 Vasumathi Kameswaran
Vasumathi Kameswaran Klaus H. Kaestner
Klaus H. Kaestner- Department of Genetics and Institute for Diabetes, Obesity and Metabolism, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
Diabetes mellitus represents a group of complex metabolic diseases that result in impaired glucose homeostasis, which includes destruction of β-cells or the failure of these insulin-secreting cells to compensate for increased metabolic demand. Despite a strong interest in characterizing the transcriptome of the different human islet cell types to understand the molecular basis of diabetes, very little attention has been paid to the role of long non-coding RNAs (lncRNAs) and their contribution to this disease. Here we summarize the growing evidence for the potential role of these lncRNAs in β-cell function and dysregulation in diabetes, with a focus on imprinted genomic loci.
Introduction
Recent technological advances in the field of genome sequencing have paved the way for a new appreciation of non-coding RNAs in gene regulation. Ultra high-throughput transcriptome analyses have revealed that the vast majority of the genome is transcribed, with two-thirds of the human genome covered by processed transcripts, of which only a small fraction (<2%) is translated into proteins (Djebali et al., 2012). The identification of several common genomic and functional features of these untranslated RNAs has led to their categorization into various classes of non-coding RNAs. One such class that has been the focus of extensive research is that of long non-coding RNAs (lncRNAs).
LncRNAs are defined as transcripts longer than 200 bp that lack protein-coding potential (Guttman et al., 2009; Derrien et al., 2012; Batista and Chang, 2013; Fatica and Bozzoni, 2014). Like messenger RNAs, lncRNAs typically have multiple exons, are processed using canonical splice sites, and may exist as several isoforms (Ponjavic et al., 2007; Cabili et al., 2011; Derrien et al., 2012). In contrast to mRNAs, lncRNAs preferentially display nuclear localization, consistent with their proposed function in chromatin organization and regulation of gene expression (Khalil et al., 2009; Zhao et al., 2010; Derrien et al., 2012; Guttman and Rinn, 2012; Rinn and Chang, 2012; Fatica and Bozzoni, 2014).
Similar to protein-coding genes, lncRNA-encoding genes are marked by chromatin signatures typical of active transcription in the cell types where they are expressed, consisting of H3K4me3 (trimethylated lysine 4 in histone H3) at the promoter, followed by H3K36me3 along the transcribed regions (so-called “K4–K36 domains”; Guttman et al., 2009; Khalil et al., 2009; Cabili et al., 2011; Guttman and Rinn, 2012; Rinn and Chang, 2012). While lncRNA exons display weaker evolutionary conservation than those of protein-coding genes, there is evidence of positive selection for a subset of lncRNAs, which may be driven by constraints to maintain secondary structure required for functional interactions with their targets (Ponjavic et al., 2007; Guttman et al., 2009; Cabili et al., 2011; Ulitsky et al., 2011; Derrien et al., 2012). In contrast, the promoters of lncRNAs are as highly conserved as those of protein-coding genes (Carninci et al., 2005; Ponjavic et al., 2007; Guttman et al., 2009; Derrien et al., 2012; Batista and Chang, 2013). Despite their overall lower expression levels, lncRNAs exhibit a higher degree of tissue specificity compared to average protein-coding genes (Mercer et al., 2008; Cabili et al., 2011; Derrien et al., 2012; Batista and Chang, 2013; Fatica and Bozzoni, 2014).
Through numerous studies, several general principles of lncRNA function have emerged. LncRNAs have been shown to function both in cis, i.e., locally close to the site of their production, and in trans, i.e., at sites on other chromosomes. LncRNAs have been proposed to act as scaffolds for chromatin modifiers, blockers of transcription, antisense RNAs, microRNA sponges, protein decoys, and enhancers (Cech and Steitz, 2014; Fatica and Bozzoni, 2014). In fact, the act of transcription of a lncRNA itself can interfere with the regulatory function of a regulatory DNA sequence, as exemplified in yeast (Martens et al., 2004) and in mammalian imprinting (Latos et al., 2012). As a result of their diverse functions in multiple tissues, mis-regulation of lncRNAs can lead to failure of normal development and, consequently, to disease. Mammalian chromatin modifiers such as the repressive polycomb complexes often lack their own specific DNA-binding domains but instead contain RNA-binding elements. LncRNAs can play critical roles in directing these repressive chromatin modifying complexes to their target regions (Bernstein and Allis, 2005; Rinn et al., 2007; Zhao et al., 2010). One such example is the Foxf1-adjacent, non-coding developmental regulatory RNA (Fendrr), a lncRNA that interacts with the polycomb repressive complex 2 (PRC2) and is critical for heart development and function (Grote et al., 2013). Similarly, the well-characterized HOTAIR lncRNA, which is transcribed from the HOXC locus, is highly upregulated in primary breast tumors and was shown to function through the silencing of tumor suppressor genes in a PRC2-dependent manner [Gupta et al., 2010; See Maass et al. (2014) for a list of lncRNAs currently implicated in human diseases]. Taken together, these features suggest that lncRNAs and other non-coding RNA species may play an essential role in defining organismal complexity (Mattick and Makunin, 2006; Taft et al., 2007).
These findings raise the possibility that lncRNAs and other non-coding RNAs may be exciting molecular candidates to account for the unresolved genetic risk in complex diseases such as diabetes (Medici et al., 1999; Hyttinen et al., 2003). Diabetes mellitus represents a group of metabolic diseases that result in impaired glucose homeostasis. In the case of type 1 diabetes (T1D), metabolic impairment is the result of autoimmune destruction of insulin-producing pancreatic β-cells. In type 2 diabetes (T2D), the most prevalent form of the disease, the defect in glucose metabolism is the result of decreased sensitivity of peripheral tissues to insulin action, accompanied by failure of β-cells to compensate for the increased metabolic demand (Zimmet et al., 2001). Together, these diseases affect over 25 million Americans and account for $176 billion in healthcare costs per year in the US alone (Association, 2013), necessitating the pursuit of more effective and personalized treatments.
Significant efforts have been made to attain a better understanding of the causes of diabetes at the molecular level. Linkage analysis of affected families led to the successful identification of causal gene mutation in several rare, Mendelian forms of the disease (Fajans et al., 2001; O’Rahilly, 2009). However, large-scale efforts to identify DNA variants associated with more common forms of diabetes through genome-wide association studies (GWAS) have predominantly identified candidate variants that lie in non-coding regions and with as yet unknown functions (McCarthy, 2010). Thus, to improve our current understanding of the molecular basis of diabetes mellitus and to develop better treatment strategies, we need to carefully characterize the transcriptome of pancreatic β-cells, with a focus on elucidating the functions of non-coding transcripts. In this review, we present a summary of recent evidence for a role of lncRNAs in the regulation of β-cell function and their potential contribution to the pathogenesis of diabetes.
β-Cell lncRNAs
The most comprehensive catalog of human lncRNAs expressed in β-cells published thus far is that by Morán et al. (2012). In this study, the authors profiled whole islet and FACS-sorted β-cells and identified 1,128 distinct transcripts that displayed many of the typical properties of lncRNAs described above, including the “K4–K36” histone modification domains, lack of protein-coding potential, and non-uniform expression levels among tissues. Most notably, the lncRNAs identified were roughly five times more islet-specific compared to general protein-coding genes, and the vast majority had orthologous genes in the mouse genome. Ku et al. (2012) similarly characterized mouse islet- and β-cell-specific transcripts and identified 1,359 high-confidence lncRNAs with several of the aforementioned properties. Using high-throughput transcriptome analysis of sorted human islets, lncRNAs expressed in α-cells have also been identified (Bramswig et al., 2013).
Of particular interest was the fact that lncRNAs were often found in proximity to critical islet-specific transcription factors (Ku et al., 2012; Morán et al., 2012). Thus, protein-coding genes adjacent to islet-enriched lncRNAs were also more likely to be islet-specific than the average protein-coding gene (Morán et al., 2012). This correlation has led to the suggestion that lncRNAs and nearby protein-coding genes share common regulatory elements. Indeed, lncRNAs were often found in large regions of open chromatin that were uniquely associated with protein-coding genes expressed highly in islets (Gaulton et al., 2010).
The temporal expression of islet lncRNAs has also been studied by Morán et al. (2012) in human embryonic pancreases as well as in a stepwise in vitro β-cell differentiation model using human embryonic stem (ES) cells (developed by Kroon et al., 2008). Unlike some lncRNAs that are known to be critical to early stages of embryonic development (Guttman et al., 2011; Grote et al., 2013), the expression of a majority of islet lncRNAs identified in this study (Morán et al., 2012) is restricted to differentiated, mature endocrine cells. The orthologous mouse lncRNAs (e.g., Mi-Lnc80) exhibit similar cell- and stage-specific expression.
The characteristics of these islet lncRNAs imply a role for these RNAs in mature β-cell function. To test this hypothesis, Morán et al. (2012) used short hairpin RNAs (shRNAs) to suppress the activity of one such lncRNA transcript in the human EndoC-βH1 β-cell line (Ravassard et al., 2011). From a panel of known islet-specific transcripts, the authors identified GLIS3 as a downstream target of HI-LNC25, a lncRNA that shares a regulatory domain with MAFB. Variants at the GLIS3 locus are associated with different risks for T1D (Barrett et al., 2009), elevated fasting glucose levels (Dupuis et al., 2010), as well as T2D (Cho et al., 2012). Loss-of function studies suggest that GLIS3 encodes a transcription factor critical for regulating the expression of insulin and several key islet-transcription factors, and may confer risk for both T1D and T2D by resulting in diminished β-cell numbers and by promoting the formation of a pro-apoptotic splice variant of the protein Bim (Kang et al., 2009; Nogueira et al., 2013; ZeRuth et al., 2013). However, the shRNA-mediated decrease in GLIS3 mRNA levels had no impact on glucose-stimulated insulin secretion or insulin transcript levels in the transduced EndoC-βH1 β-cell line, possibly because this cell line does not recapitulate all aspects of β-cell function in vivo. Additionally, only a minor fraction of β-cell expressed lncRNAs was responsive to elevated glucose levels in human islets.
As previously noted, several risk variants for common forms of diabetes identified by GWAS do not change the protein-coding potential of known genes, suggesting that they might affect as yet unidentified regulatory elements (McCarthy, 2010). Using a computational tool known as MAGENTA to search for enrichment of genetic associations in a predefined set of genes (Segrè et al., 2010), Morán et al. (2012) determined that the islet lncRNA genes identified in their study were in fact highly enriched for risk alleles associated with T2D and related phenotypes, further underscoring the need to interrogate the function of these RNAs in β-cell biology.
Overall, these studies highlighted lncRNAs as a major component of the β-cell transcriptome that is cell-type-specific, developmentally regulated, and evolutionarily conserved with strong associations to disease risk. However, it still remains to be determined how these lncRNAs may contribute to β-cell function, and if their mis-regulation may play a role in diabetes. Their expression in EndoC-βH1 cells and mouse islets provides additional platforms to evaluate their function in a systematic and comprehensive manner. Future studies will also need to address the question of whether the lncRNAs identified thus far act in cis (on neighboring islet protein-coding genes) or in trans to exert their function.
Imprinting
Some of the best characterized lncRNAs to date were first uncovered in early studies of imprinting and dosage compensation of the X-chromosome (Brannan et al., 1990; Brown et al., 1991; Fatica and Bozzoni, 2014). Imprinting refers to the biased expression of genes depending on the parental origin of the chromosome. This process is tightly regulated, typically through epigenetic modifications such as DNA methylation at cis-acting elements known as “imprinting control regions” (ICRs), to establish and maintain mono-allelic expression of specific genes (Thorvaldsen and Bartolomei, 2007). Methylation at the ICRs is maintained despite active demethylation and dynamic reprogramming in the newly formed zygote, and is only altered during establishment of methylation pattern in a sex-specific manner during primordial germ cell development (Bartolomei and Ferguson-Smith, 2011). Imprinted loci are generally found in large clusters, where both maternally- and paternally expressed genes are interspersed. Frequently, the protein-coding genes are expressed from one parental allele, while non-coding genes are expressed from the other (Barlow, 2011). LncRNAs play an essential role in the regulation of mono-allelic expression, either by acting in cis as an antisense molecule to block the transcriptional machinery, or by directly recruiting repressive chromatin modifiers to silence reciprocally expressed genes (Lee and Bartolomei, 2013).
While imprinting is most extensively studied in the context of fetal development, tissue-specific regulation in adult tissues has also been observed (Barlow, 2011; Lee and Bartolomei, 2013). As a result, several imprinted genes are also implicated in human diseases that arise from somatic tissues. One such example is that of the maternally expressed adipose tissue transcription factor, KLF14 (Parker-Katiraee et al., 2007), which is associated with risk for both T2D and high-density lipoprotein disorders (Teslovich et al., 2010; Voight et al., 2010; Small et al., 2011). Perhaps the functionally haploid nature of these loci results in their increased likelihood to be associated with susceptibility to disease, as mutations in these genes, when found on the maternal chromosome that is expressed, cannot be “covered” by the gene from the other, silenced paternal allele. This may be particularly true for metabolic disorders, as several imprinted genes encode dosage-sensitive proteins related to growth factors and energy metabolism. Interestingly, several risk variants for type 1 and type 2 diabetes identified through GWAS are located in imprinted loci including KCNQ1, MEG3, PLAGL1, and GRB10. A few of these are discussed below in the context of islet and β-cell function.
DLK1–MEG3 Locus
Recently, we identified the maternally expressed non-coding RNAs of the imprinted DLK1–MEG3 locus as down-regulated in human islets from T2D donors (Kameswaran et al., 2014). This gene cluster is located on human 14q32 (mouse chromosome 12) and contains three paternally expressed protein-coding genes, DLK1, RTL1, and DIO3. DLK1 is a non-canonical Notch ligand that is expressed in many embryonic tissues (Falix et al., 2012) and is a well-established negative regulator of adipocyte differentiation (Smas and Sul, 1993; Mitterberger et al., 2012; Abdallah et al., 2013). DLK1 is highly expressed in human and mouse β-cells (Tornehave et al., 1996; Dorrell et al., 2011; Appelbe et al., 2013). While DLK1 was demonstrated to be stimulated by growth hormone and prolactin expression in rat islets, including during pregnancy, it is not directly responsible for the mitogenic effects of these hormones on islets (Carlsson et al., 1997; Friedrichsen et al., 2003). Additionally, loss of expression of Dlk1 in unchallenged mouse β-cells does not cause any observable phenotype (Appelbe et al., 2013). Rtl1 (Retrotransposon-like 1) is critical for normal placental development and its loss results in severe developmental defects and late-fetal lethality (Sekita et al., 2008).
The maternally expressed genes are all non-coding RNAs, consisting of the lncRNA, Maternally Expressed Gene 3 (MEG3, known as Gtl2 in mice), as well as a large cluster of microRNAs (miRNAs) and snoRNAs (Schmidt et al., 2000; Seitz et al., 2004; da Rocha et al., 2008). In several tissues, including human islets, the non-coding RNAs are all derived from a single transcript that initiates from the MEG3 promoter (Tierling et al., 2006; da Rocha et al., 2008; Kameswaran et al., 2014).
Reciprocal imprinting is established by methylation of two differentially methylated regions (DMRs) on the paternal allele, one located ∼13 kb upstream of the MEG3 transcription start site (IG-DMR), and the other overlapping with the promoter of the MEG3 poly cistronic transcript (MEG3-DMR; Figure 1). While the IG-DMR is the primary ICR for this imprinted cluster, the MEG3-DMR is also critical to regulating and maintaining imprinting at this region (Kagami et al., 2010). Failure to maintain imprinting at this locus can lead to either maternal or paternal uniparental disomy (UPD) of chromosome 14, which causes distinct and severe developmental disorders (Kagami et al., 2008).
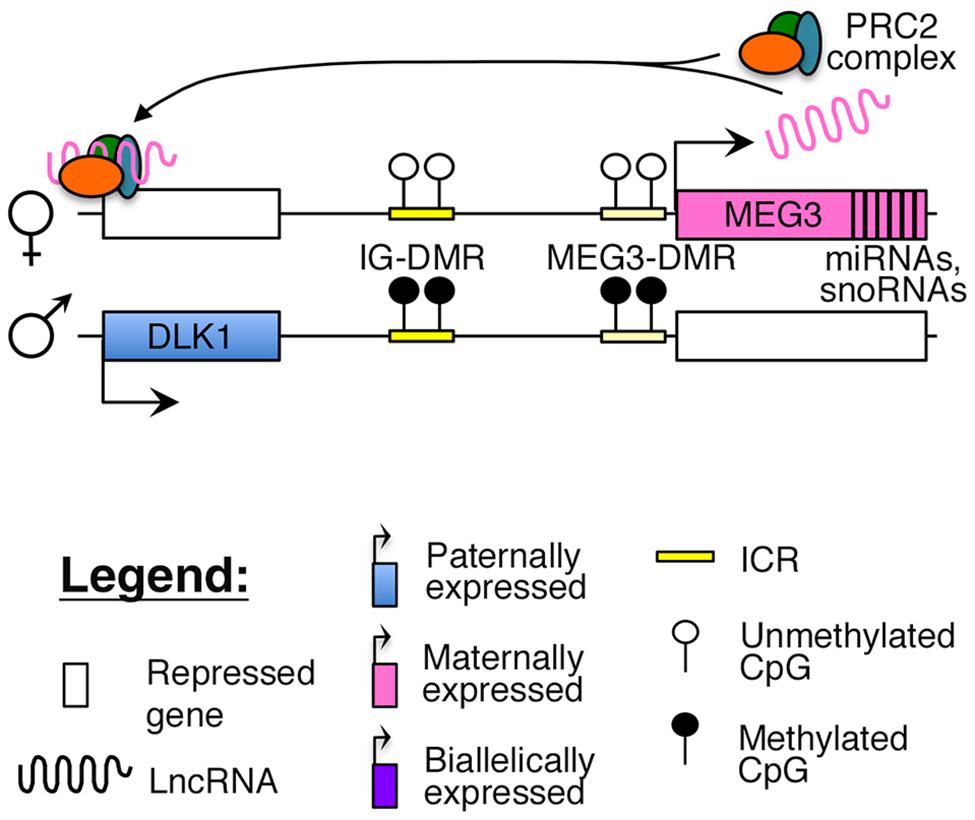
FIGURE 1. Proposed model of imprinting at the DLK1–MEG3 locus: the DLK1–MEG3 imprinted region contains a primary ICR (IG-DMR) and secondary (MEG3–DMR) ICR that overlaps with the promoter of the MEG3. Both ICRs are paternally methylated. In mouse ES cells, the Meg3 lncRNA is believed to direct PRC2 mediated silencing of Dlk1 (Zhao et al., 2010).
Increased methylation of the MEG3-DMR and related loss of MEG3 expression has been observed in several human cancers, such as pituitary and renal cell cancers and multiple myeloma (Zhao et al., 2005; Kawakami et al., 2006; Benetatos et al., 2008) to name a few (further reviewed by Benetatos et al., 2011). These studies, coupled with in vitro experiments, suggest that MEG3 functions as a tumor suppressor by activating p53, in a manner dependent upon the secondary structure of the MEG3 RNA (Zhou et al., 2007, 2012). Furthermore, decreased expression of MEG3 and hypermethylation of the DMRs may single-handedly explain the subtle phenotypic differences between induced pluripotent stem cells (iPSCs) and ES cells, such as the decreased efficiency in generating chimeric mice from iPSCs (Stadtfeld et al., 2010).
Similar to the aforementioned examples, decreased expression of MEG3 and the associated miRNAs in T2D islets strongly correlates with hypermethylation of the MEG3-DMR (Kameswaran et al., 2014). Additionally, a single nucleotide polymorphism (SNP) (rs941576) located in an intron of MEG3 was found to be associated with T1D, with the risk allele being transmitted more frequently from the father than the mother of the affected offspring (Wallace et al., 2010). Overall, these examples provide compelling evidence for the importance of MEG3 and the regulation of this imprinted region in several diseases. Despite the strong disease association of this lncRNA, and the fact that genes in this imprinted cluster are very highly expressed in human β-cells (Dorrell et al., 2011; Kameswaran et al., 2014), there are currently no postulated mechanisms for its potential role in β-cell function and diabetes pathogenesis.
Recent studies have suggested that similar to other nuclear lncRNAs, MEG3 also directly interacts with the PRC2 complex in ES cells to guide the repressive histone modification mark H3K27me3 to its target sites (Zhao et al., 2010; Kaneko et al., 2014). One study identified Dlk1 as a direct target of the Meg3-PRC2 complex in mouse ES cells (Figure 1), although this finding could not be replicated in MEG3-expressing human iPSCs, where MEG3 was found to function in trans (Zhao et al., 2010; Kaneko et al., 2014). A careful characterization of MEG3-PRC2 complex targets in adult pancreatic islets will provide better insights into the role of this lncRNA in β-cell function.
KCNQ1 Locus
The KCNQ1 gene, encoding a voltage-gated potassium channel, has been of great interest to the β-cell biology field due to its strong disease association. The gene is located in an imprinted locus on human 11p15.5, adjacent to another independently regulated imprinted locus, H19–IGF2. This region was implicated as a molecular candidate for Beckwith–Wiedemann syndrome (BWS), a disorder characterized by prenatal macrosomia, predisposition for tumor development and frequently, hyperinsulinemic hypoglycemia (Lee et al., 1997, 1999; Hussain et al., 2005). This imprinted region consists of several conserved, maternally expressed protein-coding genes, such as the cell cycle inhibitor CDKN1C, and a paternally expressed antisense lncRNA, KCNQ1 overlapping transcript1 (KCNQ1OT1; Monk et al., 2006). Loss of imprinting in this locus can lead to the suppression of CDKN1C, which is sufficient to cause re-entry of adult human β-cells into the cell cycle (Avrahami et al., 2014).
Imprinting of this region is maintained by a maternally methylated ICR, known as the KvDMR, which is also the promoter for KCNQ1OT1 (Figure 2). To maintain appropriate mono-allelic expression of imprinted genes in this locus, the KvDMR is hypomethylated on the paternal allele, leading to expression of the KCNQ1OT1 lncRNA and subsequent repression of the maternal, protein-coding genes on the same allele (Fitzpatrick et al., 2002; Ideraabdullah et al., 2008), possibly by facilitating intra-chromasomal looping to direct the repressive PRC2 complex to their promoter (Figure 2; Zhao et al., 2010; Zhang et al., 2014).
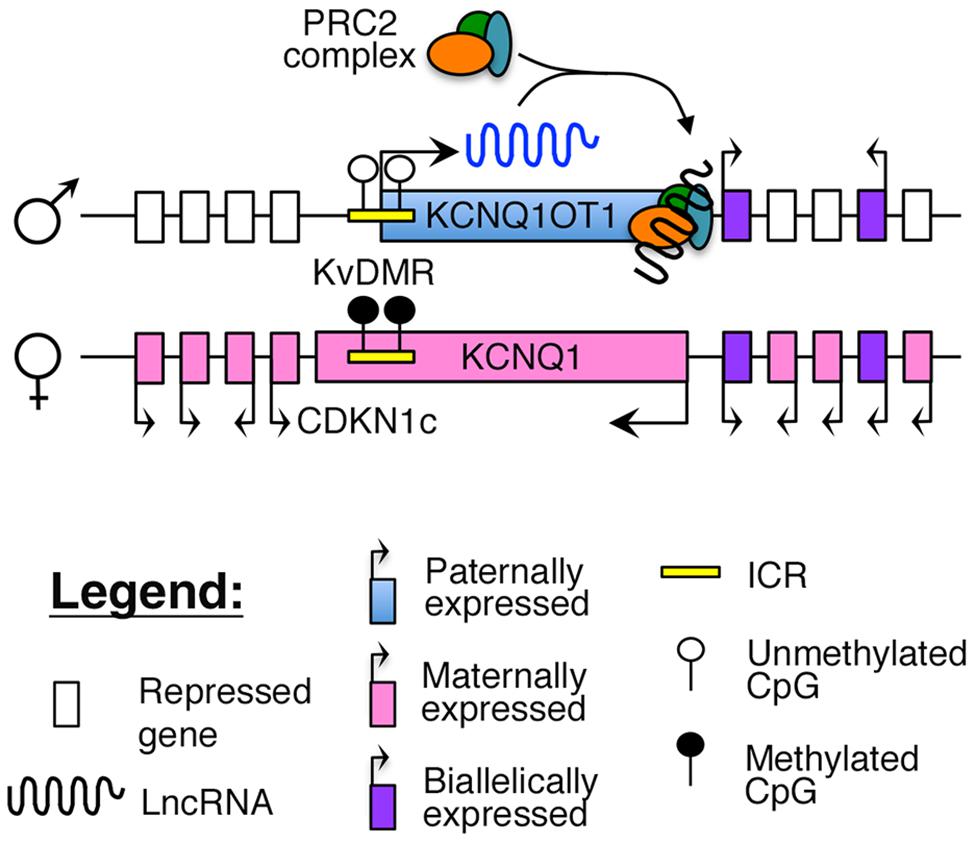
FIGURE 2. Proposed model of imprinting at the KCNQ1 locus: the KCNQ1OT1 lncRNA is expressed from the paternally unmethylated KvDMR ICR, which is methylated on the maternal allele. Recent evidence suggests that KCNQ1OT1 can directly recruit the PRC2 complex and facilitate intra-chromosomal looping to the KCNQ1 promoter (Zhang et al., 2014).
The KCNQ1 locus harbors at least two independently identified and replicated GWAS signals at SNPs located in the intron of the KCNQ1 gene (rs2237892), with one overlapping the KCNQ1OT1 lncRNA (rs231362; Unoki et al., 2008; Yasuda et al., 2008; Kong et al., 2009; Voight et al., 2010). Additional SNPs in this gene, such as rs2237895, are also reported to be associated with T2D risk in specific ethnic populations (Unoki et al., 2008). While these SNPs are predicted to confer risk for diabetes only when maternally inherited (Kong et al., 2009), the risk alleles do not correlate with each other (Kong et al., 2009; Voight et al., 2010) and have opposing effects on docking of insulin granules (Rosengren et al., 2012).
To investigate how these T2D risk variants may affect allelic expression and imprinting of this region, Travers et al. (2013) correlated the risk SNP genotypes with DNA methylation and expression patterns of the imprinted genes in human fetal pancreas and adult islets. This study revealed that fetal samples homozygous for the rs2237895 risk allele had marginally increased methylation levels at the KvDMR region. As this was not observed in the adult, these results suggest that effects of the risk allele are likely be established during early stages of islet development, as KCNQ1 and KCNQ1OT1 are only imprinted in fetal but not adult tissues (Monk et al., 2006; Travers et al., 2013). Overall, this study proposes a model whereby each risk allele for the rs2237895 SNP leads to increased methylation of the KvDMR, and consequently, decreased expression of KCNQ1OT1. However, there was no observable difference in KCNQ1 or KCNQ1OT1 expression in samples used for this study. On the contrary, KCNQ1OT1 transcript levels have been shown to be significantly elevated in T2D islets (where SNP genotype was not determined; Morán et al., 2012), which parallels an overall decrease in methylation at several tested CpGs near the KCNQ1 gene (Dayeh et al., 2014). Thus, the interpretation of variants to disease pathology at this region has been contradictory and challenging. Nevertheless, the regulation of this locus and the lncRNA KCNQ1OT1 remains relevant to β-cell biology and T2D pathogenesis.
H19–IGF2 Locus
The H19–IGF2 locus resides adjacent to the KCNQ1 region on human 11p15.5. The region consists of the paternally expressed insulin-like growth factor 2 (IGF2) gene and maternally expressed H19 lncRNA (Brannan et al., 1990; DeChiara et al., 1990; Bartolomei et al., 1991). The IGF2 protein functions as a growth factor essential for embryonic development (DeChiara et al., 1990), whereas H19 may function as a tumor suppressor (Hao et al., 1993). Imprinting at this locus is maintained by an ICR, which is selectively methylated on the paternal allele. The insulator protein, CCCTC-binding factor (CTCF), binds to critical regulatory regions in the unmethylated ICR on the maternal allele, thus blocking access of downstream enhancers to the IGF2 promoter (Figure 3; Stadnick et al., 1999; Bell and Felsenfeld, 2000; Engel et al., 2004).
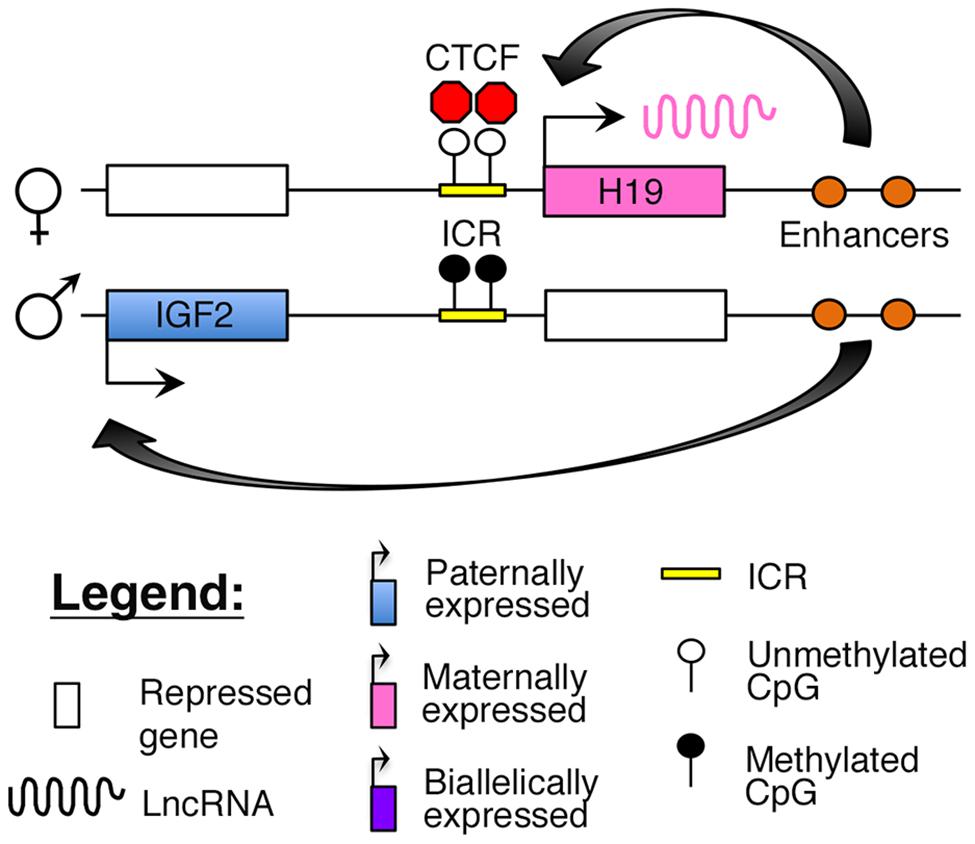
FIGURE 3. Proposed model of imprinting at the H19–IGF2 locus: the H19–IGF2 locus consists of a paternally methylated ICR. On the maternal allele, this ICR is unmethylated and is bound by the insulator protein CTCF that prevents access of the IGF2 promoter to downstream enhancers.
Loss of methylation at the H19/IGF2 ICR results in short body length and low birth weight, both in rodent models (DeChiara et al., 1990) as well as in humans, such as patients with Silver-Russell syndrome, a developmental disorder characterized by intrauterine and postnatal growth retardation (Gicquel et al., 2005). This has also been observed in humans who were periconceptually exposed to famine (Heijmans et al., 2008). There is growing evidence that intra-uterine exposure to malnutrition can predispose the offspring to metabolic complications including β-cell dysfunction and diabetes later in life (Ravelli et al., 1998; Roseboom et al., 2006). This theory is commonly referred to as the “thrifty phenotype hypothesis” (Hales, 2001) and is thought to be mediated primarily through environmentally induced epigenetic changes to key metabolic regulators (Park et al., 2008; Bramswig and Kaestner, 2012). However, first and second generation progeny of mice exposed to gestational diabetes were found to have impaired glucose tolerance with hypermethylation of the H19 ICR in islets (Ding et al., 2012). These contradicting observations may be a result of different nutrient availability that the developing fetus was exposed to, as well as the varying lengths of exposure. The above studies suggest that the H19–IGF2 locus is highly responsive to these changes in the intrauterine milieu and may represent a prognostic marker of metabolic complications later in life.
Hypermethylation of the H19–IGF2 ICR has been observed in some cases of BWS (Ohlsson et al., 1993), as well as in focal congenital hyperinsulinism (FoCHI), a glucose metabolism disorder characterized by unbridled insulin secretion from hyperplastic islet cells and consequent hypoglycemia (de Lonlay et al., 1997). Increased methylation at this ICR would be predicted to result in decreased H19 expression, loss of imprinting at this region and a concomitant increase in IGF2 expression. Although over-expression of IGF2 in mouse β-cells recapitulates the FoCHI phenotype (Devedjian et al., 2000), IGF2 expression was variable in human FoCHI lesions (Fournet et al., 2001). On the contrary, H19 transcript levels were consistently down-regulated in these cells, suggesting that H19 may have an important regulatory role in restraining islet-proliferation. This hyperproliferative phenotype, accompanied by suppression of H19 has also been reported in Wilms’ tumor (Cui et al., 1997). Taken together, the H19 lncRNA may function as a critical regulator of β-cell function and proliferation either on its own or indirectly through the regulation of IGF2 levels.
ZAC–HYMAI Locus
Transient neonatal diabetes (TNDM) is a rare form of diabetes mellitus characterized by hyperglycemia and low insulin levels within the first year of birth (Temple et al., 2000). This form of diabetes is distinct from T1D as there is no evidence for autoimmunity (Abramowicz et al., 1994; Shield et al., 1997). Although it usually resolves by 2 years of age, children with TNDM are at a higher risk of developing T2D later in life (Temple et al., 2000). The molecular cause of this disease was identified to be abnormal imprinting of chromosome 6q24, which encompasses the cell cycle regulator, ZAC/PLAGL1, and the lncRNA, HYMAI (Abramowicz et al., 1994; Arima et al., 2000; Gardner et al., 2000; Kamiya et al., 2000; Mackay et al., 2002). Both ZAC and HYMAI share a common imprinted promoter (P1 in Figure 4), which also serves as the ICR, and are expressed from the paternal allele (Arima et al., 2000; Mackay et al., 2002). However, tissue-specific usage of an alternative promoter (P2 in Figure 4) that drives biallelic expression of ZAC has also been reported (Valleley et al., 2007).
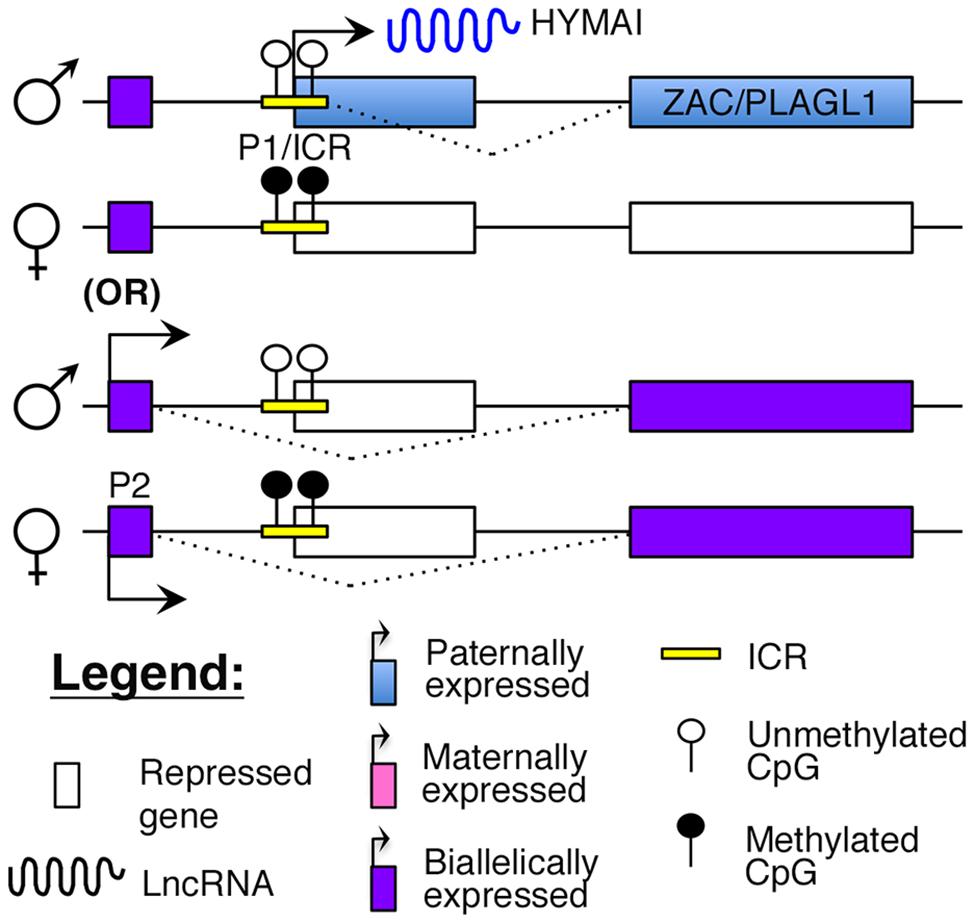
FIGURE 4. Proposed model of imprinting at the ZAC–HYMAI locus: ZAC/PLAGL1 and the lncRNA HYMAI are both paternally expressed from a common promoter that is also the ICR. However, in some tissues, ZAC is biallelically expressed from an upstream promoter.
ZAC encodes a zinc finger protein that regulates apoptosis and cell cycle arrest (Spengler et al., 1997). The protein is expressed at very high levels in insulin-producing cells in the human fetal pancreas, but not adult islets (Du et al., 2011). ZAC can also function as a transcriptional activator of CDKN1C and KCNQ1OT1 (Arima et al., 2005). ZAC is believed to control the induction of the pituitary adenylate cyclase-activating polypeptide (PACAP), a strong activator of glucose-stimulated insulin secretion (Yada et al., 1994; Ciani et al., 1999). These features of the ZAC gene make this a strong candidate for the pathogenesis of TNDM. However, the mechanism of imprinting and the function of HYMAI in the context of TNDM have yet to be established.
MALAT1, an Abundant lncRNA
The metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is a highly conserved lncRNA that is mis-regulated in several tumors (Ji et al., 2003; Gutschner et al., 2013). MALAT1 is very abundantly expressed (higher than many housekeeping genes) in multiple cell types, including the pancreas (Ji et al., 2003) and in purified human α- and β-cells (Dorrell et al., 2011). Additionally, MALAT1 is encoded within an active enhancer cluster with several binding sites for islet-transcription factors (Pasquali et al., 2014), making this is an intriguing candidate for gene regulation in human islets.
Metastasis-associated lung adenocarcinoma transcript 1 has several interacting partners through which it may mediate its function. One such interacting partner is DGCR8, a double-stranded RNA binding protein that together with Drosha mediates miRNA bioprocessing (Macias et al., 2012). MALAT1 was found to be bound to Argonaute (Ago), the primary effector of miRNA function in HeLa cells (Weinmann et al., 2009). MALAT1 was also found to be associated with Ago in human islets, suggesting that this lncRNA may be regulated by miRNAs in human cells (Kameswaran et al., 2014). In fact, we discovered several sequences that consisted of miRNAs fused to MALAT1 while assaying miRNAs and their targets that were bound to Ago in human islets. These chimeric reads were the result of ligation of two adjacent RNA species present in the RISC complex with Ago (Helwak et al., 2013), and proved that MALAT1 is regulated by several miRNAs in human islets (Kameswaran et al., 2014).
Metastasis-associated lung adenocarcinoma transcript 1 can also regulate gene expression through its association with different nuclear sub-compartments (Hutchinson et al., 2007; Yang et al., 2011; Gutschner et al., 2013). One example of this is MALAT1 localization in nuclear speckles, which are nuclear domains where splicing factors are stored and post-transcriptionally modified (Hutchinson et al., 2007; Mao et al., 2011). Through the modification of critical splicing factors, MALAT1 has been shown to contribute to alternative splicing (Tripathi et al., 2010). However, despite the abundance of this lncRNA and the early suggestions of its function from in vitro studies, mice lacking MALAT1 displayed no obvious phenotype in the absence of additional pathological stressors and exhibit largely normal nuclear speckle formation and alternative splicing patterns (Eißmann et al., 2012; Nakagawa et al., 2012; Zhang et al., 2012). Thus, the role of this lncRNA remains to be determined.
Perspective
The exciting discovery of lncRNAs and the growing recognition of their involvement in human pathogenesis have added a new level of complexity to our understanding of gene regulation. However, due to the range of sequencing and bioinformatic tools currently available, the rate of discovery of new lncRNAs has surpassed our ability to examine their function. This gap between lncRNA gene discovery and function currently holds true in the field of β-cell biology as well, necessitating the systematic analysis of mouse and human islet lncRNAs identified to date (Ku et al., 2012; Morán et al., 2012).
Factors such as overlap between the human and mouse α- and β-cell lncRNA complements (Ku et al., 2012; Morán et al., 2012; Bramswig et al., 2013), degree of conservation, expression, associated protein-coding genes, and relative distance from GWAS SNP variants may be good early predictors of important lncRNAs. However, these parameters alone may underestimate other essential candidates, as some lncRNAs exhibit low primary sequence conservation despite crucial function (Nesterova et al., 2001), or, conversely, a dispensable function despite high sequence conservation and expression (Zhang et al., 2012). These observations emphasize the need for careful loss-of-function experiments in appropriate model systems induced by metabolic and/or inflammatory challenges to clearly understand the function of these lncRNAs. Although many of the human β-cell lncRNAs are expressed in the EndoC-βH1 cell line that somewhat resembles human β-cells in vitro (Ravassard et al., 2011), targeted deletion or inhibition in mouse and human islets may be necessary in some cases to reveal their function, as seen in the example of HI-LNC25 discussed above (Morán et al., 2012).
While the loss-of-function of even abundant lncRNAs such as MALAT1 may sometimes result in a lack of phenotype (Eißmann et al., 2012; Nakagawa et al., 2012; Zhang et al., 2012), lessons from the miRNA field suggest that additional physiological and environmental stressors may be necessary to truly elucidate the function of these non-coding RNAs (Mendell and Olson, 2012). Additionally, in order to study the role of lncRNAs in the context of loss-of-function, a careful analysis of the genomic location of the lncRNAs may be required to evaluate the best method of gene silencing, as targeted recombination may result in disruption of overlapping protein-coding transcripts or their regulatory domains, further confounding data interpretation.
Given the broad range of human diseases that lncRNAs are now associated with, it is perhaps not surprising that there is growing evidence for their role in β-cell function and diabetes pathogenesis. Revealing their function will undoubtedly lead to a new wave of exciting targets to explore for therapeutic development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to colleagues whose work we could not cite due to the limited scope of this review. We thank Dr. John Le Lay and other members of the Kaestner lab for useful discussions and suggestions on this manuscript. Related work in the Kaestner lab was supported through NIDDK grant R01-DK088383.
References
Abdallah, B. M., Beck-Nielsen, H., and Gaster, M. (2013). FA1 induces pro-inflammatory and anti-adipogenic pathways/markers in human myotubes established from lean, obese, and type 2 diabetic subjects but not insulin resistance. Front. Endocrinol. (Lausanne) 4:45. doi: 10.3389/fendo.2013.00045
Abramowicz, M. J., Andrien, M., Dupont, E., Dorchy, H., Parma, J., Duprez, L.,et al. (1994). Isodisomy of chromosome 6 in a newborn with methylmalonic acidemia and agenesis of pancreatic beta cells causing diabetes mellitus. J. Clin. Invest. 94, 418–421. doi: 10.1172/JCI117339
Appelbe, O. K., Yevtodiyenko, A., Muniz-Talavera, H., and Schmidt, J. V. (2013). Conditional deletions refine the embryonic requirement for Dlk1. Mech. Dev. 130, 143–159. doi: 10.1016/j.mod.2012.09.010
Arima, T., Drewell, R. A., Oshimura, M., Wake, N., and Surani, M. A. (2000). A novel imprinted gene, HYMAI, is located within an imprinted domain on human chromosome 6 containing ZAC. Genomics 67, 248–255. doi: 10.1006/geno.2000.6266
Arima, T., Kamikihara, T., Hayashida, T., Kato, K., Inoue, T., Shirayoshi, Y.,et al. (2005). ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 33, 2650–2660. doi: 10.1093/nar/gki555
Association, A. D. (2013). Economic costs of diabetes in the U.S. in 2012. Diabetes Care 36, 1033–1046. doi: 10.2337/dc12-2625
Avrahami, D., Li, C., Yu, M., Jiao, Y., Zhang, J., Naji, A.,et al. (2014). Targeting the cell cycle inhibitor p57Kip2 promotes adult human β cell replication. J. Clin. Invest. 124, 670–674. doi: 10.1172/JCI69519
Barlow, D. P. (2011). Genomic imprinting: a mammalian epigenetic discovery model. Annu. Rev. Genet. 45, 379–403. doi: 10.1146/annurev-genet-110410–132459
Barrett, J. C., Clayton, D. G., Concannon, P., Akolkar, B., Cooper, J. D., Erlich, H. A.,et al. (2009). Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 41, 703–707. doi: 10.1038/ng.381
Bartolomei, M. S., and Ferguson-Smith, A. C. (2011). Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 3:a002592. doi: 10.1101/cshperspect.a002592
Bartolomei, M. S., Zemel, S., and Tilghman, S. M. (1991). Parental imprinting of the mouse H19 gene. Nature 351, 153–155. doi: 10.1038/351153a0
Batista, P. J., and Chang, H. Y. (2013). Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307. doi: 10.1016/j.cell.2013.02.012
Bell, A. C., and Felsenfeld, G. (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. doi: 10.1038/35013100
Benetatos, L., Dasoula, A., Hatzimichael, E., Georgiou, I., Syrrou, M., and Bourantas, K. L. (2008). Promoter hypermethylation of the MEG3 (DLK1/MEG3) imprinted gene in multiple myeloma. Clin. Lymphoma Myeloma 8, 171–175. doi: 10.3816/CLM.2008.n.021
Benetatos, L., Vartholomatos, G., and Hatzimichael, E. (2011). MEG3 imprinted gene contribution in tumorigenesis. Int. J. Cancer 129, 773–779. doi: 10.1002/ijc.26052
Bernstein, E., and Allis, C. D. (2005). RNA meets chromatin. Genes Dev. 19, 1635–1655. doi: 10.1101/gad.1324305
Bramswig, N. C., Everett, L. J., Schug, J., Dorrell, C., Liu, C., Luo, Y.,et al. (2013). Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J. Clin. Invest. 123, 1275–1284. doi: 10.1172/JCI66514
Bramswig, N. C., and Kaestner, K. H. (2012). Epigenetics and diabetes treatment: an unrealized promise? Trends Endocrinol. Metab. 23, 286–291. doi: 10.1016/j.tem.2012.02.002
Brannan, C. I., Dees, E. C., Ingram, R. S., and Tilghman, S. M. (1990). The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 10, 28–36.
Brown, C. J., Ballabio, A., Rupert, J. L., Lafreniere, R. G., Grompe, M., Tonlorenzi, R.,et al. (1991). A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349, 38–44. doi: 10.1038/349038a0
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A.,et al. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. doi: 10.1101/gad.17446611
Carlsson, C., Tornehave, D., Lindberg, K., Galante, P., Billestrup, N., Michelsen, B.,et al. (1997). Growth hormone and prolactin stimulate the expression of rat preadipocyte factor-1/delta-like protein in pancreatic islets: molecular cloning and expression pattern during development and growth of the endocrine pancreas. Endocrinology 138, 3940–3948. doi: 10.1210/endo.138.9.5408
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N.,et al. (2005). The transcriptional landscape of the mammalian genome. Science 309, 1559–1563. doi: 10.1126/science.1112014
Cech, T. R., and Steitz, J. A. (2014). The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94. doi: 10.1016/j.cell.2014.03.008
Cho, Y. S., Chen, C.-H., Hu, C., Long, J., Ong, R. T. H., Sim, X.,et al. (2012). Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat. Genet. 44, 67–72. doi: 10.1038/ng.1019
Ciani, E., Hoffmann, A., Schmidt, P., Journot, L., and Spengler, D. (1999). Induction of the PAC1-R (PACAP-type I receptor) gene by p53 and Zac. Brain Res. Mol. Brain Res. 69, 290–294. doi: 10.1016/S0169-328X(99)00116-3
Cui, H., Hedborg, F., He, L., Nordenskjöld, A., Sandstedt, B., Pfeifer-Ohlsson, S.,et al. (1997). Inactivation of H19, an imprinted and putative tumor repressor gene, is a preneoplastic event during Wilms’ tumorigenesis. Cancer Res. 57, 4469–4473.
da Rocha, S. T., Edwards, C. A., Ito, M., Ogata, T., and Ferguson-Smith, A. C. (2008). Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 24, 306–316. doi: 10.1016/j.tig.2008.03.011
Dayeh, T., Volkov, P., Salö, S., Hall, E., Nilsson, E., Olsson, A. H.,et al. (2014). Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 10:e1004160. doi: 10.1371/journal.pgen.1004160
DeChiara, T. M., Efstratiadis, A., and Robertson, E. J. (1990). A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78–80. doi: 10.1038/345078a0
de Lonlay, P., Fournet, J. C., Rahier, J., Gross-Morand, M. S., Poggi-Travert, F., Foussier, V.,et al. (1997). Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J. Clin. Invest. 100, 802–807. doi: 10.1172/JCI119594
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H.,et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
Devedjian, J. C., George, M., Casellas, A., Pujol, A., Visa, J., Pelegrín, M.,et al. (2000). Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J. Clin. Invest. 105, 731–740. doi: 10.1172/JCI5656
Ding, G.-L., Wang, F.-F., Shu, J., Tian, S., Jiang, Y., Zhang, D.,et al. (2012). Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 61, 1133–1142. doi: 10.2337/db11-1314
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A.,et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Dorrell, C., Schug, J., Lin, C. F., Canaday, P. S., Fox, A. J., Smirnova, O.,et al. (2011). Transcriptomes of the major human pancreatic cell types. Diabetologia 54, 2832–2844. doi: 10.1007/s00125-011-2283-5
Dupuis, J., Langenberg, C., Prokopenko, I., Saxena, R., Soranzo, N., Jackson, A. U.,et al. (2010). New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 42, 105–116. doi: 10.1038/ng.520
Du, X., Rousseau, M., Ounissi-Benkalha, H., Marchand, L., Jetha, A., Paraskevas, S.,et al. (2011). Differential expression pattern of ZAC in developing mouse and human pancreas. J. Mol. Histol. 42, 129–136. doi: 10.1007/s10735-011-9315-9
Eißmann, M., Gutschner, T., Hämmerle, M., Günther, S., Caudron-Herger, M., Groß, M.,et al. (2012). Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 9, 1076–1087. doi: 10.4161/rna.21089
Engel, N., West, A. G., Felsenfeld, G., and Bartolomei, M. S. (2004). Antagonism between DNA hypermethylation and enhancer-blocking activity at the H19 DMD is uncovered by CpG mutations. Nat. Genet. 36, 883–888. doi: 10.1038/ng1399
Fajans, S. S., Bell, G. I., and Polonsky, K. S. (2001). Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med. 345, 971–980. doi: 10.1056/NEJMra002168
Falix, F. A., Aronson, D. C., Lamers, W. H., and Gaemers, I. C. (2012). Possible roles of DLK1 in the Notch pathway during development and disease. Biochim. Biophys. Acta 1822, 988–995. doi: 10.1016/j.bbadis.2012.02.003
Fatica, A., and Bozzoni, I. (2014). Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 15, 7–21. doi: 10.1038/nrg3606
Fitzpatrick, G. V., Soloway, P. D., and Higgins, M. J. (2002). Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 32, 426–431. doi: 10.1038/ng988
Fournet, J. C., Mayaud, C., de Lonlay, P., Gross-Morand, M. S., Verkarre, V., Castanet, M.,et al. (2001). Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am. J. Pathol. 158, 2177–2184. doi: 10.1016/S0002-9440(10)64689-5
Friedrichsen, B. N., Carlsson, C., Møldrup, A., Michelsen, B., Jensen, C. H., Teisner, B.,et al. (2003). Expression, biosynthesis and release of preadipocyte factor-1/delta-like protein/fetal antigen-1 in pancreatic beta-cells: possible physiological implications. J. Endocrinol. 176, 257–266. doi: 10.1677/joe.0.1760257
Gardner, R. J., Mackay, D. J., Mungall, A. J., Polychronakos, C., Siebert, R., Shield, J. P.,et al. (2000). An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet. 9, 589–596. doi: 10.1093/hmg/9.4.589
Gaulton, K. J., Nammo, T., Pasquali, L., Simon, J. M., Giresi, P. G., Fogarty, M. P.,et al. (2010). A map of open chromatin in human pancreatic islets. Nat. Genet. 42, 255–259. doi: 10.1038/ng.530
Gicquel, C., Rossignol, S., Cabrol, S., Houang, M., Steunou, V., Barbu, V.,et al. (2005). Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet. 37, 1003–1007. doi: 10.1038/ng1629
Grote, P., Wittler, L., Hendrix, D., Koch, F., Währisch, S., Beisaw, A.,et al. (2013). The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 24, 206–214. doi: 10.1016/j.devcel.2012.12.012
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J.,et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi: 10.1038/nature08975
Gutschner, T., Hämmerle, M., and Diederichs, S. (2013). MALAT1 – a paradigm for long noncoding RNA function in cancer. J. Mol. Med. 91, 791–801. doi: 10.1007/s00109-013-1028-y
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D.,et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. doi: 10.1038/nature07672
Guttman, M., Donaghey, J., Carey, B. W., Garber, M., Grenier, J. K., Munson, G.,et al. (2011). lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300. doi: 10.1038/nature10398
Guttman, M., and Rinn, J. L. (2012). Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346. doi: 10.1038/nature10887
Hales, C. N. (2001). The thrifty phenotype hypothesis. Br. Med. Bull. 60, 5–20. doi: 10.1093/bmb/60.1.5
Hao, Y., Crenshaw, T., Moulton, T., Newcomb, E., and Tycko, B. (1993). Tumour-suppressor activity of H19 RNA. Nature 365, 764–767. doi: 10.1038/365764a0
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H., Blauw, G. J., Susser, E. S.,et al. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 17046–17049. doi: 10.1073/pnas.0806560105
Helwak, A., Kudla, G., Dudnakova, T., and Tollervey, D. (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. doi: 10.1016/j.cell.2013.03.043
Hussain, K., Cosgrove, K. E., Shepherd, R. M., Luharia, A., Smith, V. V., Kassem, S.,et al. (2005). Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann syndrome due to defects in the function of pancreatic beta-cell adenosine triphosphate-sensitive potassium channels. J. Clin. Endocrinol. Metab. 90, 4376–4382. doi: 10.1210/jc.2005-0158
Hutchinson, J. N., Ensminger, A. W., Clemson, C. M., Lynch, C. R., Lawrence, J. B., and Chess, A. (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8:39. doi: 10.1186/1471-2164-8-39
Hyttinen, V., Kaprio, J., Kinnunen, L., Koskenvuo, M., and Tuomilehto, J. (2003). Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes 52, 1052–1055. doi: 10.2337/diabetes.52.4.1052
Ideraabdullah, F. Y., Vigneau, S., and Bartolomei, M. S. (2008). Genomic imprinting mechanisms in mammals. Mutat. Res. 647, 77–85. doi: 10.1016/j.mrfmmm.2008.08.008
Ji, P., Diederichs, S., Wang, W., Böing, S., Metzger, R., Schneider, P. M.,et al. (2003). MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22, 8031–8041. doi: 10.1038/sj.onc.1206928
Kagami, M., O’Sullivan, M. J., Green, A. J., Watabe, Y., Arisaka, O., Masawa, N.,et al. (2010). The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 6:e1000992. doi: 10.1371/journal.pgen.1000992.s006
Kagami, M., Sekita, Y., Nishimura, G., Irie, M., Kato, F., Okada, M.,et al. (2008). Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat. Genet. 40, 237–242. doi: 10.1038/ng.2007.56
Kameswaran, V., Bramswig, N. C., McKenna, L. B., Penn, M., Schug, J., Hand, N. J.,et al. (2014). Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 19, 135–145. doi: 10.1016/j.cmet.2013.11.016
Kamiya, M., Judson, H., Okazaki, Y., Kusakabe, M., Muramatsu, M., Takada, S.,et al. (2000). The cell cycle control gene ZAC/PLAGL1 is imprinted – a strong candidate gene for transient neonatal diabetes. Hum. Mol. Genet. 9, 453–460. doi: 10.1093/hmg/9.3.453
Kaneko, S., Bonasio, R., Saldaña-Meyer, R., Yoshida, T., Son, J., Nishino, K.,et al. (2014). Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 53, 290–300. doi: 10.1016/j.molcel.2013.11.012
Kang, H. S., Kim, Y.-S., ZeRuth, G., Beak, J. Y., Gerrish, K., Kilic, G.,et al. (2009). Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol. Cell. Biol. 29, 6366–6379. doi: 10.1128/MCB.01259-09
Kawakami, T., Chano, T., Minami, K., Okabe, H., Okada, Y., and Okamoto, K. (2006). Imprinted DLK1 is a putative tumor suppressor gene and inactivated by epimutation at the region upstream of GTL2 in human renal cell carcinoma. Hum. Mol. Genet. 15, 821–830. doi: 10.1093/hmg/ddl001
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D.,et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672. doi: 10.1073/pnas.0904715106
Kong, A., Steinthorsdottir, V., Masson, G., Thorleifsson, G., Sulem, P., Besenbacher, S.,et al. (2009). Parental origin of sequence variants associated with complex diseases. Nature 462, 868–874. doi: 10.1038/nature08625
Kroon, E., Martinson, L. A., Kadoya, K., Bang, A. G., Kelly, O. G., Eliazer, S.,et al. (2008). Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443–452. doi: 10.1038/nbt1393
Ku, G. M., Kim, H., Vaughn, I. W., Hangauer, M. J., Myung Oh, C., German, M. S.,et al. (2012). Research resource: RNA-Seq reveals unique features of the pancreatic β-cell transcriptome. Mol. Endocrinol. 26, 1783–1792. doi: 10.1210/me.2012-1176
Latos, P. A., Pauler, F. M., Koerner, M. V., Şenergin, H. B., Hudson, Q. J., Stocsits, R. R.,et al. (2012). Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472. doi: 10.1126/science.1228110
Lee, J. T., and Bartolomei, M. S. (2013). X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152, 1308–1323. doi: 10.1016/j.cell.2013.02.016
Lee, M. P., DeBaun, M. R., Mitsuya, K., Galonek, H. L., Brandenburg, S., Oshimura, M.,et al. (1999). Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. U.S.A. 96, 5203–5208. doi: 10.1073/pnas.96.9.5203
Lee, M. P., Hu, R. J., Johnson, L. A., and Feinberg, A. P. (1997). Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat. Genet. 15, 181–185. doi: 10.1038/ng0297-181
Maass, P. G., Luft, F. C., and Bähring, S. (2014). Long non-coding RNA in health and disease. J. Mol. Med. 92, 337–346. doi: 10.1007/s00109-014-1131-8
Macias, S., Plass, M., Stajuda, A., Michlewski, G., Eyras, E., and Cáceres, J. F. (2012). DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 19, 760–766. doi: 10.1038/nsmb.2344
Mackay, D. J. G., Coupe, A.-M., Shield, J. P. H., Storr, J. N. P., Temple, I. K., and Robinson, D. O. (2002). Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum. Genet. 110, 139–144. doi: 10.1007/s00439-001-0671-5
Mao, Y. S., Zhang, B., and Spector, D. L. (2011). Biogenesis and function of nuclear bodies. Trends Genet. 27, 295–306. doi: 10.1016/j.tig.2011.05.006
Martens, J. A., Laprade, L., and Winston, F. (2004). Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429, 571–574. doi: 10.1038/nature02538
Mattick, J. S., and Makunin, I. V. (2006). Non-coding RNA. Hum. Mol. Genet. 15, R17–R29. doi: 10.1093/hmg/ddl046
McCarthy, M. I. (2010). Genomics, type 2 diabetes, and obesity. N. Engl. J. Med. 363, 2339–2350. doi: 10.1056/NEJMra0906948
Medici, F., Hawa, M., Ianari, A., Pyke, D. A., and Leslie, R. D. (1999). Concordance rate for type II diabetes mellitus in monozygotic twins: actuarial analysis. Diabetologia 42, 146–150. doi: 10.1007/s001250051132
Mendell, J. T., and Olson, E. N. (2012). MicroRNAs in stress signaling and human disease. Cell 148, 1172–1187. doi: 10.1016/j.cell.2012.02.005
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105, 716–721. doi: 10.1073/pnas.0706729105
Mitterberger, M. C., Lechner, S., Mattesich, M., Kaiser, A., Probst, D., Wenger, N.,et al. (2012). DLK1(PREF1) is a negative regulator of adipogenesis in CD105+/CD90+/CD34+/CD31+/FABP4- adipose-derived stromal cells from subcutaneous abdominal fat pats of adult women. Stem Cell Res 9, 35–48. doi: 10.1016/j.scr.2012.04.001
Monk, D., Arnaud, P., Apostolidou, S., Hills, F. A., Kelsey, G., Stanier, P.,et al. (2006). Limited evolutionary conservation of imprinting in the human placenta. Proc. Natl. Acad. Sci. U.S.A. 103, 6623–6628. doi: 10.1073/pnas.0511031103
Morán, I., Akerman, I., van de Bunt, M., Xie, R., Benazra, M., Nammo, T.,et al. (2012). Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 16, 435–448. doi: 10.1016/j.cmet.2012.08.010
Nakagawa, S., Ip, J. Y., Shioi, G., Tripathi, V., Zong, X., Hirose, T.,et al. (2012). Malat1 is not an essential component of nuclear speckles in mice. RNA 18, 1487–1499. doi: 10.1261/rna.033217.112
Nesterova, T. B., Slobodyanyuk, S. Y., Elisaphenko, E. A., Shevchenko, A. I., Johnston, C., Pavlova, M. E.,et al. (2001). Characterization of the genomic Xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence. Genome Res. 11, 833–849. doi: 10.1101/gr.174901
Nogueira, T. C., Paula, F. M., Villate, O., Colli, M. L., Moura, R. F., Cunha, D. A.,et al. (2013). GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 9:e1003532. doi: 10.1371/journal.pgen.1003532
Ohlsson, R., Nyström, A., Pfeifer-Ohlsson, S., Töhönen, V., Hedborg, F., Schofield, P.,et al. (1993). IGF2 is parentally imprinted during human embryogenesis and in the Beckwith-Wiedemann syndrome. Nat. Genet. 4, 94–97. doi: 10.1038/ng0593-94
O’Rahilly, S. (2009). Human genetics illuminates the paths to metabolic disease. Nature 462, 307–314. doi: 10.1038/nature08532
Parker-Katiraee, L., Carson, A. R., Yamada, T., Arnaud, P., Feil, R., Abu-Amero, S. N.,et al. (2007). Identification of the imprinted KLF14 transcription factor undergoing human-specific accelerated evolution. PLoS Genet. 3:e65. doi: 10.1371/journal.pgen.0030065
Park, J. H., Stoffers, D. A., Nicholls, R. D., and Simmons, R. A. (2008). Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Invest. 118, 2316–2324. doi: 10.1172/JCI33655
Pasquali, L., Gaulton, K. J., Rodríguez-Seguí, S. A., Mularoni, L., Miguel-Escalada, I., Akerman, I.,et al. (2014). Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 46, 136–143. doi: 10.1038/ng.2870
Ponjavic, J., Ponting, C. P., and Lunter, G. (2007). Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 17, 556–565. doi: 10.1101/gr.6036807
Ravassard, P., Hazhouz, Y., Pechberty, S., Bricout-Neveu, E., Armanet, M., Czernichow, P.,et al. (2011). A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J. Clin. Invest. 121, 3589–3597. doi: 10.1172/JCI58447
Ravelli, A. C., van der Meulen, J. H., Michels, R. P., Osmond, C., Barker, D. J., Hales, C. N.,et al. (1998). Glucose tolerance in adults after prenatal exposure to famine. Lancet 351, 173–177. doi: 10.1016/S0140-6736(97)07244-9
Rinn, J. L., and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. doi: 10.1146/annurev-biochem-051410-092902
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A.,et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. doi: 10.1016/j.cell.2007.05.022
Roseboom, T., de Rooij, S., and Painter, R. (2006). The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 82, 485–491. doi: 10.1016/j.earlhumdev.2006.07.001
Rosengren, A. H., Braun, M., Mahdi, T., Andersson, S. A., Travers, M. E., Shigeto, M.,et al. (2012). Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes 61, 1726–1733. doi: 10.2337/db11-1516
Schmidt, J. V., Matteson, P. G., Jones, B. K., Guan, X. J., and Tilghman, S. M. (2000). The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. Genes Dev. 14, 1997–2002. doi: 10.1101/gad.14.16.1997
Segrè, A. V., DIAGRAM Consortium, MAGIC investigators, Groop, L., Mootha, V. K., Daly, M. J.,et al. (2010). Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 6:e1001058. doi: 10.1371/journal.pgen.1001058
Seitz, H., Royo, H., Bortolin, M.-L., Lin, S.-P., Ferguson-Smith, A. C., and Cavaille, J. (2004). A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 14, 1741–1748. doi: 10.1101/gr.2743304
Sekita, Y., Wagatsuma, H., Nakamura, K., Ono, R., Kagami, M., Wakisaka, N.,et al. (2008). Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat. Genet. 40, 243–248. doi: 10.1038/ng.2007.51
Shield, J. P., Gardner, R. J., Wadsworth, E. J., Whiteford, M. L., James, R. S., Robinson, D. O.,et al. (1997). Aetiopathology and genetic basis of neonatal diabetes. Arch. Dis. Child. Fetal Neonatal Ed. 76, F39–F42. doi: 10.1136/fn.76.1.F39
Small, K. S., Hedman, A. K., Grundberg, E., Nica, A. C., Thorleifsson, G., Kong, A.,et al. (2011). Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 43, 561–564. doi: 10.1038/ng.833
Smas, C. M., and Sul, H. S. (1993). Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell 73, 725–734. doi: 10.1016/0092-8674(93)90252-L
Spengler, D., Villalba, M., Hoffmann, A., Pantaloni, C., Houssami, S., Bockaert, J.,et al. (1997). Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J. 16, 2814–2825. doi: 10.1093/emboj/16.10.2814
Stadnick, M. P., Pieracci, F. M., Cranston, M. J., Taksel, E., Thorvaldsen, J. L., and Bartolomei, M. S. (1999). Role of a 461-bp G-rich repetitive element in H19 transgene imprinting. Dev. Genes Evol. 209, 239–248. doi: 10.1007/s004270050248
Stadtfeld, M., Apostolou, E., Akutsu, H., Fukuda, A., Follett, P., Natesan, S.,et al. (2010). Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465, 175–181. doi: 10.1038/nature09017
Taft, R. J., Pheasant, M., and Mattick, J. S. (2007). The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 29, 288–299. doi: 10.1002/bies.20544
Temple, I. K., Gardner, R. J., Mackay, D. J., Barber, J. C., Robinson, D. O., and Shield, J. P. (2000). Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes 49, 1359–1366. doi: 10.2337/diabetes.49.8.1359
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M.,et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713. doi: 10.1038/nature09270
Thorvaldsen, J. L., and Bartolomei, M. S. (2007). SnapShot: imprinted gene clusters. Cell 130:958. doi: 10.1016/j.cell.2007.08.033
Tierling, S., Dalbert, S., Schoppenhorst, S., Tsai, C.-E., Oliger, S., Ferguson-Smith, A. C.,et al. (2006). High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics 87, 225–235. doi: 10.1016/j.ygeno.2005.09.018
Tornehave, D., Jensen, C. H., Teisner, B., and Larsson, L. I. (1996). FA1 immunoreactivity in endocrine tumours and during development of the human fetal pancreas; negative correlation with glucagon expression. Histochem. Cell Biol. 106, 535–542. doi: 10.1007/BF02473268
Travers, M. E., Mackay, D. J. G., Dekker Nitert, M., Morris, A. P., Lindgren, C. M., Berry, A.,et al. (2013). Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes 62, 987–992. doi: 10.2337/db12-0819/-/DC1
Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T.,et al. (2010). The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938. doi: 10.1016/j.molcel.2010.08.011
Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H., and Bartel, D. P. (2011). Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550. doi: 10.1016/j.cell.2011.11.055
Unoki, H., Takahashi, A., Kawaguchi, T., Hara, K., Horikoshi, M., Andersen, G.,et al. (2008). SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 40, 1098–1102. doi: 10.1038/ng.208
Valleley, E. M., Cordery, S. F., and Bonthron, D. T. (2007). Tissue-specific imprinting of the ZAC/PLAGL1 tumour suppressor gene results from variable utilization of monoallelic and biallelic promoters. Hum. Mol. Genet. 16, 972–981. doi: 10.1093/hmg/ddm041
Voight, B. F., Scott, L. J., Steinthorsdottir, V., Morris, A. P., Dina, C., Welch, R. P.,et al. (2010). Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 42, 579–589. doi: 10.1038/ng.609
Wallace, C., Smyth, D. J., Maisuria-Armer, M., Walker, N. M., Todd, J. A., and Clayton, D. G. (2010). The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 42, 68–71. doi: 10.1038/ng.493
Weinmann, L., Höck, J., Ivacevic, T., Ohrt, T., Mütze, J., Schwille, P.,et al. (2009). Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell 136, 496–507. doi: 10.1016/j.cell.2008.12.023
Yada, T., Sakurada, M., Ihida, K., Nakata, M., Murata, F., Arimura, A.,et al. (1994). Pituitary adenylate cyclase activating polypeptide is an extraordinarily potent intra-pancreatic regulator of insulin secretion from islet beta-cells. J. Biol. Chem. 269, 1290–1293.
Yang, L., Lin, C., Liu, W., Zhang, J., Ohgi, K. A., Grinstein, J. D.,et al. (2011). ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 147, 773–788. doi: 10.1016/j.cell.2011.08.054
Yasuda, K., Miyake, K., Horikawa, Y., Hara, K., Osawa, H., Furuta, H.,et al. (2008). Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 40, 1092–1097. doi: 10.1038/ng.207
ZeRuth, G. T., Takeda, Y., and Jetten, A. M. (2013). The Krüppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol. Endocrinol. 27, 1692–1705. doi: 10.1210/me.2013-1117
Zhang, B., Arun, G., Mao, Y. S., Lazar, Z., and Hung, G. (2012). The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2, 111–123. doi: 10.1016/j.celrep.2012.06.003
Zhang, H., Zeitz, M. J., Wang, H., Niu, B., Ge, S., Li, W.,et al. (2014). Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J. Cell Biol. 204, 61–75. doi: 10.1083/jcb.201304152
Zhao, J., Dahle, D., Zhou, Y., Zhang, X., and Klibanski, A. (2005). Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J. Clin. Endocrinol. Metab. 90, 2179–2186. doi: 10.1210/jc.2004-1848
Zhao, J., Ohsumi, T. K., Kung, J. T., Ogawa, Y., Grau, D. J., Sarma, K.,et al. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40, 939–953. doi: 10.1016/j.molcel.2010.12.011
Zhou, Y., Zhang, X., and Klibanski, A. (2012). MEG3 noncoding RNA: a tumor suppressor. J. Mol. Endocrinol. 48, R45–R53. doi: 10.1530/JME-12-0008
Zhou, Y., Zhong, Y., Wang, Y., Zhang, X., Batista, D. L., Gejman, R.,et al. (2007). Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 282, 24731–24742. doi: 10.1074/jbc.M702029200
Keywords: lncRNA, β-cell biology, diabetes mellitus, imprinting control region (ICR), MEG3
Citation: Kameswaran V and Kaestner KH (2014) The Missing lnc(RNA) between the pancreatic β-cell and diabetes. Front. Genet. 5:200. doi: 10.3389/fgene.2014.00200
Received: 06 May 2014; Accepted: 15 June 2014;
Published online: 01 July 2014.
Edited by:
Romano Regazzi, University of Lausanne, SwitzerlandReviewed by:
Thomas Mandrup-Poulsen, University of Copenhagen, DenmarkAnna Motterle, University of Lausanne, Switzerland
Copyright © 2014 Kameswaran and Kaestner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus H. Kaestner, Department of Genetics and Institute for Diabetes, Obesity and Metabolism, Perelman School of Medicine, University of Pennsylvania, 3400 Civic Center Boulevard, Philadelphia, PA 19104, USA e-mail:a2Flc3RuZXJAbWFpbC5tZWQ=. upenn.edu