Wilson, L., Van Eggermond, M. C., Schooten, E., Steenbergen, R. D., et al. (2007). A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J. Immunol. 179, 5317–5325.
Hoti, N., Chowdhury, W., Hsieh, J. T., Sachs, M. D., Lupold, S. E., and Rodriguez, R. (2006). Valproic acid, a histone deacetylase inhibitor, is an antagonist for oncolytic adenoviral gene therapy. Mol. Ther. 14, 768–778.
Huang, P. Y., Guo, J. H., and Hwang, L. H. (2012). Oncolytic Sindbis virus targets tumors defective in the interferon response and induces significant bystander antitumor immunity in vivo. Mol. Ther. 20, 298–305. doi: 10.1038/mt.2011.245
Hurtubise, A., Bernstein, M. L., and Momparler, R. L. (2008). Preclinical evaluation of the antineoplastic action of 5-aza-2′-deoxycytidine and different histone deacetylase inhibitors on human Ewing’s sarcoma cells. Cancer Cell Int. 8, 16. doi: 10.1186/1475-2867-8-16
Jain, R. K. (2005). Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62. doi: 10.1126/science.1104819
Janson, P. C., Marits, P., Thorn, M., Ohlsson, R., and Winqvist, O. (2008). CpG methylation of the IFNG gene as a mechanism to induce immunosuppression [correction of immunosupression] in tumor-infiltrating lymphocytes. J. Immunol. 181, 2878–2886.
Johnsen, A. K., Templeton, D. J., Sy, M., and Harding, C. V. (1999). Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J. Immunol. 163, 4224–4231.
Johnsson, A., Durand-Dubief, M., Xue-Franzen, Y., Ronnerblad, M., Ekwall, K., and Wright, A. (2009). HAT-HDAC interplay modulates global histone H3K14 acetylation in gene-coding. EMBO Rep. 10, 1009–1014.
Kanaseki, T., Ikeda, H., Takamura, Y., Toyota, M., Hirohashi, Y., Tokino, T., et al. (2003). Histone deacetylation, but not hypermethylation, modifies class II transactivator and MHC class II gene expression in squamous cell carcinomas. J. Immunol. 170, 4980–4985.
Karpf, A. R., Peterson, P. W., Rawlins, J. T., Dalley, B. K., Yang, Q., Albertson, H., et al. (1999). Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc. Natl. Acad. Sci. U.S.A. 96, 14007–14012.
Katsura, T., Iwai, S., Ota, Y., Shimizu, H., Ikuta, K., and Yura, Y. (2009). The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 16, 237–245. doi: 10.1038/cgt.2008.81
Kelly, E., and Russell, S. J. (2007). History of oncolytic viruses: genesis to genetic engineering. Mol. Ther. 15, 651–659. doi: 10.1038/sj.mt.6300108
Khan, A. N., Magner, W., and Tomasi, T. (2004). An epigenetically altered tumor cell vaccine. Cancer Immunol. Immunother. 53, 748–754. doi: 10.1007/s00262-004-0513-0
Khan, A. N. H., Magner, W. J., and Tomasi, T. B. (2007). An epigenetic vaccine model active in the prevention and treatment of melanoma. J. Transl. Med. 5, 64. doi: 10.1186/1479-5876-5-64
Klampfer, L., Huang, J., Swaby, L. A., and Augenlicht, L. (2004). Requirement of histone deacetylase activity for signaling by STAT1. J. Biol. Chem. 279, 30358–30368. doi: 10.1074/jbc.M401359200
Krecmerova, M., and Otmar, M. (2012). 5-azacytosine compounds in medicinal chemistry: current stage and future perspectives. Future Med. Chem. 4, 991–1005. doi: 10.4155/fmc.12.36
Kulaeva, O. I., Draghici, S., Tang, L., Kraniak, J. M., Land, S. J., and Tainsky, M. A. (2003). Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene 22, 4118–4127. doi: 10.1038/sj.onc.1206594
Lange, S., Lampe, J., Bossow, S., Zimmermann, M., Neubert, W., Bitzer, M., et al. (2013). A novel armed oncolytic measles vaccine virus for the treatment of cholangiocarcinoma. Hum. Gene Ther. 24, 554–564. doi: 10.1089/hum.2012.136
Lee, J., Goh, S.-H., Song, N., Hwang, J.-A., Nam, S., Choi, I. J., et al. (2012). Overexpression of IFITM1 has clinicopathologic effects on gastric cancer and is regulated by an epigenetic mechanism. Am. J. Pathol. 181, 43–52. doi: 10.1016/j.ajpath.2012.03.027
Lee, K. Y., Geng, H., Ng, K. M., Yu, J., Van Hasselt, A., Cao, Y., et al. (2008). Epigenetic disruption of interferon-γ response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal, and multiple other carcinomas. Oncogene 27, 5267–5276. doi: 10.1038/onc.2008.147
LeibundGut-Landmann, S., Waldburger, J.-M., Krawczyk, M., Otten, L. A., Suter, T., Fontana, A., et al. (2004). Mini-review: specificity and expression of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol. 34, 1513–1525. doi: 10.1002/eji.200424964
Lemay, C. G., Rintoul, J. L., Kus, A., Paterson, J. M., Garcia, V., Falls, T. J., et al. (2012). Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 20, 1791–1799. doi: 10.1038/mt.2012.128
Li, K., Markosyan, R. M., Zheng, Y. M., Golfetto, O., Bungart, B., Li, M., et al. (2013). IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 9:e1003124. doi: 10.1371/journal.ppat.1003124
Li, Q., Tang, L., Roberts, P. C., Kraniak, J. M., Fridman, A. L., Kulaeva, O. I., et al. (2008). Interferon regulatory factors IRF5 and IRF7 inhibit growth and induce senescence in immortal Li-Fraumeni fibroblasts. Mol. Cancer Res. 6, 770–784. doi: 10.1158/1541-7786.mcr-07-0114
Liikanen, I., Monsurro, V., Ahtiainen, L., Raki, M., Hakkarainen, T., Diaconu, I., et al. (2011). Induction of interferon pathways mediates in vivo resistance to oncolytic adenovirus. Mol. Ther. 19, 1858–1866. doi: 10.1038/mt.2011.144
Londhe, P., Zhu, B., Abraham, J., Keller, C., and Davie, J. (2012). CIITA is silenced by epigenetic mechanisms that prevent the recruitment of transactivating factors in rhabdomyosarcoma cells. Int. J. Cancer 131, E437–E448. doi: 10.1002/ijc.26478
Lubieniecka, J. M., De Bruijn, D. R., Su, L., Van Dijk, A. H., Subramanian, S., Van De Rijn, M., et al. (2008). Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Cancer Res. 68, 4303–4310. doi: 10.1158/0008-5472.can-08-0092
Lund, P., Kotova, I., Kedinger, V., Khanwalkar, H., Voltz, E., Hahn, W. C., et al. (2011). Transformation-dependent silencing of tumor-selective apoptosis-inducing TRAIL by DNA hypermethylation is antagonized by decitabine. Mol. Cancer Ther. 10, 1611–1623. doi: 10.1158/1535-7163.MCT-11-0140
MacTavish, H., Diallo, J. S., Huang, B., Stanford, M., Le Boeuf, F., De Silva, N., et al. (2011). Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors. PLoS ONE 5:e14462. doi: 10.1371/journal.pone.0014462
Magnani, L., Eeckhoute, J., and Lupien, M. (2011). Pioneer factors: directing transcriptional regulators within the chromatin environment. Trends Genet. 27, 465–474. doi: 10.1016/j.tig.2011.07.002
Magnani, L., Stoeck, A., Zhang, X., Lanczky, A., Mirabella, A. C., Wang, T. L., et al. (2013). Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 110, E1490–E1499. doi: 10.1073/pnas.1219992110
Maldonado, A. R., Klanke, C., Jegga, A. G., Aronow, B. J., Mahller, Y. Y., Cripe, T. P., et al. (2010). Molecular engineering and validation of an oncolytic herpes simplex virus type 1 transcriptionally targeted to midkine-positive tumors. J. Gene Med. 12, 613–623. doi: 10.1002/jgm.1479
Meissner, M., Whiteside, T. L., Van Kuik-Romein, P., Valesky, E. M., Van Den Elsen, P. J., Kaufmann, R., et al. (2008). Loss of interferon-γ inducibility of the MHC class II antigen processing pathway in head and neck cancer: evidence for post-transcriptional as well as epigenetic regulation. Br. J. Dermatol. 158, 930–940. doi: 10.1111/j.1365-2133.2008.08465.x
Merrick, A. E., Ilett, E. J., and Melcher, A. A. (2009). JX-594, a targeted oncolytic poxvirus for the treatment of cancer. Curr. Opin. Investig. Drugs 10, 1372–1382.
Morimoto, Y., Toyota, M., Satoh, A., Murai, M., Mita, H., Suzuki, H., et al. (2004). Inactivation of class II transactivator by DNA methylation and histone deacetylation associated with absence of HLA-DR induction by interferon-γ in haematopoietic tumour cells. Br. J. Cancer |90, 844–852.
Morris, A. C., Spangler, W. E., and Boss, J. M. (2000). Methylation of class II trans-activator promoter IV: a novel mechanism of MHC class II gene control. J. Immunol. 164, 4143–4149.
Muhlethaler-Mottet, A., Di Berardino, W., Otten, L. A., and Mach, B. (1998). Activation of the MHC class II transactivator CIITA by interferon-gamma requires cooperative interaction between Stat1 and USF-1. Immunity8, 157–166.
Muntean, A. G., and Hess, J. L. (2009). Epigenetic dysregulation in cancer. Am. J. Pathol. 175, 1353–1361. doi: 10.2353/ajpath.2009.081142
Naka, K., Abe, K., Takemoto, K., Dansako, H., Ikeda, M., Shimotohno, K., et al. (2006). Epigenetic silencing of interferon-inducible genes is implicated in interferon resistance of hepatitis C virus replicon-harboring cells. J. Hepatol. 44, 869–878. doi: 10.1016/j.jhep.2006.01.030
Nguyen, T. L., Abdelbary, H., Arguello, M., Breitbach, C., Leveille, S., Diallo, J. S., et al. (2008). Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc. Natl. Acad. Sci. U.S.A. 105, 14981–14986. doi: 10.1073/pnas.0803988105
Norman, K., and Lee, P. (2000). Reovirus as a novel oncolytic agent. J. Clin. Invest. 105, 1035–1038. doi: 10.1172/JCI9871
Nusinzon, I., and Horvath, C. M. (2003). Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. U.S.A. 100, 14742–14747. doi: 10.1073/pnas.2433987100
Okemoto, K., Wagner, B. P., Haseley, A. M., Price, R. L., Kaur, B., and Chiocca, A. E. (2012). 5-Azacitydine augments the anti-glioma efficacy of Herpes-based oncolytic virotherapy. AACR 103rd Annual Meeting 2012: Cancer Research, Chicago.
Otsuki, A., Patel, A., Kasai, K., Suzuki, M., Kurozumi, K., Antonio Chiocca, E., et al. (2008). Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 16, 1546–1555. doi: 10.1038/mt.2008.155
Parato, K. A., Senger, D., Forsyth, P. A., and Bell, J. C. (2005). Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 5, 965–976. doi: 10.1038/nrc1750
Patel, N., Black, J., Chen, X., Marcondes, A. M., Grady, W. M., Lawlor, E. R., et al. (2012). DNA methylation and gene expression profiling of ewing sarcoma primary tumors reveal genes that are potential targets of epigenetic inactivation. Sarcoma 2012, 498472. doi: 10.1155/2012/498472
Peserico, A., and Simone, C. (2010). Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 371832.
Pol, J., Le Boeuf, F., and Diallo, J. S. (2013). [Genetic, immunological, and pharmacological strategies to generate improved oncolytic viruses]. Med. Sci. (Paris) 29, 165–173.
Pulido, J., Kottke, T., Thompson, J., Galivo, F., Wongthida, P., Diaz, R. M., et al. (2012). Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat. Biotechnol. 30, 337–343. doi: 10.1038/nbt.2157
Radosevich, M., Jager, M., and Ono, S. J. (2007). Inhibition of MHC class II gene expression in uveal melanoma cells is due to methylation of the CIITA gene or an upstream activator. Exp. Mol. Pathol. 82, 68–76. doi: 10.1016/j.yexmp.2006.03.005
Reu, F. J., Bae, S. I., Cherkassky, L., Leaman, D. W., Lindner, D., Beaulieu, N., et al. (2006a). Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J. Clin. Oncol. 24, 3771–3779. doi: 10.1200/jco.2005.03.4074
Reu, F. J., Leaman, D. W., Maitra, R. R., Bae, S. I., Cherkassky, L., Fox, M. W., et al. (2006b). Expression of RASSF1A, an epigenetically silenced tumor suppressor, overcomes resistance to apoptosis induction by interferons. Cancer Res. 66, 2785–2793. doi: 10.1158/0008-5472.can-05-2303
Rice, J. C., and Allis, C. D. (2001). Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr. Opin. Cell Biol. 13, 263–273. doi: 10.1016/S0955-0674(00)00208-8
Russell, S. J., Peng, K. W., and Bell, J. C. (2012). Oncolytic virotherapy. Nat. Biotechnol. 30, 658–670. doi: 10.1038/nbt.2287
Sakamoto, S., Potla, R., and Larner, A. C. (2004). Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes. J. Biol. Chem. 279, 40362–40367. doi: 10.1074/jbc.M406400200
Satoh, A., Toyota, M., Ikeda, H., Morimoto, Y., Akino, K., Mita, H., et al. (2004). Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-γ-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene 23, 8876–8886. doi: 10.1038/sj.onc.1208144
Schenk, T., Chen, W. C., Gollner, S., Howell, L., Jin, L., Hebestreit, K., et al. (2012). Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611. doi: 10.1038/nm.2661
Schildhaus, H.-U., Riegel, R., Hartmann, W., Steiner, S., Wardelmann, E., Merkelbach-Bruse, S., et al. (2011). Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum. Pathol. 42, 1667–1675. doi: 10.1016/j.humpath.2010.12.025
Setiadi, A. F., David, M. D., Seipp, R. P., Hartikainen, J. A., Gopaul, R., and Jefferies, W. A. (2007). Epigenetic control of the immune escape mechanisms in malignant carcinomas. Mol. Cell. Biol. 27, 7886–7894. doi: 10.1128/mcb.01547-07
Shmulevitz, M., Marcato, P., and Lee, P. W. (2005). Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene 24, 7720–7728. doi: 10.1038/sj.onc.1209041
Sobol, P. T., Boudreau, J. E., Stephenson, K., Wan, Y., Lichty, B. D., and Mossman, K. L. (2011). Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol. Ther. 19, 335–344. doi: 10.1038/mt.2010.264
Soncini, M., Santoro, F., Gutierrez, A., Frige, G., Romanenghi, M., Botrugno, O. A., et al. (2013). The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim. Biophys. Acta 1832, 114–120. doi: 10.1016/j.bbadis.2012.10.001
Stephenson, K. B., Barra, N. G., Davies, E., Ashkar, A. A., and Lichty, B. D. (2012). Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 19, 238–246. doi: 10.1038/cgt.2011.81
Stojdl, D. F., Lichty, B., Knowles, S., Marius, R., Atkins, H., Sonenberg, N., et al. (2000). Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6, 821–825. doi: 10.1038/77558
Stojdl, D. F., Lichty, B. D., Tenoever, B. R., Paterson, J. M., Power, A. T., Knowles, S., et al. (2003). VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4, 263–275. doi: 10.1016/S1535-6108(03)00241-1
Suh, H.-S., Choi, S., Khattar, P., Choi, N., and Lee, S. C. (2010). Histone deacetylase inhibitors suppress the expression of inflammatory and innate immune response genes in human microglia and astrocytes. J. Neuroimmune Pharmacol. 5, 521–532. doi: 10.1007/s11481-010-9192-0
Suva, M. L., Riggi, N., and Bernstein, B. E. (2013). Epigenetic reprogramming in cancer. Science 339, 1567–1570. doi: 10.1126/science.1230184
Taylor, B. S., Decarolis, P. L., Angeles, C. V., Brenet, F., Schultz, N., Antonescu, C. R., et al. (2011). Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov. 1, 587–597. doi: 10.1158/2159-8290.cd-11-0181
Truax, A. D., Thakkar, M., and Greer, S. F. (2012). Dysregulated recruitment of the histone methyltransferase EZH2 to the class II transactivator (CIITA) promoter IV in breast cancer cells. PLoS ONE 7:e36013. doi: 10.1371/journal.pone.0036013
Vigneau, S., Levillayer, F., Crespeau, H., Cattolico, L., Caudron, B., Bihl, F., et al. (2001). Homology between a 173-kb region from mouse chromosome 10, telomeric to the Ifng locus, and human chromosome 12q15. Genomics 78, 206–213. doi: 10.1006/geno.2001.6656
Watanabe, T., Hioki, M., Fujiwara, T., Nishizaki, M., Kagawa, S., Taki, M., et al. (2006). Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase-specific replication-selective adenoviral agent OBP-301 in human lung cancer cells. Exp. Cell Res. 312, 256–265.
U.S. National Library of Medicine (2013). ClinicalTrials.gov [Online]. Available at: www.clinicaltrials.gov [accessed January 29, 2013].
Yamashita, M., Toyota, M., Suzuki, H., Nojima, M., Yamamoto, E., Kamimae, S., et al. (2010). DNA methylation of interferon regulatory factors in gastric cancer and non-cancerous gastric mucosae. Cancer Sci. 101, 1708–1716. doi: 10.1111/j.1349-7006.2010.01581.x
Yoo, C. B., and Jones, P. A. (2006). Epigenetic therapy of cancer: past, present, and future. Nat. Rev. Drug Discov. 5, 37–50. doi: 10.1038/nrd1930
Keywords: oncolytic virotherapy, epigenetic modulation, cancer, tumor heterogeneity, anti-viral response, antigen presentation
Citation: Forbes NE, Abdelbary H, Lupien M, Bell JC, and Diallo J-S (2013) Exploiting tumor epigenetics to improve oncolytic virotherapy. Front. Genet. 4:184. doi: 10.3389/fgene.2013.00184
Received: 03 May 2013; Paper pending published: 18 June 2013;
Accepted: 29 August 2013; Published online: 20 September 2013.
Edited by:
Alejandro García Carrancá, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México and Instituto Nacional de Cancerología, Secretaría de Salud, MexicoReviewed by:
Amancio Carnero, Instituto de Biomedicina de Sevilla, SpainAlejandro García Carrancá, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México and Instituto Nacional de Cancerología, Secretaría de Salud, Mexico
Copyright © 2013 Forbes, Abdelbary, Lupien, Bell and Diallo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John C. Bell and Jean-Simon Diallo, Center for Innovative Cancer Research, Ottawa Hospital Research Institute, 501 Smyth Road, Ottawa, ON K1H 8L6, Canada e-mail:amJlbGxAb2hyaS5jYQ==;anNkaWFsbG9AZ21haWwuY29t