 Fabrizio Ferrè
Fabrizio Ferrè Antonio Palmeri
Antonio Palmeri Manuela Helmer-Citterich
Manuela Helmer-Citterich- Centre for Molecular Bioinformatics, Department of Biology, University of Rome Tor Vergata, Rome, Italy
The central role of kinases in virtually all signal transduction networks is the driving motivation for the development of compounds modulating their activity. ATP-mimetic inhibitors are essential tools for elucidating signaling pathways and are emerging as promising therapeutic agents. However, off-target ligand binding and complex and sometimes unexpected kinase/inhibitor relationships can occur for seemingly unrelated kinases, stressing that computational approaches are needed for learning the interaction determinants and for the inference of the effect of small compounds on a given kinase. Recently published high-throughput profiling studies assessed the effects of thousands of small compound inhibitors, covering a substantial portion of the kinome. This wealth of data paved the road for computational resources and methods that can offer a major contribution in understanding the reasons of the inhibition, helping in the rational design of more specific molecules, in the in silico prediction of inhibition for those neglected kinases for which no systematic analysis has been carried yet, in the selection of novel inhibitors with desired selectivity, and offering novel avenues of personalized therapies.
Introduction
The kinome plays a predominant role in signal transduction networks and cellular responses; its involvement in a large number of pathologies is a major impulse for the identification and development of compounds modulating the activity of individual kinases or kinase families. Currently, eleven kinase inhibitors are FDA-approved for cancer treatment, and 149 inhibitors and 42 distinct kinase targets are being tested in clinical trials (Fedorov et al., 2010; Chahrour et al., 2012; see http://www.brimr.org/PKI/PKIs.htm for an updated list). In addition to their promises as therapeutical agents, kinase inhibitors are commonly used as research tools to disclose the biological consequences of the inactivation of their targets. Generally, kinase inhibitors are ATP-mimetic compounds. The majority of known inhibitors belong to the so-called type I class, and they occupy directly the ATP binding site, located in a hydrophobic cleft between the two lobes of the kinase domain, while type II inhibitors target the ATP binding site as well, but extend also to an allosteric pocket adjacent to the ATP binding site; additional non-ATP-mimetic inhibitor classes (type III, IV, and V), of which a limited number of examples is currently known, seem very promising therapeutic agents given their generally high specificity (Liu and Gray, 2006; Garuti et al., 2010; Chahrour et al., 2012; Gavrin and Saiah, 2013). An example of type I, II, and IV inhibitors is provided in Figure 1. For type I and II inhibitors, the evolutionary structural conservation of the kinase ATP-binding site can lead to off-target binding, and while similar kinases tend to show similar inhibition profiles by sharing recurring sequence and structural patterns (Chiu et al., 2013), often complex kinase/inhibitor relationships occur, where kinase bioactivity profiles cannot be reconciled to their phylogenetic relationships (Paricharak et al., 2013). While absolute specificity toward an individual kinase is not always necessary for a compound to achieve a therapeutic effect (Mencher and Wang, 2005), a detailed knowledge of target selectivity for kinase inhibitors is crucial for predicting and interpreting the effects of inhibitors, and for designing drugs with a desired selectivity. However, kinase inhibitor selectivity is generally not inclusively known for the majority of the tested compounds, as kinase research has been principally focused on a small subset of the kinome.
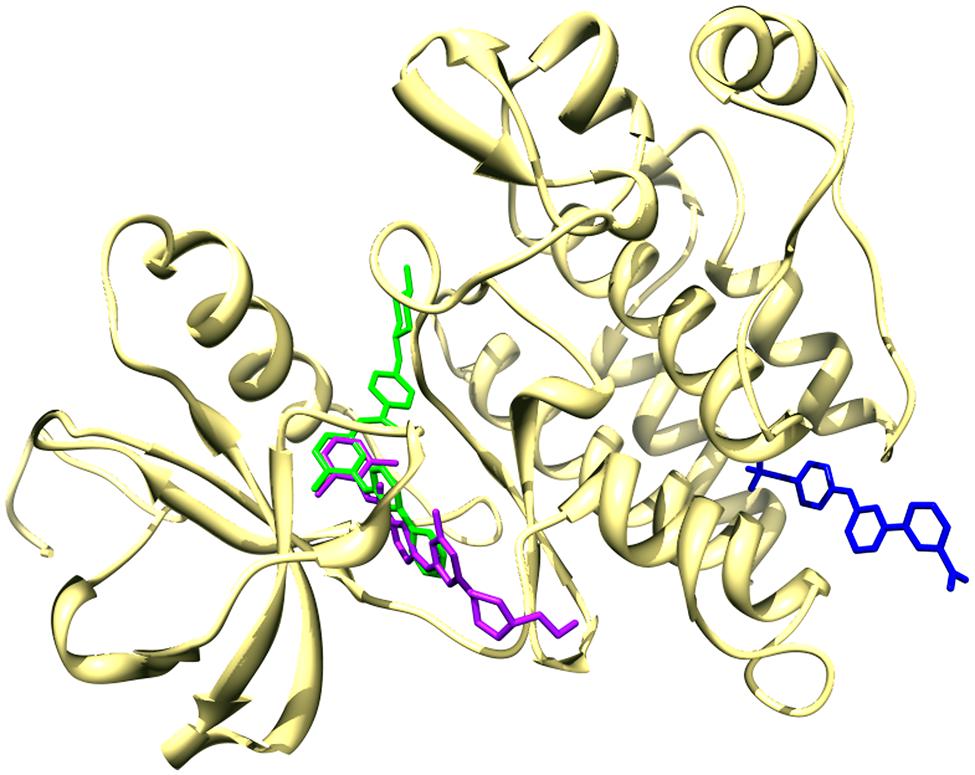
FIGURE 1. Binding of the ABL kinase with dasatinib (type I inhibitor, shown in purple), imatinib (type II inhibitor, shown in green), and GNF-2 (type IV inhibitor, shown in blue). The human ABL kinase co-crystallized with imatinib (PDB code 1IEP) was used as reference for the structural superposition of the human ABL co-crystallized with dasatinib (PDB code 2GQG) and of the mouse ABL in complex with the allosteric inhibitor GNF-2 (PDB code 3K5V). Only the ribbon representation of the human ABL kinase domain from 1IEP (chain A) is shown.
Traditional kinase inhibitor analysis is a low-throughput process in which the capability of small compounds to decrease the phosphorylation activity (usually reported as the IC50 or as the remaining or residual activity of the kinase) or their binding affinity (as its dissociation constant) is measured, but are generally not extended to the characterization of the inhibitory abilities of a given compound against the entire kinome. Such data are mined from the literature and collected in general-purpose databases such as ChEMBL (Gaulton et al., 2012) and STITCH (Kuhn et al., 2014), or in kinase-dedicated public resources such as the CheEMBL Kinase SARfari, or the commercially available Kinase Knowledgebase (KKB) by Eidogen-Sertanty (Oceanside, CA, USA) and the kinase inhibitor database provided by GVK Biosciences (Hyderabad, India). While largely populated, such databases tend to be highly heterogeneous by including evidences obtained by diverse means.
However, in recent years the results of medium- and high-throughput profiling studies became available, tackling inhibition of the phosphorylation activity for panels of widely used research compounds and clinical agents against large subsets of the human kinome (Table 1). These studies were able to identify novel inhibitor chemotypes for specific kinase targets and to reveal the target specificities of a large set of kinase inhibitors. Importantly, these panels also provide negative results, i.e., inhibitors having little or no effect on tested kinases, which are instrumental for computational learning techniques and are generally absent or scarce in low-throughput settings.
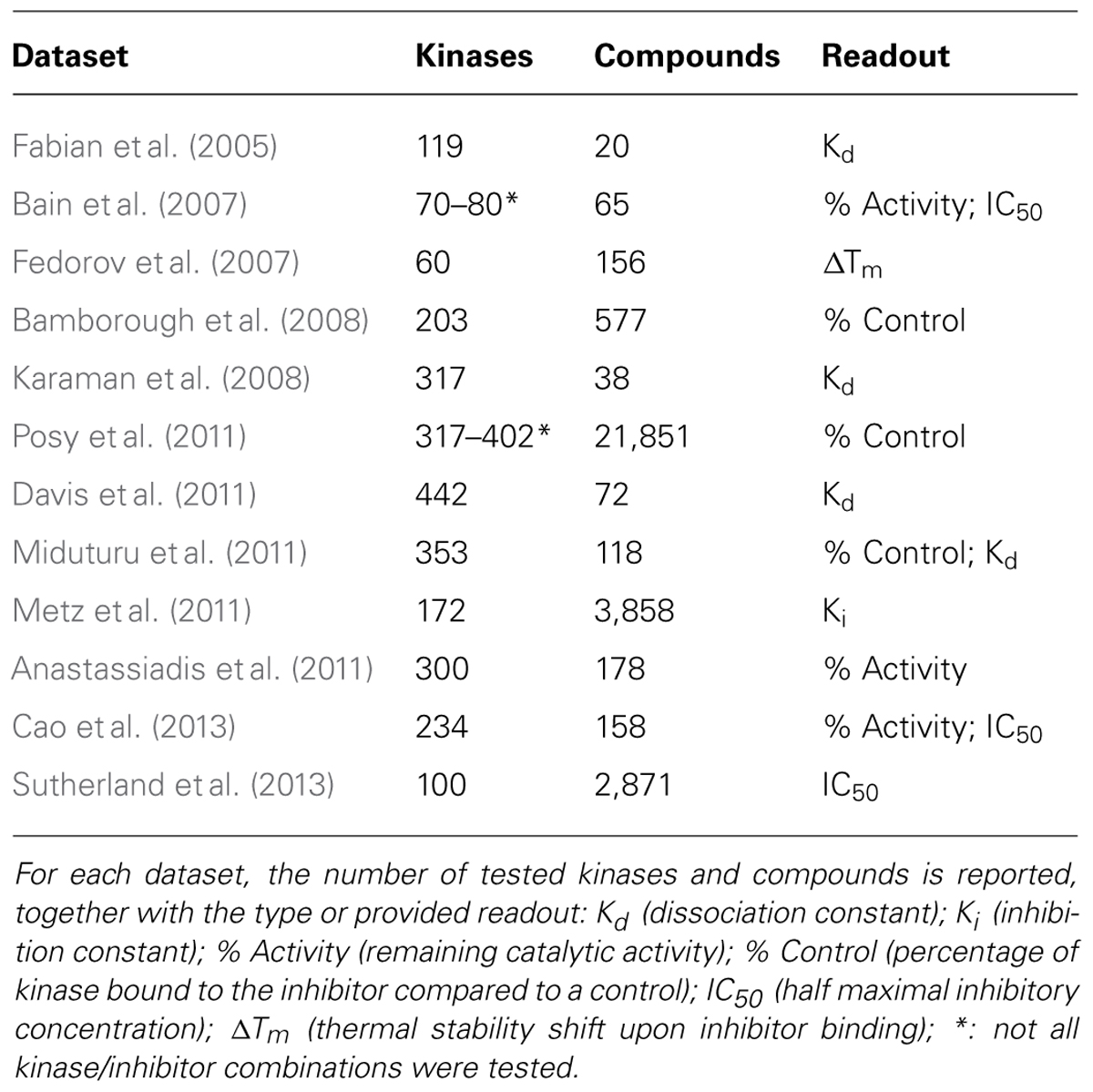
TABLE 1. Kinase/inhibitor profiling panels.
Additionally, a large and growing number of known three-dimensional (3D) structures of whole kinases or kinase domains are available in the Protein Data Bank (PDB, Berman et al., 2013), and, in few cases, the kinase was also co-crystallized with an inhibitor. These structures provide a rich background for a detailed analysis of kinase binding pockets and for a better identification of binding determinants.
Computational methods for kinase/inhibitor relationships analysis and inference were successfully attempted in the past (e.g., Manallack et al., 2002; Vieth et al., 2004; Xia et al., 2004; Chuaqui et al., 2005), but were limited by the incomplete and heterogeneous data available at the time. In this review we focused on recent computational methods and resources that employ the latest kinase inhibition profiling data but go beyond standard quantitative structure-activity relationship (QSAR) modeling approaches, which are generally specific for a single target, being instead purposely tailored toward kinase inhibition analysis and applied to the whole kinome, taking advantage from the overall kinase domain conservation and from shared binding patterns and characteristics and providing multidimensional structure-activity relationships concerning tens or hundreds of targets at the same time (Goldstein et al., 2008).
Methods for Kinase/Inhibitor Inference
Procedures that use inhibition data from panels of proteins tested against panels of compounds are generally based on numerical descriptions of physicochemical, structural and/or geometrical properties of both ligands and targets, and seek possibly non-linear relationships that explain the binding profiles. Machine learning methods are therefore particularly suited, either for classification (binds/does not bind) or regression on the measured inhibition values (e.g., IC50 or Kd). Since all information available for any kinase target and/or inhibitor is used for learning, these studies can be considered a multi-target approach. Additionally, they can be used to infer novel kinase/inhibitor relationships, also for kinases and compounds not included in the training set.
A number of recent papers explored this kind of approach, differing in the employed training dataset, in the way compounds and proteins are described and in the learning algorithm, but following similar pipelines. For example, Niijima et al. (2012) and Cao et al. (2013) both started from data extracted from Kinase SARfari [in Niijima et al. (2012) the Metz dataset was additionally used for external validation], and propose a similar kinase/inhibitor deconvolution approach, in which the whole kinase sequences, or only the kinase ATP-binding pockets, are deconstructed into residues (either described simply by amino acid type or by physicochemical characteristics) and compounds into chemical fragments or in topological Daylight fingerprints. Yabuuchi et al. (2011) developed a method, called CGBVS (chemical genomics-based virtual screening), in which compounds were represented by a large set of substructure descriptors and physicochemical properties, and protein descriptors were computed from the protein sequence dipeptide composition using a string kernel. Originally developed for G-protein-coupled receptor inhibitors, the method was also applied to kinases, using a panel of 143 kinases and 8830 inhibitors, for a total of more than 15,000 tested interactions extracted from the commercial GVK Biosciences kinase inhibitor database. In Lapins and Wikberg (2010), starting from the Karaman dataset, compounds were described by physicochemical and geometrical characteristics, while kinases were described with either alignment-independent or alignment-based methods, by building a multiple alignment of the kinase domains, excluding gap-rich positions, describing columns of the alignment with physicochemical properties, and applying principal component analysis (PCA) and partial least squares discriminant analysis to summarize descriptors. Schürer and Muskal (2013) employed the Eidogen-Sertanty KKB Q4 2009 release, including more than 430,000 tested kinase/compound pairs extracted from literature and patents. Given the heterogeneous nature of the dataset, data were subject to filtering, standardization, and clustering procedures. For each kinase in the dataset, active and inactive compounds were described using extended connectivity fingerprints, and negative instances for training were either known as inactive on a given kinase, or taken as the entire set of molecules not tested on that kinase.
Then, in these works, machine learning algorithms were trained on kinases and compounds converted into numerical descriptors, to learn associations between kinase residues and compound fragments, and for inference. Variants of a naïve Bayesian (NB) classifier or of a support vector machine (SVM) were used in Niijima et al. (2012), a random forest (RF) in Cao et al. (2013), SVM, decision trees, k-nearest neighbors, and partial least squares projections in Lapins and Wikberg (2010), an SVM in Yabuuchi et al. (2011), Laplacien-corrected NB classifiers, k-nearest neighbors, and partial least squares regression in Schürer and Muskal (2013). All these studies achieved good prediction performances: from 0.67 to 0.73 correlation coefficient in Lapins and Wikberg (2010); accuracy between 74 and 81% and matthews correlation coefficient (MCC) between 0.3 and 0.48 in different tested datasets and with different encodings and learning methods in Niijima et al. (2012); 94% accuracy and 0.98 area under the ROC curve (auROC) in Cao et al. (2013). In Schürer and Muskal (2013), the auROC for individual kinase models vary from around 0.93 to 1, and the prediction accuracy showed a positive correlation with the number of known inhibitors available for training. In Yabuuchi et al. (2011), some predicted novel inhibitors for the epidermal growth factor receptor kinase and the cyclin-dependent kinase 2 were experimentally confirmed, sometimes showing scaffold hopping (i.e., having radically different characteristics than known inhibitors).
Another class of methods includes those taking advantage of kinase 3D structures, used to obtain a more accurate representation of kinase binding sites. A reasonable assumption is that the affinity that a kinase, or a set of kinases, show toward a compound can be ascribed to set of residues that either allow or hinder the binding, and that, once identified in the 3D structures, can be looked for in other kinases to infer their binding ability, even for those kinases for which the 3D structure is unknown, by taking advantage of the kinase domain sequence conservation. Such sets of residues can additionally be converted in numerical descriptors for machine learning.
A subset of kinase/inhibitor pairs extracted from the Fabian and Karaman datasets was used in Caffrey et al. (2008). For these inhibitors the structure of the kinase/compound complex is known, and the specificity determinants can be rationalized. An algorithm was developed to predict specificity determinants given a kinase multiple sequence alignment and structural information, which was able to reproduce the known determinants and to highlight non-trivial additional factors, and can be used as basis for the design of drugs with a desired specificity.
X-ReactKIN (Brylinski and Skolnick, 2010) is a machine learning method for assessment of cross-reactivity in which each human kinase domain structure was obtained through homology modeling, and binding sites residues were predicted using computational methods. Similarity between kinases was computed by different metrics using sequence, structure, and ligand binding profiles. The system employed data from the Fabian and Karaman panels for training and validation of a NB classifier, obtaining sensitivity higher than 0.5 for around 70% of the tested compounds, and the Bamborough dataset was used for further validation, finding significant correspondence (0.53 average Pearson correlation) between predicted and experimental activity profiles. The computed cross-reactivity profiles are freely available for download.
In Huang et al. (2010), all kinase 3D structures available in the PDB at the time were superposed to obtain a fine description of a series of features known in the literature to be related to inhibitor specificity, e.g., the size of the gatekeeper residue, that affects the pocket accessibility, the hydrogen bonding and covalent bonding ability at specific positions, the flexibility of the hinge loop connecting the kinase domain small and large lobes, and others. These features were extended to kinases for which the structure in unknown via multiple alignments, converted into numerical vectors and used to estimate a similarity between each pair of kinases. Using these distances, a network of kinase binding sites was constructed, which recapitulated well a network based on the similarity between the inhibitor profiles in the Karaman dataset. Integration of the binding site similarity network with the inhibition profile network led to inference of off-target interactions, some of which were validated experimentally.
On the same lines, in Anderson et al. (2012), starting from the Karaman dataset, first kinases were clustered by similarity in binding affinity profiles for the inhibitors tested in the dataset. Kinases within the same cluster were shown to have more similar binding sites, as detected by the comparison of the binding site 3D structures extracted from the PDB. In silico docking procedures then highlighted cluster-specific residues acting as interaction hot spots, which were converted into a series of descriptors, used for RF training, achieving 76% of prediction accuracy. The RF was then used for the prediction of novel kinase/inhibitor relationships, some of which were experimentally tested, obtaining a good agreement with the predicted Ki values in 70% of the cases.
The Karaman dataset, crossed with kinase 3D structures available in the PDB, were also the starting point for the work presented in Bryant et al. (2013); the structure of a kinase bound to a known type II kinase inhibitor, imatinib, was used as template to identify contact residues, mapped to all other considered kinases using the Pfam (Punta et al., 2012) kinase family multiple alignment. A combinatorial clustering was used to find subsets of binding site residues that better correlate with the binding affinities reported in the Karaman dataset. An SVM was then trained on these data, and the prediction performance was estimated individually for each inhibitor as the auROC, which ranges from 0.5 to 1 (mean 0.8). Finally, the trained SVM was used to infer the binding ability of unlabeled kinases.
Integrative Approaches
The wealth of kinase inhibition profiling data presents great opportunities for being analyzed as a whole, by integrating data from different resources in order to provide a unified view on kinome inhibition. The whole kinase/inhibitor data can therefore be represented as a network, where binding can be treated as a binary on–off relation or weighted by the binding affinity or by the strength of the inhibitory effect. This kind of network can aid in the identification and rationalization of drugs secondary effects and facilitate drug repositioning.
KIDFamMap (Chiu et al., 2013) and K-Map (Kim et al., 2013) are free web-databases in which kinase/inhibitor relationships, retrieved from different sources, are connected and integrated with other annotations to facilitate the at-a-glance investigation of the kinome inhibition. In KIDFamMap, the Karaman, Anastassiadis and Davis profiling panels, Kinase SARfari, the PDB, and others resources, for a total of more than 186,000 kinase/compound pairs, are investigated by decomposing each interaction into a series of binding pocket sub-regions and compound fragments preferences (Chen et al., 2010), and then extending the identified rules to the whole kinome (introducing the concept of kinase/inhibitor families) and associated to known pathologies involving kinases. Queries can start from a kinase, a compound or a disease, retrieving a detailed overview of the kinase/inhibitor interaction, all the other interactions belonging to the same family, and a description of associated diseases and how allelic variants might affect the compound binding. In K-Map the Anastassiadis and Davis datasets were analyzed by building connectivity maps based on the Kolmogorov–Smirnov statistic to find correlations between inhibitors and lists of kinases. K-Map allows querying these datasets by kinase, kinase family, custom lists, or kinase-related GO terms, obtaining lists of associated inhibitors ranked by correlation significance. Similarly, the user can start from lists of inhibitors. The intent of K-Map is to provide insights for drug development and repositioning.
Caveats of integrative approaches are that to convert data into an on-off relation would require setting thresholds that might not be easy to optimize, and that data from different sources might not be directly comparable, so they must be opportunely processed. In Sutherland et al. (2013) the Anastassiadis, Metz and Davis datasets were compared to each other and to an additional profiling panel (the Sutherland dataset in Table 1), by converting each readout in an estimated IC50, testing the concordance between IC50 in different panels, and for promiscuity and selectivity measures. They found that the all panels have good agreement in assessing whether a compound is active or inactive on a given kinase, but the exact inhibition values show instead low levels of concordance, as well as measures of how much selective is a compound.
In Tang et al. (2014) the Metz, Davis, and Anastassiadis datasets were compared and integrated with data from ChEMBL and STITCH. Since these panels employed different assays and different readouts (Kd, Ki and percentage of remaining activity for the Davis, Metz and Anastassiadis datasets, respectively), a new method called KIBA (kinase inhibitor bioactivity) is introduced to obtain a single comparable activity score for each kinase/compound pair. The three panels have a relatively small number of common tested kinase/inhibitor pairs; in such cases, the Metz and Davis datasets show good degree of correlation between readouts, which is smaller when both are compared with the Anastassiadis panel. The project resulted in a kinase/inhibitor bioactivity map comprising 467 kinases and more than 50,000 compounds, which is freely available.
Conclusion
While different in methods and scope, the approaches presented here highlight the need for original and effective computational methods to unravel the rich and complex kinase/inhibitor relationships systematically measured in inhibition profiling panels, which can have significant implications in understanding the reasons of the inhibition, helping in the rational design of bioactive molecules, and can be used for the in silico prediction of inhibition for those neglected kinases for which no systematic analysis has been carried yet, and for the selection of inhibitors with desired promiscuity. Additionally, a better understanding of the kinase determinants of inhibition can help in apprehending the different response of individual patients to treatment, such as inhibitor resistance due to specific mutations, moving toward a more personalized treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by Programmi di Ricerca di rilevante Interesse Nazionale (PRIN) 2010 (prot. 20108XYHJS_006 to Manuela Helmer-Citterich).
References
Anastassiadis, T., Deacon, S. W., Devarajan, K., Ma, H., and Peterson, J. R. (2011). Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045. doi: 10.1038/nbt.2017
Anderson, P. C., De Sapio, V., Turner, K. B., Elmer, S. P., Roe, D. C., and Schoeniger, J. S. (2012). Identification of binding specificity-determining features in protein families. J. Med. Chem. 55, 1926–1939. doi: 10.1021/jm200979x
Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H.,et al. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. doi: 10.1042/BJ20070797
Bamborough, P., Drewry, D., Harper, G., Smith, G. K., and Schneider, K. (2008). Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J. Med. Chem. 51, 7898–7914. doi: 10.1021/jm8011036
Berman, H. M., Kleywegt, G. J., Nakamura, H., and Markley, J. L. (2013). How community has shaped the Protein Data Bank. Structure 21, 1485–1491. doi: 10.1016/j.str.2013.07.010
Bryant, D. H., Moll, M., Finn, P. W., and Kavraki, L. E. (2013). Combinatorial clustering of residue position subsets predicts inhibitor affinity across the human kinome. PLoS Comput. Biol. 9:e1003087. doi: 10.1371/journal.pcbi.1003087
Brylinski, M., and Skolnick, J. (2010). Cross-reactivity virtual profiling of the human kinome by X-react(KIN): a chemical systems biology approach. Mol. Pharm. 7, 2324–2333. doi: 10.1021/mp1002976
Caffrey, D. R., Lunney, E. A., and Moshinsky, D. J. (2008). Prediction of specificity-determining residues for small-molecule kinase inhibitors. BMC Bioinformatics 9:491. doi: 10.1186/1471-2105-9-491
Cao, D. S., Zhou, G. H., Liu, S., Zhang, L. X., Xu, Q. S., He, M.,et al. (2013). Large-scale prediction of human kinase/inhibitor interactions using protein sequences and molecular topological structures. Anal. Chim. Acta 792, 10–18. doi: 10.1016/j.aca.2013.07.003
Chahrour, O., Cairns, D., and Omran, Z. (2012). Small molecule kinase inhibitors as anti-cancer therapeutics. Mini Rev. Med. Chem. 12, 399–411. doi: 10.2174/138955712800493915
Chen, Y. F., Hsu, K. C., Lin, S. R., Wang, W. C., Huang, Y. C., and Yang, J. M. (2010). SiMMap: a web server for inferring site-moiety map to recognize interaction preferences between protein pockets and compound moieties. Nucleic Acids Res. 38, W424–W430. doi: 10.1093/nar/gkq480
Chiu, Y. Y., Lin, C. T., Huang, J. W., Hsu, K. C., Tseng, J. H., You, S. R.,et al. (2013). KIDFamMap: a database of kinase/inhibitor-disease family maps for kinase inhibitor selectivity and binding mechanisms. Nucleic Acids Res. 41, D430–D440. doi: 10.1093/nar/gks1218
Chuaqui, C., Deng, Z., and Singh, J. (2005). Interaction profiles of protein kinase/inhibitor complexes and their application to virtual screening. J. Med. Chem. 48, 121–133. doi: 10.1021/jm049312t
Davis, M. I., Hunt, J. P., Herrgard, S., Ciceri, P., Wodicka, L. M., Pallares, G.,et al. (2011). Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051. doi: 10.1038/nbt.1990
Fabian, M. A., Biggs W. H. III, Treiber, D. K., Atteridge, C. E., Azimioara, M. D., Benedetti, M. G.,et al. (2005). A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336. doi: 10.1038/nbt1068
Fedorov, O., Marsden, B., Pogacic, V., Rellos, P., Müller, S., Bullock, A. N.,et al. (2007). A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. U.S.A. 104, 20523–20528. doi: 10.1073/pnas.0708800104
Fedorov, O., Muller, S., and Knapp, S. (2010). The (un)targeted cancer kinome. Nat. Chem. Biol. 6, 166–169. doi: 10.1038/nchembio.297
Garuti, L., Roberti, M., and Bottegoni, G. (2010). Non-ATP competitive protein kinase inhibitors. Curr. Med. Chem. 17, 2804–2821. doi: 10.2174/092986710791859333
Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A.,et al. (2012). ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107. doi: 10.1093/nar/gkr777
Gavrin, L. K., and Saiah, E. (2013). Approaches to discover non-ATP site kinase inhibitors. Med. Chem. Commun. 4, 41–51. doi: 10.1039/c2md20180a
Goldstein, D. M., Gray, N. S., and Zarrinkar, P. P. (2008). High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug Discov. 7, 391–397. doi: 10.1038/nrd2541
Huang, D., Zhou, T., Lafleur, K., Nevado, C., and Caflisch, A. (2010). Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 26, 198–204. doi: 10.1093/bioinformatics/btp650
Karaman, M. W., Herrgard, S., Treiber, D. K., Gallant, P., Atteridge, C. E., Campbell, B. T.,et al. (2008). A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132. doi: 10.1038/nbt1358
Kim, J., Yoo, M., Kang, J., and Tan, A. C. (2013). K-Map: connecting kinases with therapeutics for drug repurposing and development. Hum. Genomics 7, 20. doi: 10.1186/1479-7364-7-20
Kuhn, M., Szklarczyk, D., Pletscher-Frankild, S., Blicher, T. H., von Mering, C., Jensen, L. J.,et al. (2014). STITCH 4: integration of protein-chemical interactions with user data. Nucleic Acids Res. 42, D401. doi: 10.1093/nar/gkt1207
Lapins, M., and Wikberg, J. E. (2010). Kinome-wide interaction modelling using alignment-based and alignment-independent approaches for kinase description and linear and non-linear data analysis techniques. BMC Bioinformatics 11:339. doi: 10.1186/1471-2105-11-339
Liu, Y., and Gray, N. S. (2006). Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364. doi: 10.1038/nchembio799
Manallack, D. T., Pitt, W. R., Gancia, E., Montana, J. G., Livingstone, D. J., Ford, M. G.,et al. (2002). Selecting screening candidates for kinase and G protein-coupled receptor targets using neural networks. J. Chem. Inf. Comput. Sci. 42, 1256–1262. doi: 10.1021/ci020267c
Mencher, S. K., and Wang, L. G. (2005). Promiscuous drugs compared to selective drugs (promiscuity can be a virtue). BMC Clin. Pharmacol. 5:3. doi: 10.1186/1472-6904-5-3
Metz, J. T., Johnson, E. F., Soni, N. B., Merta, P. J., Kifle, L., and Hajduk, P. J. (2011). Navigating the kinome. Nat. Chem. Biol. 7, 200–202. doi: 10.1038/nchembio.530
Miduturu, C. V., Deng, X., Kwiatkowski, N., Yang, W., Brault, L., Filippakopoulos, P.,et al. (2011). High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem. Biol. 18, 868–879. doi: 10.1016/j.chembiol.2011.05.010
Niijima, S., Shiraishi, A., and Okuno, Y. (2012). Dissecting kinase profiling data to predict activity and understand cross-reactivity of kinase inhibitors. J. Chem. Inf. Model. 52, 901–912. doi: 10.1021/ci200607f
Paricharak, S., Klenka, T., Augustin, M., Patel, U. A., and Bender, A. (2013). Are phylogenetic trees suitable for chemogenomics analyses of bioactivity data sets: the importance of shared active compounds and choosing a suitable data embedding method, as exemplified on Kinases. J. Cheminform. 5, 49. doi: 10.1186/1758-2946-5-49
Posy, S. L., Hermsmeier, M. A., Vaccaro, W., Ott, K. H., Todderud, G., Lippy, J. S.,et al. (2011). Trends in kinase selectivity: insights for target class-focused library screening. J. Med. Chem. 54, 54–66. doi: 10.1021/jm101195a
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C.,et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Schürer, S. C., and Muskal, S. M. (2013). Kinome-wide activity modeling from diverse public high-quality data sets. J. Chem. Inf. Model. 53, 27–38. doi: 10.1021/ci300403k
Sutherland, J. J., Gao, C., Cahya, S., and Vieth, M. (2013). What general conclusions can we draw from kinase profiling data sets? Biochim. Biophys. Acta 1834, 1425–1433. doi: 10.1016/j.bbapap.2012.12.023
Tang, J., Szwajda, A., Shakyawar, S., Xu, T., Hintsanen, P., Wennerberg, K.,et al. (2014). Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model. 54, 735–743. doi: 10.1021/ci400709d
Vieth, M., Higgs, R. E., Robertson, D. H., Shapiro, M., Gragg, E. A., and Hemmerle, H. (2004). Kinomics-structural biology and chemogenomics of kinase inhibitors and targets. Biochim. Biophys. Acta 1697, 243–257. doi: 10.1016/j.bbapap.2003.11.028
Xia, X., Maliski, E. G., Gallant, P., and Rogers, D. (2004). Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 47, 4463–4470. doi: 10.1021/jm0303195
Keywords: kinase inhibitors, kinase activity modulation, kinase/inhibitor inference, drug design and development, chemogenomics
Citation: Ferrè F, Palmeri A and Helmer-Citterich M (2014) Computational methods for analysis and inference of kinase/inhibitor relationships. Front. Genet. 5:196. doi: 10.3389/fgene.2014.00196
Received: 01 May 2014; Paper pending published: 30 May 2014;
Accepted: 13 June 2014; Published online: 30 June 2014.
Edited by:
Allegra Via, Sapienza University of Rome, ItalyCopyright © 2014 Ferrè, Palmeri and Helmer-Citterich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrizio Ferrè, Centre for Molecular Bioinformatics, Department of Biology, University of Rome Tor Vergata, Via della Ricerca Scientifica s.n.c., Rome, Italy e-mail:ZmFicml6aW8uZmVycmVAdW5pcm9tYTIuaXQ=