 Adanna G. Alexander
Adanna G. Alexander Vanessa Marfil
Vanessa Marfil Chris Li
Chris Li- 1Department of Biology, City College of New York, New York, NY, USA
- 2Department of Biology, The Graduate Center, City University of New York, New York, NY, USA
Advances in research and technology has increased our quality of life, allowed us to combat diseases, and achieve increased longevity. Unfortunately, increased longevity is accompanied by a rise in the incidences of age-related diseases such as Alzheimer’s disease (AD). AD is the sixth leading cause of death, and one of the leading causes of dementia amongst the aged population in the USA. It is a progressive neurodegenerative disorder, characterized by the prevalence of extracellular Aβ plaques and intracellular neurofibrillary tangles, derived from the proteolysis of the amyloid precursor protein (APP) and the hyperphosphorylation of microtubule-associated protein tau, respectively. Despite years of extensive research, the molecular mechanisms that underlie the pathology of AD remain unclear. Model organisms, such as the nematode, Caenorhabditis elegans, present a complementary approach to addressing these questions. C. elegans has many advantages as a model system to study AD and other neurodegenerative diseases. Like their mammalian counterparts, they have complex biochemical pathways, most of which are conserved. Genes in which mutations are correlated with AD have counterparts in C. elegans, including an APP-related gene, apl-1, a tau homolog, ptl-1, and presenilin homologs, such as sel-12 and hop-1. Since the neuronal connectivity in C. elegans has already been established, C. elegans is also advantageous in modeling learning and memory impairments seen during AD. This article addresses the insights C. elegans provide in studying AD and other neurodegenerative diseases. Additionally, we explore the advantages and drawbacks associated with using this model.
Introduction to Alzheimer’s Disease
Alzheimer’s disease (AD) is the 6th leading cause of death in the US and affects more than 35 million people worldwide (Alzheimer’s Disease International, 2014). AD is a neurodegenerative disease characterized by a progressive loss of memory. Most cases of AD occur sporadically in aged people (>60 years, late-onset AD) without a clear inheritance pattern. However, in 5% of the cases (familial or early onset AD) AD symptoms appear earlier and are linked with gene mutations. Both forms of AD have two main neuropathologic features: the presence of extra-neuronal amyloid plaques, often referred to as senile plaques, and intraneuronal neurofibrillary tangles (Kidd, 1964; Luse and Smith, 1964; Terry et al., 1964; Krigman et al., 1965). Amyloid plaques are aggregates of the beta-amyloid peptide (Aβ), a cleavage product of the amyloid precursor protein (APP; Glenner and Wong, 1984; Masters et al., 1985; Kang et al., 1987). Hyperphosphorylation of the microtubule associated protein tau causes its polymerization into paired helical filaments (PHFs) and, presumably, its formation into neurofibrillary tangles (Goedert et al., 1989a).
Mutations in the APP gene and/or the enzymes involved in APP processing (γ-secretase components presenilins, PSEN1 and PSEN2; Chartier-Harlin et al., 1991; Goate et al., 1991; Murrell et al., 1991; Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995) are correlated with early onset AD. These mutations increase the levels of toxic Aβ species and promote neurodegeneration. By contrast, a recently identified mutation in APP affects cleavage of APP, causing less Aβ production and conferring neuroprotective benefits (Jonsson et al., 2012). Despite the significant advances made using APP transgenic and knockout models in mammals, unraveling the cellular role of APP has been difficult. Alternative animal models provide complementary approaches to dissecting the function of APP and tau. In this review, we discuss the latest uses of the nematode Caenorhabditis elegans as a model system for the study of AD. We also include a brief review of a few representative examples of how C. elegans is being utilized to model other neurodegenerative diseases.
C. elegans as a Model for Alzheimer’s Disease
Caenorhabditis elegans is a free-living, non-parasitic nematode that was first introduced as a model organism by Sydney Brenner in 1963 (Brenner, 1974). It is a small (1 mm in length), transparent roundworm, which makes it easy for manipulation, and has a short life cycle of 3 days from egg to adult at 25°C (Brenner, 1974). Under suitable growing conditions, hatched animals develop through four larval stages (L1–L4), each punctuated by a molt, to arise as an adult hermaphrodite with 959 somatic cells (Sulston and Horvitz, 1977). Its life span is between 2 and 3 weeks, which facilitates the study of its biology. Completion of the C. elegans genome sequence in 1998 (C. elegans Sequencing Consortium, 1998) demonstrated that roughly 38% of worm genes have a human ortholog, such as APP and tau (Shaye and Greenwald, 2011). Hence, C. elegans has many excellent advantages as an in vivo model for the study of AD and other neurodegenerative diseases.
Molecular Pathways of APP. Similarities and Differences between Mammals and C. elegans
Function and Processing of APP: Non-Amyloidogenic and Amyloidogenic Aβ Pathway
The APP family of proteins contains three members, APP, APLP1, and APLP2 (Wasco et al., 1992, 1993; Sprecher et al., 1993; Sandbrink et al., 1994; Slunt et al., 1994), which are characterized by a large extracellular region containing conserved E1 and E2 domains, a single transmembrane domain, and a small cytosolic domain (Kang et al., 1987). APLP1 and APLP2 do not contain the Aβ sequence and, hence, do not produce Aβ (Figure 1A; Wasco et al., 1992, 1993). The APP gene family is required for viability and brain development. APP mouse knockouts were viable, but had behavioral and cognitive defects (Ring et al., 2007). While knockout of APLP1 resulted in postnatal growth defects (Heber et al., 2000), mice in which APLP2 was inactivated appeared wild type (von Koch et al., 1997). Double knockouts of APLP2 and either APP or APLP1, however, resulted in postnatal lethality (von Koch et al., 1997; Heber et al., 2000); the lethality of APP/APLP2 double knockouts could be rescued by knock-in of an APP extracellular fragment, sAPPα (Weyer et al., 2011), suggesting that sAPPα is sufficient for viability. The triple knockout caused lethality and a type II lissencephaly and cortical disorganization (Herms et al., 2004). Collectively, these results suggest that APP family members have essential and redundant functions during development, including proper brain development, and these functions do not require Aβ.
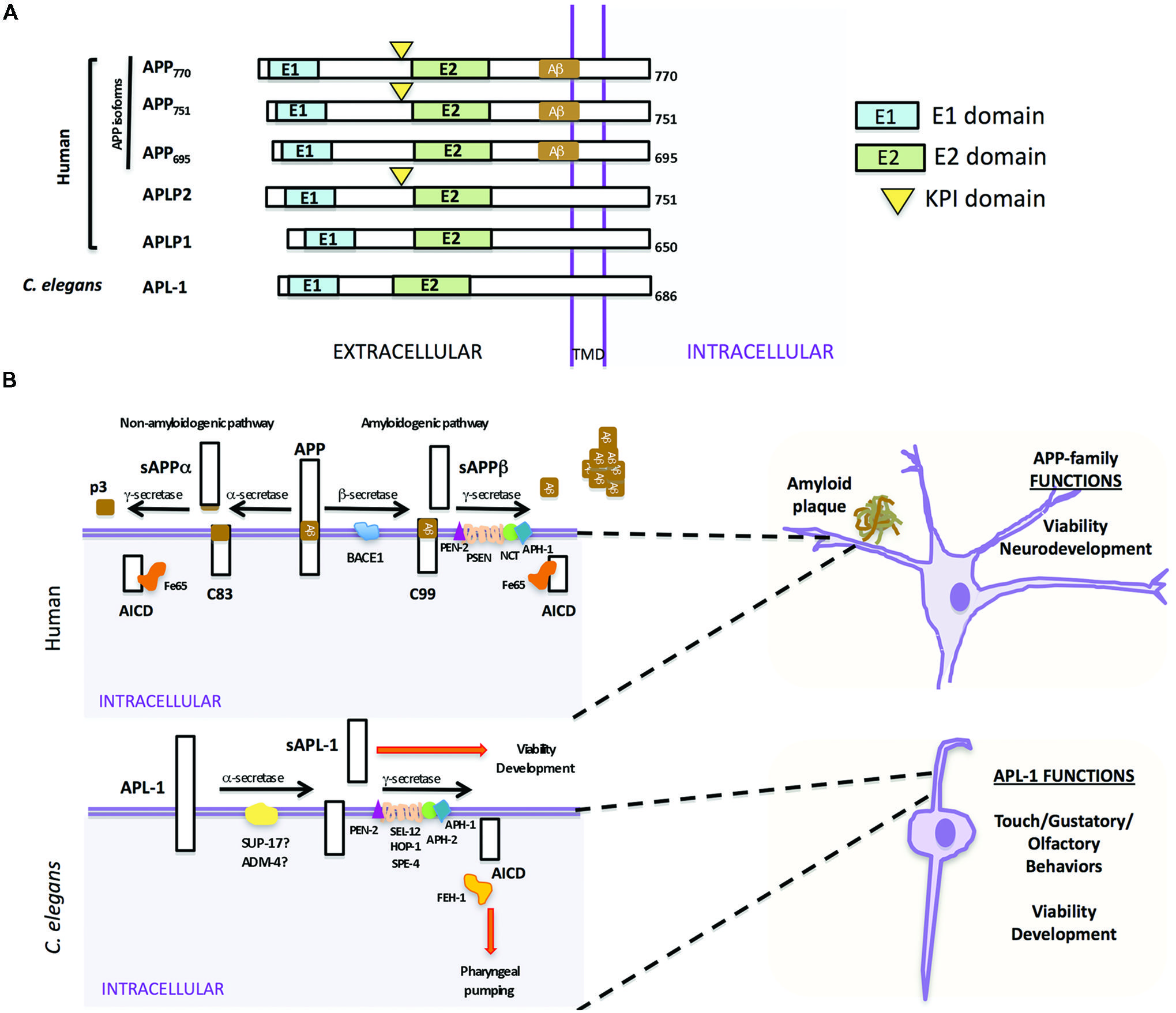
FIGURE 1. Similarities and differences between human APP and Caenorhabditis elegans APL-1. (A) Schematic representation of human APP isoforms, other members of the APP family (APLP1 and APLP2), and C. elegans APL-1. (B) Comparison between human APP proteolytic pathways (top) and C. elegans APL-1 proteolytic pathway (bottom). Top. APP can be cleaved by two different pathways. In the anti-amyloidogenic pathway, APP is first cleaved by the α-secretase to release an extracellular fragment sAPPα. The remaining APP fragment (APP-CTFα or C83) is then cleaved by the γ-secretase complex to release p3 extracellularly and the APP intracellular domain (AICD) to the cytosol. In the amyloidogenic pathway, β-secretase first cleaves APP, releasing the sAPPβ fragment. The APP-CTFβ (C99) fragment is subsequently cleaved by the γ-secretase complex, liberating the AICD to the cytosol and Aβ to the lumen. Aβ will aggregate to form amyloid plaques. Bottom. In C. elegans, APL-1 is first cleaved by the α-secretase homologs SUP-17/ADM-4, liberating the extracellular sAPL-1 that is known to regulate worm viability and development. The γ-secretase complex will then cleave the remaining APL-1-CTF to release AICD into the cytosol. General functions of the APP family and APL-1 are indicated.
In mammals, APP is processed through two proteolytic pathways, only one of which produces Aβ (Figure 1B; Haass et al., 1992, 1994a,b). In the non-amyloidogenic pathway, APP is first cleaved by an α-secretase within the Aβ sequence to release an extracellular fragment, sAPPα (Figure 1B). The remaining APP fragment (known as APP-CTFα or C83) is then cleaved by the γ-secretase complex to release the APP intracellular domain (AICD) to the cytosol. By contrast, in the amyloidogenic pathway, after cleavage by the β-secretase (BACE) to release sAPPβ, the remaining APP fragment (known as APP-CTFβ or C99) is then cleaved by the γ-secretase complex, liberating Aβ to the lumen and AICD to the cytosol (Gu et al., 2001; Sastre et al., 2001; Weidemann et al., 2002). This latter pathway is likely favored in AD patients.
Mammalian γ-secretase is a protease complex consisting of several components: presenilins 1 and 2 (PSEN1 and PSEN2), nicastrin (NCT), anterior pharynx defective (APH-1), and the presenilin enhancer (PEN-2; Yu et al., 2000; Francis et al., 2002). PSEN1 and PSEN2 are the catalytic components of the γ-secretase complex. NCT works as a stabilizing cofactor required for γ-secretase complex assembly and trafficking (Li et al., 2003; Zhang et al., 2005) and PEN-2 and APH-1 have a role in the maturation process of PSEN1 and PSEN2 (Luo et al., 2003). Besides APP, the γ-secretase complex is also involved in the proteolysis of Notch receptors, and the first identification of any PEN-2 or APH-1 ortholog was in C. elegans as the result of a genetic screen for modifiers of the Notch pathway (Francis et al., 2002; Goutte et al., 2002). Within the γ-secretase complex, only mutations in PSEN1 and PSEN2 have been associated with early onset AD (Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995).
Processing of C. elegans APL-1
In C. elegans there is only one APP-related gene, apl-1. Like human APP (Kang et al., 1987), APL-1 contains a large extracellular region whose conserved E1 and E2 domains share 46 and 49% sequence similarity to human APP, respectively, a transmembrane domain, and a relatively small cytosolic domain, which shares 71% sequence similarity to human APP (Figure 1A; Daigle and Li, 1993). Notably, unlike APP but similar to APLP1 and APLP2, APL-1 does not contain the Aβ sequence (Daigle and Li, 1993).
Two α-secretase proteins are present in C. elegans, SUP-17 and ADM-4 (Jarriault and Greenwald, 2005). They work redundantly in the cleavage of the C. elegans Notch homologs, LIN-12 and GLP-1 (Jarriault and Greenwald, 2005). However, no experiments thus far have tested whether SUP-17 or ADM-4 cleaves APL-1. No BACE ortholog has been identified by bioinformatic searches and no β-secretase activity that cleaves human APP has been detected in C. elegans, suggesting that APL-1 is only processed by the α/γ-secretase processing pathway (Link, 2006). α-secretase cleavage of APL-1 releases the extracellular fragment, sAPL-1; subsequent cleavage of APL-1-CTFα by the γ-secretase complex liberates the intracellular domain (AICD; Figure 1B).
The initial characterizations of human PSEN1 (then called S182) and PSEN2 (first named E5-1) described them as novel proteins with multiple transmembrane domains (Rogaev et al., 1995; Sherrington et al., 1995). The cellular functions of the presenilins were determined by their homology to the C. elegans protein, SEL-12 (Levitan and Greenwald, 1995). The two C. elegans Notch genes, lin-12 and glp-1, are involved in many cell fate decisions during development, including vulval cell specification and germline development (Greenwald et al., 1983; Lambie and Kimble, 1991; Newman et al., 1995; Levitan et al., 1996) sel-12/PSEN was identified in a genetic screen to isolate suppressors of a dominant Lin-12/Notch multivulva phenotype (Levitan and Greenwald, 1995). Loss of sel-12/PSEN suppressed the Lin-12/Notch multivulva phenotype and produced a defect in egg laying that was rescued by introducing human PSEN1 or PSEN2, suggesting a conserved function between human and C. elegans presenilins (Levitan and Greenwald, 1995; Levitan et al., 1996). Like human PSENs (Thinakaran et al., 1996), SEL-12/PSEN is cleaved to attain its final topology (Li and Greenwald, 1996). A C. elegans PSEN gene family was identified and includes sel-12, hop-1, and spe-4 (L’Hernault and Arduengo, 1992; Levitan and Greenwald, 1995; Li and Greenwald, 1997; Westlund et al., 1999); spe-4 is exclusively expressed in male gonadal cells and will not be further discussed (Arduengo et al., 1998). Knockdown of hop-1/PSEN in sel-12/PSEN mutants showed maternal effect lethality, germline defects, and missing anterior pharynx, defects associated with loss of glp-1/Notch function, suggesting that sel-12 and hop-1 function redundantly in the LIN-12 and GLP-1/Notch pathways (Westlund et al., 1999). Similarly, mice carrying a null mutation in PSEN1 showed embryonic lethality, skeletal defects, and disrupted somite boundaries (Shen et al., 1997), similar to the phenotypes seen in Notch1 knockouts (Krebs et al., 2000, 2003, 2004; Duarte et al., 2004; Gale et al., 2004).
In screening for novel mutants showing the glp-1/Notch phenotype of defective anterior pharynx, Goutte et al. (2000, 2002) identified two genes, aph-1 and aph-2, which encodes the C. elegans NCT ortholog. Independently, Francis et al. (2002) screened for enhancers of sel-12/PSEN activity and identified pen-2. aph-2/NCT, pen-2, and aph-1 are all required for proper Notch signaling. Human PSEN, NCT, Aph1α2, and PEN-2 were subsequently shown to physically associate and cooperatively regulate the maturation of individual components to form a proteolytically active γ-secretase complex (Kimberly et al., 2003).
Function and Regulation of APL-1
Like the mammalian APP family (Slunt et al., 1994; Lorent et al., 1995; Thinakaran et al., 1995), apl-1 is expressed in multiple tissue types. apl-1 expression is observed in neurons, supporting cells, and head muscles throughout development, while expression in vulval muscles, vulval cells, and hypodermal seam cells is not detected until the L4 stage to adult (Hornsten et al., 2007; Niwa et al., 2008).
Inactivation of apl-1, such as with the yn10 null allele, results in a completely penetrant lethality during the first to second larval (L1–L2) transition due to a molting defect (Hornsten et al., 2007). apl-1 activity is also necessary for later larval transitions, as RNAi knockdown of apl-1 in an RNAi-sensitized background showed animals with molting defects during the L3–L4 and L4 to adult transitions (Wiese et al., 2010). This lethality was rescued by microinjection of an apl-1 genomic fragment or cDNA. Hence, similar to the mammalian APP family, apl-1 has an essential function. High levels of apl-1 expression caused an incompletely penetrant L1 lethality (70% lethality), shortened body length, and morphogenetic, reproductive, and locomotory defects (Hornsten et al., 2007; Ewald et al., 2012b). These results indicate that levels of APL-1 must be tightly regulated as loss of APL-1 as well as high levels of APL-1 result in lethality. When sel-12/PSEN activity was reduced in transgenic animals with APL-1 overexpression, the 70% lethality was partially rescued, suggesting that SEL-12/PSEN regulates APL-1 cleavage and/or trafficking (Hornsten et al., 2007). The underlying basis of the loss- and gain-of-function apl-1 lethality is still unclear, but is not dependent on activation of CED-3/ caspase or necrotic cell death pathways (Hornsten et al., 2007). Characterization of apl-1 function may provide insights into the general function and pathways of APP, of which much is still unknown.
The apl-1(yn5) mutant, which contains a deletion of the region encoding the APL-1 transmembrane and cytosolic domains, produces only the extracellular domain of APL-1 (APL-1EXT) and is viable. Because APL-1EXT is not further cleaved by α-secretase, APL-1EXT is slightly larger than sAPL-1 and is expressed at high levels in apl-1(yn5) mutants (Hornsten et al., 2007). Hence, the APL-1 extracellular domain is sufficient for viability, similar to the rescue of APP/APLP2 double mutants by the knock-in of sAPPα (Weyer et al., 2011). However, although apl-1(yn5) mutants are viable, they display several phenotypes, including a slower developmental progression, decreased body length, reproductive defects, and temperature-sensitive lethality (Hornsten et al., 2007; Ewald et al., 2012b). Because these defects can be phenocopied by microinjection of APL-1EXT transgenes into wild-type animals, the phenotypes are due to overexpression of APL-1EXT and not due to loss of APL-1 signaling through its cytoplasmic domain (Ewald et al., 2012b). Interestingly, pan-neuronal expression of APL-1EXT, but not expression from muscle or hypodermal cells, is sufficient to rescue the lethality observed in apl-1 null mutants (Hornsten et al., 2007), suggesting that the cells (i.e., neurons) from which sAPL-1 is released as well as the extracellular milieu in which sAPL-1 travels is functionally relevant. We suggest that high levels of sAPP may also contribute to the pathology seen in AD patients. Down’s syndrome patients, whose chromosome 21 trisomy includes trisomy of APP, display a high incidence of AD and intellectual disability (Zigman, 2013), perhaps contributed in small part by the high levels of APP expression.
Decreasing apl-1 activity by RNAi resulted in hypersensitivity to aldicarb, an acetylcholinesterase inhibitor (Wiese et al., 2010). Using apl-1 knockouts to test different apl-1 deletion constructs, Wiese et al. (2010) determined that lack of sAPL-1 is responsible for the aldicarb hypersensitivity. These findings are consistent with mammalian studies, which show that a lack of APP and APLP2 impairs synaptic function at cholinergic neuromuscular junctions (Wang et al., 2005).
Heterochronic genes, whose differential spatiotemporal expression ensures proper progression through larval stages and transition into adulthood (Chalfie et al., 1981; Ambros and Horvitz, 1984), regulate expression of apl-1 in hypodermal seam cells (Niwa et al., 2008). Loss of let-7 microRNA (miRNA) function caused precocious seam cell development and vulval bursting at the adult stage, leading to death (Reinhart et al., 2000). These let-7 phenotypes can be rescued by knockdown of apl-1 (Niwa et al., 2008). apl-1, however, is not a direct target of let-7 miRNA. NHR-25/Ftz-F1, which is a nuclear hormone receptor (NHR) that is required for completion of larval molts (Asahina et al., 2000; Gissendanner and Sluder, 2000), binds an enhancer element in the promoter of apl-1 to regulate apl-1 expression in seam cells (Niwa and Hada, 2010). nhr-25/Ftz-F1 transcripts are possible targets of the let-7 family of miRNAs for downregulation (Hayes et al., 2006). Regulation of continued apl-1 expression in adult seam cells and other cell types is unknown.
Pathways through which apl-1 functions
The apl-1(yn5) phenotypes require activity of the DAF-16/FOXO transcription factor, which is negatively regulated by the insulin pathway. C. elegans has only one insulin/IGF-1 receptor, DAF-2 (Kimura et al., 1997). Under favorable environmental conditions, such as when adequate food is present, signaling through the insulin pathway activates a conserved PI 3-kinase/AKT cascade (Morris et al., 1996; Kimura et al., 1997; Paradis and Ruvkun, 1998; Paradis et al., 1999), which causes phosphorylation of DAF-16/FOXO, thereby allowing reproductive development (Larsen et al., 1995; Gems et al., 1998; Henderson and Johnson, 2001). Phosphorylation of DAF-16/FOXO causes its sequestration in the cytoplasm (Lin et al., 2001), thereby preventing it from entering the nucleus to activate its target genes (Henderson and Johnson, 2001; Lee et al., 2001), which regulate longevity (Lin et al., 1997; Ogg et al., 1997), stress resistance (McElwee et al., 2003, 2004; Murphy et al., 2003), and dauer formation (Riddle et al., 1981; Vowels and Thomas, 1992). Environmental conditions also affect other metabolic functions, such as reproductive behavior, which is inhibited under starvation conditions (Seidel and Kimble, 2011), and body size. Starvation survival behavior is regulated by DAF-16/FOXO activity (Lee and Ashrafi, 2008) and the insulin (So et al., 2011) and DAF-7/TGFβ (Savage-Dunn et al., 2003) pathways work in parallel to regulate body length via daf-16/FOXO activity.
The slowed development, decreased body size, and decreased reproductive rates of apl-1(yn5) mutants are dependent on daf-16/FOXO activity. At 20°C, mutants with decreased insulin signaling or apl-1(yn5) mutants showed a delayed developmental progression and shorter body length, which were enhanced when insulin signaling was decreased in apl-1(yn5) mutants [i.e., daf-2(e1370); apl-1(yn5) double mutants]; at 25°C, the apl-1(yn5) mutants with decreased insulin signaling went into L1 arrest (Ewald et al., 2012b). By contrast, when daf-16/FOXO activity was removed from apl-1(yn5) mutants, the delayed developmental progression, decreased reproductive rate, and smaller body length of apl-1(yn5) single mutants were suppressed. Furthermore, loss of daf-16/FOXO activity in apl-1(yn5) mutants with decreased insulin signaling rescued the short body length and L1 arrest phenotypes (Ewald et al., 2012b). These results suggest that sAPL-1 signals in a parallel pathway to the insulin pathway or modulates the DAF-2/insulin/IGF-1 pathway to activate daf-16/FOXO activity to affect developmental progression, reproductive rates, and body length (Figure 2). Mammalian sAPP may have similar roles in development.
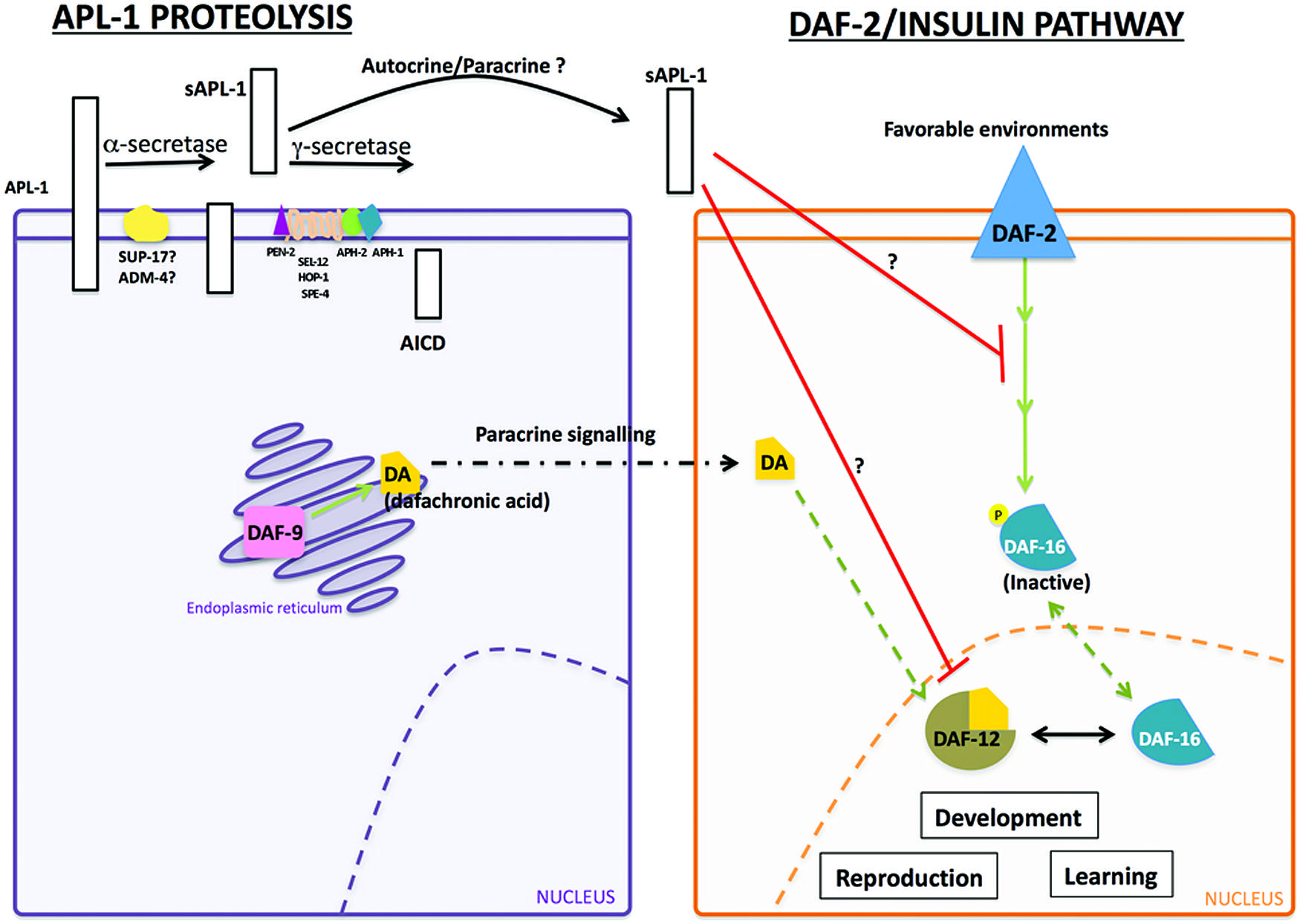
FIGURE 2. Interaction between sAPL-1 and DAF-2 insulin/IGF-1 receptor and DAF-12/NHR pathways. Schematic representation of APL-1 proteolytic pathway and how sAPL-1 may modulate DAF-2 insulin/IGF-1 receptor and DAF-12/NHR signaling pathways. APL-1 is cleaved by the α/γ-secretase pathway in C. elegans. Released sAPL-1 could act as a signaling molecule in the same cell (autocrine regulation) or in neighboring cells (paracrine regulation) to inhibit daf-2 insulin/IGF-1 receptor and daf-12/NHR pathways to affect worm viability and development. The exact mechanism by which sAPL-1inhibits daf-2 insulin/IGF-1 receptor and daf-12/NHR is still unknown and labeled as a question mark (see Ewald et al., 2012b).
Activity of the daf-12/NHR signals in multiple pathways to integrate environmental stimuli with metabolic needs and can modulate the insulin pathway as well as function in an independent pathway (Gerisch et al., 2001; Dumas et al., 2010). Decreasing daf-12/NHR activity in apl-1(yn5) mutants rescued the slow development, low reproductive rate, and decreased body length phenotypes (Ewald et al., 2012b). Hence, decreased insulin signaling and signaling through a parallel daf-12/NHR pathway converge to activate daf-16/FOXO for the phenotypes seen in apl-1(yn5) mutants. Noteworthy, levels of insulin/IGF-1 receptors are decreased in AD brains (Steen et al., 2005), and APP processing and Aβ production in vitro was modulated by insulin signaling (Gasparini et al., 2001). Analogous to C. elegans, sAPP may also act to modulate the insulin pathway.
Pan-neuronal APL-1 expression affects learning
In transgenic mice expressing human or mouse APP, animals showed an increased lethality and learning defects that were not correlated with Aβ deposition (Hsiao et al., 1995); similarly, doubly transgenic mice carrying transgenes with APP and PSEN1 mutations showed learning defects that were not correlated with the number of Aβ plaques (Holcomb et al., 1999). The mechanisms underlying these defects are unclear. Use of the C. elegans model could be informative. C. elegans has many sensory modalities, including smell and taste. They respond to volatile and water soluble chemicals by moving toward or away from chemoattractive or chemorepulsive stimuli, respectively. Many chemoattractants and chemorepellants have been identified and the neural circuits mediating the chemosensory response identified (Bargmann and Horvitz, 1991). For instance, when C. elegans is given the choice between a neutral compound and a chemoattractant, such as benzaldehyde, animals will move toward benzaldehyde; this response is mediated by the AWC neurons (Bargmann et al., 1993); similarly, ASEL, a gustatory neuron, mediates chemoattraction to sodium acetate (Bargmann and Horvitz, 1991; Pierce-Shimomura et al., 2001).
Although apl-1 is not expressed in AWC neurons and the morphology of sensory neurons appears wild type with GFP markers, the overall chemoattractive response to benzaldyhyde and sodium acetate was decreased in apl-1(yn10/+) heterozygotes and transgenic animals that overexpress APL-1 [e.g., ynIs79 (Papl-1::apl-1::GFP)] (Ewald et al., 2012a). The chemotaxis response was restored in APL-1 overexpression lines [e.g., ynIs79 (Papl-1::apl-1::GFP)] when insulin signaling was decreased, but not when daf-16/FOXO activity was decreased, suggesting that daf-16/FOXO activity is needed for normal chemotaxis in transgenic lines overexpressing APL-1 (Figure 2). Pan-neuronal expression of APL-1 or targeted overexpression of APL-1 in the AWC or ASEL neurons resulted in wild-type chemotaxis responses (Ewald et al., 2012a). By contrast, ectopic expression of apl-1 with the snb-1 promoter, which drives pan-neuronal and multi-cell type expression, resulted in no chemotaxis response to benzaldehyde or sodium acetate (Ewald et al., 2012a). When signaling through the DAF-2/insulin/IGF-1 receptor, DAF-12/NHR, or DAF-7/TGFβ was decreased, the chemotaxis response toward benzaldehyde and sodium acetate in these transgenic lines was restored, indicating that the loss of the chemotaxis response due to ectopic apl-1 signaling in cells outside the nervous system is dependent on insulin and TGFβ signaling.
In addition to chemosensory responses, C. elegans is also capable of associative chemosensory plasticity (Wen et al., 1997). For example, when benzaldehyde was paired with starvation for as short as 30 min, C. elegans showed a significant reduction in preference for benzaldehyde; persistence of this plasticity was positively correlated with the length of pairing time (Colbert and Bargmann, 1995; Tomioka et al., 2006; Lin et al., 2010), suggestive of stable memory formation. Both chemotaxis and associative plasticity are dependent on insulin signaling (Tomioka et al., 2006; Lin et al., 2010). Little associative plasticity, however, was observed after pairing benzaldehyde with starvation for 60 min in animals with pan-neuronal APL-1 expression (Ewald et al., 2012a). The plasticity could be restored when daf-16/FOXO, daf-12/NHR, or daf-7/TGFβ activity was decreased, indicating that the impaired associative plasticity with pan-neuronal APL-1 expression requires daf-16/FOXO, daf-12/NHR, and daf-7/TGFβ activity (Ewald et al., 2012a).
Touch habituation is another sensory characteristic affected by pan-neuronal APL-1 expression. When a gentle touch is applied to the head of the animal, C. elegans responds by moving backward; conversely, when touched on the tail, the animal moves forward (Chalfie and Sulston, 1981). This response to gentle body touch is mediated by six mechanosensory touch neurons (Chalfie et al., 1985). After six cycles of alternating head/tail touches, wild-type animals habituated and became unresponsive (Ewald et al., 2012a). apl-1(yn10/+) heterozygotes or transgenic animals that overexpress APL-1 showed touch habituation. By contrast, animals with pan-neuronal APL-1 expression were slow to habituate and required more alternating head/tail touch cycles before becoming habituated (Ewald et al., 2012a). Collectively, these results indicate that pan-neuronal overexpression of APL-1 causes learning defects. These results parallel those seen in mammalian models in which overexpression of APP leads to cognitive defects, independent of Aβ aggregates (Hsiao et al., 1995; Simón et al., 2009), thereby suggesting that sAPP activity, in addition to Aβ aggregates, contributes to cognitive defects. Whether these cognitive defects depend on the DAF-16/FOXO transcription factor and/or TGFβ signaling remains to be tested.
APL-1 Trafficking is Important for Synaptic Transmission
APL-1, like APP (Koo et al., 1990), is transported from the cell body to synapses (Wiese et al., 2010). UNC-108, which is a neuronally expressed GTPase and localizes to the Golgi complex and early endosomes (Mangahas et al., 2008; Edwards et al., 2009), is involved in the maturation of dense core vesicles (Borgonovo et al., 2006; Mangahas et al., 2008; Edwards et al., 2009; Sumakovic et al., 2009) and the packaging of APL-1 into mature dense core vesicles (Wiese et al., 2010). Both UNC-116/kinesin-1 and UNC-104/KIF1A/kinesin-3 are involved in the anterograde transport of APL-1 (Wiese et al., 2010; Arimoto et al., 2011), but only UNC-116/kinesin-1 and dynein motors are responsible for the retrograde transport of APL-1 back to the cell body (Arimoto et al., 2011). The rates of anterograde and retrograde transport of APL-1 vesicles are 1.1 μm/s and 1.6 μm/s, respectively (Arimoto et al., 2011). Hence, APL-1 is transported similarly as in mammalian models where kinesin-1 is responsible for APP axonal transport (Koo et al., 1990). Surprisingly, mutations in unc-116/kinesin-1 and unc-104/KIF1A/kinesin-3 both caused decreased levels of APL-1 expression without affecting transcript levels, suggesting that without transport motors, APL-1 does not accumulate in cell bodies because of protein degradation (Wiese et al., 2010; Arimoto et al., 2011). APL-1 is also internalized from the cell surface of neurons via a RAB-5-dependent endocytosis (Wiese et al., 2010).
Aicd Intracellular Trafficking
The Fe65 family of proteins binds the cytoplasmic YENPTY sequence of APP, APLP1, and APLP2, via their PTB2 domain (Guénette et al., 1996; Zambrano et al., 1997; Duilio et al., 1998; Russo et al., 1998). Likewise, the sole family member ortholog in C. elegans, FEH-1, has a WW domain and PTB1 and PTB2 domains, which closely resemble those of the Fe65 family, and the PTB2 domain of FEH-1 interacts with APL-1 (Zambrano et al., 2002).
FEH-1 is expressed in pharyngeal muscle and neuronal processes and is necessary for survival. Inactivation of feh-1 caused an incompletely penetrant embryonic lethality. Survivors showed little pharyngeal pumping and were unable to feed, thereby resulting in L1 arrest (Zambrano et al., 2002). Decreasing feh-1 activity or decreasing feh-1 dosage caused pharyngeal pumping rates to increase, suggesting that the rate of pharyngeal pumping is feh-1 dosage dependent. However, the functional significance of FEH-1 and APL-1 interactions is unclear as apl-1(yn5) mutants, which do not have an AICD domain, and apl-1(yn10/+) heterozygotes do not have defective pumping rates (Ewald et al., 2012b).
Investigating the Amyloid Hypothesis of AD in C. elegans
Aβ peptide, the cleavage product of APP believed to underlie the pathology of AD (Glenner and Wong, 1984; Masters et al., 1985; Gorevic et al., 1986; Selkoe et al., 1986), is not present in APL-1 (Daigle and Li, 1993) nor does C. elegans possess β-secretase activity to produce Aβ (Link, 2006). Nevertheless, C. elegans provides a powerful in vivo genetic system to study the effects of neurotoxic Aβ through transgene analysis (Shankar et al., 2008). Many transgenic strains have been generated in which a signal sequence followed by the human Aβ sequence is expressed in all cells, in all neurons, in specific subsets of neurons, or in muscle cells (Figure 4). These strains produce either Aβ1-42 or Aβ3-42.
AD is a late onset neurodegenerative disease. C. elegans expressing human Aβ3-42 in muscle tissue (Link, 1995; Link et al., 2001) showed an age-dependent paralysis at 20°C (Cohen et al., 2006; McColl et al., 2009); paralysis occurred more rapidly and more severely when Aβ1-42 was produced at 25°C (McColl et al., 2009). The level of muscle paralysis was significantly decreased when insulin signaling was decreased (Cohen et al., 2006). Furthermore, inhibiting daf-16/FOXO and hsf-1, which encodes a heat shock protein transcription factor (Hsu et al., 2003; Morley and Morimoto, 2004), reversed the effects of decreased insulin signaling (Cohen et al., 2006). Hence, the paralysis effects of Aβ correlates with age and is dependent on insulin signaling.
Since aggregated Aβ is toxic to neurons (Glenner and Wong, 1984; Masters et al., 1985; Gorevic et al., 1986; Selkoe et al., 1986) and causes muscle paralysis in C. elegans, molecules and pathways that can prevent the formation or promote the disassembly of Aβ aggregates can be screened for in C. elegans. For instance, when C. elegans extracts are incubated with aggregated human Aβ3-42 in the presence or absence of protease inhibitors, disaggregation occurred, but disaggregation did not occur when extracts were either heated to denature proteins or incubated with proteinases (Bieschke et al., 2009). Hence, an unidentified protein or protein complex in C. elegans extracts can disaggregate Aβ3-42 aggregates.
Several orthologs to human heat shock (HSP) chaperone proteins were found to interact directly with Aβ3-42 in C. elegans. C. elegans HSP-16 proteins, HSP-16-1, HSP-16-2, and HSP-16–48, orthologs of αB-crystallin, bound intracellular Aβ3-42 monomers and soluble Aβ3-42 oligomers, but not fibrillar Aβ3-42 (Fonte et al., 2002). Moreover, hsp-16 transcript levels were upregulated in Aβ3-42 transgenic lines, but whether these chaperone proteins protect against or promote Aβ paralysis is unclear (Fonte et al., 2002). By contrast, increased expression of the HSP70 chaperones had a protective role by suppressing paralysis (Fonte et al., 2008). These results are consistent with human studies showing that HSP70 and αB-crystallin were upregulated in AD brains (Hamos et al., 1991; Perez et al., 1991; Shinohara et al., 1993; Renkawek et al., 1994; Yoo et al., 2001) and binds Aβ (Liang, 2000).
Transgenic lines in which Aβ is expressed in glutamatergic neurons showed age-dependent neurodegeneration, whereby 7-day adults showed 75% glutamatergic neurodegeneration (Treusch et al., 2011). This degeneration was suppressed when genes involved in clathrin-mediated endocytosis, such as unc-11, unc-26, Y44E3A.4, C. elegans RTS1 ortholog, C. elegans ADE12 ortholog, and human CRMI, were co-expressed with Aβ, and enhanced when a PBS2/MAP2K4 mitogen-activated protein kinase transgene was co-expressed with Aβ in glutamatergic neurons (Treusch et al., 2011). Interestingly, mutations in the C. elegans human REST ortholog spr-4, which suppressed the sel-12/PSEN egg-laying defect (Lakowski et al., 2003), also enhanced the degeneration seen in the transgenic animals expressing Aβ in glutamatergic neurons (Lu et al., 2014). Modifying clathrin-mediated endocytosis in rat cortical neurons was similarly neuroprotective against Aβ aggregates (Treusch et al., 2011). In addition, early stage AD brains showed higher expression of REST target genes, while late stage AD and frontotemporal dementia (FTD) brains showed lower expression (Lu et al., 2014). Hence, REST may confer neuroprotective benefits in C. elegans and in humans (Lu et al., 2014).
The C. elegans Aβ model also proves useful in screens to identify drugs that disaggregate Aβ. The drug PBT2, an 8-hydroxy quinoline analog, reversed AD phenotypes in mice within days (Adlard et al., 2008). Similarly, C. elegans expressing inducible Aβ1-42, which become paralyzed within 48 h after induction, were protected against paralysis when exposed to PBT2 (McColl et al., 2012).
C. elegans lrp-1 Functions Similarly to LRP2/Megalin, an LDL Receptor Family Member
In mammals, the LDL receptor family is responsible for many functions, including binding ligands for internalization and degradation and cholesterol metabolism (Brown and Goldstein, 1986; Mahley, 1988; Herz et al., 1992; Willnow et al., 1994). Binding of LRP1 to sAPP770 or full-length APP770, one of the APP isoforms (Figure 1), causes its internalization and degradation (Kounnas et al., 1995; Knauer et al., 1996); disrupting cell surface APP internalization with an LRP-antagonist increases sAPPα processing and full-length APP at the cell surface and decreases Aβ formation, suggesting that LRP1-APP interactions favor APP processing through the amyloidogenic pathway (Ulery et al., 2000). Apolipoprotein E and LRP2/megalin have also been implicated in Aβ clearance (Zlokovic et al., 1996; Deane et al., 2004; Carro et al., 2005).
C. elegans LRP-1 most closely resembles mammalian LRP2/megalin (Yochem and Greenwald, 1993; Yochem et al., 1999). C. elegans does not have the ability to synthesize cholesterol and, therefore, must rely on dietary sources (Hieb and Rothstein, 1968). Inactivation of lrp-1 resulted in late larval lethality due to molting defects during the L3–L4 transition (Yochem et al., 1999). When wild-type C. elegans were grown in the absence of cholesterol, the molting defects of the lrp-1 knockouts were phenocopied (Yochem et al., 1998; Wiese et al., 2012), suggesting that LRP-1 is involved in cholesterol uptake from the environment. LRP-1 is expressed in the epithelial hypodermal cells, hyp6 and hyp7, where it localizes to their apical surface (Yochem et al., 1999) and where apl-1 is also expressed in adults (Niwa et al., 2008). Similarly, its mammalian counterpart, LRP2/megalin, is mainly expressed at the apical surface of epithelial cells (Cui et al., 1996; Morales et al., 1996; Willnow et al., 1996; Nielsen et al., 1998; Zheng et al., 1998; Hermo et al., 1999; Mizuta et al., 1999).
LRP2/megalin interacts with different domains of APP (Zlokovic et al., 1996; Pietrzik et al., 2004; Carro et al., 2005; Yoon et al., 2005; Cam and Bu, 2006). A physical interaction between APL-1 and LRP-1 has not been determined. Expression of LRP-1 with an N-terminal domain truncation did not rescue the lethality of apl-1 null mutants, suggesting that sAPL-1 is not activating an lrp-1 pathway (Hornsten et al., 2007). When lrp-1 expression is decreased or when wild-type animals are deprived of dietary cholesterol, neurotransmission is affected (Wiese et al., 2012). However, apl-1 null mutants die at an earlier stage in development than lrp-1 null mutants, suggesting that apl-1 functions in earlier developmental pathways that are necessary for survival.
C. elegans as a Model for Other Neurodegenerative Diseases
PTL-1 as a Tau Model
Accumulation of neurofibrillary tangles in cell bodies is another hallmark characteristic of AD and other neurodegenerative disorders. The major component of these tangles is tau, which belongs to the family of microtubule-associated proteins (MAPs) that includes MAP2 and MAP4 (Lee et al., 1988; Lewis et al., 1988; Goedert et al., 1989b; Chapin and Bulinski, 1991). MAPs share characteristic homology domains, including a proline-rich domain and a region of a variable number of tandem amino acid repeats (Figure 3A; Goedert et al., 1988, 1989a,b, 1992a; Lee et al., 1988; Lewis et al., 1988; Aizawa et al., 1990). Tau is the predominant MAP expressed in axons, while MAP2 is expressed in dendrites (Matus et al., 1981; Binder et al., 1985) and MAP4 is expressed in dividing cells (Bulinski and Borisy, 1980). MAPs bind microtubules and are responsible for promoting microtubule assembly and stability (reviewed in Amos and Schlieper, 2005). MAP family members appear to have redundant functions; mice in which tau was knocked out were viable, but showed increased levels of MAP1A (Harada et al., 1994), suggesting that upregulation of MAP1A can compensate for the lack of tau in vivo. Tau phosphorylation affects its ability to bind microtubules and can cause a conformational change that favors tubulin assembly (Figure 3B; Feijoo et al., 2005). Aberrant hyperphosphorylation of tau, however, impairs its ability to bind microtubules, thus resulting in their disassembly (Lindwall and Cole, 1984; Biernat et al., 1993; Bramblett et al., 1993). In addition, phosphorylated tau self-aggregates into PHFs and presumably generates the intracellular neurofibrillary tangles characteristic of AD patients (Figure 3B; Goedert et al., 1992b; Alonso et al., 1996, 1997, 2001; Billingsley and Kincaid, 1997).
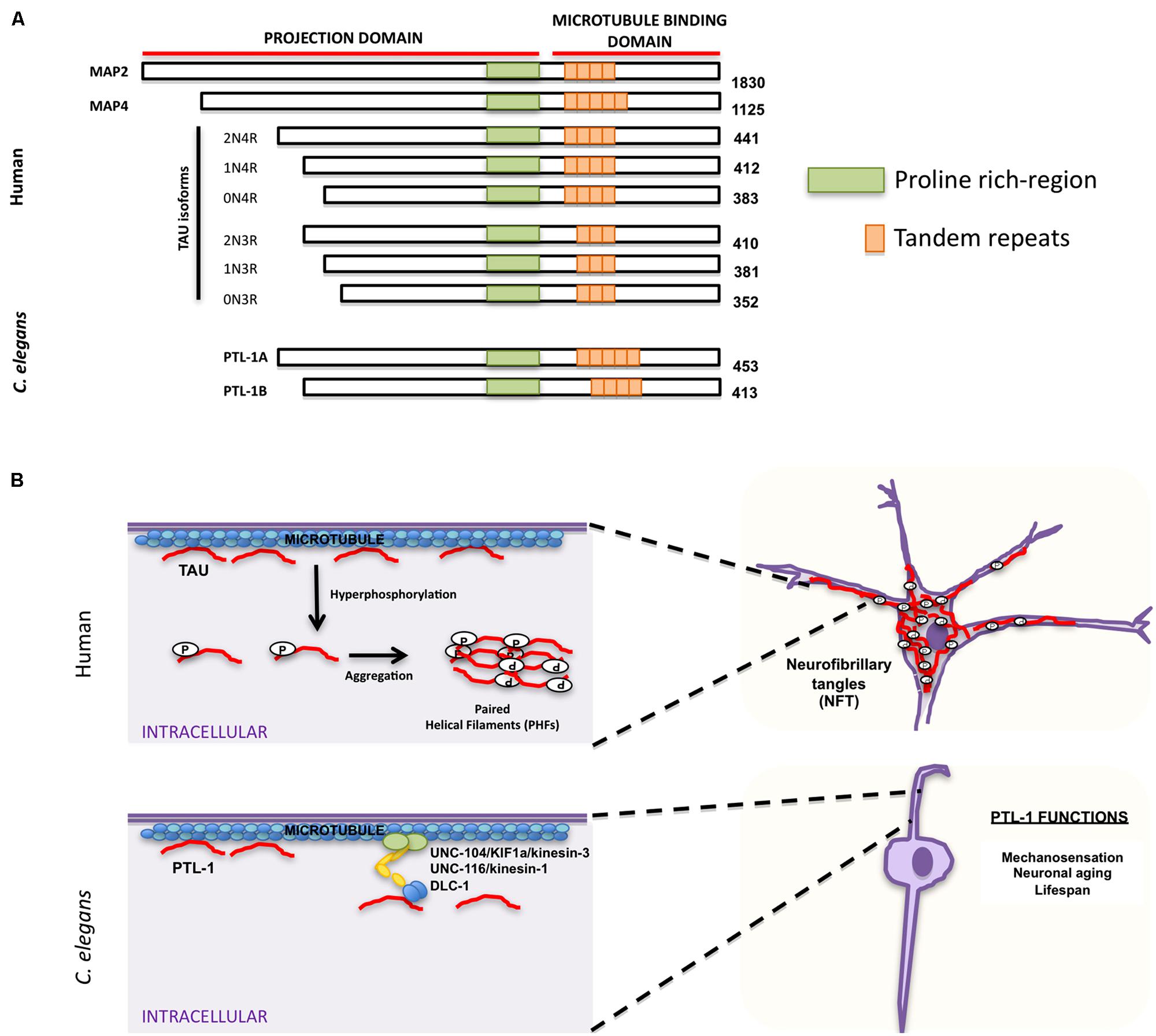
FIGURE 3. Similarities and differences between human tau and Caenorhabditis elegans PTL-1. (A) Schematic representation of human microtubule binding proteins (MAPs) family, including tau isoforms and C. elegans PTL-1 isoforms. (B) Comparison between tau functions in humans (top) and C. elegans PTL-1/tau functions (bottom). Top. Tau has a physiological role in promoting and maintaining microtubule stability. In pathological conditions tau is hyperphosphorylated and self-aggregates into paired helical filaments (PHFs) that can form intracellular neurofibrillary tangles (NFT). Bottom. C. elegans PTL-1/tau binds microtubules and induces microtubule assembly. It also affects synaptic transport through motor proteins UNC-104/KIF1a/kinesin-3, UNC-116/kinesin-1, and DLC-1/dynein. PTL-1/tau is also important for C. elegans mechanosensation and aging.
Because of the functional redundancy of MAPs, their specific functions have been difficult to determine. C. elegans has only one tau homolog protein with tau-like repeats 1 (PTL-1; Goedert et al., 1996; McDermott et al., 1996). PTL-1 exists as two isoforms, PTL-1A and PTL-1B, with five or four tandem repeats, respectively (Goedert et al., 1996; Figure 3A). They have a high level of sequence homology with mammalian tau, especially in the C-terminal microtubule binding region (Goedert et al., 1996; McDermott et al., 1996). Both PTL-1A and PTL-1B bound microtubules in vitro and induced tubulin polymerization (Goedert et al., 1996; McDermott et al., 1996). PTL-1 is initially expressed embryonically in the epidermis of elongating embryos and in head neurons; in larval and adult animals PTL-1 is expressed mainly in the mechanosensory neurons mediating gentle body touch (Figure 3B; Goedert et al., 1996; Gordon et al., 2008), although transcriptional fusions show a wider expression pattern in neurons and stomatointestinal cells (Gordon et al., 2008).
Loss of ptl-1/tau results in an incompletely penetrant lethality at the same stage of embryogenesis (Gordon et al., 2008) at which PTL-1/tau expression is first observed (Goedert et al., 1996). ptl-1/tau mutants that escaped lethality showed normal development, but had a shortened lifespan, and although the overall integrity of microtubule structure appeared unaffected at the light microscopic level, there was a significant reduction in gentle touch sensitivity as compared to wild-type (Gordon et al., 2008; Chew et al., 2013). These touch defects were enhanced in mutants with defects in β- and α-tubulin, indicating that the absence of full-length PTL-1/tau disrupts mechanosensation, but it does so independently of tubulin (Gordon et al., 2008). In mutants in which only the C-terminal microtubule binding repeats of PTL-1/tau are deleted, touch sensitivity was identical to that of wild-type (Chew et al., 2013), suggesting that the N-terminal domain of PTL-1/tau is sufficient for gentle touch responses.
As C. elegans ages, mechanosensory touch neurons exhibit age-related morphological changes: cell bodies initially elaborate branches and axons subsequently display blebbing and branching (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). Strikingly, mechanosensory touch neurons in ptl-1/tau mutants displayed these aging characteristics at higher incidences and at an earlier stage than wild-type animals; GABAergic neurons also showed age-related phenotypes, such as ectopic branching (Chew et al., 2013). Expression of a human tau isoform (htau40) in ptl-1/tau mutants rescued the touch insensitivity, but not the morphological aging defects, indicating that htau40 shares some functional conservation with PTL-1/tau (Chew et al., 2013). Interestingly, when ptl-1/tau was expressed in Sf9 cells, cells projected neurite-like processes that were positive for PTL-1/tau immunoreactivity (Goedert et al., 1996) and that were indistinguishable from those visualized when htau40 was expressed in Sf9 cells (Knops et al., 1991; Chen et al., 1992). Although wild-type PTL-1/tau has not been reported to aggregate into fibrils, PTL-1/tau, like human tau, clearly has an essential role in maintaining neuronal integrity, controlling neuronal aging, and affecting lifespan (Goedert et al., 1996; Gordon et al., 2008). While there are no known mutations in tau that are associated with AD, tau mutations are associated with FTD with parkinsonism (FTPD-17), another form of dementia (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998; see Section “Frontotemporal Dementia”).
Frontotemporal Dementia
Frontotemporal dementia (FTD) is a group of neurodegenerative disorders characterized by severe brain frontotemporal lobar degeneration (reviewed in Rabinovici and Miller, 2010). In some cases it may be hard to distinguish between FTD and AD; however, FTD usually develops earlier in life and is more likely to have a genetic component (Lindau et al., 2000; Pasquier, 2005). Many mutations can cause FTD with or without motor neuron disease (Cruts et al., 2012). Two mutations have been well characterized and are associated with specific types of FTD: tau-positive FTD linked to chromosome 17 (FTD-17) and FTD caused by TDP43 proteinopathy (FTD-TDP43). Patients with FTD-17 suffer behavioral changes and often Parkinson-like motor problems. While mutations in the tau gene MAPT are not described in familial or sporadic AD, MAPT tau mutations are linked with FTD-17 (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998).
Several transgenic lines expressing human tau harboring FTD-17 mutations (htau-FTD-17) have been generated in C. elegans (Figure 4B) (see also Section “PTL-1 as a Tau Model”; Kraemer et al., 2003; Miyasaka et al., 2005; Brandt et al., 2009; Fatouros et al., 2012). Pan-neuronal transgene expression of wild type or htau-FTD-17 caused an uncoordinated phenotype that progressively worsened with age, an accumulation of insoluble tau, and neurodegeneration (Kraemer et al., 2003). Similarly, expression of htau-FTD-17 in touch neurons resulted in a decrease in the touch response due to neuritic abnormalities and tau accumulation (Miyasaka et al., 2005). Using these C. elegans models of tauopathy in forward genetic screens, Kraemer and co-workers identified two new factors, SUT-1 and SUT-2, that may participate in the pathological pathway activated by tau (Kraemer et al., 2003; Kraemer and Schellenberg, 2007; Guthrie et al., 2009). Moreover, down-regulation of the human SUT-2 homolog (MSUT-2) in mammalian cell lines caused a marked decrease in tau aggregation, suggesting that MSUT-2 may be a good candidate target for FTD therapies (Wheeler et al., 2010; Guthrie et al., 2011). More recently, Fatouros et al. (2012) generated two htau-FTD-17 transgenic models: one with a pro-aggregant mutated form of human tau (deletion of K280) and a second with mutated forms of human tau (I277P and I308P) that prevented tau aggregation. The tau (ΔK280) transgenic line had high levels of tau aggregation, which caused uncoordinated movement in adults, axonal defects, and alterations in presynaptic structures (Fatouros et al., 2012); the locomotory defects could be partially suppressed by a compound of the aminothioenopyridazine (ATPZ) class cmp16, suggesting that this compound may be neuroprotective (Fatouros et al., 2012). The tau (I277P and I308P) transgenic lines had low levels of tau aggregates and displayed only mild phenotypes with significantly less morphological abnormalities.
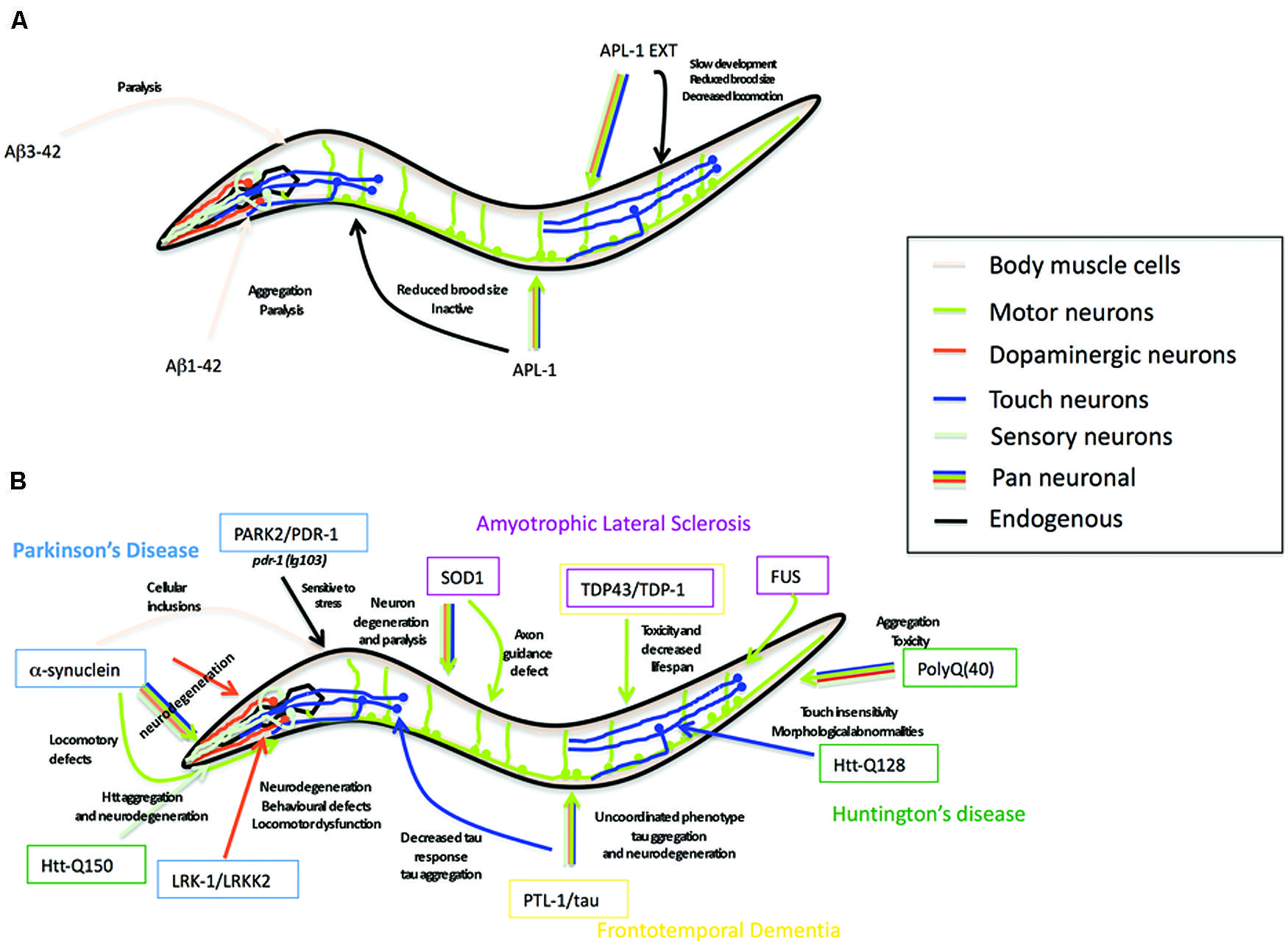
FIGURE 4. Caenorhabditis elegans as a transgenic model for AD and other neurodegenerative diseases. (A) Summary of the Alzheimer’s disease models in C. elegans expressing human Aβ peptide or C. elegans full-length APL-1 or APL-1 extracellular domain (APL-1 EXT). Arrow color represents the tissues where transgenes were expressed. Phenotypes observed are next to the arrow. (B) Summary of the C. elegans models for Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Huntington’s disease (HD). Genes modeling PD are shown in blue boxes, ALS in pink boxes, FTD in yellow boxes, and HD in green boxes. Arrow color represents tissues where transgenes were expressed. Phenotypes observed are written close to the arrow.
Accumulation of TDP-43 [transactive response (TAR) DNA-binding protein] is found in ∼50% of the cases of FTD (The Association for Frontotemporal Degeneration, 2014) and has numerous genetic causes. However, only one case has been reported with mutations in the TDP-43 gene (Borroni et al., 2009). C. elegans models overexpressing human TDP-43 or its C. elegans ortholog TDP-1 recapitulates some of the FTD phenotypes, including neurotoxicity and protein aggregation (see also Section “Amyotrophic Lateral Sclerosis”).
C9orf72 encodes a protein that regulates endosomal trafficking and autophagy in primary neurons and neuronal cells (Farg et al., 2014). It is expressed in multiple tissue types, including cerebellar cortex and spinal cord (DeJesus-Hernandez et al., 2011). Hexanucleotide (GGGGCC) repeat expansions in a non-coding region of C9orf72 are found in patients with amyotrophic lateral sclerosis (ALS) and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012), providing the first genetic link between the two diseases, although it remains unclear how C9orf72 hexanucleotide expansion triggers ALS and FTD pathology. Mutations in the C. elegans C9ORF72 ortholog alfa-1 caused age-dependent motility defects, leading to paralysis and degeneration of GABAergic motoneurons (Therrien et al., 2013), suggesting that a loss-of-function mechanism is involved in the C9ORF72-dependent pathogenesis.
Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects the control of body movements. The impaired motor control in PD is the result of the death of dopaminergic (DA) neurons (Hughes et al., 1992; Fahn and Sulzer, 2004). The disease is characterized by the accumulation of α-synuclein into neuronal inclusions called Lewy bodies (Lewy, 1912; Tretiakoff, 1919). Most PD cases are of unknown cause. However, ∼5–10% of PD cases are familial (Wood-Kaczmar et al., 2006) and include mutations in the following genes: α-synuclein, parkin (PRKN), leucine-rich repeat kinase 2 (LRRK2), PTEN-induced putative kinase 1 (PINK1), and ATP13A2 (Hardy, 2010).
α-synuclein is a presynaptic neuronal protein whose cellular function is not well understood, but may include controlling the supply of synaptic vesicles in neuronal terminals and regulating dopamine release. It is a small acidic protein (14 kDa) whose sequence can be divided into three domains: the N-terminal α-helical domain (amino acids 1–65), the central hydrophobic domain (residues 66–95), and the acidic carboxyl-terminal domain (residues 96–140; Recchia et al., 2004). Three mutations in the α-helical domain (A53T, A30P, E46K) are linked with autosomal dominant early onset PD, suggesting that these mutations can predispose to oligomer and fibril formation (Polymeropoulos et al., 1997; Krüger et al., 1998; Conway et al., 2000a,b,c; Zarranz et al., 2004). Because C. elegans has no α-synuclein ortholog, C. elegans models are based on transgenic worms overexpressing wild-type or mutant forms of human α-synuclein (Figure 4B). Although different transgenic lines showed some differences, most lines with pan-neuronal or DA neuronal expression of wild-type or mutated α-synuclein (A53T and/or A30P mutations) displayed locomotory defects and degeneration of dopamine neurons (Lakso et al., 2003; Kuwahara et al., 2006; Cao et al., 2010). Furthermore, downregulating the activity of the nuclease EndoG decreased α-synuclein toxicity in DA neurons (Büttner et al., 2013). EndoG is a mitochondria-specific endonuclease that mediates cellular death by apoptosis (Li et al., 2001). When cell death is induced, EndoG translocates from the mitochondria to the nucleus to fragment DNA (Li et al., 2001). Approximately 50% of the dopamine neurons expressing α-synuclein degenerate, whereas a mutation in cps-6, which encodes the C. elegans EndoG ortholog, rescues this degeneration (Büttner et al., 2013). Similar results were found in yeast, flies, and human cells suggesting that EndoG is a conserved requirement for α-synuclein toxicity (Büttner et al., 2013).
C. elegans models of α-synuclein overexpression-induced toxicity have also been examined by whole genome RNAi knockdown and microarray screenings (Vartiainen et al., 2006; Hamamichi et al., 2008; Kuwahara et al., 2008; van Ham et al., 2008). These screens highlighted the importance of endocytosis for ameliorating α-synuclein-dependent neurotoxicity (Kuwahara et al., 2008). Transgenic lines expressing α-synuclein specifically in body wall muscle cells produced inclusions as animals aged, resembling a feature of neurons in patients with PD; the number of inclusions decreased when genes affecting different biological processes, such as vesicle and lysosomal trafficking (W08D2.5/ATP13A2), lipid metabolism, and lifespan control (sir-2.1,lagr-1), were knocked down (van Ham et al., 2008). In a genome-wide microarray analysis to identify genes that were modulated in C. elegans overexpressing wild-type or A53T human α-synuclein, seven genes encoding components of the ubiquitin-proteasome machinery and 35 mitochondrial function genes were found to be upregulated, while nine genes encoding histones H1, H2B, and H4 were down regulated (Vartiainen et al., 2006). These data provide support for the role of the proteasome complex and mitochondrial proteins in mediating neurotoxicity.
Parkin (human PARK2) is a component of an ubiquitin E3 ligase that is part of the proteasome complex (Shimura et al., 2000). Mutations in PARK2 have been associated with early onset recessive forms of PD (Kitada et al., 1998; Poorkaj et al., 2004). C. elegans has a parkin ortholog, PDR-1. A truncated form of PDR-1(Δaa24–247) encoded by the in-frame deletion null allele pdr-1(lg103) had altered solubility and propensity to aggregate when expressed in cell lines, resembling parkin mutant proteins in PD (Springer et al., 2005). Furthermore, pdr-1 mutants were hypersensitive to different proteotoxic stress conditions, suggesting that PDR-1/PARK2 mutations act to block the proteostasis machinery, thereby making it easier for proteins to abnormally fold and aggregate (Springer et al., 2005).
Mutations in LRRK2/leucine-rich repeat kinase 2 are the most common known cause of late-onset PD. LRRK2 belongs to the LRRK family; gain-of-function LRRK2 mutations interfere with chaperone-mediated autophagic functions and presumably decrease levels of α-synuclein degradation (Orenstein et al., 2013). Transgenic worms overexpressing pathogenic mutant forms of LRRK2 in DA neurons caused DA neurodegeneration (Liu et al., 2011; Yao et al., 2013). Interestingly, treatment of the transgenic worms with kinase inhibitors resulted in arrested neurodegeneration, suggesting that LRRK2 kinase activity is important for its pathogenesis (Liu et al., 2011; Yao et al., 2013).
Many studies have reported a link between toxin exposure and increased risk of PD. C. elegans has been used to test different toxins and help elucidate the mechanism by which they produce neurotoxicity. Administration of the 6-hydroxydopamine (6-OHDA) neurotoxin to C. elegans produced specific degeneration of dopamine neurons (Nass et al., 2002). By performing forward genetic and high-throughput chemical screens, mutations within the dopamine transporter dat-1 were found to suppress 6-OHDA sensitivity (Nass et al., 2005) and bromocriptine, quinpirole, and acetaminophen, and plant extracts from Bacopa monnieri and Uncaria tomentosa were found to be neuroprotective (Marvanova and Nichols, 2007; Locke et al., 2008; Ruan et al., 2010; Jadiya et al., 2011; Shi et al., 2013). These data demonstrate that pathological characteristics of PD can be recapitulated in C. elegans models and used to investigate the mechanism by which α-synuclein and other PD proteins produce neurotoxicity and cause motor defects.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is a neurodegenerative disease characterized by the death of motor neurons in brain and spinal cord and progressive paralysis of the body (Hardiman et al., 2011). Approximately 10% of ALS cases are familial and associated with mutations in several genes. The most common mutation in familial ALS is found in the superoxide dismutase enzyme (SOD1) with more than 160 different mutations identified (Wroe et al., 2008). SOD1 is a ubiquitously expressed protein that converts the toxic radical superoxide anion to hydrogen peroxide. Although it is not clear yet how SOD1 mutations causes motor neuron degeneration, toxicity is likely generated by a gain-of-function mechanism (Valentine et al., 2005) and associated with misfolding and aggregation of the enzyme (Pasinelli and Brown, 2006).
Transgenic lines expressing mutant human SOD1 proteins have been successfully generated in C. elegans and recapitulate the motor neuron degeneration and paralysis characteristic of ALS patients (Figure 4B) (Witan et al., 2008; Gidalevitz et al., 2009; Wang et al., 2009; Li et al., 2014). The locomotion defect caused by pan-neuronal expression of the SOD1(G85R) mutant isoform was reduced when insulin signaling was decreased (Boccitto et al., 2012), suggesting that decreased insulin signaling increases the capacity of cells to prevent the accumulation of toxic non-soluble proteins and opening the possibility of finding new therapeutic targets. Similarly, when wild-type or mutant SOD1(G93A) was expressed exclusively in GABAergic motor neurons, animals showed an age-dependent paralysis and accumulation of wild-type and mutant SOD1(G93A), although defects were more severe in the mutant lines; interestingly, the SOD1 aggregates were soluble in the wild-type SOD1 lines and insoluble in the SOD1(G93A) lines (Li et al., 2014). In addition, motor neurons showed axonal guidance defects during development and caspase-independent cell death in adulthood in the wild-type and SOD1(G93A) lines (Li et al., 2014).
Other genes associated with ALS have also been modeled using C. elegans. TDP-43 [TAR DNA-binding protein] is a 43 kDa RNA binding protein identified as the main component of ubiquitinated protein aggregates (Tsuda et al., 2008; Murakami et al., 2012; Vaccaro et al., 2012a,b; Han et al., 2013; Therrien et al., 2013) found in patients with sporadic ALS (Neumann et al., 2006) and also in some cases of FTD (see Section “Frontotemporal Dementia”). TDP-43 is normally located in the nucleus of neurons, but dominant mutations in TDP-43 cause aberrant localization of TDP-43 in the cytoplasm, thereby preventing it from functioning in the nucleus (Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008; Ash et al., 2010). C. elegans has one TDP-43 ortholog, TDP-1. TDP-1 controls longevity and oxidative stress in the worm by regulating the insulin pathway (Vaccaro et al., 2012b). Overexpression of tdp-1/TDP-43 resulted in toxicity and decreased lifespan, analogous to the phenotypes found in ALS patients (Vaccaro et al., 2012b). In transgenic worms expressing TDP-43 harboring ALS-associated mutations, proteotoxicity affecting neuronal functions was induced. Similar results were found when the RNA binding protein FUS with ALS-related mutations was expressed in the nematode (Murakami et al., 2012; Vaccaro et al., 2012b).
Excess exposure to some pesticides and chemicals, such as the metalloid selenium, have been implicated in the etiology of ALS (Vinceti et al., 2009; Kamel et al., 2012; Malek et al., 2012). Exposure to high levels of sodium selenite in the worm induced neurodegeneration and resulted in paralysis (Estevez et al., 2012, 2014). When insulin pathway activity was reduced, the adverse effects of environmental selenium exposure was altered (Estevez et al., 2014). Overall, the C. elegans models have highlighted the possible importance of the insulin and autophagy pathways in the generation of ALS.
Huntington’s Disease
Huntington’s disease (HD) is a progressive neurodegenerative disorder inherited through autosomal dominant mutations of the IT15 gene. ITI5 encodes the huntingtin protein, whose functions remain unknown (The Huntington’s Disease Collaborative Research Group, 1993). The mutations result in an N-terminal polyglutamine (polyQ) expansion (Goldberg et al., 1996; Mangiarini et al., 1996). In normal individuals, up to 34 repeats have been reported, whereas in HD afflicted individuals, up to 100 polyQ repeats have been recorded (The Huntington’s Disease Collaborative Research Group, 1993). The huntingtin-polyQ (HdhQ) proteins form aggregates, whose toxicity is determined by the length of the polyQ expansion and which cause swollen, disorganized, and ribosome-deficient endoplasmic reticulum and chromatin irregularities (Martindale et al., 1998). Eventually, cellular defects caused by the aggregates culminate in HD symptoms, which include involuntary movement, cognitive impairment, and loss of neurons in the striatum and deep layers of the frontal cortex (Martin and Gusella, 1986).
Although C. elegans does not have a huntingtin homolog, transgenic C. elegans models that express an N-terminal human huntingtin (htt) fragment with different numbers of CAG repeats have been used to model HD and identify genes that prevent polyQ aggregates (Figure 4B). The models generally express the repeats in specific neurons, such as the ASH sensory neurons, which are multi-modal sensory neurons that mediate avoidance to chemo- and mechanosensory stimuli. In transgenic animals expressing htt171 with 150 CAG repeats (htt171-Q150), 13% of the ASH neurons began to lose function after 8 days, suggesting an age-dependent degeneration (Faber et al., 1999). This loss of ASH function was reversed in a ced-3/caspase (Faber et al., 1999) or hda-3/HDAC (Bates et al., 2006) mutant background, suggesting that processes characteristic of apoptotic cell death and histone deacetylases play a role in HD (Dragunow et al., 1995). By contrast, the number of htt171-Q150 aggregates and neurodegeneration were enhanced when genes mediating autophagy, CREB, CREB binding proteins, and pqe-1 were disrupted, suggesting that autophagy and activation of CREB target genes decreases htt171-Q150 aggregation and are neuroprotective (Faber et al., 2002; Bates et al., 2006; Jia et al., 2007). Transgenic animals expressing fewer CAG repeats (2, 23, and 95 polyQ) showed normal ASH function (Faber et al., 1999). The onset of behavioral defects are consistent with most cases of HD, in which symptoms usually appear during midlife (Vonsattel et al., 1985; Martin and Gusella, 1986; Strong et al., 1993; The Huntington’s Disease Collaborative Research Group, 1993; Gusella and MacDonald, 1995) and fewer than 10% of reported cases occur before the age of 21 (Farrer and Conneally, 1985; van Dijk et al., 1986; Nance, 1997; Siesling et al., 1997).
In a different HD model, htt57-Q128 was expressed in the touch mechanosensory neurons (Parker et al., 2001). These transgenic animals did not show neurodegeneration, but had a significantly reduced response to posterior touch and a milder defect in anterior touch response (Parker et al., 2001). The touch neurons contained polyQ aggregates and morphological abnormalities primarily along axonal processes (Parker et al., 2001). The touch insensitivity could be rescued by activating Sir2 sirtuins (Parker et al., 2005), which act through the DAF-16/FOXO transcription factor to promote longevity (Tissenbaum and Guarente, 2001). Similarly, in neuronal cell lines derived from knockin HdhQ111 mice, activation of sirtuins reduced the level of cell death (Parker et al., 2005). Additionally, in an RNAi based screen for genes that suppressed htt57-Q128 defects, identified C. elegans genes were also upregulated in the striatum of mouse HD models (Lejeune et al., 2012). Thus, C. elegans is a useful model to identify additional genes that may protect against or contribute to defects caused by polyQ expansions.
RNAi knockdown of dnj-27/ERdj5, an ER luminal protein upregulated in response to ER stress, exacerbated the impaired mobility observed when a Q40 transgene is expressed in body wall muscles, suggesting that dnj-27 interacts with polyQ and protects against polyQ induced paralysis (Muñoz-Lobato et al., 2014).
C. elegans has the advantage that it is transparent, allowing visualization of the formation of that aggregates, including aggregates made by shorter polyQ tracts, whereas only longer tracts are visible in mammals (Brignull et al., 2006). To determine the threshold number of polyQ repeats needed to elicit a morphological and behavioral response, varying lengths of polyQ repeats were tested in C. elegans. Pan-neuronal expression of more than 40 polyQ led to variable protein aggregation and paralysis (Brignull et al., 2006). These data suggest that 40 polyQ may be the critical number of repeats to elicit HD symptoms and are consistent with unaffected humans who have up to 34 polyQ repeats and HD patients who have as few as 42 repeats (The Huntington’s Disease Collaborative Research Group, 1993).
Overall, C. elegans HD models illustrate that human huntingtin polyQs disrupt the morphology and function of sensory neurons. The genetic and RNAi screens highlight candidate genes that may be involved in HD pathogenesis in mammalian models and provide insights into genes that may serve a protective role against polyQ toxicity. In addition to HD, other diseases caused by polyQ repeats include spinocerebellar ataxias and spinal and bulbar muscular atrophy (Orr and Zoghbi, 2007). Hence, using C. elegans provides another approach toward determining how polyQ pathogenicity contributes to neurodegeneration.
Advantages and Limitations of the C. elegans Model
The use of C. elegans to study AD and other neurodegenerative diseases has, as many other models, many advantages as well as some drawbacks. Major advantages of C. elegans include the ability to perform forward genetic, RNAi, and high throughput chemical screens and the ease of generating transgenic lines. These benefits have been effective in informing the role of APP and the presenilins and identifying components of the γ-secretase complex. The function of APP and the pathways in which it acts are still unclear. C. elegans presents a complementary system to understand the function and pathways of an APP-related protein, APL-1. Furthermore, overexpression of APL-1 by mutation or by transgene induces phenotypes that converge on the insulin/DAF-16/FOXO pathways, similar to what has been found in mammals. Although APL-1 does not contain the Aβ sequence and C. elegans does not have β-secretase activity, transgenic lines that produce Aβ expression pan-neuronally or in muscle are being used to identify pathways that detoxify the Aβ aggregates, some of which also involve the insulin/DAF-16/FOXO pathways. Whether these models are relevant to human pathology or whether the pathways will be conserved in humans are unknown; however, they present alternative approaches to understanding neurodegenerative diseases for which there are currently few effective therapies. Human tau, as well as mutant tau isoforms, have also been expressed in the worm to recapitulate AD and FTD phenotypes. Recent findings have shown that PTL-1 regulates neuronal aging in the worm. These findings may be important to link aging and tau pathology in AD and FTD patients. Although C. elegans transgene models have many advantages, they also have several disadvantages. In C. elegans, transgenes are present as extrachromosomal arrays and are not integrated into the genome as they are in other systems; a few copies to several hundred copies of the transgene are present in the arrays, so the level of overexpression can be much higher than what is found in vivo. Fortunately, methods for single copy insertions have now been developed (Frøkjaer-Jensen et al., 2008).
AD is considered a multifactorial disease in which other risk factors, such as neuroinflammation, head trauma, and diabetes, may be important in the development of the disease. The C. elegans nervous system is simple compared to the human nervous system. This simplicity allows researchers to study neuronal function and neural circuits in a tractable system. However, the complex network of connections and cell interactions found in humans is not mimicked in C. elegans and this complexity may underlie some of the pathology of neurodegenerative diseases. Nevertheless, most of the pathways and signaling molecules in C. elegans are conserved between worms and mammals. The goal is to translate some of the C. elegans insights into understanding the pathology of AD and other neurodegenerative diseases and designing effective strategies to treat the diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Collin Ewald for reviewing our manuscript. This work was supported by grants from the NIH and NSF.
References
Adlard, P. A., Cherny, R. A., Finkelstein, D. I., Gautier, E., Robb, E., Cortes, M.,et al. (2008). Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59, 43–55. doi: 10.1016/j.neuron.2008.06.018
Aizawa, H., Emori, Y., Murofushi, H., Kawasaki, H., Sakai, H., and Suzuki, K. (1990). Molecular cloning of a ubiquitously distributed microtubule-associated protein with Mr 190,000. J. Biol. Chem. 265, 13849–13855.
Alonso, A., Zaidi, T., Novak, M., Grundke-Iqbal, I., and Iqbal, K. (2001). Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928. doi: 10.1073/pnas.121119298
Alonso, A. C., Grundke-Iqbal, I., and Iqbal, K. (1996). Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787. doi: 10.1038/nm0796-783
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303. doi: 10.1073/pnas.94.1.298
Alzheimer’s Disease International. (2014). World Alzheimer report 2013 [Online]. Available at: http://www.alz.co.uk/ (accessed March 05, 2014).
Ambros, V., and Horvitz, H. R. (1984). Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226, 409–416. doi: 10.1126/science.6494891
Amos, L. A., and Schlieper, D. (2005). Microtubules and maps. Adv. Protein Chem. 71, 257–298. doi: 10.1016/S0065-3233(04)71007-4
Arduengo, P. M., Appleberry, O. K., Chuang, P., and L’hernault, S. W. (1998). The presenilin protein family member SPE-4 localizes to an ER/Golgi derived organelle and is required for proper cytoplasmic partitioning during Caenorhabditis elegans spermatogenesis. J. Cell Sci. 111, 3645–3654.
Arimoto, M., Koushika, S. P., Choudhary, B. C., Li, C., Matsumoto, K., and Hisamoto, N. (2011). The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. J. Neurosci. 31, 2216–2224. doi: 10.1523/JNEUROSCI.2653-10.2011
Asahina, M., Ishihara, T., Jindra, M., Kohara, Y., Katsura, I., and Hirose, S. (2000). The conserved nuclear receptor Ftz-F1 is required for embryogenesis, moulting and reproduction in Caenorhabditis elegans. Genes Cells 5, 711–723. doi: 10.1046/j.1365-2443.2000.00361.x
Ash, P. E., Zhang, Y. J., Roberts, C. M., Saldi, T., Hutter, H., Buratti, E.,et al. (2010). Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 19, 3206–3218. doi: 10.1093/hmg/ddq230
Bargmann, C. I., Hartwieg, E., and Horvitz, H. R. (1993). Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell 74, 515–527. doi: 10.1016/0092-8674(93)80053-H
Bargmann, C. I., and Horvitz, H. R. (1991). Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 7, 729–742. doi: 10.1016/0896-6273(91)90276-6
Bates, E. A., Victor, M., Jones, A. K., Shi, Y., and Hart, A. C. (2006). Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 26, 2830–2838. doi: 10.1523/JNEUROSCI.3344-05.2006
Biernat, J., Gustke, N., Drewes, G., Mandelkow, E. M., and Mandelkow, E. (1993). Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11, 153–163. doi: 10.1016/0896-6273(93)90279-Z
Bieschke, J., Cohen, E., Murray, A., Dillin, A., and Kelly, J. W. (2009). A kinetic assessment of the C. elegans amyloid disaggregation activity enables uncoupling of disassembly and proteolysis. Protein Sci. 18, 2231–2241. doi: 10.1002/pro.234
Billingsley, M. L., and Kincaid, R. L. (1997). Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem. J. 323, 577–591.
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1985). The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378. doi: 10.1083/jcb.101.4.1371
Boccitto, M., Lamitina, T., and Kalb, R. G. (2012). Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS ONE 7:e33494. doi: 10.1371/journal.pone.0033494
Borgonovo, B., Ouwendijk, J., and Solimena, M. (2006). Biogenesis of secretory granules. Curr. Opin. Cell Biol. 18, 365–370. doi: 10.1016/j.ceb.2006.06.010
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S.,et al. (2009). Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30, E974–E983. doi: 10.1002/humu.21100
Bramblett, G. T., Goedert, M., Jakes, R., Merrick, S. E., Trojanowski, J. Q., and Lee, V. M. (1993). Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 10, 1089–1099. doi: 10.1016/0896-6273(93)90057-X
Brandt, R., Gergou, A., Wacker, I., Fath, T., and Hutter, H. (2009). A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 30, 22–33. doi: 10.1016/j.neurobiolaging.2007.05.011
Brignull, H. R., Moore, F. E., Tang, S. J., and Morimoto, R. I. (2006). Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J. Neurosci. 26, 7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006
Brown, M. S., and Goldstein, J. L. (1986). A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47. doi: 10.1126/science.3513311
Bulinski, J. C., and Borisy, G. G. (1980). Microtubule-associated proteins from cultured HeLa cells. Analysis of molecular properties and effects on microtubule polymerization. J. Biol. Chem. 255, 11570–11576.
Büttner, S., Habernig, L., Broeskamp, F., Ruli, D., Vögtle, F. N., Vlachos, M.,et al. (2013). Endonuclease G mediates α-synuclein cytotoxicity during Parkinson’s disease. EMBO J. 32, 3041–3054. doi: 10.1038/emboj.2013.228
C. elegans Sequencing Consortium. (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018.
Cam, J. A., and Bu, G. (2006). Modulation of beta-amyloid precursor protein trafficking and processing by the low density lipoprotein receptor family. Mol. Neurodegener. 1:8. doi: 10.1186/1750-1326-1-8
Cao, P., Yuan, Y., Pehek, E. A., Moise, A. R., Huang, Y., Palczewski, K.,et al. (2010). Alpha-synuclein disrupted dopamine homeostasis leads to dopaminergic neuron degeneration in Caenorhabditis elegans. PLoS ONE 5:e9312. doi: 10.1371/journal.pone.0009312
Carro, E., Spuch, C., Trejo, J. L., Antequera, D., and Torres-Aleman, I. (2005). Choroid plexus megalin is involved in neuroprotection by serum insulin-like growth factor I. J. Neurosci. 25, 10884–10893. doi: 10.1523/JNEUROSCI.2909-05.2005
Chalfie, M., Horvitz, H. R., and Sulston, J. E. (1981). Mutations that lead to reiterations in the cell lineages of C. elegans. Cell 24, 59–69. doi: 10.1016/0092-8674(81)90501-8
Chalfie, M., and Sulston, J. (1981). Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev. Biol. 82, 358–370. doi: 10.1016/0012-1606(81)90459-0
Chalfie, M., Sulston, J. E., White, J. G., Southgate, E., Thomson, J. N., and Brenner, S. (1985). The neural circuit for touch sensitivity in Caenorhabditis elegans. J. Neurosci. 5, 956–964.
Chapin, S. J., and Bulinski, J. C. (1991). Non-neuronal 210 × 103 Mr microtubule-associated protein (MAP4) contains a domain homologous to the microtubule-binding domains of neuronal MAP2 and tau. J. Cell Sci. 98, 27–36.
Chartier-Harlin, M. C., Crawford, F., Houlden, H., Warren, A., Hughes, D., Fidani, L.,et al. (1991). Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844–846. doi: 10.1038/353844a0
Chen, J., Kanai, Y., Cowan, N. J., and Hirokawa, N. (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677. doi: 10.1038/360674a0
Chew, Y. L., Fan, X., Götz, J., and Nicholas, H. R. (2013). PTL-1 regulates neuronal integrity and lifespan in C. elegans. J. Cell Sci. 126, 2079–2091. doi: 10.1242/jcs.jcs124404
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610. doi: 10.1126/science.1124646
Colbert, H. A., and Bargmann, C. I. (1995). Odorant-specific adaptation pathways generate olfactory plasticity in C. elegans. Neuron 14, 803–812. doi: 10.1016/0896-6273(95)90224-4
Conway, K. A., Harper, J. D., and Lansbury, P. T. (2000a). Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39, 2552–2563. doi: 10.1021/bi991447r
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Harper, J. D., Williamson, R. E.,et al. (2000b). Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann. N. Y. Acad. Sci. 920, 42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., and Lansbury, P. T. (2000c). Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576. doi: 10.1073/pnas.97.2.571
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. doi: 10.1002/humu.22117
Cui, S., Verroust, P. J., Moestrup, S. K., and Christensen, E. I. (1996). Megalin/gp330 mediates uptake of albumin in renal proximal tubule. Am. J. Physiol. 271, F900–F907.
Daigle, I., and Li, C. (1993). apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc. Natl. Acad. Sci. U.S.A. 90, 12045–12049. doi: 10.1073/pnas.90.24.12045
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K.,et al. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J.,et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dragunow, M., Faull, R. L., Lawlor, P., Beilharz, E. J., Singleton, K., Walker, E. B.,et al. (1995). In situ evidence for DNA fragmentation in Huntington’s disease striatum and Alzheimer’s disease temporal lobes. Neuroreport 6, 1053–1057. doi: 10.1097/00001756-199505090-00026
Duarte, A., Hirashima, M., Benedito, R., Trindade, A., Diniz, P., Bekman, E.,et al. (2004). Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 18, 2474–2478. doi: 10.1101/gad.1239004
Duilio, A., Faraonio, R., Minopoli, G., Zambrano, N., and Russo, T. (1998). Fe65L2: a new member of the Fe65 protein family interacting with the intracellular domain of the Alzheimer’s beta-amyloid precursor protein. Biochem. J. 330, 513–519.
Dumas, K. J., Guo, C., Wang, X., Burkhart, K. B., Adams, E. J., Alam, H.,et al. (2010). Functional divergence of dafachronic acid pathways in the control of C. elegans development and lifespan. Dev. Biol. 340, 605–612. doi: 10.1016/j.ydbio.2010.02.022
Edwards, S. L., Charlie, N. K., Richmond, J. E., Hegermann, J., Eimer, S., and Miller, K. G. (2009). Impaired dense core vesicle maturation in Caenorhabditis elegans mutants lacking Rab2. J. Cell Biol. 186, 881–895. doi: 10.1083/jcb.200902095
Estevez, A. O., Morgan, K. L., Szewczyk, N. J., Gems, D., and Estevez, M. (2014). The neurodegenerative effects of selenium are inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like growth factor signaling in Caenorhabditis elegans. Neurotoxicology 41C, 28–43. doi: 10.1016/j.neuro.2013.12.012
Estevez, A. O., Mueller, C. L., Morgan, K. L., Szewczyk, N. J., Teece, L., Miranda-Vizuete, A.,et al. (2012). Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans. Neurotoxicology 33, 1021–1032. doi: 10.1016/j.neuro.2012.04.019
Ewald, C. Y., Cheng, R., Tolen, L., Shah, V., Gillani, A., Nasrin, A.,et al. (2012a). Pan-neuronal expression of APL-1, an APP-related protein, disrupts olfactory, gustatory, and touch plasticity in Caenorhabditis elegans. J. Neurosci. 32, 10156–10169. doi: 10.1523/JNEUROSCI.0495-12.2012
Ewald, C. Y., Raps, D. A., and Li, C. (2012b). APL-1, the Alzheimer’s Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics 191, 493–507. doi: 10.1534/genetics.112.138768
Faber, P. W., Alter, J. R., Macdonald, M. E., and Hart, A. C. (1999). Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. U.S.A. 96, 179–184. doi: 10.1073/pnas.96.1.179
Faber, P. W., Voisine, C., King, D. C., Bates, E. A., and Hart, A. C. (2002). Glutamine/proline-rich PQE-1 proteins protect Caenorhabditis elegans neurons from huntingtin polyglutamine neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 17131–17136. doi: 10.1073/pnas.262544899
Fahn, S., and Sulzer, D. (2004). Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx 1, 139–154. doi: 10.1602/neurorx.1.1.139
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A., Levina, V.,et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Farrer, L. A., and Conneally, P. M. (1985). A genetic model for age at onset in Huntington disease. Am. J. Hum. Genet. 37, 350–357.
Fatouros, C., Pir, G. J., Biernat, J., Koushika, S. P., Mandelkow, E., Mandelkow, E. M.,et al. (2012). Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 21, 3587–3603. doi: 10.1093/hmg/dds190
Feijoo, C., Campbell, D. G., Jakes, R., Goedert, M., and Cuenda, A. (2005). Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J. Cell Sci. 118, 397–408. doi: 10.1242/jcs.01655
Fonte, V., Kapulkin, W. J., Kapulkin, V., Taft, A., Fluet, A., Friedman, D.,et al. (2002). Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 9439–9444. doi: 10.1073/pnas.152313999
Fonte, V., Kipp, D. R., Yerg, J., Merin, D., Forrestal, M., Wagner, E.,et al. (2008). Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J. Biol. Chem. 283, 784–791. doi: 10.1074/jbc.M703339200
Francis, R., Mcgrath, G., Zhang, J., Ruddy, D. A., Sym, M., Apfeld, J.,et al. (2002). aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 3, 85–97. doi: 10.1016/S1534-5807(02)00189-2
Frøkjaer-Jensen, C., Davis, M. W., Hopkins, C. E., Newman, B. J., Thummel, J. M., Olesen, S. P.,et al. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375–1383. doi: 10.1038/ng.248
Gale, N. W., Dominguez, M. G., Noguera, I., Pan, L., Hughes, V., Valenzuela, D. M.,et al. (2004). Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. U.S.A. 101, 15949–15954. doi: 10.1073/pnas.0407290101
Gasparini, L., Gouras, G. K., Wang, R., Gross, R. S., Beal, M. F., Greengard, P.,et al. (2001). Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 21, 2561–2570.
Gems, D., Sutton, A. J., Sundermeyer, M. L., Albert, P. S., King, K. V., Edgley, M. L.,et al. (1998). Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150, 129–155.
Gerisch, B., Weitzel, C., Kober-Eisermann, C., Rottiers, V., and Antebi, A. (2001). A hormonal signaling pathway influencing C. elegans metabolism, reproductive development, and life span. Dev. Cell 1, 841–851. doi: 10.1016/S1534-5807(01)00085-5
Gidalevitz, T., Krupinski, T., Garcia, S., and Morimoto, R. I. (2009). Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 5:e1000399. doi: 10.1371/journal.pgen.1000399
Gissendanner, C. R., and Sluder, A. E. (2000). nhr-25, the Caenorhabditis elegans ortholog of ftz-f1, is required for epidermal and somatic gonad development. Dev. Biol. 221, 259–272. doi: 10.1006/dbio.2000.9679
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D.,et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ana.21344
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. doi: 10.1016/0006-291X(84)91209-9
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L.,et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Goedert, M., Baur, C. P., Ahringer, J., Jakes, R., Hasegawa, M., Spillantini, M. G.,et al. (1996). PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J. Cell Sci. 109, 2661–2672.
Goedert, M., Cohen, E. S., Jakes, R., and Cohen, P. (1992a). p42 MAP kinase phosphorylation sites in microtubule-associated protein tau are dephosphorylated by protein phosphatase 2A1. Implications for Alzheimer’s disease [corrected]. FEBS Lett. 312, 95–99. doi: 10.1016/0014-5793(92)81418-L
Goedert, M., Spillantini, M. G., Cairns, N. J., and Crowther, R. A. (1992b). Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168. doi: 10.1016/0896-6273(92)90117-V
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989a). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989b). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399.
Goedert, M., Wischik, C. M., Crowther, R. A., Walker, J. E., and Klug, A. (1988). Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U.S.A. 85, 4051–4055. doi: 10.1073/pnas.85.11.4051
Goldberg, Y. P., Kalchman, M. A., Metzler, M., Nasir, J., Zeisler, J., Graham, R.,et al. (1996). Absence of disease phenotype and intergenerational stability of the CAG repeat in transgenic mice expressing the human Huntington disease transcript. Hum. Mol. Genet. 5, 177–185. doi: 10.1093/hmg/5.2.177
Gordon, P., Hingula, L., Krasny, M. L., Swienckowski, J. L., Pokrywka, N. J., and Raley-Susman, K. M. (2008). The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev. Genes Evol. 218, 541–551. doi: 10.1007/s00427-008-0250-z
Gorevic, P. D., Goñi, F., Pons-Estel, B., Alvarez, F., Peress, N. S., and Frangione, B. (1986). Isolation and partial characterization of neurofibrillary tangles and amyloid plaque core in Alzheimer’s disease: immunohistological studies. J. Neuropathol. Exp. Neurol. 45, 647–664. doi: 10.1097/00005072-198611000-00004
Goutte, C., Hepler, W., Mickey, K. M., and Priess, J. R. (2000). aph-2 encodes a novel extracellular protein required for GLP-1-mediated signaling. Development 127, 2481–2492.
Goutte, C., Tsunozaki, M., Hale, V. A., and Priess, J. R. (2002). APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U.S.A. 99, 775–779. doi: 10.1073/pnas.022523499
Greenwald, I. S., Sternberg, P. W., and Horvitz, H. R. (1983). The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell 34, 435–444. doi: 10.1016/0092-8674(83)90377-X
Gu, Y., Misonou, H., Sato, T., Dohmae, N., Takio, K., and Ihara, Y. (2001). Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J. Biol. Chem. 276, 35235–35238. doi: 10.1074/jbc.C100357200
Gusella, J. F., and MacDonald, M. E. (1995). Huntington’s disease: CAG genetics expands neurobiology. Curr. Opin. Neurobiol. 5, 656–662. doi: 10.1016/0959-4388(95)80072-7
Guthrie, C. R., Greenup, L., Leverenz, J. B., and Kraemer, B. C. (2011). MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 20, 1989–1999. doi: 10.1093/hmg/ddr079
Guthrie, C. R., Schellenberg, G. D., and Kraemer, B. C. (2009). SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 18, 1825–1838. doi: 10.1093/hmg/ddp099
Guénette, S. Y., Chen, J., Jondro, P. D., and Tanzi, R. E. (1996). Association of a novel human FE65-like protein with the cytoplasmic domain of the beta-amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 93, 10832–10837. doi: 10.1073/pnas.93.20.10832
Haass, C., Hung, A. Y., Selkoe, D. J., and Teplow, D. B. (1994a). Mutations associated with a locus for familial Alzheimer’s disease result in alternative processing of amyloid beta-protein precursor. J. Biol. Chem. 269, 17741–17748.
Haass, C., Koo, E. H., Teplow, D. B., and Selkoe, D. J. (1994b). Polarized secretion of beta-amyloid precursor protein and amyloid beta-peptide in MDCK cells. Proc. Natl. Acad. Sci. U.S.A. 91, 1564–1568. doi: 10.1073/pnas.91.4.1564
Haass, C., Koo, E. H., Mellon, A., Hung, A. Y., and Selkoe, D. J. (1992). Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357, 500–503. doi: 10.1038/357500a0
Hamamichi, S., Rivas, R. N., Knight, A. L., Cao, S., Caldwell, K. A., and Caldwell, G. A. (2008). Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. U.S.A. 105, 728–733. doi: 10.1073/pnas.0711018105
Hamos, J. E., Oblas, B., Pulaski-Salo, D., Welch, W. J., Bole, D. G., and Drachman, D. A. (1991). Expression of heat shock proteins in Alzheimer’s disease. Neurology 41, 345–350. doi: 10.1212/WNL.41.3.345
Han, S. M., El Oussini, H., Scekic-Zahirovic, J., Vibbert, J., Cottee, P., Prasain, J. K.,et al. (2013). VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in Caenorhabditis elegans. PLoS Genet. 9:e1003738. doi: 10.1371/journal.pgen.1003738
Harada, A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Ohshima, T.,et al. (1994). Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491. doi: 10.1038/369488a0
Hardiman, O., Van Den Berg, L. H., and Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649. doi: 10.1038/nrneurol.2011.153
Hardy, J. (2010). Genetic analysis of pathways to Parkinson disease. Neuron 68, 201–206. doi: 10.1016/j.neuron.2010.10.014
Hayes, G. D., Frand, A. R., and Ruvkun, G. (2006). The mir-84 and let-7 paralogous microRNA genes of Caenorhabditis elegans direct the cessation of molting via the conserved nuclear hormone receptors NHR-23 and NHR-25. Development 133, 4631–4641. doi: 10.1242/dev.02655
Heber, S., Herms, J., Gajic, V., Hainfellner, J., Aguzzi, A., Rülicke, T.,et al. (2000). Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 20, 7951–7963.
Henderson, S. T., and Johnson, T. E. (2001). daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 11, 1975–1980. doi: 10.1016/S0960-9822(01)00594-2
Hermo, L., Lustig, M., Lefrancois, S., Argraves, W. S., and Morales, C. R. (1999). Expression and regulation of LRP-2/megalin in epithelial cells lining the efferent ducts and epididymis during postnatal development. Mol. Reprod. Dev. 53, 282–293. doi: 10.1002/(SICI)1098-2795(199907)53:3<282::AID-MRD4>3.0.CO;2-A
Herms, J., Anliker, B., Heber, S., Ring, S., Fuhrmann, M., Kretzschmar, H.,et al. (2004). Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23, 4106–4115. doi: 10.1038/sj.emboj.7600390
Herz, J., Clouthier, D. E., and Hammer, R. E. (1992). LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71, 411–421. doi: 10.1016/0092-8674(92)90511-A
Hieb, W. F., and Rothstein, M. (1968). Sterol requirement for reproduction of a free-living nematode. Science 160, 778–780. doi: 10.1126/science.160.3829.778
Holcomb, L. A., Gordon, M. N., Jantzen, P., Hsiao, K., Duff, K., and Morgan, D. (1999). Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav. Genet. 29, 177–185. doi: 10.1023/A:1021691918517
Hornsten, A., Lieberthal, J., Fadia, S., Malins, R., Ha, L., Xu, X.,et al. (2007). APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc. Natl. Acad. Sci. U.S.A. 104, 1971–1976. doi: 10.1073/pnas.0603997104
Hsiao, K. K., Borchelt, D. R., Olson, K., Johannsdottir, R., Kitt, C., Yunis, W.,et al. (1995). Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15, 1203–1218. doi: 10.1016/0896-6273(95)90107-8
Hsu, A. L., Murphy, C. T., and Kenyon, C. (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145. doi: 10.1126/science.1083701
Hughes, A. J., Ben-Shlomo, Y., Daniel, S. E., and Lees, A. J. (1992). What features improve the accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology 42, 1142–1146. doi: 10.1212/WNL.42.6.1142
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H.,et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Jadiya, P., Khan, A., Sammi, S. R., Kaur, S., Mir, S. S., and Nazir, A. (2011). Anti-Parkinsonian effects of Bacopa monnieri: insights from transgenic and pharmacological Caenorhabditis elegans models of Parkinson’s disease. Biochem. Biophys. Res. Commun. 413, 605–610. doi: 10.1016/j.bbrc.2011.09.010
Jarriault, S., and Greenwald, I. (2005). Evidence for functional redundancy between C. elegans ADAM proteins SUP-17/Kuzbanian and ADM-4/TACE. Dev. Biol. 287, 1–10. doi: 10.1016/j.ydbio.2005.08.014
Jia, K., Hart, A. C., and Levine, B. (2007). Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3, 21–25. doi: 10.4161/auto.3528
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S.,et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C.,et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kamel, F., Umbach, D. M., Bedlack, R. S., Richards, M., Watson, M., Alavanja, M. C.,et al. (2012). Pesticide exposure and amyotrophic lateral sclerosis. Neurotoxicology 33, 457–462. doi: 10.1016/j.neuro.2012.04.001
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H.,et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Kidd, M. (1964). Alzheimer’s disease–An electron microscopical study. Brain 87, 307–320. doi: 10.1093/brain/87.2.307
Kimberly, W. T., Lavoie, M. J., Ostaszewski, B. L., Ye, W., Wolfe, M. S., and Selkoe, D. J. (2003). Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. U.S.A. 100, 6382–6387. doi: 10.1073/pnas.1037392100
Kimura, K. D., Tissenbaum, H. A., Liu, Y., and Ruvkun, G. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946. doi: 10.1126/science.277.5328.942
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S.,et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Knauer, M. F., Orlando, R. A., and Glabe, C. G. (1996). Cell surface APP751 forms complexes with protease nexin 2 ligands and is internalized via the low density lipoprotein receptor-related protein (LRP). Brain Res. 740, 6–14. doi: 10.1016/S0006-8993(96)00711-1
Knops, J., Kosik, K. S., Lee, G., Pardee, J. D., Cohen-Gould, L., and Mcconlogue, L. (1991). Overexpression of tau in a nonneuronal cell induces long cellular processes. J. Cell Biol. 114, 725–733. doi: 10.1083/jcb.114.4.725
Koo, E. H., Sisodia, S. S., Archer, D. R., Martin, L. J., Weidemann, A., Beyreuther, K.,et al. (1990). Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. U.S.A. 87, 1561–1565. doi: 10.1073/pnas.87.4.1561
Kounnas, M. Z., Moir, R. D., Rebeck, G. W., Bush, A. I., Argraves, W. S., Tanzi, R. E.,et al. (1995). LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted beta-amyloid precursor protein and mediates its degradation. Cell 82, 331–340. doi: 10.1016/0092-8674(95)90320-8
Kraemer, B. C., and Schellenberg, G. D. (2007). SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 16, 1959–1971. doi: 10.1093/hmg/ddm143
Kraemer, B. C., Zhang, B., Leverenz, J. B., Thomas, J. H., Trojanowski, J. Q., and Schellenberg, G. D. (2003). Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. U.S.A. 100, 9980–9985. doi: 10.1073/pnas.1533448100
Krebs, L. T., Shutter, J. R., Tanigaki, K., Honjo, T., Stark, K. L., and Gridley, T. (2004). Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 18, 2469–2473. doi: 10.1101/gad.1239204
Krebs, L. T., Xue, Y., Norton, C. R., Shutter, J. R., Maguire, M., Sundberg, J. P.,et al. (2000). Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 14, 1343–1352.
Krebs, L. T., Xue, Y., Norton, C. R., Sundberg, J. P., Beatus, P., Lendahl, U.,et al. (2003). Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis 37, 139–143. doi: 10.1002/gene.10241
Krigman, M. R., Feldman, R. G., and Bensch, K. (1965). Alzheimer’s presenile dementia. A histochemical and electron microscopic study. Lab. Invest. 14, 381–396.
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S.,et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kuwahara, T., Koyama, A., Gengyo-Ando, K., Masuda, M., Kowa, H., Tsunoda, M.,et al. (2006). Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J. Biol. Chem. 281, 334–340. doi: 10.1074/jbc.M504860200
Kuwahara, T., Koyama, A., Koyama, S., Yoshina, S., Ren, C. H., Kato, T.,et al. (2008). A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum. Mol. Genet. 17, 2997–3009. doi: 10.1093/hmg/ddn198
L’Hernault, S. W., and Arduengo, P. M. (1992). Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J. Cell Biol. 119, 55–68. doi: 10.1083/jcb.119.1.55
Lakowski, B., Eimer, S., Göbel, C., Böttcher, A., Wagler, B., and Baumeister, R. (2003). Two suppressors of sel-12 encode C2H2 zinc-finger proteins that regulate presenilin transcription in Caenorhabditis elegans. Development 130, 2117–2128. doi: 10.1242/dev.00429
Lakso, M., Vartiainen, S., Moilanen, A. M., Sirviö, J., Thomas, J. H., Nass, R.,et al. (2003). Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 86, 165–172. doi: 10.1046/j.1471-4159.2003.01809.x
Lambie, E. J., and Kimble, J. (1991). Two homologous regulatory genes, lin-12 and glp-1, have overlapping functions. Development 112, 231–240.
Larsen, P. L., Albert, P. S., and Riddle, D. L. (1995). Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics 139, 1567–1583.
Lee, B. H., and Ashrafi, K. (2008). A TRPV channel modulates C. elegans neurosecretion, larval starvation survival, and adult lifespan. PLoS Genet. 4:e1000213. doi: 10.1371/journal.pgen.1000213
Lee, R. Y., Hench, J., and Ruvkun, G. (2001). Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 11, 1950–1957. doi: 10.1016/S0960-9822(01)00595-4
Lee, V. M., Otvos, L., Carden, M. J., Hollosi, M., Dietzschold, B., and Lazzarini, R. A. (1988). Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc. Natl. Acad. Sci. U.S.A. 85, 1998–2002. doi: 10.1073/pnas.85.6.1998
Lejeune, F. X., Mesrob, L., Parmentier, F., Bicep, C., Vazquez-Manrique, R. P., Parker, J. A.,et al. (2012). Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genomics 13:91. doi: 10.1186/1471-2164-13-91.
Levitan, D., Doyle, T. G., Brousseau, D., Lee, M. K., Thinakaran, G., Slunt, H. H.,et al. (1996). Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 93, 14940–14944. doi: 10.1073/pnas.93.25.14940
Levitan, D., and Greenwald, I. (1995). Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 377, 351–354. doi: 10.1038/377351a0
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H.,et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Lewis, S. A., Wang, D. H., and Cowan, N. J. (1988). Microtubule-associated protein MAP2 shares a microtubule binding motif with tau protein. Science 242, 936–939. doi: 10.1126/science.3142041
Li, J., Li, T., Zhang, X., Tang, Y., Yang, J., and Le, W. (2014). Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration. Neurobiol. Aging 35, 837–846. doi: 10.1016/j.neurobiolaging.2013.09.003
Li, L. Y., Luo, X., and Wang, X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–99. doi: 10.1038/35083620
Li, T., Ma, G., Cai, H., Price, D. L., and Wong, P. C. (2003). Nicastrin is required for assembly of presenilin/gamma-secretase complexes to mediate Notch signaling and for processing and trafficking of beta-amyloid precursor protein in mammals. J. Neurosci. 23, 3272–3277.
Li, X., and Greenwald, I. (1996). Membrane topology of the C. elegans SEL-12 presenilin. Neuron 17, 1015–1021. doi: 10.1016/S0896-6273(00)80231-7
Li, X., and Greenwald, I. (1997). HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc. Natl. Acad. Sci. U.S.A. 94, 12204–12209. doi: 10.1073/pnas.94.22.12204
Liang, J. J. (2000). Interaction between beta-amyloid and lens alphaB-crystallin. FEBS Lett. 484, 98–101. doi: 10.1016/S0014-5793(00)02136-0
Lin, C. H., Tomioka, M., Pereira, S., Sellings, L., Iino, Y., and Van Der Kooy, D. (2010). Insulin signaling plays a dual role in Caenorhabditis elegans memory acquisition and memory retrieval. J. Neurosci. 30, 8001–8011. doi: 10.1523/JNEUROSCI.4636-09.2010
Lin, K., Dorman, J. B., Rodan, A., and Kenyon, C. (1997). daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319–1322. doi: 10.1126/science.278.5341.1319
Lin, K., Hsin, H., Libina, N., and Kenyon, C. (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139–145. doi: 10.1038/88850
Lindau, M., Almkvist, O., Kushi, J., Boone, K., Johansson, S. E., Wahlund, L. O.,et al. (2000). First symptoms–frontotemporal dementia versus Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 11, 286–293. doi: 10.1159/000017251
Lindwall, G., and Cole, R. D. (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305.
Link, C. D. (1995). Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 92, 9368–9372. doi: 10.1073/pnas.92.20.9368
Link, C. D. (2006). C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp. Gerontol. 41, 1007–1013. doi: 10.1016/j.exger.2006.06.059
Link, C. D., Johnson, C. J., Fonte, V., Paupard, M., Hall, D. H., Styren, S.,et al. (2001). Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol. Aging 22, 217–226. doi: 10.1016/S0197-4580(00)00237-2
Liu, Z., Hamamichi, S., Lee, B. D., Yang, D., Ray, A., Caldwell, G. A.,et al. (2011). Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 20, 3933–3942. doi: 10.1093/hmg/ddr312
Locke, C. J., Fox, S. A., Caldwell, G. A., and Caldwell, K. A. (2008). Acetaminophen attenuates dopamine neuron degeneration in animal models of Parkinson’s disease. Neurosci. Lett. 439, 129–133. doi: 10.1016/j.neulet.2008.05.003
Lorent, K., Overbergh, L., Moechars, D., De Strooper, B., Van Leuven, F., and Van Den Berghe, H. (1995). Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the alpha-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, alpha-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience 65, 1009–1025. doi: 10.1016/0306-4522(94)00555-J
Lu, T., Aron, L., Zullo, J., Pan, Y., Kim, H., Chen, Y.,et al. (2014). REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454. doi: 10.1038/nature13163
Luo, W. J., Wang, H., Li, H., Kim, B. S., Shah, S., Lee, H. J.,et al. (2003). PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 278, 7850–7854. doi: 10.1074/jbc.C200648200
Luse, S. A., and Smith, K. R. (1964). The ultrastructure of senile plaques. Am. J. Pathol. 44, 553–563.
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
Majounie, E., Abramzon, Y., Renton, A. E., Keller, M. F., Traynor, B. J., and Singleton, A. B. (2012). Large C9orf72 repeat expansions are not a common cause of Parkinson’s disease. Neurobiol. Aging 33, e2521–e2522. doi: 10.1016/j.neurobiolaging.2012.05.007
Malek, A. M., Barchowsky, A., Bowser, R., Youk, A., and Talbott, E. O. (2012). Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: a meta-analysis of epidemiological studies: pesticide exposure as a risk factor for ALS. Environ. Res. 117, 112–119. doi: 10.1016/j.envres.2012.06.007
Mangahas, P. M., Yu, X., Miller, K. G., and Zhou, Z. (2008). The small GTPase Rab2 functions in the removal of apoptotic cells in Caenorhabditis elegans. J. Cell Biol. 180, 357–373. doi: 10.1083/jcb.200708130
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C.,et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/S0092-8674(00)81369-0
Martin, J. B., and Gusella, J. F. (1986). Huntington’s disease. Pathogenesis and management. N. Engl. J. Med. 315, 1267–1276. doi: 10.1056/NEJM198611133152006
Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., Mccutcheon, K.,et al. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154. doi: 10.1038/ng0298-150
Marvanova, M., and Nichols, C. D. (2007). Identification of neuroprotective compounds of Caenorhabditis elegans dopaminergic neurons against 6-OHDA. J. Mol. Neurosci. 31, 127–137.
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., Mcdonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Matus, A., Bernhardt, R., and Hugh-Jones, T. (1981). High molecular weight microtubule-associated proteins are preferentially associated with dendritic microtubules in brain. Proc. Natl. Acad. Sci. U.S.A. 78, 3010–3014. doi: 10.1073/pnas.78.5.3010
McColl, G., Roberts, B. R., Gunn, A. P., Perez, K. A., Tew, D. J., Masters, C. L.,et al. (2009). The Caenorhabditis elegans A beta 1-42 model of Alzheimer disease predominantly expresses A beta 3-42. J. Biol. Chem. 284, 22697–22702. doi: 10.1074/jbc.C109.028514
McColl, G., Roberts, B. R., Pukala, T. L., Kenche, V. B., Roberts, C. M., Link, C. D.,et al. (2012). Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 7:57. doi: 10.1186/1750-1326-7-57
McDermott, J. B., Aamodt, S., and Aamodt, E. (1996). ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau microtubule-associated proteins. Biochemistry 35, 9415–9423. doi: 10.1021/bi952646n
McElwee, J., Bubb, K., and Thomas, J. H. (2003). Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2, 111–121. doi: 10.1046/j.1474-9728.2003.00043.x
McElwee, J. J., Schuster, E., Blanc, E., Thomas, J. H., and Gems, D. (2004). Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 279, 44533–44543. doi: 10.1074/jbc.M406207200
Miyasaka, T., Ding, Z., Gengyo-Ando, K., Oue, M., Yamaguchi, H., Mitani, S.,et al. (2005). Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 20, 372–383. doi: 10.1016/j.nbd.2005.03.017
Mizuta, K., Saito, A., Watanabe, T., Nagura, M., Arakawa, M., Shimizu, F.,et al. (1999). Ultrastructural localization of megalin in the rat cochlear duct. Hear. Res. 129, 83–91. doi: 10.1016/S0378-5955(98)00221-4
Morales, C. R., Igdoura, S. A., Wosu, U. A., Boman, J., and Argraves, W. S. (1996). Low density lipoprotein receptor-related protein-2 expression in efferent duct and epididymal epithelia: evidence in rats for its in vivo role in endocytosis of apolipoprotein J/clusterin. Biol. Reprod. 55, 676–683. doi: 10.1095/biolreprod55.3.676
Morley, J. F., and Morimoto, R. I. (2004). Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 15, 657–664. doi: 10.1091/mbc.E03-07-0532
Morris, J. Z., Tissenbaum, H. A., and Ruvkun, G. (1996). A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382, 536–539. doi: 10.1038/382536a0
Murakami, T., Yang, S. P., Xie, L., Kawano, T., Fu, D., Mukai, A.,et al. (2012). ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 21, 1–9. doi: 10.1093/hmg/ddr417
Murphy, C. T., Mccarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J.,et al. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283. doi: 10.1038/nature01789
Murrell, J., Farlow, M., Ghetti, B., and Benson, M. D. (1991). A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254, 97–99. doi: 10.1126/science.1925564
Muñoz-Lobato, F., Rodríguez-Palero, M. J., Naranjo-Galindo, F. J., Shephard, F., Gaffney, C. J., Szewczyk, N. J.,et al. (2014). Protective role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases. Antioxid. Redox. Signal. 20, 217–235. doi: 10.1089/ars.2012.5051
Nance, M. A. (1997). Genetic testing of children at risk for Huntington’s disease. US Huntington Disease Genetic Testing Group. Neurology 49, 1048–1053. doi: 10.1212/WNL.49.4.1048
Nass, R., Hahn, M. K., Jessen, T., Mcdonald, P. W., Carvelli, L., and Blakely, R. D. (2005). A genetic screen in Caenorhabditis elegans for dopamine neuron insensitivity to 6-hydroxydopamine identifies dopamine transporter mutants impacting transporter biosynthesis and trafficking. J. Neurochem. 94, 774–785. doi: 10.1111/j.1471-4159.2005.03205.x
Nass, R., Hall, D. H., Miller, D. M., and Blakely, R. D. (2002). Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 99, 3264–3269. doi: 10.1073/pnas.042497999
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T.,et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Newman, A. P., White, J. G., and Sternberg, P. W. (1995). The Caenorhabditis elegans lin-12 gene mediates induction of ventral uterine specialization by the anchor cell. Development 121, 263–271.
Nielsen, R., Birn, H., Moestrup, S. K., Nielsen, M., Verroust, P., and Christensen, E. I. (1998). Characterization of a kidney proximal tubule cell line, LLC-PK1, expressing endocytotic active megalin. J. Am. Soc. Nephrol. 9, 1767–1776.
Niwa, R., and Hada, K. (2010). Identification of a spatio-temporal enhancer element for the Alzheimer’s amyloid precursor protein-like-1 gene in the nematode Caenorhabditis elegans. Biosci. Biotechnol. Biochem. 74, 2497–2500. doi: 10.1271/bbb.100450
Niwa, R., Zhou, F., Li, C., and Slack, F. J. (2008). The expression of the Alzheimer’s amyloid precursor protein-like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans. Dev. Biol. 315, 418–425. doi: 10.1016/j.ydbio.2007.12.044
Ogg, S., Paradis, S., Gottlieb, S., Patterson, G. I., Lee, L., Tissenbaum, H. A.,et al. (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999. doi: 10.1038/40194
Orenstein, S. J., Kuo, S. H., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I.,et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi: 10.1038/nn.3350
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621. doi: 10.1146/annurev.neuro.29.051605.113042
Pan, C. L., Peng, C. Y., Chen, C. H., and Mcintire, S. (2011). Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 9274–9279. doi: 10.1073/pnas.1011711108
Paradis, S., Ailion, M., Toker, A., Thomas, J. H., and Ruvkun, G. (1999). A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev. 13, 1438–1452. doi: 10.1101/gad.13.11.1438
Paradis, S., and Ruvkun, G. (1998). Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 12, 2488–2498. doi: 10.1101/gad.12.16.2488
Parker, J. A., Arango, M., Abderrahmane, S., Lambert, E., Tourette, C., Catoire, H.,et al. (2005). Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 37, 349–350. doi: 10.1038/ng1534
Parker, J. A., Connolly, J. B., Wellington, C., Hayden, M., Dausset, J., and Neri, C. (2001). Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. U.S.A. 98, 13318–13323. doi: 10.1073/pnas.231476398
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi: 10.1038/nrn1971
Pasquier, F. (2005). Telling the difference between frontotemporal dementia and Alzheimer’s disease. Curr. Opin. Psychiatry 18, 628–632. doi: 10.1097/01.yco.0000185988.05741.2a
Perez, N., Sugar, J., Charya, S., Johnson, G., Merril, C., Bierer, L.,et al. (1991). Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer’s disease. Mol. Brain Res. 11, 249–254. doi: 10.1016/0169-328X(91)90033-T
Pierce-Shimomura, J. T., Faumont, S., Gaston, M. R., Pearson, B. J., and Lockery, S. R. (2001). The homeobox gene lim-6 is required for distinct chemosensory representations in C. elegans. Nature 410, 694–698. doi: 10.1038/35070575
Pietrzik, C. U., Yoon, I. S., Jaeger, S., Busse, T., Weggen, S., and Koo, E. H. (2004). FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J. Neurosci. 24, 4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A.,et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Poorkaj, P., Bird, T. D., Wijsman, E., Nemens, E., Garruto, R. M., Anderson, L.,et al. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825. doi: 10.1002/ana.410430617
Poorkaj, P., Nutt, J. G., James, D., Gancher, S., Bird, T. D., Steinbart, E.,et al. (2004). parkin mutation analysis in clinic patients with early-onset Parkinson [corrected] disease. Am. J. Med. Genet. A 129A, 44–50. doi: 10.1002/ajmg.a.30157
Rabinovici, G. D., and Miller, B. L. (2010). Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24, 375–398. doi: 10.2165/11533100-000000000-00000
Recchia, A., Debetto, P., Negro, A., Guidolin, D., Skaper, S. D., and Giusti, P. (2004). Alpha-synuclein and Parkinson’s disease. FASEB J. 18, 617–626. doi: 10.1096/fj.03-0338rev
Reinhart, B. J., Slack, F. J., Basson, M., Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E.,et al. (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906. doi: 10.1038/35002607
Renkawek, K., Voorter, C. E., Bosman, G. J., Van Workum, F. P., and De Jong, W. W. (1994). Expression of alpha B-crystallin in Alzheimer’s disease. Acta Neuropathol. 87, 155–160. doi: 10.1007/BF00296185
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R.,et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riddle, D. L., Swanson, M. M., and Albert, P. S. (1981). Interacting genes in nematode dauer larva formation. Nature 290, 668–671. doi: 10.1038/290668a0
Ring, S., Weyer, S. W., Kilian, S. B., Waldron, E., Pietrzik, C. U., Filippov, M. A.,et al. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007
Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y.,et al. (1995). Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778. doi: 10.1038/376775a0
Ruan, Q., Harrington, A. J., Caldwell, K. A., Caldwell, G. A., and Standaert, D. G. (2010). VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson’s disease. Neurobiol. Dis. 37, 330–338. doi: 10.1016/j.nbd.2009.10.011
Russo, T., Faraonio, R., Minopoli, G., De Candia, P., De Renzis, S., and Zambrano, N. (1998). Fe65 and the protein network centered around the cytosolic domain of the Alzheimer’s beta-amyloid precursor protein. FEBS Lett. 434, 1–7. doi: 10.1016/S0014-5793(98)00941-7
Sandbrink, R., Masters, C. L., and Beyreuther, K. (1994). Similar alternative splicing of a non-homologous domain in beta A4-amyloid protein precursor-like proteins. J. Biol. Chem. 269, 14227–14234.
Sastre, M., Steiner, H., Fuchs, K., Capell, A., Multhaup, G., Condron, M. M.,et al. (2001). Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2, 835–841. doi: 10.1093/embo-reports/kve180
Savage-Dunn, C., Maduzia, L. L., Zimmerman, C. M., Roberts, A. F., Cohen, S., Tokarz, R.,et al. (2003). Genetic screen for small body size mutants in C. elegans reveals many TGFbeta pathway components. Genesis 35, 239–247. doi: 10.1002/gene.10184
Seidel, H. S., and Kimble, J. (2011). The oogenic germline starvation response in C. elegans. PLoS ONE 6:e28074. doi: 10.1371/journal.pone.0028074
Selkoe, D. J., Abraham, C. R., Podlisny, M. B., and Duffy, L. K. (1986). Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer’s disease. J. Neurochem. 46, 1820–1834. doi: 10.1111/j.1471-4159.1986.tb08501.x
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I.,et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shaye, D. D., and Greenwald, I. (2011). OrthoList: a compendium of C. elegans genes with human orthologs. PLoS ONE 6:e20085. doi: 10.1371/journal.pone.0020085
Shen, J., Bronson, R. T., Chen, D. F., Xia, W., Selkoe, D. J., and Tonegawa, S. (1997). Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89, 629–639. doi: 10.1016/S0092-8674(00)80244-5
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M.,et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shi, Z., Lu, Z., Zhao, Y., Wang, Y., Zhao-Wilson, X., Guan, P.,et al. (2013). Neuroprotective effects of aqueous extracts of Uncaria tomentosa: insights from 6-OHDA induced cell damage and transgenic Caenorhabditis elegans model. Neurochem. Int. 62, 940–947. doi: 10.1016/j.neuint.2013.03.001
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S.,et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shinohara, H., Inaguma, Y., Goto, S., Inagaki, T., and Kato, K. (1993). Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J. Neurol. Sci. 119, 203–208. doi: 10.1016/0022-510X(93)90135-L
Siesling, S., Vegter-Van Der Vlis, M., and Roos, R. A. (1997). Juvenile Huntington disease in the Netherlands. Pediatr. Neurol. 17, 37–43. doi: 10.1016/S0887-8994(97)00069-6
Simón, A. M., Schiapparelli, L., Salazar-Colocho, P., Cuadrado-Tejedor, M., Escribano, L., López De Maturana, R.,et al. (2009). Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol. Dis. 33, 369–378. doi: 10.1016/j.nbd.2008.11.005
Slunt, H. H., Thinakaran, G., Von Koch, C., Lo, A. C., Tanzi, R. E., and Sisodia, S. S. (1994). Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J. Biol. Chem. 269, 2637–2644.
So, S., Miyahara, K., and Ohshima, Y. (2011). Control of body size in C. elegans dependent on food and insulin/IGF-1 signal. Genes Cells 16, 639–651. doi: 10.1111/j.1365-2443.2011.01514.x
Spillantini, M. G., Crowther, R. A., Kamphorst, W., Heutink, P., and Van Swieten, J. C. (1998). Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am. J. Pathol. 153, 1359–1363. doi: 10.1016/S0002-9440(10)65721-5
Sprecher, C. A., Grant, F. J., Grimm, G., O’hara, P. J., Norris, F., Norris, K.,et al. (1993). Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry 32, 4481–4486. doi: 10.1021/bi00068a002
Springer, W., Hoppe, T., Schmidt, E., and Baumeister, R. (2005). A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet. 14, 3407–3423. doi: 10.1093/hmg/ddi371
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B.,et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R.,et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80.
Strong, T. V., Tagle, D. A., Valdes, J. M., Elmer, L. W., Boehm, K., Swaroop, M.,et al. (1993). Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 5, 259–265. doi: 10.1038/ng1193-259
Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156. doi: 10.1016/0012-1606(77)90158-0
Sumakovic, M., Hegermann, J., Luo, L., Husson, S. J., Schwarze, K., Olendrowitz, C.,et al. (2009). UNC-108/RAB-2 and its effector RIC-19 are involved in dense core vesicle maturation in Caenorhabditis elegans. J. Cell Biol. 186, 897–914. doi: 10.1083/jcb.200902096
Tank, E. M., Rodgers, K. E., and Kenyon, C. (2011). Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. 31, 9279–9288. doi: 10.1523/JNEUROSCI.6606-10.2011
Terry, R. D., Gonatas, N. K., and Weiss, M. (1964). Ultrastructural studies in Alzheimer’s presenile dementia. Am. J. Pathol. 44, 269–297.
The Association for Frontotemporal Degeneration. (2014). The Association for Frontotemporal Degeneration [Online]. Available at: http://www.theaftd.org (accessed March 18, 2014)
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Therrien, M., Rouleau, G. A., Dion, P. A., and Parker, J. A. (2013). Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE 8:e83450. doi: 10.1371/journal.pone.0083450
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G.,et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. doi: 10.1016/S0896-6273(00)80291-3
Thinakaran, G., Kitt, C. A., Roskams, A. J., Slunt, H. H., Masliah, E., Von Koch, C.,et al. (1995). Distribution of an APP homolog, APLP2, in the mouse olfactory system: a potential role for APLP2 in axogenesis. J. Neurosci. 15, 6314–6326.
Tissenbaum, H. A., and Guarente, L. (2001). Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230. doi: 10.1038/35065638
Tomioka, M., Adachi, T., Suzuki, H., Kunitomo, H., Schafer, W. R., and Iino, Y. (2006). The insulin/PI 3-kinase pathway regulates salt chemotaxis learning in Caenorhabditis elegans. Neuron 51, 613–625. doi: 10.1016/j.neuron.2006.07.024
Toth, M. L., Melentijevic, I., Shah, L., Bhatia, A., Lu, K., Talwar, A.,et al. (2012). Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 32, 8778–8790. doi: 10.1523/JNEUROSCI.1494-11.2012
Tretiakoff, K. (1919). Contribution a l’Etude de L’Anatomie pathologique du Locus Niger de Soemmering avec quelques déductions relatives à la pathogénie des troubles du tonus musculaire et De La Maladie de Parkinson. University of Paris: Paris.
Treusch, S., Hamamichi, S., Goodman, J. L., Matlack, K. E., Chung, C. Y., Baru, V.,et al. (2011). Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 334, 1241–1245. doi: 10.1126/science.1213210
Tsuda, H., Han, S. M., Yang, Y., Tong, C., Lin, Y. Q., Mohan, K.,et al. (2008). The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133, 963–977. doi: 10.1016/j.cell.2008.04.039
Ulery, P. G., Beers, J., Mikhailenko, I., Tanzi, R. E., Rebeck, G. W., Hyman, B. T.,et al. (2000). Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer’s disease. J. Biol. Chem. 275, 7410–7415. doi: 10.1074/jbc.275.10.7410
Vaccaro, A., Patten, S. A., Ciura, S., Maios, C., Therrien, M., Drapeau, P.,et al. (2012a). Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS ONE 7:e42117. doi: 10.1371/journal.pone.0042117
Vaccaro, A., Tauffenberger, A., Ash, P. E., Carlomagno, Y., Petrucelli, L., and Parker, J. A. (2012b). TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 8:e1002806. doi: 10.1371/journal.pgen.1002806
Valentine, J. S., Doucette, P. A., and Zittin Potter, S. (2005). Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593. doi: 10.1146/annurev.biochem.72.121801.161647
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B.,et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. doi: 10.1016/S1474-4422(08)70071-1
van Dijk, J. G., Van Der Velde, E. A., Roos, R. A., and Bruyn, G. W. (1986). Juvenile Huntington disease. Hum. Genet. 73, 235–239. doi: 10.1007/BF00401235
van Ham, T. J., Thijssen, K. L., Breitling, R., Hofstra, R. M., Plasterk, R. H., and Nollen, E. A. (2008). C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 4:e1000027. doi: 10.1371/journal.pgen.1000027
Vartiainen, S., Pehkonen, P., Lakso, M., Nass, R., and Wong, G. (2006). Identification of gene expression changes in transgenic C. elegans overexpressing human alpha-synuclein. Neurobiol. Dis. 22, 477–486. doi: 10.1016/j.nbd.2005.12.021
Vinceti, M., Maraldi, T., Bergomi, M., and Malagoli, C. (2009). Risk of chronic low-dose selenium overexposure in humans: insights from epidemiology and biochemistry. Rev. Environ. Health 24, 231–248. doi: 10.1515/REVEH.2009.24.3.231
von Koch, C. S., Zheng, H., Chen, H., Trumbauer, M., Thinakaran, G., Van Der Ploeg, L. H.,et al. (1997). Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 18, 661–669. doi: 10.1016/S0197-4580(97)00151-6
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577. doi: 10.1097/00005072-198511000-00003
Vowels, J. J., and Thomas, J. H. (1992). Genetic analysis of chemosensory control of dauer formation in Caenorhabditis elegans. Genetics 130, 105–123.
Wang, J., Farr, G. W., Hall, D. H., Li, F., Furtak, K., Dreier, L.,et al. (2009). An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 5:e1000350. doi: 10.1371/journal.pgen.1000350
Wang, P., Yang, G., Mosier, D. R., Chang, P., Zaidi, T., Gong, Y. D.,et al. (2005). Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J. Neurosci. 25, 1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005
Wasco, W., Bupp, K., Magendantz, M., Gusella, J. F., Tanzi, R. E., and Solomon, F. (1992). Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc. Natl. Acad. Sci. U.S.A. 89, 10758–10762. doi: 10.1073/pnas.89.22.10758
Wasco, W., Gurubhagavatula, S., Paradis, M. D., Romano, D. M., Sisodia, S. S., Hyman, B. T.,et al. (1993). Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat. Genet. 5, 95–100. doi: 10.1038/ng0993-95
Weidemann, A., Eggert, S., Reinhard, F. B., Vogel, M., Paliga, K., Baier, G.,et al. (2002). A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41, 2825–2835. doi: 10.1021/bi015794o
Wen, J. Y., Kumar, N., Morrison, G., Rambaldini, G., Runciman, S., Rousseau, J.,et al. (1997). Mutations that prevent associative learning in C. elegans. Behav. Neurosci. 111, 354–368. doi: 10.1037/0735-7044.111.2.354
Westlund, B., Parry, D., Clover, R., Basson, M., and Johnson, C. D. (1999). Reverse genetic analysis of Caenorhabditis elegans presenilins reveals redundant but unequal roles for sel-12 and hop-1 in Notch-pathway signaling. Proc. Natl. Acad. Sci. U.S.A. 96, 2497–2502. doi: 10.1073/pnas.96.5.2497
Weyer, S. W., Klevanski, M., Delekate, A., Voikar, V., Aydin, D., Hick, M.,et al. (2011). APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280. doi: 10.1038/emboj.2011.119
Wheeler, J. M., Guthrie, C. R., and Kraemer, B. C. (2010). The role of MSUT-2 in tau neurotoxicity: a target for neuroprotection in tauopathy? Biochem. Soc. Trans. 38, 973–976. doi: 10.1042/BST0380973
Wiese, M., Antebi, A., and Zheng, H. (2010). Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans. PLoS ONE 5:e12790. doi: 10.1371/journal.pone.0012790
Wiese, M., Antebi, A., and Zheng, H. (2012). Regulation of neuronal APL-1 expression by cholesterol starvation. PLoS ONE 7:e32038. doi: 10.1371/journal.pone.0032038
Willnow, T. E., Hilpert, J., Armstrong, S. A., Rohlmann, A., Hammer, R. E., Burns, D. K.,et al. (1996). Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. U.S.A. 93, 8460–8464. doi: 10.1073/pnas.93.16.8460
Willnow, T. E., Sheng, Z., Ishibashi, S., and Herz, J. (1994). Inhibition of hepatic chylomicron remnant uptake by gene transfer of a receptor antagonist. Science 264, 1471–1474. doi: 10.1126/science.7515194
Witan, H., Kern, A., Koziollek-Drechsler, I., Wade, R., Behl, C., and Clement, A. M. (2008). Heterodimer formation of wild-type and amyotrophic lateral sclerosis-causing mutant Cu/Zn-superoxide dismutase induces toxicity independent of protein aggregation. Hum. Mol. Genet. 17, 1373–1385. doi: 10.1093/hmg/ddn025
Wood-Kaczmar, A., Gandhi, S., and Wood, N. W. (2006). Understanding the molecular causes of Parkinson’s disease. Trends Mol. Med. 12, 521–528. doi: 10.1016/j.molmed.2006.09.007
Wroe, R., Wai-Ling Butler, A., Andersen, P. M., Powell, J. F., and Al-Chalabi, A. (2008). ALSOD: the Amyotrophic Lateral Sclerosis Online Database. Amyotroph. Lateral Scler. 9, 249–250. doi: 10.1080/17482960802146106
Yao, C., Johnson, W. M., Gao, Y., Wang, W., Zhang, J., Deak, M.,et al. (2013). Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum. Mol. Genet. 22, 328–344. doi: 10.1093/hmg/dds431
Yochem, J., and Greenwald, I. (1993). A gene for a low density lipoprotein receptor-related protein in the nematode Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 90, 4572–4576. doi: 10.1073/pnas.90.10.4572
Yochem, J., Gu, T., and Han, M. (1998). A new marker for mosaic analysis in Caenorhabditis elegans indicates a fusion between hyp6 and hyp7, two major components of the hypodermis. Genetics 149, 1323–1334.
Yochem, J., Tuck, S., Greenwald, I., and Han, M. (1999). A gp330/megalin-related protein is required in the major epidermis of Caenorhabditis elegans for completion of molting. Development 126, 597–606.
Yokoseki, A., Shiga, A., Tan, C. F., Tagawa, A., Kaneko, H., Koyama, A.,et al. (2008). TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 63, 538–542. doi: 10.1002/ana.21392
Yoo, B. C., Kim, S. H., Cairns, N., Fountoulakis, M., and Lubec, G. (2001). Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 280, 249–258. doi: 10.1006/bbrc.2000.4109
Yoon, I. S., Pietrzik, C. U., Kang, D. E., and Koo, E. H. (2005). Sequences from the low density lipoprotein receptor-related protein (LRP) cytoplasmic domain enhance amyloid beta protein production via the beta-secretase pathway without altering amyloid precursor protein/LRP nuclear signaling. J. Biol. Chem. 280, 20140–20147. doi: 10.1074/jbc.M413729200
Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A.,et al. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54. doi: 10.1038/35024009
Zambrano, N., Bimonte, M., Arbucci, S., Gianni, D., Russo, T., and Bazzicalupo, P. (2002). feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J. Cell Sci. 115, 1411–1422.
Zambrano, N., Buxbaum, J. D., Minopoli, G., Fiore, F., De Candia, P., De Renzis, S.,et al. (1997). Interaction of the phosphotyrosine interaction/phosphotyrosine binding-related domains of Fe65 with wild-type and mutant Alzheimer’s beta-amyloid precursor proteins. J. Biol. Chem. 272, 6399–6405. doi: 10.1074/jbc.272.10.6399
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I.,et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zhang, Y. W., Luo, W. J., Wang, H., Lin, P., Vetrivel, K. S., Liao, F.,et al. (2005). Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma-secretase components. J. Biol. Chem. 280, 17020–17026. doi: 10.1074/jbc.M409467200
Zheng, G., Marino’, M., Zhao, J., and Mccluskey, R. T. (1998). Megalin (gp330): a putative endocytic receptor for thyroglobulin (Tg). Endocrinology 139, 1462–1465. doi: 10.1210/endo.139.3.5978
Zigman, W. B. (2013). Atypical aging in Down syndrome. Dev. Disabil. Res. Rev. 18, 51–67. doi: 10.1002/ddrr.1128
Zlokovic, B. V., Martel, C. L., Matsubara, E., Mccomb, J. G., Zheng, G., Mccluskey, R. T.,et al. (1996). Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood–brain and blood–cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. U.S.A. 93, 4229–4234. doi: 10.1073/pnas.93.9.4229
Keywords: apl-1, C. elegans, Alzheimer’s disease, ALS, Parkinson disease, model systems
Citation: Alexander AG, Marfil V and Li C (2014) Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 5:279. doi: 10.3389/fgene.2014.00279
Received: 08 April 2014; Accepted: 31 July 2014;
Published online: 05 September 2014.
Edited by:
Shin Murakami, Touro University-California, USAReviewed by:
Patrick Hu, University of Michigan, USAYelena V. Budovskaya, University of Amsterdam, Netherlands
Copyright © 2014 Alexander, Marfil and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris Li, Department of Biology, City College of New York, New York, NY, USA e-mail:Y2xpQHNjaS5jY255LmN1bnkuZWR1
†Adanna G. Alexander and Vanessa Marfil have contributed equally to this work.