 Huiping Zhou
Huiping Zhou Runping Liu
Runping Liu- 1Department of Microbiology and Immunology, School of Medicine, Virginia Commonwealth University, Richmond, VA, USA
- 2McGuire Veterans Affairs Medical Center, Richmond, VA, USA
The endoplasmic reticulum (ER) is an important player in regulating protein synthesis and lipid metabolism. Perturbation of ER homeostasis, referred as “ER stress,” has been linked to numerous pathological conditions, such as inflammation, cardiovascular diseases, and metabolic disorders. The liver plays a central role in regulating nutrient and lipid metabolism. Accumulating evidence implicates that ER stress disrupts lipid metabolism and induces hepatic lipotoxicity. Here, we review the major ER stress signaling pathways, how ER stress contributes to the dysregulation of hepatic lipid metabolism, and the potential causative mechanisms of ER stress in hepatic lipotoxicity. Understanding the role of ER stress in hepatic metabolism may lead to the identification of new therapeutic targets for metabolic diseases.
Introduction
Endoplasmic reticulum is an important intracellular organelle responsible for protein synthesis, folding, modification, and trafficking. In addition, the ER also plays a crucial role in calcium homeostasis and in regulating the biosynthesis of steroids, lipids and carbohydrates (Borgese et al., 2006). In the ER, millions of proteins are synthesized, but not all of them are able to be properly folded and processed. Under normal physiological conditions, the unfolded or misfolded proteins are directed to degradation pathways through activating an evolutionally conserved signaling pathway, the UPR. Activation of the UPR can either help eliminate the unfolded proteins and restore cellular homeostasis, or activate a cascade of intracellular events resulting in cell death (Shen et al., 2004; Merksamer and Papa, 2010). The UPR is of particular importance in hepatocytes, which are rich in ER content and responsible for the synthesis of proteins, cholesterol, bile acids, and phospholipids. The UPR and its contribution to hepatic injury have been investigated in various liver diseases including ALD, NAFLD, DILD, cholestatic liver disease, and viral hepatitis (Zhou et al., 2006; Kaplowitz et al., 2007; Colgan et al., 2011; Dara et al., 2011; Jo et al., 2013).
ER Stress and the UPR
The ER is a membranous network of cisternae responsible for the synthesis and export of proteins and lipids. It is also crucial for cellular calcium homeostasis (Borgese et al., 2006; Gorlach et al., 2006). The ability of the ER to adapt to the metabolic changes, such as an increase in protein synthesis and accumulation of unfolded proteins and cholesterol in the ER lumen, is of paramount importance for the cell. When the misfolded or unfolded proteins accumulate in the ER, the ER stress and its related signaling pathways, UPR, are activated (Ron and Walter, 2007; Schroder, 2008).
Three main branches of the UPR-mediated signaling pathways have been identified so far: the IRE1 pathway, protein kinase RNA-like ER kinase (PERK) pathway, and ATF6 pathway. As illustrated in Figure 1, IRE1, PERK, and ATF6 are associated with the ER membrane. Under non-stressed condition, these transmembrane proteins are bound to a chaperone protein, BiP/GRP78, which is also known as the master regulator of the UPR. The binding of BiP/GRP78 to these UPR transducers prevents them from activation. When the ER is stressed by accumulation of misfolded or unfolded proteins, depletion of ER calcium content, or increase of free cholesterol in the ER lumen, BiP/GRP78 is released from the UPR transducers. The disassociation of BiP/GRP78 from the UPR transducers results in the activation of IRE1-, PERK-, and ATF6-mediated signaling pathways. PERK activation results in a rapid down-regulation of protein synthesis via phosphorylation of eIF-2α and inhibition of the formation of the translation initiation complex (DuRose et al., 2009). The phosphorylated eIF-2α further promotes the translation of ATF4, a member of the basic leucine zipper (bZIP)-containing protein subfamily. IRE1α has both protein kinase and endoribonuclease activities. Under ER stress conditions, IRE1α is oligomerized and autophosphorylated. The activated IRE1α removes a 26-bp intron from the XBP1 mRNA, resulting in the production of spliced XBP1 protein (XBP1s). XBP1s, which is also a bZIP transcription factor, regulates the expression of several genes involved in UPR and ER-assisted degradation (ERAD) to help restore ER homeostasis (Acosta-Alvear et al., 2007). In addition, IRE1α also induces the activation of stress kinases, JNK and p38 MAPK, that promote apoptosis (Ron and Hubbard, 2008). ATF6 is the third branch of the UPR. Dissociation of BiP/GRP78 from ATF6 leads to its translocation to the Golgi, where it is processed into its active form by cleavage of its N-terminal domain by S1P and S2P (Chen et al., 2002). The activated ATF6 (ATF-6 N terminal domain) is translocated to the nucleus and functions as a transcription factor, promoting the expression of downstream target genes involved in ER stress including XBP1, GADD153 (also known as CHOP), and ER chaperones (Oyadomari and Mori, 2004). CHOP is a proapoptotic transcription factor that plays a critical role in ER stress-mediated apoptosis (Marciniak et al., 2004).
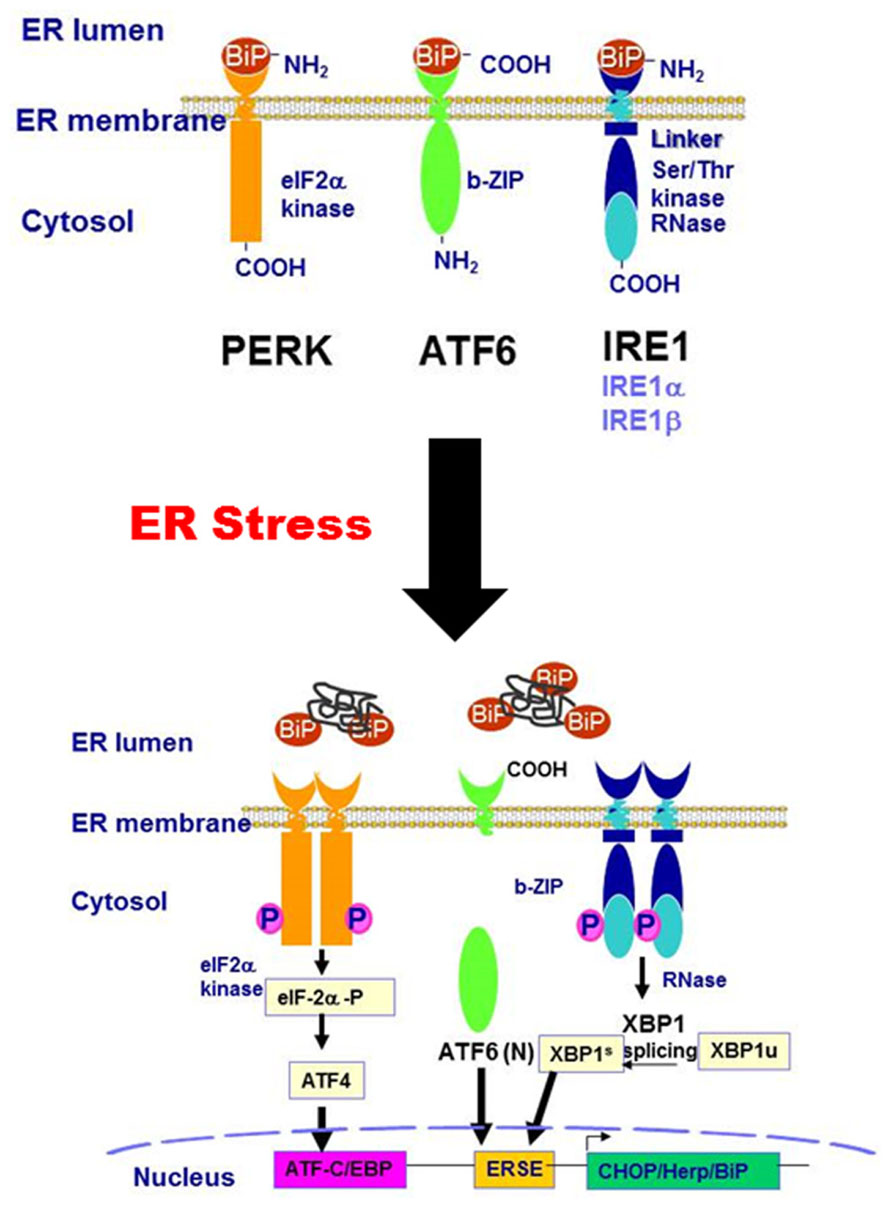
FIGURE 1. Schematic diagram of the ER stress-activated signaling pathways.
Hepatic Lipid Metabolism
Liver is the central metabolic organ and plays a critical role in fatty acid and cholesterol metabolism (Hylemon et al., 2001). Several inter-dependent pathways are involved in hepatic lipid metabolism. The hepatic fatty acids can be derived from de novo lipogenesis, hydrolysis of triglyceride from cytoplasmic lipid droplets or direct uptake of non-esterified fatty acids from circulation (Bechmann et al., 2012). The liver is also a major organ in the processing of lipids into the various lipoproteins, in particular VLDL and LDL. Fatty acids synthesized by the liver are converted into triglyceride and exported as constituents of VLDL into the blood circulation. The VLDLs absorbed into peripheral tissues are partially digested by lipoprotein lipase into LDL and free fatty acids. LDL is transported into the cell via LDL receptors for its conversion into free fatty acids, cholesterol, and other components of LDL. Similarly, cholesterol also can be derived from de novo synthesis or absorbed from the diet, and are transported into circulation as lipoprotein particles (Horton et al., 2002a; Babin and Gibbons, 2009; van Heyningen, 2009; Musso et al., 2013; Faust and Kovacs, 2014). The cholesterol can be stored in cells as cholesterol esters or metabolized into bile acids. The hepatic triglycerides and cholesterol contents are tightly regulated by multiple interrelated signaling pathways. Under normal physiological conditions, lipid input is equal to lipid output from the body. Disruption of either the input or output pathways will result in dysregulation of lipid metabolism (Hylemon et al., 2001). Tremendous studies have been done to elucidate the extremely complex regulation network of hepatic lipid homeostasis (Bradbury and Berk, 2004; Bradbury, 2006; Weickert and Pfeiffer, 2006; Nguyen et al., 2008; Musso et al., 2009; Sparks and Dong, 2009; van Heyningen, 2009; Trauner et al., 2010; Jump, 2011; Fu et al., 2012). Here, we will focus on the current understanding regarding the role of ER stress in hepatic lipid metabolism.
UPR and Hepatic Lipid Homeostasis
The ER is the primary site of lipid metabolism. Many enzymes and regulatory proteins of lipid metabolism reside in the ER. Perturbation of ER homeostasis contributes to hepatic steatosis, inflammation and insulin resistance in the liver (Kaplowitz et al., 2007; Hotamisligil, 2010; Rohrl et al., 2014). Although the UPR was originally identified as a conserved signaling pathway, functioning to maintain essential ER homeostasis, numerous studies indicate that the UPR has broader functions and plays an essential role in maintaining hepatic lipid homeostasis. Recent studies have shown that the pharmacologic ER stress inducers increase de novo lipogenesis and lipid droplet formation in hepatocytes by up-regulating a subset of genes encoding key lipogenic trans-activators and enzymes (Lee et al., 2012).
Hepatic lipid homeostasis is controlled by numerous transcription factors and nuclear receptors. The SREBPs are master regulators of lipid homeostasis (Eberle et al., 2004) and play a critical role in de novo lipid biosynthesis (Amemiya-Kudo et al., 2002). SREBP-1 controls fatty acid and triglyceride biosynthesis, while SREBP-2 controls cholesterol metabolism and LDL receptor expression. SREBPs, are basic-helix-loop-helix-leucine zipper (bHLHLZ) transcription factors bound to the ER membranes as an inactive precursor (Brown and Goldstein, 1997). The regulation of SREBP activity is controlled within the ER by the interaction of SCAP with insulin regulated proteins (Insigs; Yang et al., 2002). The Insigs can cause ER retention of the SREBP-SCAP complex and prevent the activation of SREBPs. When the sterol level is low, Insigs are disassociated with SCAP, which allows the SREBP-SCAP complex to migrate to the Golgi, where SREBPs are processed into active forms by S1P and S2P (Rawson, 2003; Lee and Ye, 2004). The activated SREBPs are subsequently translocated into nucleus, where SREBPs regulate the expression of various genes involved in lipid metabolism by binding to the sterol regulatory element of their target genes (Horton et al., 2002a,b; Radhakrishnan et al., 2007). It has been shown that ER stress induces proteolytic activation of SREBPs by increasing the turnover of Insig-1 (Lee and Ye, 2004). The expression of Insigs is regulated by insulin and FXRα (Yabe et al., 2003; Hubbert et al., 2007). Overexpression of hepatic Insigs has been shown to reduce hepatic lipogenesis (Engelking et al., 2004; Takaishi et al., 2004).
ER stress and lipid metabolism are tightly intertwined. The chronic ER stress is the most important contributor to metabolic diseases. Unresolved ER stress induces dysregulation of hepatic lipid metabolism (Fu et al., 2012). It is also well-documented that excess saturated fatty acids and cholesterol can induce ER stress and disrupt lipid metabolism in hepatocytes, macrophages and adipocytes (Fribley et al., 2009; Colgan et al., 2011; Anderson et al., 2012; Fu et al., 2012; Wang et al., 2013; Zha et al., 2013). The various components of the UPR signaling pathways play a role in the regulation of lipid metabolism.
IRE1α-XBP1 Pathway
IRE1α-XBP1 pathway is one of three main branches of UPR, which has been identified as a critical regulator of hepatic lipid metabolism. Hepatic-specific deletion of IRE1α increased hepatic lipid levels and reduced plasma lipid by altering several genes involved in hepatic lipid metabolism under ER stress conditions such as C/EBPβ, C/EBPδ, peroxisome proliferator-activated receptor γ (PPARγ), and enzymes involved in triglyceride biosynthesis (Zhang et al., 2011). Although, these results suggest a plausible protective role of IRE1α from hepatic steatosis, the deletion of IRE1α blocks the basal level of the UPR in liver which may lead to an unresolved ER stress. Thus, it is still unclear whether the induction of lipogenesis genes in IRE1α-/- mice is due to the loss of IRE1α function or elevated ER stress. Studies done by Lee et al. (2008) reported that disruption of hepatic XBP-1 significantly reduced serum triglyceride, cholesterol and fatty acids levels by decreasing de novo hepatic lipogenesis in mice. In addition, the IRE1α-XBP1 pathway is also involved in regulation of hepatic VLDL assembly and secretion (Wang et al., 2012). IRE1α is required for efficient secretion of VLDL and LDL from hepatocytes under the condition of ER stress (Zhang et al., 2011). A most recent study done by Rohrl et al. (2014) identified novel links between ER stress and hepatic cholesterol metabolism. Activation of acute ER stress reduced ABCA1 expression and induced ABCA1 redistribution to tubular perinuclear compartments in hepatocytes, which significantly diminished cholesterol efflux to apoA-I and HDL formation (Rohrl et al., 2014).
PERK-ATF-4 Pathway
Protein kinase RNA-like ER kinase activation induces eIF2α phosphorylation, which causes translation attenuation that is required to protect against apoptosis in response to ER stress. Although the exact role of PERK in hepatic steatosis is still not completely understood, a recent report also suggests that antipsychotic drugs (APDs)-induced activation of PERK-p-eIF2α signaling pathway increases intracellular lipid accumulation through activation of SREBP-1c and SREBP-2 in hepatocytes (Lauressergues et al., 2012). Attenuation of eIF2α in GADD34 transgenic mice significantly altered the metabolism profile and reduced high fat diet-induced hepatic steatosis (Oyadomari et al., 2008). As a downstream transcriptional factor of the UPR, ATF4 avoids global suppression of protein expression induced by p-eIF2α due to a different upstream signaling pathway. It has been shown that ATF4 deficiency preferentially attenuated hepatic lipogenesis via down-regulation of PPARγ, SREBP-1c, ACC and SCD expression without affecting hepatic triglyceride production and fatty acid oxidation (Li et al., 2011; Xiao et al., 2013). A recent study further identified that activation of PERK-eIF2α-ATF4 pathway under ER stress condition is required for hepatic VLDL receptor up-regulation in hepatocytes, which is responsible for intracellular accumulation of triglycerides and hepatic steatosis (Jo et al., 2013). Furthermore, attenuation of global translation by activation of PERK-eIF2α pathway also decreases ApoB expression, which further promotes hepatic steatosis.
ATF6 Pathway
Both ATF6 and SREBPs are activated by the same proteases (S1P and S2P) in the Golgi (Sakai et al., 1998; Ye et al., 2000; Horton et al., 2002b). Several independent studies have shown that ATF6 and XBP-1 share similar DNA binding specificities (Yoshida et al., 2000, 2001; Acosta-Alvear et al., 2007; Misiewicz et al., 2013). In addition, ATF6 and XBP-1 are able to form a heterodimer and regulate down-stream target genes (Yamamoto et al., 2007). Several recent studies in ATF6α knockout mice indicate that ATF6 also plays an important role in regulating hepatic lipid homeostasis (Rutkowski et al., 2008; Yamamoto et al., 2010). Similar to IRE1α, deletion of ATF6α does not result in an apparent phenotype under physiological conditions. However, under ER stress conditions, ATF6α knockout mice exhibited severe liver injury and hepatic steatosis caused by inhibition of fatty acid β-oxidation and VLDL formation (Yamamoto et al., 2010). In addition, the CHOP expression is significantly up-regulated while PPARα expression and ApoB-100 protein levels are decreased in the livers of ATF6α knockout mice (Rutkowski et al., 2008; Yamamoto et al., 2010). A recent study in zebrafish with fatty liver disease demonstrated that ER stress induces fatty liver disease. During chronic ER stress, ATF6 promotes steatosis. However, ATF6 prevents acute ER stress-induced steatosis. This study suggest that ATF6 can play both protective and pathological roles in fatty liver disease (Cinaroglu et al., 2011).
ER Stress Master Regulator-GRP78/BiP
GRP78/BiP is a glucose-regulated protein that functions as a molecular chaperone in the ER (Mote et al., 1998). As described in previous section, GRP78 acts as a master regulator of the activation of UPR signaling pathways. Numerous studies have indicated that ER stress is an important component of the hepatic steatosis and insulin resistance in obese rodent models (Kammoun et al., 2009; Chen et al., 2013; Teodoro-Morrison et al., 2013). GRP78 plays a critical role in maintaining hepatic lipid homeostasis. Overexpression of GRP78 prevents ER stress-induced SREBP-1c proteolytic cleavage and reduced hepatic steatosis (Kammoun et al., 2009). It also has been reported that overproduction of GRP78 prevents palmitate-induced ER stress and cytotoxicity in human HepG2 cells (Gu et al., 2010) and high fat diet-induced type 2 diabetes in mouse models (Teodoro-Morrison et al., 2013). A most recent study further indicated that GRP78 is able to prevent oxidative stress-induced injury by inhibiting lipid peroxidation (Suyama et al., 2014).
C/EBP Homologous Protein
C/EBP homologous protein is a proapoptotic transcriptional factor downstream of all three UPR signaling pathways. Numerous evidences suggest that CHOP activation promotes cell apoptosis and induces tissue injury. In absence of CHOP, both cells and animals are protected against various pharmacological and physiological insults (Tabas and Ron, 2011). It has been shown that CHOP deficiency attenuates cholestasis-induced liver fibrosis and methionine-choline-deficient (MCD) diet-induced steatohepatitis, fibrosis, and carcinogenesis in mice (Tamaki et al., 2008; Toriguchi et al., 2013). In a murine model of intragastric ethanol feeding, CHOP null mice have remarkable absence of hepatocellular apoptosis, but no protection against alcohol-induced steatosis (Ji et al., 2005). In human liver cell lines, saturated fatty acids induce ER stress and apoptosis via the PERK/ATF4/CHOP (Cao et al., 2012). Our recent studies suggest that CHOP is a major player in human immunodeficiency virus protease inhibitor-induced hepatic lipotoxicity in mice (Wang et al., 2013). Studies done by Chikka et al. (2013) further suggest CHOP has a non-apoptotic role in regulating hepatic metabolic genes during ER stress. In addition, a recent study reported that CHOP expression is up-regulated in human hepatocellular carcinoma (HCC) and two mouse HCC models. CHOP expression contributes to hepatic carcinogenesis by promoting inflammation and cell apoptosis (DeZwaan-McCabe et al., 2013). These studies indicate CHOP is a common contributing factor in ER stress-induced liver injury.
Therapeutic Potential Targeting ER Stress in Metabolic Diseases
Chronic ER stress has been implicated in the pathogenesis of metabolic diseases such as a diabetes, obesity, cardiovascular diseases as well as fatty liver disease (Colgan et al., 2011; Malhi and Kaufman, 2011; Mollica et al., 2011; Bechmann et al., 2012; Cnop et al., 2012). Targeting the specific UPR signaling pathways to attenuate ER stress and UPR activation would provide opportunities in developing new therapeutic strategies in a wide array of diseases. Several studies have shown the promising effects of small chemical chaperones on alleviating the UPR activation in animal models, such as 4-phenylbutyric acid (4-PBA) and tauroursodeoxycholic acid (TUDCA; Heubi et al., 2002; Ozcan et al., 2006; Basseri et al., 2009; Engin and Hotamisligil, 2010; Lee et al., 2010). Both 4-PBA and TUDCA have been approved by US Food and Drug Administration (FDA) for treating children with urea-cycle disorders and cholestatic liver disease, respectively. However, more clinical studies are needed to validate the potential application of these chemical chaperones in treating ER stress-associated metabolic diseases.
Conclusion
Hepatic lipid homeostasis requires integration of multiple signals. A significant amount of evidence indicates that activation of ER stress signaling pathways play a critical role in various diseases associated with dysregulation of hepatic lipid metabolism. Although acute ER stress response helps restore ER homeostasis, prolonged or chronic ER stress activation contributes to development of various metabolic diseases including NAFLD, type 2 diabetes, and atherosclerosis by inducing widespread pathologic apoptosis (Tabas and Ron, 2011). The balance of the UPR signaling pathways, such as ATF6, IRE1/XBP1, and PERK/ATF4, is critical for maintaining cellular homeostasis. However, the exact mechanisms underlying ER stress-induced disruption of hepatic lipid homeostasis remains to be fully identified. Recent studies have shown that ER stress is also closely linked to inflammation and autophagy, which are two important players in regulating hepatic lipid metabolism (Ogata et al., 2006; Yorimitsu et al., 2006; Hoyer-Hansen and Jaattela, 2007; Yorimitsu and Klionsky, 2007; Hotamisligil, 2010; Hummasti and Hotamisligil, 2010; Mollica et al., 2011; Qiu et al., 2011; Adolph et al., 2012; Hasnain et al., 2012; Kolattukudy and Niu, 2012). The contribution of autophagy to lipid metabolism has been reviewed in several excellent reviews (Singh, 2010; Amir and Czaja, 2011; Czaja, 2011; Ding et al., 2011; Lavallard et al., 2012). Elucidating the signaling pathways of ER stress and its intertwining with other intracellular signaling components not only furthers our current understanding of lipid metabolism in a central metabolic organ, but also helps develop an effective approach that can be used to treat patients with metabolic diseases.
A growing body of evidence links ER stress and UPR activation to diseases associated with lipid metabolism. The UPR signaling pathways and activation of transcription factors such as XBP1 and ATF6 have novel roles in controlling the transcriptional regulation of lipogenesis. While IRE1α itself is protective against ER-stress-induced lipogenesis and hepatic steatosis, its downstream mediator XBP1 promotes transcription of genes involved in fatty acid and cholesterol biosynthesis. Phosphorylation of eIF2α downstream of PERK affects the transcriptional activity of C/EBPs, PPARγ, and SREBP-1c thereby leading to lipid accumulation and hepatic steatosis under high-fat-diet conditions. Similar to IRE1α, ATF6α also protects against ER stress-induced steatosis and lipid droplet formation in mice. Furthermore, nuclear ATF6 attenuates SREBP2-mediated lipogenesis. The exact mechanisms by which ER stress signaling pathways affect lipid homeostasis are incompletely understood. Given the temporal differences in the activation of the three arms of the UPR, a closer examination of each branch of the UPR will allow for a better understanding of how various components of this signaling network impact lipogenesis and disease progression. Such studies will further enhance our understanding of the biological and pharmacological tools needed to effectively treat ER-associated diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Yunzhou Li for editing and proofreading the manuscript. Huiping Zhou is supported by National Institutes of Health (NIH) Grant R01 DK-057543, VA Merit Award 1I01BX001390, and National Science Foundation of China Grant 81070245 and 81270489.
Abbreviations
ABCA1, ATP-binding cassette, sub-family A, member 1; ACC, acetyl-CoA carboxylase; ALD, alcoholic liver disease; ATF, activating transcription factor; C/EBP, CCAAT/enhancer-binding protein; CHOP, C/EBP homologous protein; DILD, drug-induced liver disease; eIF2α, eukaryotic translation initiation factor; ER, endoplasmic reticulum; FAS, fatty acid synthase; FXRα, farnesold X receptor α; GADD, growth arrest and DNA damage-inducible protein; HDL, high density lipoprotein; Insig, insulin-induced gene; IRE1, inositol requiring enzyme 1; JNK, c-Jun N-terminal kinase; LDL, low density lipoprotein; MCD, methionine-choline-deficient diet; NAFLD, non-alcoholic fatty liver disease; PERK, protein kinase RNA-like ER kinase; S1P, site 1 protease; S2P, site 2 protease; SCAP, SREBP cleavage activating protein; SCD, stearoyl-CoA desaturase; SREBP, sterol regulatory element binding protein; UPR, unfolded protein response; VLDL, very low density lipoprotein; VLDLR, VLDL receptor; XBP, X-box binding protein.
References
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66. doi: 10.1016/j.molcel.2007.06.011
Adolph, T. E., Niederreiter, L., Blumberg, R. S., and Kaser, A. (2012). Endoplasmic reticulum stress and inflammation. Dig. Dis. 30, 341–346. doi: 10.1159/000338121
Amemiya-Kudo, M., Shimano, H., Hasty, A. H., Yahagi, N., Yoshikawa, T., Matsuzaka, T., et al. (2002). Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J. Lipid Res. 43, 1220–1235.
Amir, M., and Czaja, M. J. (2011). Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 5, 159–166. doi: 10.1586/egh.11.4
Anderson, E. K., Hill, A. A., and Hasty, A. H. (2012). Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler. Thromb. Vasc. Biol. 32, 1687–1695. doi: 10.1161/ATVBAHA.112.250142
Babin, P. J., and Gibbons, G. F. (2009). The evolution of plasma cholesterol: direct utility or a “spandrel” of hepatic lipid metabolism? Prog. Lipid Res. 48, 73–91. doi: 10.1016/j.plipres.2008.11.002
Basseri, S., Lhotak, S., Sharma, A. M., and Austin, R. C. (2009). The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid Res. 50, 2486–2501. doi: 10.1194/jlr.M900216-JLR200
Bechmann, L. P., Hannivoort, R. A., Gerken, G., Hotamisligil, G. S., Trauner, M., and Canbay, A. (2012). The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 56, 952–964. doi: 10.1016/j.jhep.2011.08.025
Borgese, N., Francolini, M., and Snapp, E. (2006). Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358–364. doi: 10.1016/j.ceb.2006.06.008
Bradbury, M. W. (2006). Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. Am. J. Physiol. Gastroint. Liver Physiol. 290, G194–G198. doi: 10.1152/ajpgi.00413.2005
Bradbury, M. W., and Berk, P. D. (2004). Lipid metabolism in hepatic steatosis. Clin. Liver Dis. 8, 639–671. doi: 10.1016/j.cld.2004.04.005
Brown, M. S., and Goldstein, J. L. (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. doi: 10.1016/S0092-8674(00)80213-5
Cao, J., Dai, D. L., Yao, L., Yu, H. H., Ning, B., Zhang, Q., et al. (2012). Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 364, 115–129. doi: 10.1007/s11010-011-1211-9
Chen, W. T., Zhu, G., Pfaffenbach, K., Kanel, G., Stiles, B., and Lee, A. S. (2013). GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene doi: 10.1038/onc.2013.437 [Epub ahead of print].
Chen, X., Shen, J., and Prywes, R. (2002). The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 277, 13045–13052. doi: 10.1074/jbc.M110636200
Chikka, M. R., McCabe, D. D., Tyra, H. M., and Rutkowski, D. T. (2013). C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. J. Biol. Chem. 288, 4405–4415. doi: 10.1074/jbc.M112.432344
Cinaroglu, A., Gao, C., Imrie, D., and Sadler, K. C. (2011). Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology 54, 495–508. doi: 10.1002/hep.24396
Cnop, M., Foufelle, F., and Velloso, L. A. (2012). Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18, 59–68. doi: 10.1016/j.molmed.2011.07.010
Colgan, S. M., Hashimi, A. A., and Austin, R. C. (2011). Endoplasmic reticulum stress and lipid dysregulation. Expert Rev. Mol. Med. 13, e4. doi: 10.1017/S1462399410001742
Czaja, M. J. (2011). Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 140, 1895–1908. doi: 10.1053/j.gastro.2011.04.038
Dara, L., Ji, C., and Kaplowitz, N. (2011). The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 53, 1752–1763. doi: 10.1002/hep.24279
DeZwaan-McCabe, D., Riordan, J. D., Arensdorf, A. M., Icardi, M. S., Dupuy, A. J., and Rutkowski, D. T. (2013). The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 9:e1003937. doi: 10.1371/journal.pgen.1003937
Ding, W. X., Manley, S., and Ni, H. M. (2011). The emerging role of autophagy in alcoholic liver disease. Exp. Biol. Med. 236, 546–556. doi: 10.1258/ebm.2011.010360
DuRose, J. B., Scheuner, D., Kaufman, R. J., Rothblum, L. I., and Niwa, M. (2009). Phosphorylation of eukaryotic translation initiation factor 2α coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol. Cell. Biol. 29, 4295–4307. doi: 10.1128/MCB.00260-09
Eberle, D., Hegarty, B., Bossard, P., Ferre, P., and Foufelle, F. (2004). SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848. doi: 10.1016/j.biochi.2004.09.018
Engelking, L. J., Kuriyama, H., Hammer, R. E., Horton, J. D., Brown, M. S., Goldstein, J. L., et al. (2004). Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J. Clin. Invest. 113, 1168–1175. doi: 10.1172/JCI20978
Engin, F., and Hotamisligil, G. S. (2010). Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes. Metab. 12(Suppl. 2), 108–115. doi: 10.1111/j.1463-1326.2010.01282.x
Faust, P. L., and Kovacs, W. J. (2014). Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 98, 75–85. doi: 10.1016/j.biochi.2013
Fribley, A., Zhang, K., and Kaufman, R. J. (2009). Regulation of apoptosis by the unfolded protein response. Methods Mol. Biol. 559, 191–204. doi: 10.1007/978-1-60327-017-5_14
Fu, S., Watkins, S. M., and Hotamisligil, G. S. (2012). The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 15, 623–634. doi: 10.1016/j.cmet.2012.03.007
Gorlach, A., Klappa, P., and Kietzmann, T. (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391–1418. doi: 10.1089/ars.2006.8.1391
Gu, X., Li, K., Laybutt, D. R., He, M. L., Zhao, H. L., Chan, J. C., et al. (2010). Bip overexpression, but not CHOP inhibition, attenuates fatty-acid-induced endoplasmic reticulum stress and apoptosis in HepG2 liver cells. Life Sci. 87, 724–732. doi: 10.1016/j.lfs.2010.10.012
Hasnain, S. Z., Lourie, R., Das, I., Chen, A. C., and McGuckin, M. A. (2012). The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 90, 260–270. doi: 10.1038/icb.2011.112
Heubi, J. E., Wiechmann, D. A., Creutzinger, V., Setchell, K. D., Squires, R. Jr., Couser, R., et al. (2002). Tauroursodeoxycholic acid (TUDCA) in the prevention of total parenteral nutrition-associated liver disease. J. Pediatr. 141, 237–242. doi: 10.1067/mpd.2002.125802
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002a). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131. doi: 10.1172/JCI0215593
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002b). SREBPs: transcriptional mediators of lipid homeostasis. Cold Spring Harb. Symp. Quant. Biol. 67, 491–498. doi: 10.1101/sqb.2002.67.491
Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. doi: 10.1016/j.cell.2010.02.034
Hoyer-Hansen, M., and Jaattela, M. (2007). Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 14, 1576–1582. doi: 10.1038/sj.cdd.4402200
Hubbert, M. L., Zhang, Y., Lee, F. Y., and Edwards, P. A. (2007). Regulation of hepatic Insig-2 by the farnesoid X receptor. Mol. Endocrinol. 21, 1359–1369. doi: 10.1210/me.2007-0089
Hummasti, S., and Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 107, 579–591. doi: 10.1161/CIRCRESAHA.110.225698
Hylemon, P., Pandak, W., and Vlahcevic, Z. (2001). Liver: Biology and Pathobiology. Philadelphia, PA: Lippincott Williams & Wilkins.
Ji, C., Mehrian-Shai, R., Chan, C., Hsu, Y. H., and Kaplowitz, N. (2005). Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 29, 1496–1503. doi: 10.1097/01.alc.0000174691.03751.11
Jo, H., Choe, S. S., Shin, K. C., Jang, H., Lee, J. H., Seong, J. K., et al. (2013). Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 57, 1366–1377. doi: 10.1002/hep.26126
Jump, D. B. (2011). Fatty acid regulation of hepatic lipid metabolism. Curr. Opin. Clin. Nutr. Metab. Care 14, 115–120. doi: 10.1097/MCO.0b013e328342991c
Kammoun, H. L., Chabanon, H., Hainault, I., Luquet, S., Magnan, C., Koike, T., et al. (2009). GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119, 1201–1215. doi: 10.1172/JCI37007
Kaplowitz, N., Than, T. A., Shinohara, M., and Ji, C. (2007). Endoplasmic reticulum stress and liver injury. Semin. Liver Dis. 27, 367–377. doi: 10.1055/s-2007-991513
Kolattukudy, P. E., and Niu, J. (2012). Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ. Res. 110, 174–189. doi: 10.1161/CIRCRESAHA.111.243212
Lauressergues, E., Bert, E., Duriez, P., Hum, D., Majd, Z., Staels, B., et al. (2012). Does endoplasmic reticulum stress participate in APD-induced hepatic metabolic dysregulation? Neuropharmacology 62, 784–796. doi: 10.1016/j.neuropharm.2011.08.048
Lavallard, V. J., Meijer, A. J., Codogno, P., and Gual, P. (2012). Autophagy, signaling and obesity. Pharmacol. Res. 66, 513–525. doi: 10.1016/j.phrs.2012.09.003
Lee, A. H., Scapa, E. F., Cohen, D. E., and Glimcher, L. H. (2008). Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496. doi: 10.1126/science.1158042
Lee, J. N., and Ye, J. (2004). Proteolytic activation of sterol regulatory element-binding protein induced by cellular stress through depletion of Insig-1. J. Biol. Chem. 279, 45257–45265. doi: 10.1074/jbc.M408235200
Lee, J. S., Zheng, Z., Mendez, R., Ha, S. W., Xie, Y., and Zhang, K. (2012). Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 211, 29–38. doi: 10.1016/j.toxlet.2012.02.017
Lee, Y. Y., Hong, S. H., Lee, Y. J., Chung, S. S., Jung, H. S., Park, S. G., et al. (2010). Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem. Biophys. Res. Commun. 397, 735–739. doi: 10.1016/j.bbrc.2010.06.022
Li, H., Meng, Q., Xiao, F., Chen, S., Du, Y., Yu, J., et al. (2011). ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem. J. 438, 283–289. doi: 10.1042/BJ20110263
Malhi, H., and Kaufman, R. J. (2011). Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809. doi: 10.1016/j.jhep.2010.11.005
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. doi: 10.1101/gad.1250704
Merksamer, P. I., and Papa, F. R. (2010). The UPR and cell fate at a glance. J. Cell Sci. 123, 1003–1006. doi: 10.1242/jcs.035832
Misiewicz, M., Dery, M. A., Foveau, B., Jodoin, J., Ruths, D., and LeBlanc, A. C. (2013). Identification of a novel endoplasmic reticulum stress response element regulated by XBP1. J. Biol. Chem. 288, 20378–20391. doi: 10.1074/jbc.M113.457242
Mollica, M. P., Lionetti, L., Putti, R., Cavaliere, G., Gaita, M., and Barletta, A. (2011). From chronic overfeeding to hepatic injury: role of endoplasmic reticulum stress and inflammation. Nutrition, metabolism, and cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. 21, 222–230. doi: 10.1016/j.numecd.2010.10.012
Mote, P. L., Tillman, J. B., and Spindler, S. R. (1998). Glucose regulation of GRP78 gene expression. Mech. Ageing Dev. 104, 149–158. doi: 10.1016/S0047-6374(98)00064-5
Musso, G., Gambino, R., and Cassader, M. (2009). Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid Res. 48, 1–26. doi: 10.1016/j.plipres.2008.08.001
Musso, G., Gambino, R., and Cassader, M. (2013). Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 52, 175–191. doi: 10.1016/j.plipres.2012.11.002
Nguyen, P., Leray, V., Diez, M., Serisier, S., Le Bloc’h, J., Siliart, B., et al. (2008). Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. (Berl.) 92, 272–283. doi: 10.1111/j.1439-0396.2007.00752.x
Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231. doi: 10.1128/MCB.01453-06
Oyadomari, S., Harding, H. P., Zhang, Y., Oyadomari, M., and Ron, D. (2008). Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 7, 520–532. doi: 10.1016/j.cmet.2008.04.011
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389. doi: 10.1038/sj.cdd.4401373
Ozcan, U., Yilmaz, E., Ozcan, L., Furuhashi, M., Vaillancourt, E., Smith, R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. doi: 10.1126/science.1128294
Qiu, W., Zhang, J., Dekker, M. J., Wang, H., Huang, J., Brumell, J. H., et al. (2011). Hepatic autophagy mediates endoplasmic reticulum stress-induced degradation of misfolded apolipoprotein B. Hepatology 53, 1515–1525. doi: 10.1002/hep.24269
Radhakrishnan, A., Ikeda, Y., Kwon, H. J., Brown, M. S., and Goldstein, J. L. (2007). Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. U.S.A. 104, 6511–6518. doi: 10.1073/pnas.0700899104
Rawson, R. B. (2003). The SREBP pathway – insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 4, 631–640. doi: 10.1038/nrm1174
Rohrl, C., Eigner, K., Winter, K., Korbelius, M., Obrowsky, S., Kratky, D., et al. (2014). Endoplasmic reticulum stress impairs cholesterol efflux and synthesis in hepatic cells. J. Lipid Res. 55, 94–103. doi: 10.1194/jlr.M043299
Ron, D., and Hubbard, S. R. (2008). How IRE1 reacts to ER stress. Cell 132, 24–26. doi: 10.1016/j.cell.2007.12.017
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Rutkowski, D. T., Wu, J., Back, S. H., Callaghan, M. U., Ferris, S. P., Iqbal, J., et al. (2008). UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 15, 829–840. doi: 10.1016/j.devcel.2008.10.015
Sakai, J., Nohturfft, A., Goldstein, J. L., and Brown, M. S. (1998). Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J. Biol. Chem. 273, 5785–5793. doi: 10.1074/jbc.273.10.5785
Schroder, M. (2008). Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 65, 862–894. doi: 10.1007/s00018-007-7383-5
Shen, X., Zhang, K., and Kaufman, R. J. (2004). The unfolded protein response – a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 28, 79–92. doi: 10.1016/j.jchemneu.2004.02.006
Singh, R. (2010). Autophagy and regulation of lipid metabolism. Results Probl. Cell Differ. 52, 35–46. doi: 10.1007/978-3-642-14426-4_4
Sparks, J. D., and Dong, H. H. (2009). FoxO1 and hepatic lipid metabolism. Curr. Opin. Lipidol. 20, 217–226. doi: 10.1097/MOL.0b013e32832b3f4c
Suyama, K., Watanabe, M., Sakabe, K., Otomo, A., Okada, Y., Terayama, H., et al. (2014). GRP78 suppresses lipid peroxidation and promotes cellular antioxidant levels in glial cells following hydrogen peroxide exposure. PLoS ONE 9:e86951. doi: 10.1371/journal.pone.0086951
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. doi: 10.1038/ncb0311-184
Takaishi, K., Duplomb, L., Wang, M. Y., Li, J., and Unger, R. H. (2004). Hepatic insig-1 or -2 overexpression reduces lipogenesis in obese Zucker diabetic fatty rats and in fasted/refed normal rats. Proc. Natl. Acad. Sci. U.S.A. 101, 7106–7111. doi: 10.1073/pnas.0401715101
Tamaki, N., Hatano, E., Taura, K., Tada, M., Kodama, Y., Nitta, T., et al. (2008). CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. American journal of physiology. Gastrointest. Liver Physiol. 294, G498–G505. doi: 10.1152/ajpgi.00482.2007
Teodoro-Morrison, T., Schuiki, I., Zhang, L., Belsham, D. D., and Volchuk, A. (2013). GRP78 overproduction in pancreatic beta cells protects against high-fat-diet-induced diabetes in mice. Diabetologia 56, 1057–1067. doi: 10.1007/s00125-013-2855-7
Toriguchi, K., Hatano, E., Tanabe, K., Takemoto, K., Nakamura, K., Koyama, Y., et al. (2013). CHOP deficiency attenuates steatohepatitis, fibrosis and carcinogenesis in mice fed an MCD diet. J. Gastroenterol. Hepatol. doi: 10.1111/jgh.12481 [Epub ahead of print].
Trauner, M., Claudel, T., Fickert, P., Moustafa, T., and Wagner, M. (2010). Bile acids as regulators of hepatic lipid and glucose metabolism. Dig. Dis. 28, 220–224. doi: 10.1159/000282091
van Heyningen, C. (2009). Lipid metabolism: reviews of HDL and cholesterol efflux, ethanol and hepatic lipid metabolism and lipid disorders during childhood. Curr. Opin. Lipidol. 20, 77–78. doi: 10.1097/MOL.0b013e328321e5a5
Wang, S., Chen, Z., Lam, V., Han, J., Hassler, J., Finck, B. N., et al. (2012). IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 16, 473–486. doi: 10.1016/j.cmet.2012.09.003
Wang, Y., Zhang, L., Wu, X., Gurley, E. C., Kennedy, E., Hylemon, P. B., et al. (2013). The role of CCAAT enhancer-binding protein homologous protein in human immunodeficiency virus protease-inhibitor-induced hepatic lipotoxicity in mice. Hepatology 57, 1005–1016. doi: 10.1002/hep.26107
Weickert, M. O., and Pfeiffer, A. F. (2006). Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia 49, 1732–1741. doi: 10.1007/s00125-006-0295-3
Xiao, G., Zhang, T., Yu, S., Lee, S., Calabuig-Navarro, V., Yamauchi, J., et al. (2013). ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J. Biol. Chem. 288, 25350–25361. doi: 10.1074/jbc.M113.470526
Yabe, D., Komuro, R., Liang, G., Goldstein, J. L., and Brown, M. S. (2003). Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc. Natl. Acad. Sci. U.S.A. 100, 3155–3160. doi: 10.1073/pnas.0130116100
Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., et al. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365–376. doi: 10.1016/j.devcel.2007.07.018
Yamamoto, K., Takahara, K., Oyadomari, S., Okada, T., Sato, T., Harada, A., et al. (2010). Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 21, 2975–2986. doi: 10.1091/mbc.E09-02-0133
Yang, T., Espenshade, P. J., Wright, M. E., Yabe, D., Gong, Y., Aebersold, R., et al. (2002). Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500. doi: 10.1016/S0092-8674(02)00872-3
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. doi: 10.1016/S1097-2765(00)00133-7
Yorimitsu, T., and Klionsky, D. J. (2007). Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 3, 160–162.
Yorimitsu, T., Nair, U., Yang, Z., and Klionsky, D. J. (2006). Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299–30304. doi: 10.1074/jbc.M607007200
Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M., et al. (2000). ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000
Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M., et al. (2001). Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6α and 6β that activates the mammalian unfolded protein response. Mol. Cell. Biol. 21, 1239–1248. doi: 10.1128/MCB.21.4.1239-1248.2001
Zha, B. S., Wan, X., Zhang, X., Zha, W., Zhou, J., Wabitsch, M., et al. (2013). HIV protease inhibitors disrupt lipid metabolism by activating endoplasmic reticulum stress and inhibiting autophagy activity in adipocytes. PLoS ONE 8:e59514. doi: 10.1371/journal.pone.0059514
Zhang, K., Wang, S., Malhotra, J., Hassler, J. R., Back, S. H., Wang, G., et al. (2011). The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 30, 1357–1375. doi: 10.1038/emboj.2011.52
Keywords: ER stress, UPR, hepatic, lipid metabolism, liver diseases
Citation: Zhou H and Liu R (2014) ER stress and hepatic lipid metabolism. Front. Genet. 5:112. doi: 10.3389/fgene.2014.00112
Received: 22 February 2014; Accepted: 15 April 2014;
Published online: 09 May 2014.
Edited by:
Kezhong Zhang, Wayne State University, USAReviewed by:
Chunying Li, Wayne State University School of Medicine, USAAshok Kumar, Wayne State University, USA
Copyright © 2014 Zhou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiping Zhou, Department of Microbiology and Immunology, School of Medicine, Virginia Commonwealth University, P.O. Box 980678, Richmond, VA 23298-0678, USA e-mail:aHpob3VAdmN1LmVkdQ==