 Martine Therrien
Martine Therrien J. Alex Parker
J. Alex Parker- 1Départment de Pathologie et Biologie Cellulaire, CRCHUM-Centre Hospitalier de l'Université de Montréal, Université de Montréal, Montréal, QC, Canada
- 2Départment de Pathologie et Biologie Cellulaire, Départment de Neurosciences, CRCHUM-Centre Hospitalier de l'Université de Montréal, Université de Montréal, Montréal, QC, Canada
Neurodegenerative diseases share pathogenic mechanisms at the cellular level including protein misfolding, excitotoxicity and altered RNA homeostasis among others. Recent advances have shown that the genetic causes underlying these pathologies overlap, hinting at the existence of a genetic network for neurodegeneration. This is perhaps best illustrated by the recent discoveries of causative mutations for amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD). Once thought to be distinct entities, it is now recognized that these diseases exist along a genetic spectrum. With this wealth of discoveries comes the need to develop new genetic models of ALS and FTD to investigate not only pathogenic mechanisms linked to causative mutations, but to uncover potential genetic interactions that may point to new therapeutic targets. Given the conservation of many disease genes across evolution, Caenorhabditis elegans is an ideal system to investigate genetic interactions amongst these genes. Here we review the use of C. elegans to model ALS and investigate a putative genetic network for ALS/FTD that may extend to other neurological disorders.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting 1–2/100,000 individuals. Most cases of ALS are sporadic, but 10% of cases are familial (Turner et al., 2013b). Mutations in the gene superoxide dismutase 1 (SOD1) were identified in 1993 (Rosen et al., 1993) as the first cause of familial ALS. Thanks to the recent advances in genetics, more than 20 genes are now linked to ALS (Chen et al., 2013) (Table 1). Genes recently shown to be mutated in ALS include the DNA/RNA binding proteins TAR DNA binding protein 43 (TARDBP) and Fused-in-sarcoma (FUS) (Kabashi et al., 2008; Sreedharan et al., 2008; Vance et al., 2009). More recently, mutations in C9ORF72 have turned out to be a major cause of familial and sporadic ALS (DeJesus-Hernandez et al., 2011; Renton et al., 2011).

Table 1. ALS genes and their C. elegans orthologs.
ALS is characterized by the selective loss of motor neurons in the motor cortex, the brainstem and the spinal cord, the loss of myelin in the spinal cord, and the presence of neuroinflammation (Robberecht and Philips, 2013). Onset of the disease usually begins in the lower limb and spreads toward the upper motor neurons leading to muscle weakness, fasciculation, and wasting. Death occurs 3–5 years after the beginning of the symptoms (Kiernan et al., 2011) and is caused by respiratory failure due to denervation of the respiratory muscles.
50% of ALS patients show cognitive impairment, of which 15% met the criteria of frontotemporal dementia (FTD) (Ringholz et al., 2005). FTD is a group of non-Alzheimer dementias characterized by atrophy of the frontal and/or temporal lobes causing mid-life behavioral changes or language impairment (Warren et al., 2013). Over the past few years, the identification of TDP-43, C9ORF72 and UBQLN2 as genes causing ALS and FTD has suggested a similarity for both diseases (Morris et al., 2012). Similar pathogenic mechanisms have been suggested for ALS and FTD (Van Langenhove et al., 2012; Ling et al., 2013) but so far it is unclear how patients with the same genetic mutations can have either ALS, FTD or both.
The genes involved in ALS have diverse functions and we still do not know how they interact to cause motor neuron degeneration. Most of the research over the past 20 years has focused on the toxicity caused by mutant SOD1. Among the proposed mechanisms of toxicity are mitochondrial dysfunction, axonal dysfunction, excitotoxicity and neuroinflammation (Turner et al., 2013b). However, TDP-43, FUS, and C9ORF72 proteins seem to point toward RNA toxicity (Ling et al., 2013). Most importantly, only one drug, riluzole, is used to slow disease progression and has only modest effects (Kiernan et al., 2011). Diagnosis is difficult and requires an experienced neurologist to differentiate between ALS and other neurological diseases (Turner et al., 2013a). It is thought that the clinical manifestations of ALS are downstream events that occur much later after the initial insult to the nervous system (Turner et al., 2013a). Therefore, the identification of biomarkers is essential for the rapid, early diagnosis of ALS, and the identification of new drugs limiting the degeneration of motor neuron is an essential unmet need for ALS patients.
To understand better the impact of the genetic mutations on the function of the different proteins involved in ALS, in vivo models have proved to be essential. Ever since the first SOD1 mouse was developed in 1994 (Gurney et al., 1994), several groups have tried to investigate ALS pathogenesis by expressing different ALS related mutations in mice, an approach that has recently been extended to other genes including TDP-43 and FUS for example. While the over expression of wild type SOD1 causes a mild denervation of neurons (Epstein et al., 1987), the over expression of SOD1G93A causes a loss of motor neurons, neuroinflammation, and reduces life span (Gurney et al., 1994; Guo et al., 2009). One model expressing mutant TDP-43Q331K or M337V in the mouse central nervous system has shown selectivity for large caliber motor neuron neurodegeneration (Arnold et al., 2013), while others over expressing mutant TDP-43G348C, A315T and FUSR512C, 14Δ have limited neuronal loss (Swarup et al., 2011; Verbeeck et al., 2012). Some rodent models display relevant ALS pathology, but given the time and expense to develop models for many of the recently discovered ALS genes, not to mention the difficulty of manipulating several genes at once, some laboratories have turned to simpler organisms to model ALS toxicity.
One model showing increasing popularity is the nematode Caenorhabditis elegans. This 1 mm long worm has a painstakingly characterized, invariant cell lineage that includes 302 neurons. The nervous system, its interconnections and its synapses are well studied which makes it an ideal model to study mechanisms of neuronal toxicity. The C. elegans genome was the first to be fully sequenced in 1998 and includes more than 19,000 genes on 6 chromosomes (C. elegans Sequencing Consortium, 1998). Since then, deletion mutants have been produced for many C. elegans genes and approximately 80% of C. elegans genes have a human homolog (Lai et al., 2000) (Shaye and Greenwald, 2011). C. elegans behavior is well studied and many experimental assays are available, including for worm locomotion. Worms initiate movement by bending their body to advance forward in a sinusoidal pattern, a process that is orchestrated by GABAergic and cholinergic neurons. Cholinergic neurons initiate the contraction along the dorsal or ventral body wall muscles while the GABAergic neurons send an inhibitory signal on the opposite side (Jorgensen, 2005).
C. elegans has been an important tool for the characterization of many neurodegenerative disorders (Li and Le, 2013). Many protein-misfolding disorders have been modeled in worms including Alzheimer's disease, Parkinson's disease, Huntington's disease and different spinocerebellar ataxias. Also, the toxicity of non-coding mutations in C. elegans resemble the toxicity in mammalian tissues (Wang et al., 2011b). Since many cellular stress and survival pathways are conserved in worms, our group and others have used C. elegans to model ALS. This review aims to summarize the work done modeling ALS in C. elegans and highlights the future possibilities and applications.
Use of C. elegans to Model SOD1 Toxicity
SOD1 is an enzyme that catalyzes the conversion of O2- into O2 and H2O2. More than 160 mutations causative for ALS have been found in SOD1 since 1993 (Al-Chalabi et al., 2012). Phenotypic heterogeneity is observed among SOD1 mutation carriers where SOD1A4V seems to cause an aggressive form of ALS while SOD1D90A causes a milder, long duration ALS (Renton et al., 2014). It is hypothesized that SOD1 mutations cause toxicity through a gain of function, even though a loss of enzyme activity have been observed in patients and some models (Saccon et al., 2013). Many pathogenic mechanisms have been hypothesized but no consensus has been reached, although it is thought that the misfolding of mutant SOD1, and sometimes wild type SOD1, may be an important first step of the pathogenesis observed in patients (Pickles and Vande Velde, 2012). Based on pathological evidence, it is now acknowledged that ALS caused by mutations in SOD1 is a distinctive form of ALS (Mackenzie et al., 2007).
Several groups have used C. elegans to model SOD1 toxicity (Table 2) starting with Oeda and colleagues who showed that the ubiquitous expression of human mutant SOD1 impairs the worm's response to oxidative stress and causes protein aggregates (Oeda et al., 2001). It was later shown that expression of mutant SOD1 throughout the worm's entire nervous system resulted in locomotion defects and impaired neuronal transmission (Wang et al., 2009). Interestingly, the formation of aggregates seemed to be restricted to certain mechanosensory neurons despite the pan neuronal expression of SOD1. Other models are non-neuronal in nature and have relied on the expression of SOD1 proteins in the body wall muscles where it was observed that distinct SOD1 mutations have varying propensities to aggregate (Gidalevitz et al., 2009). More recently a C. elegans model was generated based on the expression of SOD1 in the worm's motor neurons showing neurodegeneration in the absence of caspases (Li et al., 2013a), an intriguing finding since the motor neuron loss observed in mouse models is associated with caspase activation (Pasinelli et al., 2000). Whether this reflects a difference between invertebrate and vertebrate systems, or reflects a novel mechanism of neurodegeneration remains to be determined.
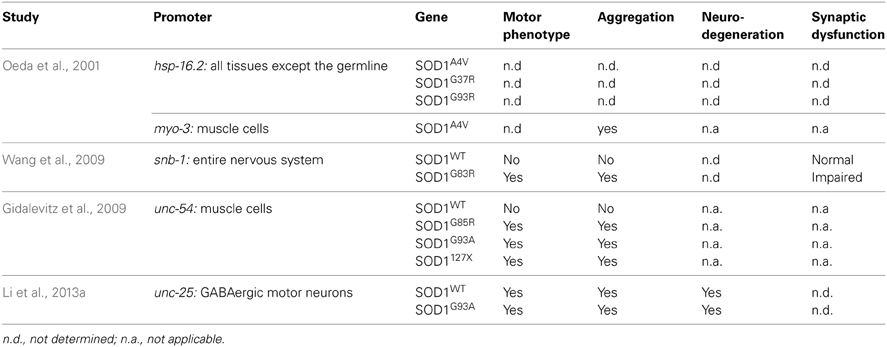
Table 2. Summary of transgenic SOD1 models.
The C. elegans sod-1 gene has a similar function to human SOD1. sod-1 loss of function mutants have increased O2- levels, shorter lifespan and are sensitive to some environmental stresses (Yanase et al., 2009). Inversely, overexpression of the worm sod-1 increases lifespan and increases the level of H202, the by-product of the catalase reaction of SOD1. However, the increased lifespan seems to be independent of SOD-1 catalase activity, but may be due to altered endoplasmic reticulum (ER) stress signaling (Cabreiro et al., 2011). Interestingly, Van Raamsdonk et al. have generated a sod null worm, where all five C. elegans sod genes have been mutated and these worms have a normal lifespan and response to oxidative damage but are sensitive to many acute environmental stresses (Van Raamsdonk and Hekimi, 2012).
In summary, many aspects of SOD1 function and toxicity are conserved in worms, but some questions remain. It is known that mutant SOD1 is found in association with the mitochondria in SOD1 mouse model and ALS patients (Pickles and Vande Velde, 2012). To our knowledge, no group has yet investigated the effects of human mutant SOD1 in worm mitochondria. However, it was recently shown that a cleavage product of vpr-1, the ortholog of VAPB also involved in ALS, affects mitochondrial organization in muscle cells (Han et al., 2013). A similar analysis of the different SOD1 transgenic models would be interesting and could help identify pathways and drugs that act specifically on this important aspect of ALS pathogenesis.
Use of C. elegans to Model TDP-43 Toxicity
TDP-43 is a protein encoded by the TARDBP gene on chromosome 1. The protein contains two RNA binding domains, a glycine rich domain and nuclear export and import signals. TDP-43 is similar to the members of the ribonucleoprotein family. TDP-43 was identified in 2006 as the main constituents of sporadic and familial ALS/FTD aggregates (Neumann et al., 2006). In patients, the ubiquitinated aggregates are present in the most affected regions of the brain and spinal cord. These aggregates contain a hyperphosphorylated form of TDP-43 and the C terminus cleaved fragment (Neumann et al., 2006). In 2008, mutations in the TARDBP gene were linked to familial and sporadic ALS/FTD cases (Kabashi et al., 2008; Sreedharan et al., 2008; Sreedharan and Brown, 2013). So far, more than 40 mutations in TARDBP have been linked to ALS/FTD and most of them are found in the C terminus region of the protein, a region involved in protein-protein interactions (Al-Chalabi et al., 2012).
Under normal cellular conditions, TDP-43 protein shuttles from the nucleus to the cytoplasm. The normal function of TDP-43 is still unclear but the protein participates in transcription, miRNA processing, mRNA splicing, RNA transport and stress granule formation (Ling et al., 2013). The pathogenic effect of the mutant proteins is not well understood and it is still unclear if the toxicity is a gain of function, a loss of function, or both (Ling et al., 2013; Vanden Broeck et al., 2014). An important aspect of TDP-43 toxicity was discovered when characterizing TDP-43 wild type mice. Mice with elevated expression of wild type TDP-43 also have characteristics of TDP-43 mutant proteins (Xu et al., 2010). Therefore, expression level is important and should be considered when generating different transgenic models.
To clarify the toxicity caused by the expression of mutant TDP-43, several groups have developed C. elegans models (Table 3). In 2010, Ash and colleagues developed the first TDP-43 overexpression model in C. elegans. The pan neuronal expression of human TDP-43 and C. elegans TDP-1 resulted in worms with uncoordinated, slow movements and defasciculation of the GABAergic motor neurons (Ash et al., 2010). The results regarding the expression of human TDP-43 were confirmed by Liachko and colleagues who also observed motility defects and degeneration phenotypes from the expression of mutant TDP-43 proteins throughout the worms nervous system (Liachko et al., 2010). These phenotypes also highly correlated with protein phosphorylation levels where hyperphosphorylation increased the toxicity of mutant TDP-43 proteins similarly to what is observed in ALS patients (Liachko et al., 2010). The TDP-43 C terminus fragment shows another similarity with patients. Zhang and colleagues showed that the pan neuronal expression of human TDP-43 C′ fragment caused a phenotype similar to the expression of wild type or mutant TDP-43 (Zhang et al., 2011). Even though no neuronal loss was observed in the latter model, all strains displayed synaptic transmission abnormalities. In worms, GABAergic neurons seem to be particularly sensitive to the expression of TDP-43 (Liachko et al., 2010). To evaluate if the effect of TDP-43 expression in these neurons could cause locomotor defects, our group developed models in which human wild type or mutant TDP-43 were expressed specifically in GABAergic motor neurons as directed by an unc-47 promoter (McIntire et al., 1997). Interestingly, the overexpression of mutant TDP-43, but not wild type TDP-43, caused an adult-onset, progressive paralysis phenotype accompanied by GABAergic neurodegeneration and synaptic transmission impairment (Vaccaro et al., 2012b). Finally, some of these models showed aggravation of the phenotypes during aging recapitulating an important feature of ALS and neurodegeneration (Liachko et al., 2010; Vaccaro et al., 2012b).
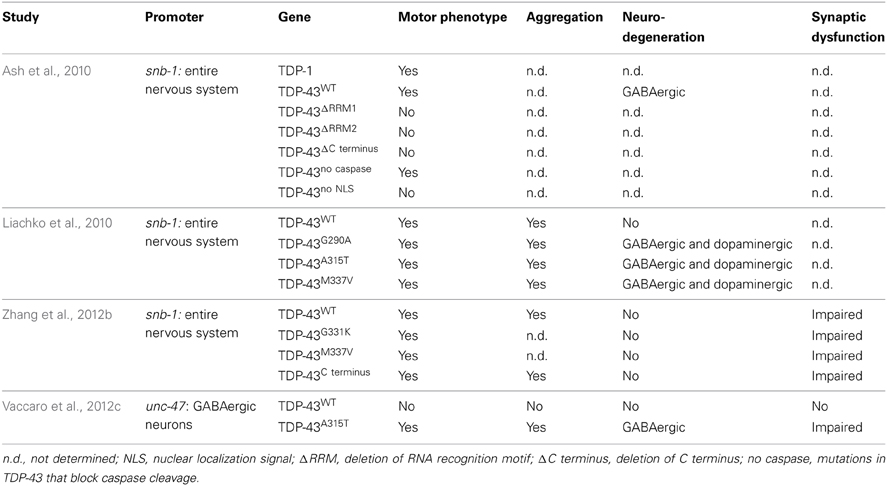
Table 3. Summary of transgenic TDP-43 models.
It is still unclear if the toxicity of mutant TDP-43 proteins in ALS patients arises from a gain of function, a loss of function or if both mechanisms are employed. The transgenic C. elegans models of TDP-43 are based on the overexpression of TDP-43 in worms and likely represent a gain of function rather than a loss of function. However, the C. elegans genome has an ortholog of TDP-43 called TDP-1. TDP-1 is a primarily nuclear protein expressed in most tissues including body wall muscles, pharynx and neurons (Vaccaro et al., 2012c; Zhang et al., 2012b). TDP-1 contains two RNA binding motifs, a nuclear localization signal and an export signal but lacks the glycine rich domain found in human TDP-43. TDP-1 seems to be functionally conserved because the expression of human TDP-43 can rescue the toxicity of a loss of function of a tdp-1 mutant (Zhang et al., 2012b).
Mutant tdp-1 animals show numerous phenotypes including slow development, and locomotion defects (Liachko et al., 2010; Zhang et al., 2012b). TDP-1 was also shown to be involved in lifespan and the cellular stress response. Somewhat paradoxically, worms lacking tdp-1 have a longer lifespan but are more sensitive to oxidative and osmotic stresses (Vaccaro et al., 2012c; Zhang et al., 2012b). The expression of tdp-1 can be induced by oxidative stress, either chemically or from activation of the ER stress response, and it is thought that chronic induction of tdp-1 by stress is ultimately cytotoxic and reduces the worms lifespan (Vaccaro et al., 2012c). Furthermore, several studies have shown that wild type TDP-1 protein may contribute to the neurodegeneration elicited by mutant protein in C. elegans. Neurodegeneration was suppressed by deleting tdp-1 from worms in several ALS models (Vaccaro et al., 2012c; Zhang et al., 2012b) as well as in a C. elegans model of Huntington's disease (Tauffenberger et al., 2013a) suggesting there may be genetic interactions amongst genes linked to neurodegeneration. Interestingly, a transcriptome analysis of tdp-1(ok803) showed that one of the biological process that was highly affected in the mutant worms was the ER unfolded protein response (Zhang et al., 2012b). ER stress and proteostasis have been a recurrent theme in ALS research (Matus et al., 2013; Musarò, 2013) which is of interest since sporadic and familial cases of ALS are known to have an abnormal ER stress response (Ilieva et al., 2007; Atkin et al., 2008; Hetz et al., 2009; Ito et al., 2009).
Use of C. elegans to Model FUS Toxicity
After the identification of TDP-43, several groups examined related RNA-binding proteins for their potential contributions to ALS. In 2009, a protein with a similar function, FUS, was identified as causative of ALS (Kwiatkowski et al., 2009; Vance et al., 2009). Similar to TDP-43, FUS contains a RNA binding domain and a glycine rich domain but also has a two arginine glycine rich regions and one large glutamine, glycine, serine, tyrosine domain in N terminus. Because of their high degree of structural similarity, it was hypothesized that FUS and TDP-43 share common functions. It is known that FUS can bind DNA and RNA and is involved in many of the same RNA processing activities of TDP-43 (Ling et al., 2013). FUS transgenic models are relatively recent additions to the research field and much remains to be learned about the function of FUS and the implication of the mutant protein in neurodegeneration.
Two transgenic models have been developed in C. elegans for FUS (Table 4). Murakami et al. (2012) expressed several FUS mutations and two truncated FUS proteins throughout the worm's nervous system. Interestingly, only the mutations that caused aggregation resulted in motor phenotypes in worms. The motor phenotype could not be rescued by the expression of wild type FUS suggesting a gain of function mechanism. Our group confirmed a similar toxicity mechanism in models expressing FUS in the worm motor neurons. Expression of FUS wild type did not cause aggregation but expression of mutant FUS caused aggregation accompanied by paralysis, neuronal synaptic impairment and neurodegeneration (Vaccaro et al., 2012b).
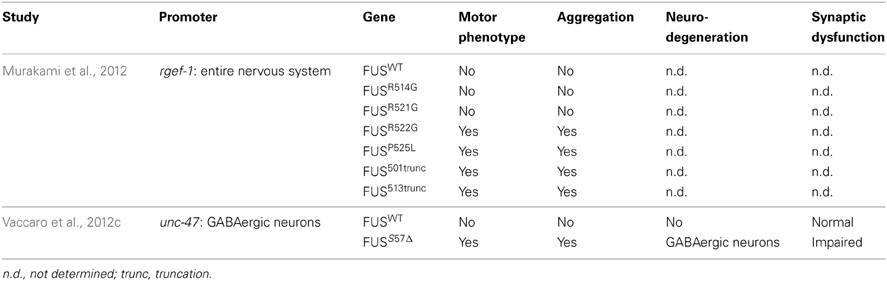
Table 4. Summary of transgenic FUS models.
FUS is well conserved and the C. elegans ortholog is named fust-1. In contrast to tdp-1, a fust-1 deletion mutant could not alleviate the toxicity induced by the expression of C′ TDP-43 fragment (Zhang et al., 2012b), suggesting a different role in proteotoxicity. In Drosophila, Cabeza (Caz), the Drosophila ortholog of FUS, is expressed in motor neurons and a decreased expression of Caz causes a motor phenotype and motor neuron degeneration (Wang et al., 2011a; Sasayama et al., 2012). These results suggest a link between the expression and function of FUS, and the specificity of ALS neurodegeneration and we await further investigations of fust-1 in C. elegans.
Use of C. elegans to Model C9ORF72 Toxicity
A region of chromosome 9 had been linked to ALS for several years (Morita et al., 2006; Vance et al., 2006; van Es et al., 2009; Shatunov et al., 2010) but the gene was only identified in 2011 (DeJesus-Hernandez et al., 2011; Renton et al., 2011) and has since been shown to be a major cause of sporadic and familial ALS (Turner et al., 2013b). The basis of the mutation is a GGGGCC repeat expansion within the first intron of C9ORF72. Many questions remain to be answered about the role of C9ORF72 in the pathogenesis of ALS. It is still not clear whether the GGGGCC repeat expansion results in a loss of function, a gain of function or both, or if the size of the repeat has differential effects on these potential mechanisms. Recent reports have observed decreased expression of C9ORF72 when the GGGGCC repeat reaches pathogenic length (DeJesus-Hernandez et al., 2011; Ciura et al., 2013; Xi et al., 2013). Whether decreased expression contributes to ALS pathogenesis is unknown since very little is known about the biological role of C9ORF72 other than its sequence similarity to the GDP/GTP exchange factor “Differentially Expressed in Normal and Neoplasia” (DENN) (Zhang et al., 2012a; Levine et al., 2013). DENN proteins are involved in the regulation of Rab-GTPases and endocytosis. Recently, C9ORF72 was shown to be implicated in endosomal trafficking (Farg et al., 2014), confirming its role as a DENN protein. In C. elegans, work has been previously done regarding some Rab proteins using deletion mutants and GFP reporters (Sato et al., 2008) to investigate endocytosis (Fares and Grant, 2002). C. elegans would be an ideal model to confirm the involvement of C9ORF72 in this pathway. The C. elegans homolog of C9ORF72 is named alfa-1 (ALS/FTD associated gene homolog). Our group characterized the loss of function mutant alfa-1(ok3062) where we observed that decreased expression of alfa-1 causes a motility defect, neurodegeneration specifically of the motor neurons and sensitivity to osmotic stress (Therrien et al., 2013). Further characterization still remains to be done but it is interesting that loss of alfa-1 is linked to neuronal integrity specifically for GABAergic motor neurons in worm.
GGGGCC repeat expansions are found in the first intron of C9ORF72 and the presence of such long non-coding repeats is suggestive of a toxic gain of function mechanism driving neurodegeneration as seen in many of the trinucleotide repeat expansion diseases. In patients, the repeat was shown to induce abnormal translation (non-ATG translation of the repeat, also called RAN translation) leading to the production of different dipeptides(Ash et al., 2013; Mori et al., 2013b). Also of interest were the presence of RNA foci containing the expanded GGGGCC repeat in patients (DeJesus-Hernandez et al., 2011). It is unknown whether a toxic gain of function is caused by the presence of toxic RNA or the presence of toxic protein, or both. So far, no groups have generated transgenic worms to model this aspect of the toxicity however the expression of the non-coding GGGGCC repeat in Drosophila causes neurodegeneration (Xu et al., 2013). C. elegans may be useful to model non-coding repeats based on previous efforts studying the expression of non-coding CUG repeats that were toxic to worms (Chen et al., 2007) and recapitulated aspects of RNA foci toxicity (Wang et al., 2011b).
Stress Response and Age-Dependent Neurodegeneration in C. elegans
With the identification of TDP-43 in most ALS aggregates and later the identification of mutations affecting TARDBP and FUS genes, RNA metabolism has become an important area of investigation in ALS research. Under normal conditions, both proteins are mainly observed in the nucleus but the mutant proteins are also found in the cytoplasm. FUS and TDP-43 contain a low-complexity prion-like domain and a RNA binding domain suggesting a role in RNA metabolism (Li et al., 2013b). High throughput RNA-sequencing experiments have been used to identify targets of TDP-43 and FUS in normal or disease states. In worms, the transcriptome of the tdp-1(ok803) mutant has been studied (Zhang et al., 2012b) and showed the involvement of TDP-1 in various aspects of development.
Under cellular stress, wild type and mutant TDP-43 and FUS proteins form RNA granules (Bosco et al., 2010; Dormann et al., 2010; Liu-Yesucevitz et al., 2010; Gal et al., 2011; McDonald et al., 2011). These granules are usually formed in order to protect RNA from degradation under stress conditions. In worms, a variety of different RNA granules exist: P granules, P bodies and stress granules. P granules are the most characterized RNA granules in worms and are highly involved in cellular development (Updike and Strome, 2010). However, human proteins found in P bodies and stress granules, such as TIA1, the decapping enzymes and polyA binding proteins, have C. elegans ortholog and their role seem conserved regarding RNA granules (Jud et al., 2008; Sun et al., 2011). An active area of research concerns whether mutant TDP-43 and/or FUS proteins interfere with stress granule homeostasis. In a transgenic model of FUS, wild type and mutant FUS were shown to colocalize to stress granules after a heat shock but only the recruitment of mutant FUS to the stress granules caused persistent motility defects in the worms (Murakami et al., 2012). Most work done in C. elegans to study stress granules have used thermal stress as an inducer of the granule (Sun et al., 2011). In cells, formation of granules containing FUS is also initiated by other environmental stresses such as osmotic stress (Baron et al., 2013) and oxidative stress (Vance et al., 2013), thus the effect of these other stresses would be interesting to evaluate. Since most of the components of the granules are conserved in worms, C. elegans could be a powerful system to investigate stress biology in the context of aging, an aspect not easily studied in cellular systems.
Since TDP-43 and FUS are components of stress granules, this has led to the hypothesis that both proteins may be involved in the cellular stress response. The genetic pathways governing cellular stress signaling have been studied to great success in C. elegans. The different stress response pathways are highly characterized in worms with the insulin/IGF-1 pathway being a major, conserved signaling axis (Lau and Chalasani, 2014). In worms, the insulin/IGF-1 pathway has a sole insulin/IGF-1 receptor, DAF-2, that acts through the kinases AGE-1, PDK and AKT to phosphorylate the FOXO transcription factor DAF-16, and regulate stress resistance and longevity (Lapierre and Hansen, 2012). The most common stresses applied to worms in laboratory settings include exposure to thermal, oxidative, osmotic or hypoxic stresses (Rodriguez et al., 2013). While each is a damaging stress, they can elicit distinct genetic signaling pathways with diverse outcomes. An open question in the field of late-onset neurodegeneration is whether environmental components exist to account for the range in disease onset and progression for what are many highly penetrant, monogenic, dominantly acting disorders. A stress intrinsic to ALS and many neurodegenerative diseases is proteotoxicity. Here mutant proteins misfold leading to a diverse range of proteotoxic consequences. Thus, cells maintain an extensive network of mechanisms, including the insulin/IGF-1 pathway, to maintain protein homeostasis in the face of environmentally derived damage or genetically encoded misfolded proteins.
Work from C. elegans directly linked tdp-1 to the insulin/IGF-1 pathway and proteotoxicity. In C. elegans tdp-1 is required for the stress resistance of daf-2 mutants and the stress-induced expression of tdp-1 was dependent on daf-16. These data suggest a role for TDP-1/TDP-43 in the insulin/IGF-1 pathway and it remains to see if insulin/IGF-1 signaling is altered by disease-associated TDP-43 mutations.
Interestingly, in Vaccaro et al., mutant TDP-43 and mutant FUS proteins were only expressed in the 26 GABAergic motor neurons but activated the ER unfolded protein response chaperone HSP-4 in intestinal tissue (Vaccaro et al., 2012c). This observation suggests that proteotoxic insults can induce stress-signaling pathways in other tissues. It is not known if this is due to a diffusible signaling molecule, or if the mutant proteins make their way from the nervous system to adjacent tissue. TDP-43, FUS, HNRNPA1, HNRNAP2B and TAF15 all contain a prion-like domain (Couthouis et al., 2011; Polymenidou and Cleveland, 2011; Kim et al., 2013) and misfolded SOD1 protein may be able to self propagate (Grad and Cashman, 2014). Thus, these proteins could share properties with toxic prion protein (PrPsc) that misfolds, become infectious, and spreads from cell to cell (Kabir and Safar, 2014). The development of ALS symptoms, starting usually in the lower limb and spreading upward, also suggests a propagation mechanism. Little is known about the propagation potential of the ALS associated misfolded proteins in C. elegans transgenic models. Mutant TDP-43 and FUS proteins in the worm's motor neurons were shown to induce the expression of HSP-4 in the intestine, but the proteins were not visualized outside of the neurons where they were expressed (Vaccaro et al., 2012c). A prion model was however characterized expressing Sup35NM, a yeast prion protein, in the body wall muscle of the worm. The most toxic form of the protein was shown between muscle cells, in the intestine and the coelomocytes, and the toxic fibrils were able to induce protein misfolding (Nussbaum-Krammer et al., 2013). Also, proteostasis, ER stress resistance and longevity, all major ALS research topics, have been recently shown to have important cell-non-autonomous components (Taylor and Dillin, 2013; van Oosten-Hawle et al., 2013). Since C. elegans is transparent, direct visualization of tagged proteins during development and aging is possible. The development of additional tools should help establish if a propagation mechanism exists for mutant TDP-43, FUS and SOD1 proteins.
Identification of Genetic Interactions
Recent genetic advances have identified many new causative genes for familial cases of ALS (Table 1). Moreover, genome-wide association studies (GWAS) have also been done in sporadic ALS cohorts to identify potential genes (Renton et al., 2014). With the increasing number of genes linked to ALS along with the diverse functions of these genes, it is essential to identify common pathological pathways relevant to ALS. Genetic interactions amongst genes can refer to functional relationships amongst a group of genes (Boucher and Jenna, 2013). However, genetic interactions are not always easy to interpret and do not necessarily point toward genes that function in the same pathway but rather identify functional similarity between genes that could be in the same pathway or in compensatory pathways (Boucher and Jenna, 2013). Therefore, identification of genetic interactions between ALS genes could point toward potential therapeutic avenues for ALS patients (Figure 1).
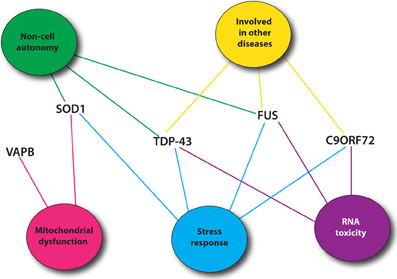
Figure 1. Most common ALS genes and pathogenic pathways. Shown are mutant proteins associated with ALS and putative, shared toxicity mechanisms including RNA toxicity, the cellular stress response, mitochondrial impairment and cell autonomy/non-autonomy. Many of these pathways can be easily investigated using C. elegans.
Among the proteins identified, TDP-43, SOD1, FUS, OPTN, UBQLN2, and NEFH proteins are found in familial and sporadic ALS inclusions (Al-Chalabi et al., 2012). In zebrafish and Drosophila, FUS, and TDP-43 were shown to interact genetically together but independently of SOD1 (Kabashi et al., 2011; Lanson et al., 2011; Wang et al., 2011a). The rapid development of phenotypes and the availability of multiple mutants or RNAi clones make C. elegans an expedient model to study genetic interactions. In worms, TDP-1 was shown to participate in the neurotoxicity observed in motor neuron caused by human TDP-43 and human FUS (Vaccaro et al., 2012c). However, FUS and TDP-43 seem to interact differently with PGRN, a gene involved in FTD, and C9ORF72/ALFA-1 (Tauffenberger et al., 2013a; Therrien et al., 2013). Those results provide an interesting start to the identification of common pathological pathways in ALS. Finally, the characterization of the loss of function mutant of pgrn-1, the C. elegans ortholog of progranulin, showed that PGRN-1 is involved in apoptotic cell clearance (Kao et al., 2011). Understanding how pgrn-1 interacts with the different genes involved in ALS/FTD could help to better understand the variation observed along ALS/FTD continuum (Mackenzie et al., 2011).
At a broader level, screening for potential genetic modifiers using RNAi has brought a new understanding of the pathogenesis. For example, SOD1 aggregation was linked to motor dysfunction in worms, but upon decreasing the expression of chaperone proteins, the worms exhibited larger aggregates and increased locomotion deficits (Wang et al., 2009). A non-biased screening approach has recently demonstrated that targeting proteins that affect the phosphorylation levels of TDP-43 greatly affects its neuronal toxicity, setting the stage for novel therapeutic approaches (Liachko et al., 2013). Thus far, only a handful of genetic screens have been described for C. elegans ALS models but future studies may help uncover pathogenic mechanisms and therapeutic strategies.
TDP-43 aggregates were shown to be the main protein found in non-SOD1 ALS cases (Neumann et al., 2006). However, the presence of TDP-43 aggregates is not exclusive to ALS. TDP-43 aggregates are observed in other neurodegenerative diseases such as Huntington's disease, Parkinson's disease, Alzheimer's disease, and FTD (Mackenzie et al., 2010). FUS is also found in the aggregates of polyglutamine disorders (Woulfe et al., 2010) and mutations in FUS were linked to essential tremor (Merner et al., 2012). Recently, FIG4 and VCP were also identified in different neurodegenerative aggregates (Mori et al., 2013a; Kon et al., 2014). Uncovering specific genetic interactions that involve these proteins could also help our understanding of their recruitment to the aggregates of so many neurodegenerative disorders. Using other models, groups have shown that intermediate polyglutamine repeat of ATXN2 gene and EPHA4 are potent modulators of ALS toxicity (Elden et al., 2010; Van Hoecke et al., 2012). Therefore, a genetic interaction map may extend the role of these genes beyond ALS and perhaps into other neurodegenerative disorders.
Using C. elegans Model for ALS Drug Discovery
The small size of C. elegans, its rapid life cycle, its ease of cultivation and ability to obtain large numbers of animals makes it an attractive model for drug discovery. Furthermore, worms can be grown on solid media or in liquid culture, the latter being relatively easy to adapt for drug screening purposes often through use of multiwell plates and/or automated screening methods (O'Reilly et al., 2013). The transparency of C. elegans makes it an ideal model for neurodegeneration applications since protein aggregation and neuronal morphology can be easily assayed as a complement to behavioral phenotypes.
Boyd and colleagues have shown that drugs identified from cell based systems often have relevance in C. elegans (Boyd et al., 2013). A screen to identify compounds that decrease TDP-43 aggregation was performed in cell lines and many of the molecules identified were able to suppress the impaired motility phenotype of worms expressing mutant human TDP-43 (Boyd et al., 2013).
Our group has also developed a high-throughput drug-screening assay. We observed that the paralysis phenotype that typically manifests over 5–12 days on solid media can be observed after just hours when the worms are placed in liquid culture (Vaccaro et al., 2012b; Therrien et al., 2013). Using this technique, more than 4000 FDA approved compounds were screened in our laboratory. From this screen, we identified a number of molecules including methylene blue and others acting on the ER stress response that decrease the toxicity of TDP-43 (Vaccaro et al., 2012a, 2013). Interestingly, these drugs were also confirmed in zebrafish ALS models confirming that these compounds can be effective across species. These compounds are therefore promising leads for testing in mammalian models.
Even though in the disease state, aging and neurodegeneration seem to go hand in hand, we have shown that the drugs that act on neurodegeneration can be separated from those that broadly affect lifespan (Tauffenberger et al., 2013b) suggesting that lifespan extension is not a strong predictor of neuroprotection.
Other Motor Neuron Diseases
ALS is part of the neurological group of disorders called motor neuron diseases. This group also includes spinal muscular atrophy (SMA), primary lateral sclerosis (PLS), hereditary spastic paraplegia (HSP) and many others affecting the upper and/or lower motor neurons. The causative genes of these diseases are involved in many cellular functions, however they all share a common toxic pathways since they mainly affect motor neurons. Finding similarities and differences among those diseases could highly increase our understanding of motor neuron toxicity. C. elegans has been used to study two of these, SMA and HSP.
HSP is a group of disorders affecting mainly the lower motor neurons. More than 40 loci have been linked to HSP and the genes identify are involved in axon pathfinding and myelination, mitochondrial maintenance and membrane trafficking (Blackstone, 2012). Recently, a large network including many of these genes have been identify and this network is highly similar to Parkinson's, ALS and Alzheimer's diseases (Novarino et al., 2014). Using C. elegans, the function and toxicity of two HSP genes have been investigated. First, spas-1, the C. elegans ortholog of spastin, also called SPG4, was shown to be involved in the development of microtubules. SPAS-1 is expressed in the cytoskeleton and is involved specifically in the disassembly of microtubules (Matsushita-Ishiodori et al., 2007). Then, the pan neuronal expression of NIPA-1 associated mutations led to motor deficits and shortened the lifespan of transgenic worms probably through the activation of caspases and increased ER toxicity (Zhao et al., 2008). With the rapid discovery of new HSP genes, more models are surely to come and will help unravel similarities between these diseases.
SMA is a rare autosomal recessive disorder and a leading genetic cause of infant death. All genetic causes of SMA lead to a decreased expression of the proteins survival of motor neuron (SMN) 1 and 2 (Arnold and Burghes, 2013). It mainly affects the lower motor neurons, but recent evidences suggest that it can be a systemic disease affecting the vascular, cardiac and hepatic functions as well as affecting bone formation (Hamilton and Gillingwater, 2013). C. elegans possesses one ortholog of the SMN gene, smn-1. In 1999, Miguel-Aliaga and colleagues showed that decreased expression of smn-1 in worms resulted in severe locomotion defects and sterility (Miguel-Aliaga et al., 1999). Then SMN-1 was shown to interact with SMI-1, a known interactor of SMN in humans (Burt et al., 2006). Briese and colleagues characterized the first smn-1 deletion mutant observing that the mutants displayed early developmental arrest, which could be rescued by reintroducing expression of smn-1 in the nervous system, while expression in muscle cells was ineffective (Briese et al., 2009). Little is known about any downstream targets of SMN and no drugs are available. Thus, several groups have used C. elegans to identify modifiers of the smn-1 phenotypes. In a cross-species study, it was shown that proteins involved in endocytosis and mRNA regulation could modify the toxicity (Dimitriadi et al., 2010). Also, knowing that the ubiquitin-proteasome pathway degrades SMN, decreased expression of Mibl, an E3 ligase, was shown to ameliorate smn-1 phenotypes (Kwon et al., 2013). Since the smn-1 deletion allele ok355 is an early larval lethal phenotype, to aid the development of drug screening Sleigh and colleagues identified a less severe mutant allele that more closely resembles the severity of SMA (Sleigh et al., 2011). Using this mutant, they identified several small molecules that alleviate smn-1 phenotypes of the worms, therefore, being highly promising compounds for SMA drug development (Sleigh et al., 2011).
Perspectives
With the discovery of many new ALS genes comes the need to better understand their functions, expression patterns and their modes of toxicity. C. elegans has proven to be an informative model to study neurodegeneration mechanisms arising from multiple ALS related proteins. We envision that the introduction of new transgenic and genetic models will help unravel important questions about the normal and pathogenic roles of these proteins.
Most models explained here recapitulate some if not many, important features of ALS, however, phenotypic variations are seen amongst the different models, for a number of reasons. First, the models do not all use the same mutations, thus the resulting mutant proteins may not all be equally toxic, or display the same interactions with other proteins. Also, the level of expression is important to consider as for example, there is considerable evidence that TDP-43 levels are tightly regulated (Budini and Buratti, 2011), and elevated expression is toxic in nearly every system studied (Ash et al., 2010; Xu et al., 2010; Estes et al., 2011). The most common method to generate transgenic worms is by microinjection to create stable lines followed by radiation to integrate the transgene in the genome. This procedure typically produces transgenics with multiple copies of the gene inserted in the genome, thus some of the toxicity observed may be due to overexpression. Aware of this issue, a new generation of ALS transgenic worms should be constructed based on single copy integration (Frøkjaer-Jensen et al., 2008) or with the CRISPR-Cas9 method (Friedland et al., 2013) instead to ensure that transgenic lines have a similar level of expression from the same genomic location. Finally, the phenotypic variance may also be due to the promoter used. Some models have used pan neuronal expression constructs, while others have targeted transgenic expression to specific neuronal populations. In humans, most of these proteins are expressed ubiquitously but only specific neuronal populations are sensitive to degeneration. Thus, worm models based on motor neuron transgenics could be ideal model to uncover conserved mechanisms of motor neuron degeneration. To confirm the specificity of each phenotype, mutant and wild type proteins should be carefully compared and similar changes should be confirmed in higher eukaryotes. For example, mutant TDP-43 and FUS proteins induce an ER stress response in worms which is not observed when the wild type proteins are expressed (Vaccaro et al., 2012b). Also, the ER stress response was shown to be activated in other ALS models and in patients (see section above).
These models are setting the stage for novel toxicity hypothesis. The immune system seems to play an important role in the neurodegeneration observed in ALS. Protein aggregation could activate the immune response and neuroinflammation actively contributes to disease progression (McCombe and Henderson, 2011). C. elegans relies on an evolutionary conserved, innate immune response (Engelmann and Pujol, 2010) that coordinates its activity with the insulin/IGF-1 pathway (Singh and Aballay, 2009) suggesting these may be pathways worth investigating. Also, in the past year, a convergence of data has suggested a role for glial cells in ALS neurodegeneration (Parisi et al., 2013; Valori et al., 2013; Chiu et al., 2014). The worm has 56 glial cells and some are found at the neuromuscular junction (Oikonomou and Shaham, 2011). Characterization of the cross talk between the neurons and the glial cells would also be an interesting area of investigation.
An important topic related to ALS and to other neurodegenerative disorders is aging. The risk of ALS increases with age, peaking between 70 and 80 years old (Gordon, 2013). Aging pathways are well characterized in worms and among others, include the insulin/IGF-1, the target of rapamycin (TOR) and germline signaling pathways. There is a strong overlap between protetotoxicity and aging where autophagy and lipid metabolism are major targets (Lapierre and Hansen, 2012). Evaluating the toxic impact of mutant proteins during aging is not feasible in many models, but is easily accomplished using C. elegans. The development of models with age-related toxicity is essential and could help understand the link between the proteotoxicity and aging.
When using C. elegans or other animal models, most studies have focused on the toxicity of known ALS genes. It is important to note that almost 90% of ALS cases are sporadic ALS with no link to known genetic abnormalities. Therefore, we still do not know how most patients develop ALS. However, it is important to know that sporadic and familial cases of ALS are clinically indistinguishable (Al-Chalabi and Hardiman, 2013). Given that ALS patients can live between 6 months and 6 years after diagnosis, it has been hypothesized that environmental factors may influence disease onset and progression (Al-Chalabi and Hardiman, 2013). Many environmental factors have been examined in relation to ALS but there is no consensus for their contribution to the disease (Al-Chalabi and Hardiman, 2013). C. elegans could be useful to study some of the environmental risks hypothesized. In fact, several groups have identified compounds that could cause specific degeneration of motor neurons (Du and Wang, 2009; Negga et al., 2012; Estevez et al., 2014) opening the door to identifying environmental modifiers of degeneration in ALS models.
However, how relevant are any of these findings to humans? Will any of the drugs identified in C. elegans translate to mammalian models let alone ALS patients? So far, many drugs identified using rodent models focusing mainly on protein aggregation and cell death mechanisms have failed in subsequent clinical trials. Using C. elegans to identify drugs acting on early neuronal dysfunction mechanisms could be an effective way to prevent ensuing cellular decline and death. From a liquid culture screen, our group has identified a compound with this property (unpublished results). The compound is effective in vertebrate ALS animal models and is now being tested in ALS patients. Therefore, large screens using C. elegans targeting specific early aspects of neurodegeneration seem promising and show relevance in higher organisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding from ALS Canada, the Canadian Institutes of Health Research, the Congressionally Directed Medical Research Program (CDMRP) Amyotrophic Lateral Sclerosis Research Program (USA), and the Muscular Dystrophy Association (USA) supported this work.
References
Al-Chalabi, A., and Hardiman, O. (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617–628. doi: 10.1038/nrneurol.2013.203
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and van den Berg, L. H. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352. doi: 10.1007/s00401-012-1022-4
Arnold, E. S., Ling, S.-C., Huelga, S. C., Lagier-Tourenne, C., Polymenidou, M., Ditsworth, D., et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. U.S.A. 110, E736–E745. doi: 10.1073/pnas.1222809110
Arnold, W. D., and Burghes, A. H. M. (2013). Spinal muscular atrophy: development and implementation of potential treatments. Ann. Neurol. 74, 348–362. doi: 10.1002/ana.23995
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W.-L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Ash, P. E. A., Zhang, Y. J., Roberts, C. M., Saldi, T., Hutter, H., Buratti, E., et al. (2010). Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 19, 3206–3218. doi: 10.1093/hmg/ddq230
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Baron, D. M., Kaushansky, L. J., Ward, C. L., Sama, R. R. K., Chian, R.-J., Boggio, K. J., et al. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 8:30. doi: 10.1186/1750-1326-8-30
Blackstone, C. (2012). Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 35, 25–47. doi: 10.1146/annurev-neuro-062111-150400
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175. doi: 10.1093/hmg/ddq335
Boucher, B., and Jenna, S. (2013). Genetic interaction networks: better understand to better predict. Front. Genet. 4:290. doi: 10.3389/fgene.2013.00290
Boyd, J. D., Lee-Armandt, J. P., Feiler, M. S., Zauur, N., Liu, M., Kraemer, B., et al. (2013). A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J. Biomol. Screen. 19, 44–56. doi: 10.1177/1087057113501553
Briese, M., Esmaeili, B., Fraboulet, S., Burt, E. C., Christodoulou, S., Towers, P. R., et al. (2009). Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum. Mol. Genet. 18, 97–104. doi: 10.1093/hmg/ddn320
Budini, M., and Buratti, E. (2011). TDP-43 autoregulation: implications for disease. J. Mol. Neurosci. 45, 473–479. doi: 10.1007/s12031-011-9573-8
Burt, E. C., Towers, P. R., and Sattelle, D. B. (2006). Caenorhabditis elegans in the study of SMN-interacting proteins: a role for SMI-1, an orthologue of human Gemin2 and the identification of novel components of the SMN complex. Invert. Neurosci. 6, 145–159. doi: 10.1007/s10158-006-0027-x
C. elegans Sequencing Consortium. (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. doi: 10.1126/science.282.5396.2012
Cabreiro, F., Ackerman, D., Doonan, R., Araiz, C., Back, P., Papp, D., et al. (2011). Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic. Biol. Med. 51, 1575–1582. doi: 10.1016/j.freeradbiomed.2011.07.020
Chen, K.-Y., Pan, H., Lin, M.-J., Li, Y.-Y., Wang, L.-C., Wu, Y.-C., et al. (2007). Length-dependent toxicity of untranslated CUG repeats on Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 352, 774–779. doi: 10.1016/j.bbrc.2006.11.102
Chen, S., Sayana, P., Zhang, X., and Le, W. (2013). Genetics of amyotrophic lateral sclerosis: an update. Mol. Neurodegener. 8:28. doi: 10.1186/1750-1326-8-28
Chiu, H., Alqadah, A., and Chang, C. (2014). The role of microRNAs in regulating neuronal connectivity. Front. Cell. Neurosci. 7:283. doi: 10.3389/fncel.2013.00283
Ciura, S., Lattante, S., Le Ber, I., Latouche, M., Tostivint, H., Brice, A., et al. (2013). Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol. 74, 180–187. doi: 10.1002/ana.23946
Couthouis, J., Hart, M. P., Shorter, J., DeJesus-Hernandez, M., Erion, R., Oristano, R., et al. (2011). A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci.U.S.A. 108, 20881–20890. doi: 10.1073/pnas.1109434108
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dimitriadi, M., Sleigh, J. N., Walker, A., Chang, H. C., Sen, A., Kalloo, G., et al. (2010). Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet. 6:e1001172. doi: 10.1371/journal.pgen.1001172
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
Du, M., and Wang, D. (2009). The neurotoxic effects of heavy metal exposure on GABAergic nervous system in nematode Caenorhabditis elegans. Environ. Toxicol. Pharmacol. 27, 314–320. doi: 10.1016/j.etap.2008.11.011
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Engelmann, I., and Pujol, N. (2010). Innate immunity in C. elegans. Adv. Exp. Med. Biol. 708, 105–121. doi: 10.1007/978-1-4419-8059-5_6
Epstein, C. J., Avraham, K. B., Lovett, M., Smith, S., Elroy-Stein, O., Rotman, G., et al. (1987). Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 84, 8044–8048. doi: 10.1073/pnas.84.22.8044
Estes, P. S., Boehringer, A., Zwick, R., Tang, J. E., Grigsby, B., and Zarnescu, D. C. (2011). Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321. doi: 10.1093/hmg/ddr124
Estevez, A. O., Morgan, K. L., Szewczyk, N. J., Gems, D., and Estevez, M. (2014). The neurodegenerative effects of selenium are inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like growth factor signaling in Caenorhabditis elegans. Neurotoxicology 41, 28–43. doi: 10.1016/j.neuro.2013.12.012
Fares, H., and Grant, B. (2002). Deciphering endocytosis in Caenorhabditis elegans. Traffic 3, 11–19. doi: 10.1034/j.1600-0854.2002.30103.x
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A. K., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. doi: 10.1093/hmg/ddu068. [Epub ahead of print].
Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., and Calarco, J. A. (2013). Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Meth. 10, 741–743. doi: 10.1038/nmeth.2532
Frøkjaer-Jensen, C., Davis, M. W., Hopkins, C. E., Newman, B. J., Thummel, J. M., Olesen, S.-P., et al. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375–1383. doi: 10.1038/ng.248
Gal, J., Zhang, J., Kwinter, D. M., Zhai, J., Jia, H., Jia, J., et al. (2011). Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 32, 2323.e27–e40. doi: 10.1016/j.neurobiolaging.2010.06.010
Gidalevitz, T., Krupinski, T., Garcia, S., and Morimoto, R. I. (2009). Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 5:e1000399. doi: 10.1371/journal.pgen.1000399
Gordon, P. H. (2013). Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 4, 295–310. doi: 10.14336/AD.2013.0400295
Grad, L. I., and Cashman, N. R. (2014). Prion-like activity of Cu/Zn superoxide dismutase: implications for amyotrophic lateral sclerosis. Prion 8, 35–43. doi: 10.4161/pri.27602
Guo, Y.-S., Wu, D.-X., Wu, H.-R., Wu, S.-Y., Yang, C., Li, B., et al. (2009). Sensory involvement in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Exp. Mol. Med. 41, 140–150. doi: 10.3858/emm.2009.41.3.017
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Hamilton, G., and Gillingwater, T. H. (2013). Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 19, 40–50. doi: 10.1016/j.molmed.2012.11.002
Han, S. M., Oussini, E. L. H., Scekic-Zahirovic, J., Vibbert, J., Cottee, P., and Miller, M. A. (2013). VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in Caenorhabditis elegans. PLoS Genet. 9:e1003738. doi: 10.1371/journal.pgen.1003738
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Ilieva, E. V., Ayala, V., Jové, M., Dalfó, E., Cacabelos, D., Povedano, M., et al. (2007). Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130, 3111–3123. doi: 10.1093/brain/awm190
Ito, Y., Yamada, M., Tanaka, H., Aida, K., Tsuruma, K., Shimazawa, M., et al. (2009). Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 36, 470–476. doi: 10.1016/j.nbd.2009.08.013
Jud, M. C., Czerwinski, M. J., Wood, M. P., Young, R. A., Gallo, C. M., Bickel, J. S., et al. (2008). Large P body-like RNPs form in C. elegans oocytes in response to arrested ovulation, heat shock, osmotic stress, and anoxia and are regulated by the major sperm protein pathway. Dev. Biol. 318, 38–51. doi: 10.1016/j.ydbio.2008.02.059
Kabashi, E., Bercier, V., Lissouba, A., Liao, M., Brustein, E., Rouleau, G. A., et al. (2011). FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 7:e1002214. doi: 10.1371/journal.pgen.1002214
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kabir, M. E., and Safar, J. G. (2014). Implications of prion adaptation and evolution paradigm for human neurodegenerative diseases. Prion 8, 113–118. doi: 10.4161/pri.27661
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. 108, 4441–4446. doi: 10.1073/pnas.1100650108
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, H. J., Kim, N. C., Wang, Y.-D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Kon, T., Mori, F., Tanji, K., Miki, Y., Toyoshima, Y., Yoshida, M., et al. (2014). ALS-associated protein FIG4 is localized in Pick and Lewy bodies, and also neuronal nuclear inclusions, in polyglutamine and intranuclear inclusion body diseases. Neuropathology 34, 19–26. doi: 10.1111/neup.12056
Kwiatkowski, T. J., Bosco, D. A., LeClerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Kwon, D. Y., Dimitriadi, M., Terzic, B., Cable, C., Hart, A. C., Chitnis, A., et al. (2013). The E3 ubiquitin ligase mind bomb 1 ubiquitinates and promotes the degradation of survival of motor neuron protein. Mol. Biol. Cell 24, 1863–1871. doi: 10.1091/mbc.E13-01-0042
Lai, C. H., Chou, C. Y., Ch'ang, L. Y., Liu, C. S., and Lin, W. (2000). Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 10, 703–713. doi: 10.1101/gr.10.5.703
Lanson, N. A., Maltare, A., King, H., Smith, R., Kim, J. H., Taylor, J. P., et al. (2011). A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 20, 2510–2523. doi: 10.1093/hmg/ddr150
Lapierre, L. R., and Hansen, M. (2012). Lessons from C. elegans: signaling pathways for longevity. Trends Endocrinol. Metab. 23, 637–644. doi: 10.1016/j.tem.2012.07.007
Lau, H. E., and Chalasani, S. H. (2014). Divergent and convergent roles for insulin-like peptides in the worm, fly and mammalian nervous systems. Invert. Neurosci. doi: 10.1007/s10158-013-0166-9. [Epub ahead of print].
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Li, J., and Le, W. (2013). Modeling neurodegenerative diseases in Caenorhabditis elegans. Exp. Neurol. 250, 94–103. doi: 10.1016/j.expneurol.2013.09.024
Li, J., Li, T., Zhang, X., Tang, Y., Yang, J., and Le, W. (2013a). Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration. Neurobiol. Aging 35, 837–846. doi: 10.1016/j.neurobiolaging.2013.09.003
Li, Y. R., King, O. D., Shorter, J., and Gitler, A. D. (2013b). Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372. doi: 10.1083/jcb.201302044
Liachko, N. F., Guthrie, C. R., and Kraemer, B. C. (2010). Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans Model of TDP-43 proteinopathy. J. Neurosci. 30, 16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010
Liachko, N. F., McMillan, P. J., Guthrie, C. R., Bird, T. D., Leverenz, J. B., and Kraemer, B. C. (2013). CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann Neurol. 74, 39–52. doi: 10.1002/ana.23870
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y.-J., Vanderweyde, T., Vanderwyde, T., Citro, A., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE 5:e13250. doi: 10.1371/journal.pone.0013250
Mackenzie, I. R. A., Ansorge, O., Strong, M., Bilbao, J., Zinman, L., Ang, L. C., et al. (2011). Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 122, 87–98. doi: 10.1007/s00401-011-0838-7
Mackenzie, I. R. A., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Matsushita-Ishiodori, Y., Yamanaka, K., and Ogura, T. (2007). The C. elegans homologue of the spastic paraplegia protein, spastin, disassembles microtubules. Biochem. Biophys. Res. Commun. 359, 157–162. doi: 10.1016/j.bbrc.2007.05.086
Matus, S., Valenzuela, V., Medinas, D. B., and Hetz, C. (2013). ER dysfunction and protein folding stress in ALS. Int. J. Cell Biol. 2013, 674751. doi: 10.1155/2013/674751
McCombe, P. A., and Henderson, R. D. (2011). The role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med. 11, 246–254. doi: 10.2174/156652411795243450
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McIntire, S. L., Reimer, R. J., Schuske, K., Edwards, R. H., and Jorgensen, E. M. (1997). Identification and characterization of the vesicular GABA transporter. Nature 389, 870–876. doi: 10.1038/39908
Merner, N. D., Girard, S. L., Catoire, H., Bourassa, C. V., Belzil, V. V., Rivière, J.-B., et al. (2012). Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet. 91, 313–319. doi: 10.1016/j.ajhg.2012.07.002
Miguel-Aliaga, I., Culetto, E., Walker, D. S., Baylis, H. A., Sattelle, D. B., and Davies, K. E. (1999). The Caenorhabditis elegans orthologue of the human gene responsible for spinal muscular atrophy is a maternal product critical for germline maturation and embryonic viability. Hum. Mol. Genet. 8, 2133–2143. doi: 10.1093/hmg/8.12.2133
Mori, F., Tanji, K., Toyoshima, Y., Sasaki, H., Yoshida, M., Kakita, A., et al. (2013a). Valosin-containing protein immunoreactivity in tauopathies, synucleinopathies, polyglutamine diseases and intranuclear inclusion body disease. Neuropathology 33, 637–644. doi: 10.1111/neup.12050
Mori, K., Weng, S.-M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morita, M., Al-Chalabi, A., Andersen, P. M., Hosler, B., Sapp, P., Englund, E., et al. (2006). A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66, 839–844. doi: 10.1212/01.wnl.0000200048.53766.b4
Morris, H. R., Waite, A. J., Williams, N. M., Neal, J. W., and Blake, D. J. (2012). Recent advances in the genetics of the ALS-FTLD complex. Curr. Neurol. Neurosci. Rep. 12, 243–250. doi: 10.1007/s11910-012-0268-5
Murakami, T., Yang, S.-P., Xie, L., Kawano, T., Fu, D., Mukai, A., et al. (2012). ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 21, 1–9. doi: 10.1093/hmg/ddr417
Musarò, A. (2013). Understanding ALS: new therapeutic approaches. FEBS J. 280, 4315–4322. doi: 10.1111/febs.12087
Negga, R., Stuart, J. A., Machen, M. L., Salva, J., Lizek, A. J., Richardson, S. J., et al. (2012). Exposure to glyphosate- and/or Mn/Zn-ethylene-bis-dithiocarbamate-containing pesticides leads to degeneration of γ-aminobutyric acid and dopamine neurons in Caenorhabditis elegans. Neurotox. Res. 21, 281–290. doi: 10.1007/s12640-011-9274-7
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D., et al. (2014). Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511. doi: 10.1126/science.1247363
Nussbaum-Krammer, C. I., Park, K.-W., Li, L., Melki, R., and Morimoto, R. I. (2013). Spreading of a prion domain from cell-to-cell by vesicular transport in Caenorhabditis elegans. PLoS Genet. 9:e1003351. doi: 10.1371/journal.pgen.1003351
Oeda, T., Shimohama, S., Kitagawa, N., Kohno, R., Imura, T., Shibasaki, H., et al. (2001). Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 10, 2013–2023. doi: 10.1093/hmg/10.19.2013
Oikonomou, G., and Shaham, S. (2011). The glia of Caenorhabditis elegans. Glia 59, 1253–1263. doi: 10.1002/glia.21084
O'Reilly, L. P., Luke, C. J., Perlmutter, D. H., Silverman, G. A., and Pak, S. C. (2013). C. elegans in high-throughput drug discovery. Adv. Drug Deliv. Rev. doi: 10.1016/j.addr.2013.12.001. [Epub ahead of print].
Parisi, C., Arisi, I., D'Ambrosi, N., Storti, A. E., Brandi, R., D'Onofrio, M., et al. (2013). Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 4, e959. doi: 10.1038/cddis.2013.491
Pasinelli, P., Houseweart, M. K., Brown, R. H., and Cleveland, D. W. (2000). Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 97, 13901–13906. doi: 10.1073/pnas.240305897
Pickles, S., and Vande Velde, C. (2012). Misfolded SOD1 and ALS: zeroing in on mitochondria. Amyotroph Lateral Scler 13, 333–340. doi: 10.3109/17482968.2012.648645
Polymenidou, M., and Cleveland, D. W. (2011). The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147, 498–508. doi: 10.1016/j.cell.2011.10.011
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. doi: 10.1212/01.wnl.0000172911.39167.b6
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rodriguez, M., Snoek, L. B., De Bono, M., and Kammenga, J. E. (2013). Worms under stress: C. elegans stress response and its relevance to complex human disease and aging. Trends Genet. 29, 367–374. doi: 10.1016/j.tig.2013.01.010
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Saccon, R. A., Bunton-Stasyshyn, R. K. A., Fisher, E. M. C., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. doi: 10.1093/brain/awt097
Sasayama, H., Shimamura, M., Tokuda, T., Azuma, Y., Yoshida, T., Mizuno, T., et al. (2012). Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE 7:e39483. doi: 10.1371/journal.pone.0039483
Sato, M., Sato, K., Liou, W., Pant, S., Harada, A., and Grant, B. D. (2008). Regulation of endocytic recycling by C. elegans Rab35 and its regulator RME-4, a coated-pit protein. EMBO J. 27, 1183–1196. doi: 10.1038/emboj.2008.54
Shatunov, A., Mok, K., Newhouse, S., Weale, M. E., Smith, B., Vance, C., et al. (2010). Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 9, 986–994. doi: 10.1016/S1474-4422(10)70197-6
Shaye, D. D., and Greenwald, I. (2011). OrthoList: a compendium of C. elegans genes with human orthologs. PLoS ONE 6:e20085. doi: 10.1371/journal.pone.0020085
Singh, V., and Aballay, A. (2009). Regulation of DAF-16-mediated innate immunity in Caenorhabditis elegans. J. Biol. Chem. 284, 35580–35587. doi: 10.1074/jbc.M109.060905
Sleigh, J. N., Buckingham, S. D., Esmaeili, B., Viswanathan, M., Cuppen, E., Westlund, B. M., et al. (2011). A novel Caenorhabditis elegans allele, smn-1(cb131), mimicking a mild form of spinal muscular atrophy, provides a convenient drug screening platform highlighting new and pre-approved compounds. Hum. Mol. Genet. 20, 245–260. doi: 10.1093/hmg/ddq459
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Sreedharan, J., and Brown, R. H. (2013). Amyotrophic lateral sclerosis: problems and prospects. Ann. Neurol. 74, 309–316. doi: 10.1002/ana.2401
Sun, Y., Yang, P., Zhang, Y., Bao, X., Li, J., Hou, W., et al. (2011). A genome-wide RNAi screen identifies genes regulating the formation of P bodies in C. elegans and their functions in NMD and RNAi. Protein Cell 2, 918–939. doi: 10.1007/s13238-011-1119-x
Swarup, V., Phaneuf, D., Bareil, C., Robertson, J., Rouleau, G. A., Kriz, J., et al. (2011). Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 134, 2610–2626. doi: 10.1093/brain/awr159
Tauffenberger, A., Chitramuthu, B. P., Bateman, A., Bennett, H. P. J., and Parker, J. A. (2013a). Reduction of polyglutamine toxicity by TDP-43, FUS and progranulin in Huntington's disease models. Hum. Mol. Genet. 22, 782–794. doi: 10.1093/hmg/dds485
Tauffenberger, A., Julien, C., and Parker, J. A. (2013b). Evaluation of longevity enhancing compounds against transactive response DNA-binding protein-43 neuronal toxicity. Neurobiol. Aging 34, 2175–2182. doi: 10.1016/j.neurobiolaging.2013.03.014
Taylor, R. C., and Dillin, A. (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. doi: 10.1016/j.cell.2013.05.042
Therrien, M., Rouleau, G. A., Dion, P. A., and Parker, J. A. (2013). Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE 8:e83450. doi: 10.1371/journal.pone.0083450
Turner, M. R., Bowser, R., Bruijn, L., Dupuis, L., Ludolph, A., McGrath, M., et al. (2013a). Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14(Suppl. 1), 19–32. doi: 10.3109/21678421.2013.778554
Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chiò, A., de Carvalho, M., et al. (2013b). Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322. doi: 10.1016/S1474-4422(13)70036-X
Updike, D., and Strome, S. (2010). P granule assembly and function in Caenorhabditis elegans germ cells. J. Androl. 31, 53–60. doi: 10.2164/jandrol.109.008292
Vaccaro, A., Patten, S. A., Aggad, D., Julien, C., Maios, C., Kabashi, E., et al. (2013). Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 55, 64–75. doi: 10.1016/j.nbd.2013.03.015
Vaccaro, A., Patten, S. A., Ciura, S., Maios, C., Therrien, M., Drapeau, P., et al. (2012a). Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS ONE 7:e42117. doi: 10.1371/journal.pone.0042117
Vaccaro, A., Tauffenberger, A., Aggad, D., Rouleau, G., Drapeau, P., and Parker, J. A. (2012b). Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS ONE 7:e31321. doi: 10.1371/journal.pone.0031321
Vaccaro, A., Tauffenberger, A., Ash, P. E. A., Carlomagno, Y., Petrucelli, L., and Parker, J. A. (2012c). TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 8:e1002806. doi: 10.1371/journal.pgen.1002806
Valori, C. F., Brambilla, L., Martorana, F., and Rossi, D. (2013). The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 71, 287–297. doi: 10.1007/s00018-013-1429-7
Vance, C., Al-Chalabi, A., Ruddy, D., Smith, B. N., Hu, X., Sreedharan, J., et al. (2006). Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain 129, 868–876. doi: 10.1093/brain/awl030
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vance, C., Scotter, E. L., Nishimura, A. L., Troakes, C., Mitchell, J. C., Kathe, C., et al. (2013). ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 22, 2676–2688. doi: 10.1093/hmg/ddt117
Vanden Broeck, L., Callaerts, P., and Dermaut, B. (2014). TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends Mol. Med. 20, 66–71. doi: 10.1016/j.molmed.2013.11.003
van Es, M. A., Veldink, J. H., Saris, C. G. J., Blauw, H. M., van Vught, P. W. J., Birve, A., et al. (2009). Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087. doi: 10.1038/ng.442
Van Hoecke, A., Schoonaert, L., Lemmens, R., Timmers, M., Staats, K. A., Laird, A. S., et al. (2012). EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422. doi: 10.1038/nm.2901
Van Langenhove, T., van der Zee, J., and Van Broeckhoven, C. (2012). The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann. Med. 44, 817–828. doi: 10.3109/07853890.2012.665471
van Oosten-Hawle, P., Porter, R. S., and Morimoto, R. I. (2013). Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153, 1366–1378. doi: 10.1016/j.cell.2013.05.015
Van Raamsdonk, J. M., and Hekimi, S. (2012). Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. U.S.A. 109, 5785–5790. doi: 10.1073/pnas.1116158109
Verbeeck, C., Deng, Q., DeJesus-Hernandez, M., Taylor, G., Ceballos-Diaz, C., Kocerha, J., et al. (2012). Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol. Neurodegener. 7:53. doi: 10.1186/1750-1326-7-53
Wang, J., Farr, G. W., Hall, D. H., Li, F., Furtak, K., Dreier, L., et al. (2009). An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet 5:e1000350. doi: 10.1371/journal.pgen.1000350
Wang, J.-W., Brent, J. R., Tomlinson, A., Shneider, N. A., and McCabe, B. D. (2011a). The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest. 121, 4118–4126. doi: 10.1172/JCI57883
Wang, L.-C., Chen, K.-Y., Pan, H., Wu, C.-C., Chen, P.-H., Liao, Y.-T., et al. (2011b). Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell. Mol. Life Sci. 68, 1255–1267. doi: 10.1007/s00018-010-0522-4
Warren, J. D., Rohrer, J. D., and Rossor, M. N. (2013). Clinical review. Frontotemporal dementia. BMJ 347, f4827. doi: 10.1136/bmj.f4827
Woulfe, J., Gray, D. A., and Mackenzie, I. R. A. (2010). FUS-immunoreactive intranuclear inclusions in neurodegenerative disease. Brain Pathol. 20, 589–597. doi: 10.1111/j.1750-3639.2009.00337.x
Xi, Z., Zinman, L., Moreno, D., Schymick, J., Liang, Y., Sato, C., et al. (2013). Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 92, 981–989. doi: 10.1016/j.ajhg.2013.04.017
Xu, Y.-F., Gendron, T. F., Zhang, Y.-J., Lin, W.-L., D'Alton, S., Sheng, H., et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
Xu, Z., Poidevin, M., Li, X., Li, Y., Shu, L., Nelson, D. L., et al. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 7778–7783. doi: 10.1073/pnas.1219643110
Yanase, S., Onodera, A., Tedesco, P., Johnson, T. E., and Ishii, N. (2009). SOD-1 deletions in Caenorhabditis elegans alter the localization of intracellular reactive oxygen species and show molecular compensation. J. Gerontol. A Bio. Sci. Med. Sci. 64, 530–539. doi: 10.1093/gerona/glp020
Zhang, D., Iyer, L. M., He, F., and Aravind, L. (2012a). Discovery of Novel DENN Proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 3:283. doi: 10.3389/fgene.2012.00283
Zhang, T., Hwang, H. Y., Hao, H., Talbot, C., and Wang, J. (2012b). Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem. 287, 8371–8382. doi: 10.1074/jbc.M111.311977
Zhang, T., Mullane, P. C., Periz, G., and Wang, J. (2011). TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum. Mol. Genet. 20, 1952–1965. doi: 10.1093/hmg/ddr076
Zhao, J., Matthies, D. S., Botzolakis, E. J., Macdonald, R. L., Blakely, R. D., and Hedera, P. (2008). Hereditary spastic paraplegia-associated mutations in the NIPA1 gene and its Caenorhabditis elegans homolog trigger neural degeneration in vitro and in vivo through a gain-of-function mechanism. J. Neurosci. 28, 13938–13951. doi: 10.1523/JNEUROSCI.4668-08.2008
Keywords: C. elegans, ALS (Amyotrophic lateral sclerosis), TDP-43, FUS, C9orf72, SOD1, genetic networks, motor neuron disease
Citation: Therrien M and Parker JA (2014) Worming forward: amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 5:85. doi: 10.3389/fgene.2014.00085
Received: 14 February 2014; Paper pending published: 28 February 2014;
Accepted: 30 March 2014; Published online: 17 April 2014.
Edited by:
Shin Murakami, Touro University-California, USAReviewed by:
Eirini Lionaki, Foundation for Research and Technology-Hellas, GreeceMaria Markaki, Foundation for Research and Technology -Hellas, Greece
Marta Artal Sanz, Pablo de Olavide University, Spain
Copyright © 2014 Therrien and Parker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Alex Parker, CRCHUM- Centre Hospitalier de l'Université de Montréal, Université de Montréal, 900 St-Denis, Tour Viger, R09.440, Montréal, QC H2X 0A9, Canada e-mail:amEucGFya2VyQHVtb250cmVhbC5jYQ==