 Steve Meaney1,2*
Steve Meaney1,2*
- 1School of Biological Sciences, College of Sciences and Health, Dublin Institute of Technology, Dublin, Ireland
- 2Environmental Sustainability and Health Institute, Dublin Institute of Technology, Dublin, Ireland
Although best known as a risk factor for cardiovascular disease, cholesterol is a vital component of all mammalian cells. In addition to key structural roles, cholesterol is a vital biochemical precursor for numerous biologically important compounds including oxysterols and bile acids, as well as acting as an activator of critical morphogenic systems (e.g., the Hedgehog system). A variety of sophisticated regulatory mechanisms interact to coordinate the overall level of cholesterol in cells, tissues and the entire organism. Accumulating evidence indicates that in additional to the more “traditional” regulatory schemes, cholesterol homeostasis is also under the control of epigenetic mechanisms such as histone acetylation and DNA methylation. The available evidence supporting a role for these mechanisms in the control of cholesterol synthesis, elimination, transport and storage are the focus of this review.
Introduction
Cholesterol (cholest-5-ene-3β-ol) is an ubiquitous 27-carbon steroid and a vital component of cellular membranes in vertebrates (Vance and Vance, 2002). It is essential for life, and defects in the ability to synthesize it leads to severe clinical conditions such as the Smith–Lemli–Opitz syndrome and desmosterolosis (Porter, 2003). In man the total pool of cholesterol is 120–130 g and as it is a neutral, hydrophobic lipid the vast majority is found as the free sterol in the membrane (Cook, 1958). The distribution of cholesterol across organs, cells, and subcellular membranes is not even, with some organs (e.g., the brain) and cell types (e.g., oligodendroglia) highly enriched in cholesterol. Within the cell, the low cholesterol content in intracellular membranes facilitates cholesterol sensing and homeostatic feedback (Steck and Lange, 2010; Jeon and Osborne, 2012). In addition to this essential structural role cholesterol is also a precursor for numerous biomolecules of physiological importance including bile acids, steroid hormones, and oxysterols1.
All nucleated cells can synthesize cholesterol. As a consequence, under normal circumstances there is no requirement to ingest cholesterol. However, a typical western diet contains approximately 300–500 mg of cholesterol per day and so cholesterol originates from either de novo synthesis or dietary intake (Wang, 2007). This cholesterol is absorbed in the small intestine and is distributed throughout the body via the action of lipoproteins to most, but not all, tissues. The brain is a notable exception – cholesterol is unable to pass the blood–brain barrier and cholesterol in the brain is due to local synthesis (Björkhem and Meaney, 2004). Cholesterol in excess of requirements may either be stored as cholesteryl esters or eliminated via oxidation and/or conversion into bile acids (Björkhem, 2013). Thus, in both the cell and the intact organism, the overall cholesterol level depends on the contribution of cholesterol synthesis, absorption, elimination, and storage. This review presents a review of the current evidence of the role of epigenetic mechanisms in each of these processes.
Fundamentals of Lipoprotein Homeostasis
Due to its insoluble nature, cholesterol cannot be solubilized in the plasma and is transported as cargo in proteolipid complexes known as lipoprotein particles. These particles consist of (i) a major structural apolipoprotein, (ii) peripheral apolipoproteins, (iii) structural lipids (phospholipids and cholesterol), and (iv) cargo lipids (highly hydrophobic lipid species such as triacylglycerols (TAG), steryl esters and fat soluble drugs, and vitamins). Lipoproteins can be divided into two broad classes – those containing Apolipoprotein B (APOB) and those containing Apolipoprotein A1 (APOA1) as core structural components. In combination with other peripheral apolipoproteins such as APOE, APOCII, and APOCI, these proteins define the function and metabolic fates of lipoproteins in the body (Vance and Vance, 2008).
Dietary lipids are absorbed via the small intestine and packaged into large particles known as chylomicrons (CM), which contain the intestine-specific APOB48, which is a truncated form of APOB. CM are essentially TAG delivery systems that distribute dietary lipids throughout the body for direct use and/or storage (Vance and Vance, 2008). CM remnant particles eventually return to the liver where their remaining lipid cargo, now relatively enriched in cholesterol, integrates into endogenous hepatic lipid pathways. The liver produces a second class of APOB containing lipoproteins, very low density lipoprotein (VLDL), which contain the full-length APOB and are more enriched with cholesterol than CM. In similarity with CM, VLDL participates in TAG distribution from the liver to the periphery. However, as core TAG are delivered to the tissues of the body VLDL shrink and shed peripheral apolipoproteins. During this process, the proportion of cholesterol in the particle increases progressively, ultimately leading to the production of low-density lipoprotein (LDL) particles. By binding to the LDL-receptor (LDLR) on the outer surface of expressing cells, LDL can be internalized into the cell and its cholesterol containing cargo delivered inside the cell. As an individual, LDLR is reused multiple times, LDL acts as an efficient cholesterol delivery system.
In contrast to APOB pathways, which are responsible for lipid transport to the periphery, APOA1 containing lipoproteins (i.e., high-density lipoproteins, HDL) are the mediators for reverse cholesterol transport (RCT). APOA1 is mainly synthesized by the liver and intestine, although other cell such as macrophages can also produce APOA1. Nascent HDL particles are lipid poor and collect free cholesterol and phospholipids from peripheral tissues in an ABCA1-dependent process. Esterification of cholesterol by lecithin-cholesterol acyl transferase (LCAT) leads to an increase in core cholesteryl esters and the formation of HDL2 particles. HDL2 may be further enriched with lipids by the action of the ATP-binding cassette subfamily G member 4 (ABCG4). This lipid cargo may then be delivered to the liver via Scavenger Receptor B1 (SCARB1). As the HDL particle is not destroyed during lipid delivery to the liver, it can participate in RCT several times, enhancing the efficiency of the RCT process.
For more details on the details of lipoprotein homeostasis the reader is referred to Kingwell (Kingwell et al., 2014) and chapters 17–20 of Vance and Vance (2008).
Cholesterol Synthesis
The biosynthesis of cholesterol is a highly complex process involving more than 30 different reactions and over 15 enzymes present in many different subcellular compartments (Sharpe and Brown, 2013). Conceptually, this pathway may be divided into two stages (i) condensation of isoprenoid units to yield the 30-carbon molecule squalene and (ii) cyclization of squalene to produce lanosterol, which is converted to cholesterol.
Cholesterol biosynthesis begins with the formation of the six-carbon 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) from one molecule of acetyl-CoA (2C) and one of acetoacetyl-CoA. This reaction is catalyzed by the soluble enzyme 3-hydroxy-3-methylglutaryl-Co-enzyme A synthase (HMGCS). HMG-CoA is then converted to mevalonate by the action of the membrane bound enzyme 3-hydroxy-3-methylglutaryl-Co-enzyme A reductase (HMGCR) in a reaction considered to be the primary flux governing step in the cholesterol biosynthesis pathway and the target of the statin class of cholesterol lowering drugs. Recently, Gill et al. (2011) proposed that squalene epoxidase (SQLE), which catalyzes the addition of the oxygen moiety to squalene, may be a second flux governing step.
Regulation of cholesterol biosynthesis is achieved via an elegant system of feedback inhibition which senses intracellular levels of cholesterol and subsequently modulates the expression of the key proteins involved in cholesterol homeostasis (Ye and DeBose-Boyd, 2011). The master regulators of this process are the sterol response element binding proteins (SREBF1 and SREBF2, also known as SREBP1 and SREBP2), which are hairpin-shaped membrane-anchored transcription factors localized to the endoplasmic reticulum (ER). Gene regulation occurs when the transcriptionally active part of the molecule (a member of the basic loop helix leucine zipper family) is released from its membrane anchor and translocates to the nucleus. This process is dependent on the levels sterols and fatty acids, although only cholesterol will be discussed here.
SREBF is retained in the ER due to cholesterol-dependent interactions with two proteins: SREBP cleavage activating protein (SCAP) and insulin sensitive gene 1 (INSIG-1). When cellular cholesterol levels are reduced, the amount bound to SCAP is reduced, leading to a conformational change, which allows INSIG-1 to dissociate from the SREBF–SCAP complex and is tagged for degradation by the proteosome (Ye and DeBose-Boyd, 2011). In the absence of INSIG-1, the complex then translocates to the Golgi apparatus, where is subjected to two sequential proteolytic cleavages, first by site-1 protease (S1P) and then by site-2 protease (S2P). The net result of these cleavages is that the transcriptionally active domain is released from SREBF and is free to translocate to the nucleus.
Recent evidence has highlighted a role for microRNAs (miRNAs) in cholesterol homeostasis. miRNA 33a and miR-33b are encoded by introns in the SREBF1 and SREBF2 genes, respectively, and are highly similar. miR-33a is co-regulated with SREBF2, with low cholesterol levels promoting an increase and high sterol levels promoting the opposite (Marquart et al., 2010). Very recently Yang et al. (2014) identified a potential miRNA response element for miR-185 in the promoter of Srebf2 and demonstrated that lentiviral overexpression of this miRNA resulted in a decrease in hepatic Hmgcr and Ldlr. In vivo, miR-185 is regulated by Srebp1c, thus suggesting the presence of a complex feedback loop integrating classical mechanisms with miRNA-dependent processes. In addition, miR-520d and miR-224 both significantly suppress the mRNA expression of Hmgcr. Notably, these miRNAs also target various genes in cholesterol transport and lipoprotein homeostasis (cf below), indicating that they have a key role in the overall coordination of cholesterol homeostasis.
The SREBF proteins are known to be acetylated at conserved lysine residues by the histone acetylase cAMP responsive element binding protein 1-binding protein (CBP)/p300 (EP300, more commonly known as p300 or KAT3B; Giandomenico et al., 2003). This impedes the ubiquitination and degradation of SREBF and so prolongs its residence time in the nucleus, thus promoting its transcriptional activity (ibid). Recent evidence indicates that sirtuin 1 (SIRT1) can counteract this by directly deacetylating SREBF (Walker et al., 2010). SREBF may also be be phosphorylated, which creates a recognition site for the F-Box and WD Repeat Domain Containing 7, E3 Ubiquitin Protein (FBXW7) and allows the multiprotein SCF complex to ubiquitinate it (Sundqvist et al., 2005). Thus, the steady-state amount of transcriptionally active SREBF is under complex regulation at multiple levels and maintenance of active SREBF mediated gene transcription requires continuous cleavage of the transcriptionally active domain.
The rate of cholesterol synthesis varies considerably between different organs and tissues, and even between the cells of the same organ (Dietschy and Turley, 2002). Unsurprisingly, the expression of cholesterogenic genes varies in parallel with the rate of synthesis. A further source of variability is the age-dependency of cholesterol synthesis (Dietschy, 2009) with some cell types being very active during some periods of life and then becoming effectively quiescent afterwards (e.g., oligodendroglia). The combination of these factors leads to a unique synthesis profile in each cell. Although, the mechanism(s) underpinning this regulation has not been elucidated it is likely that epigenetic mechanisms play an important role.
Several investigators have reported that the genes involved in cholesterol biosynthesis are under epigenetic regulation, using a variety of in vivo and in vitro approaches (summarized in Table 1). Using an in vitro approach, Villagra et al. (2007) demonstrated that overexpression of histone deacetylase 3 (HDAC3) in CHO cells resulted in a 10-fold decrease of the rate of cholesterol synthesis. To rule out a confounding effect of SREBF mediated effects the authors used the SRD-13A cell line, which cannot cleave SREBF and are cholesterol auxotrophs (Rawson et al., 1999). Compared to a control cell line there was only a slight difference following HDAC overexpression, indicating the effect is primarily mediated by epigenetic mechanisms. Intriguingly, the HDAC3 effect was observed even though HMGCR was slightly up- rather than downregulated indicating that the effect was not at the classical flux controlling step in cholesterol synthesis. Instead the authors presented evidence that lanosterol synthase (LSS) which catalyses the cyclisation of the isoprenoid 2,3-oxidosqualene to the steroid lanosterol, was markedly downregulated by HDAC3 expression or treatment with the HDACi trichostatin A (TSA). Subsequently, Knutson et al. (2008) used an Hdac3 knockout mouse to show that loss of Hdac3 in vivo resulted in a de-repression of cholesterol synthesis and secretion by the liver, with several genes of the cholesterol synthesis pathway increased significantly. This lead to an age dependent increase in liver cholesterol content (fivefold increase in cholesteryl esters and a twofold increase in total cholesterol at P56) and a corresponding increase in serum total (threefold) and LDL-cholesterol (fivefold). Although there are differences in these reports, most likely due to the different experimental approaches, they are consistent in that an increase in HDAC3 leads to a decrease in cholesterol synthesis and content, while loss of Hdac3 leads to the opposite situation.
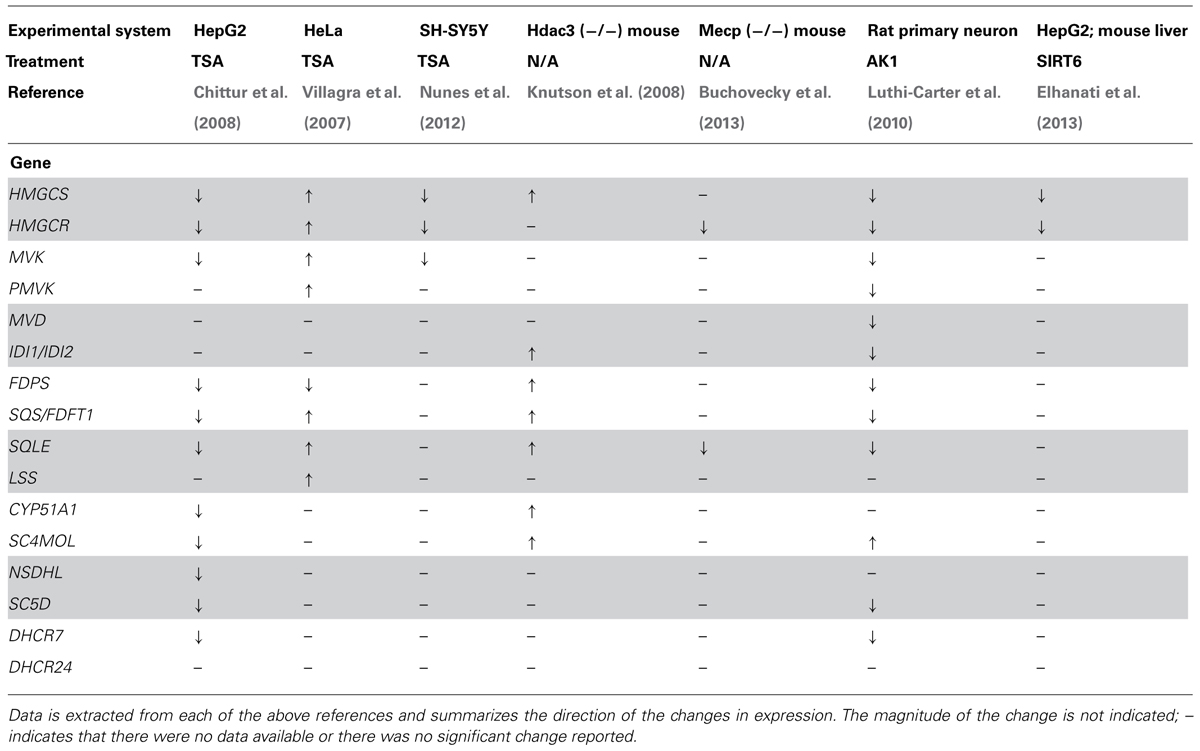
TABLE 1. Reported epigenetic changes in the expression of genes involved in cholesterol biosynthesis.
In a more broad array based survey, Chittur et al. (2008) carried out transcriptional profiling on HepG2 hepatoma and F9 mouse embyronic carcinoma cells following treatment with TSA. They reported that numerous genes of cholesterol synthesis were downregulated by TSA and confirmed the change in expression using qPCR. SREBF2 was also decreased, which may contribute to the changes observed in the cholesterol synthesis pathway in the cell systems studied. They did not report if cholesterol levels were altered by the treatment. Drzewinska et al. (2011) reported that 24-dehydrocholseterol reductase (DHCR24), which is not reported in the abovementioned studies, was induced by treatment with either TSA or 5-azacytidine (5AZA) in some, but not all, of the cell types tested. Taken together, the data available in connection with these studies indicates that epigenetic mechanisms are important for the regulation of the cholesterol synthesis pathway. Moreover, they also provide concrete evidence of cell-type sensitivity to epigenetic regulation.
As described above, members of the SIRT family are protein deaceylases that have been shown to have effects on cholesterol homeostasis. SIRT2 has also been reported to have effects on cholesterol biosynthesis, although the data are somewhat variable. Transcriptional profiling of of primary rat neuronal cultures following treatment with small molecule SIRT2 inhibitors (AK-1 or AGK2) for 24 h resulted in the decreased expression of many genes involved in cholesterol biosynthesis, while total sterol levels were also decreased to about two-thirds of the vehicle treated controls (Luthi-Carter et al., 2010). These investigators also reported that abundance of SREBF in the nucleus was reduced following SIRT inhibition, an effect that they speculated was due to a trafficking defect. However, given the sensitivity of SREBF to acetylation, this observation may be explained by increased degradation of nuclear SREBF rather than decreased trafficking. In contrast, studies using a Sirt2 knock-out mouse failed to recapitulate these data, with no changes in the mRNA of cholesterol synthesis genes (Bobrowska et al., 2012). However, as Bobrowska et al. analyzed brain regions with mixed cell types rather than primary cultures of a single cell type, any differences (e.g., in the neuronal compartment) may have been “diluted” by more abundant cell types in which there were no change. A further confounder is the known age-dependent changes in the expression of genes involved in sterol homeostasis in neurons, with significant changes occurring in the immediate postnatal period (Ohyama et al., 2006). Extrapolation of results concerning cholesterol homeostasis from primary cultures based on embryonic cells to the adult situation should therefore be done with caution.
SIRT6 has also been shown to be involved in cholesterol homeostasis. Elhanati et al. (2013) reported that cholesterol content in HepG2 cells was reduced (by approximately one-third) following ectopic overexpression of SIRT6 while RNAi mediated knockdown resulted in an increase in cellular cholesterol content of the same magnitude. The mRNA expression of both HMGCS and HMGCS changed in parallel with the changes in cholesterol content. Importantly similar results were found in the livers of a mouse model overexpressing Sirt6. The authors provided evidence that this regulation was due to effects of SIRT6 on SREBF1 and SREBF2 via a combination of direct transcriptional regulation, inhibition of the proteolytic release of the transcriptionally active form of SREBF and promotion of phosphorylation of SREBF by AMP activated protein kinase (AMPK).
The expression of miR-33a and miR-33b, causes a decrease in the expression of SIRT6 (Elhanati et al., 2013). Effects related to mIR-dependent regulation may account for the magnitude of the equal but opposite regulation by SIRT6 overexpression an SIRT6 knockdown, respectively. Subsequently, Tao et al. (2013) described additional mechanistic details and highlighted a key role for the forkhead box O3 (FOXO3) protein in recruiting SIRT6 to the SREBF2 promoter, leading to histone deacetylation and reduced expression of cholesterol biosynthetic genes. Importantly, these investigators also provided independent replication of the effect of Sirt6 overexpression on cholesterol concentration in vivo.
Very recently Poirier presented evidence that the commonly used DNA methylation inhibitor regulator 5AZA is capable of reducing the impeding cleavage of SREBF, thus reducing the expression of HMGCR (Poirier et al., 2014). Critically these effects were independent of effects on DNA methylation. These results add an extra layer to the interpretation of the results of studies on DNA methylation and cholesterol homeostasis using 5AZA.
Cholesterol Absorption
The absorption of cholesterol from the intestinal lumen is a complex and incompletely understood process (van der Wulp et al., 2013). As a very hydrophobic molecule cholesterol requires the presence of bile salts and phospholipids to form mixed micelles which permit its uptake into the enterocytes. Various proteins have been associated with inward flux of cholesterol into the enterocyte, most notably the Niemann Pick Disease Type C1 Like-1 (NPC1L1) protein. The heterodimer formed by the ATP binding cassette sub-family members G5 and G8 (ABCG5 and ABCG8, respectively) is associated with an outward sterol flux back into the intestinal lumen. There is very limited data on the involvement of role of epigenetic mechanisms in the regulation of these key genes. Very recently, Malhotra et al. (2014) described a key role for promoter methylation in the regulation of Npc1l1 expression in the mouse gastrointestinal tract. It is well established that Npc1l1 expression is restricted in the gastrointestinal tract with expression in the small but not the large intestine (Davis and Altmann, 2009). The low colonic expression appears to be a result of extensive methylation of the promoter in the colon but not in the ileum or jejunum, and 5AZA treatment resulted in the derepression of Npc1l1 expression in the colon (Malhotra et al., 2014). Furthermore, using a gene knockdown approach it was shown that both DNA methyltransferases 1 and 3B (DNMT1 and DNMT3B, respectively) are involved in maintaining the hypermethylated state in the colon. Simultaneous knockdown of both was required for the depression effect which is consistent with redundancy in the regulatory process. In similarity with NPC1L1 there are very limited data on epigenetic regulation of ABCG5/ABCG8, and none in the context of the intestine as far as it has been possible to determine. Studies on mouse liver carried out by Imai et al. (2009), however, indicate that the common promoter is acetylated and unmethylated, in the liver at least.
Cholesterol Elimination
Cholesterol is the obligate precursor for the formation of bile acids, which are essential for the absorption of dietary lipids. Formation of bile is the major route of cholesterol elimination from the intact organism and although the pathways terminate in the liver, bile acid synthesis can be initiated in almost all cell types (Björkhem, 2013; van der Wulp et al., 2013). Two interconnected pathways which differ at the point in which the C27 carboxylic acid group is added are recognized – the classical (or neutral) pathway and the alternative (or acidic) pathway. The key structural modifications which occur are the saturation of the sterol nucleus, introduction of 2–3 hydroxyl groups in the alpha configuration and the oxidation of the side chain to yield a 24-carbon molecule. As part of these pathways a class of compounds called oxysterols (or cholesterol oxidation products) are generated, each of which contains one or more additional oxygen containing functional groups. The enzymes responsible for the formation of these oxysterols (i.e., genes responsible for oxysterol synthesis, GROS) are unevenly distributed across different cells and tissues in the body, e.g., CYP7A1 is liver specific while CYP46A1 is found in CNS neurons only (Meaney, 2013). In similarity with the situation for ABCG5/ABCG8 and NPC1L1 above, epigenetic mechanisms have been shown to have a role in the regulation of these genes. Only those genes where there is data indicating a role for epigenetic regulation will be discussed.
Cholesterol 7α-hydroxylase (CYP7A1) is the rate limiting step in the classical pathway of bile acid biosynthesis. Its regulation has been studied in detail and the reader is referred to Chiang (2013) for a recent review. In brief, bile acids, by acting as ligands for the nuclear hormone receptor FXR, lead to the suppression of CYP7A1 expression via an indirect negative feedback cycle dependent on the small heterodimer partner (SHP) protein. From an epigenetic view point, the work of Kemper and colleagues has highlighted a key role for the farnesoid X-receptor (FXR) dependent events at the promoter of SHP – binding of ligand activated FXR to the promoter leads to the recruitment of p300 and a subsequent increase in both histone and FXR acetylation. Numerous chromatin remodeling factors – Brahma-related gene-1 (BRG1), Brg-1 associated factors (BAF) and activating signal cointegrator-2 containing co-activator complex (ASCOM) – are then recruited to the promoter, promoting significant chromatin remodeling, which triggers upregulation of SHP. SHP interacts with liver receptor homolog-1 (LRH1) at the CYP7A1 promoter and recruits SIN3 transcription regulator family member A (SIN3A), HDACs1 and 2 and euchromatic histone-lysine N-methyltransferase 2 (EHMT2, also known as G9a). Ultimately a SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 2 (SMARCA2, also known as BRM) complex remodels the chromatin to suppresses CYP7A1 expression (Smith et al., 2013). SHP has also been shown to recruit SIRT1 to LRH1 target genes (including SHP itself), leading to SIRT1 dependent histone deacetylation and inhibition of transcription of the CYP7A1 gene and providing a self-regulating feedback loop (Chanda et al., 2010).
In vivo studies indicate that Hdac3 may also play a role in regulation of Cyp7a1 – knockout of Hdac3 in the mouse leads to increased histone acetylation at the Cyp7a1 promoter and an increase in mRNA expression (Knutson et al., 2008). Treatment of mice with TSA or valproic acid (VPA) resulted in a large increase in Cyp7a1 expression and prevented normal feedback regulation by bile acids, probably by interfering with the sophisticated mechanisms described above. In addition data has been presented that Hdac7 was critical for the HDACi mediated increase. There is limited evidence for a role of miRNA in the regulation of CYP7A1. Song et al. (2010) reported that CYP7A1 was a target for the liver enriched miR-122 as well as miR-422a and that silencing of these miRNAs induced gene expression.
Taken together this evidence indicates that epigenetic mechanisms are critical for the regulation of CYP7A1 in many species, although there is clearly more work to be done to elaborate additional details. For example, the basis for the hepatospecificity has yet to be established and the role of SIRTs requires further scrutiny.
Sterol 27-hydroxylase (CYP27A1) catalyzes the formation of both 27-hydroxycholesterol (27-OHC) and 3β-hydroxy-5-cholestenoic acid (CA) and is the initial step in the alternative pathway of bile acid biosynthesis. Although certain cell types (i.e., hepatocytes and cells of the monocytic lineage) express high levels of the enzyme it is considered relatively ubiquitous. Studies on epigenetic regulation of CYP27A1 are limited and are descriptive only – 5AZA was shown to have no effect in macrophages (Escher et al., 2005) while TSA treatment of HepG2 cells resulted in a six-fold decrease of CYP27A1 mRNA at 24 h followed by a return to basal levels by 48 h (Chittur et al., 2008). In contrast, treatment of SH-SY5Y neuroblastoma cells with TSA caused a five-fold increase in expression (Shafaati et al., 2009). While it is difficult to reconcile these findings in the absence of additional mechanistic details, it is highly likely that cell specific factors are involved.
Cholesterol 24S-hydroxylase (CYP46A1) catalyzes the formation of 24S-hydroxycholesterol (24S-OHC), a key process for maintenance of overall cholesterol homeostasis within the brain of vertebrates. It is well established that CYP46A1 is regulated by histone acetylation status – treatment with TSA in vitro results in a large increase in mRNA expression (Ballas et al., 2005; Milagre et al., 2008; Shafaati et al., 2009). Under in vivo conditions, treatment of mice with TSA leads to a modestly increased level of Cyp46a1 in the liver but not the brain (Shafaati et al., 2009). Rodrigues et al. have carried out an elegant series of experiments designed to uncover the details of the mechanism underpinning this effect. Specificity proteins (SP) 1, 3, 4 appear to be critical for the effect of HDACi – loss of SP3 from the promoter triggers a decrease in the occupancy of HDACs 1 and 2, with subsequent recruitment of CBP and p300 (Milagre et al., 2012). In a subsequent study these authors identified a potential role for MEK-ERK signaling in the regulatory process and provided evidence that pERK is present at the CYP46A1 promoter where it phosphorylates SP3, thus triggering recruitment of corepressors (Nunes et al., 2012). Given the highly restricted expression of CYP46A1 – in healthy brain it is only present in neurons – this mechanism may represent a supression mechanism important for cell-specificity of CYP46A1. In addition, the RE1-silencing transcription factor (REST, also known as NRSF) may play a role in the suppression of CYP46A1 in non-neuronal cells (Meaney, 2013). Although the CYP46A1 promoter is GC rich and contains several predicted CpG islands it does not appear that these are methylated. However, treatment with DNMT inhibitors lead to an increase in the expression of CYP46A1, by a mechanism involving an SP3 dependent loss of SIN3A and HDAC(s) (Milagre et al., 2010). In the Mecp2 null mouse, expression of CYP46A1 was markedly decreased with a concomitant reduction in the rate of cholesterol synthesis rate (Buchovecky et al., 2013). These data indicate that epigenetic mechanisms are critical for the regulation of brain cholesterol balance and may indeed represent critical coordination point between brain cholesterol synthesis and eliminations.
Cholesterol 25-hydroxylase (CH25H) catalyses the synthesis of 25-hydroxycholesterol (25-OHC), a molecule which has very recently been described to have potent anti-viral and paracrine immunomodulatory effects (Bauman et al., 2009; McDonald and Russell, 2010; Liu et al., 2013). It has been shown that the signal transducers and activators of transcription 1 (STAT1) pathway can regulate Ch25h in mice (Blanc et al., 2013). As HDAC activity is required for STAT1 signaling, epigenetic mechanisms likely contribute to this process. Gold et al. (2012) demonstrated that deletion of the activating transcription factor-3 (Atf3) gene lead to increased Ch25h promoter acetylation in bone marrow derived macrophages, with the consequence of increased mRNA and 25-OHC.
In contrast to the other genes responsible for the formation of oxysterols, CYP3A4 encodes an enzyme with a primary responsibility for the metabolism of pharmaceuticals and xenobiotics. In addition, it has been reported that CYP3A4 can also produce both 4β- and 25-hydroxycholesterol (Honda et al., 2011; Diczfalusy, 2013). CYP3A4 is hepatospecific and its expression can be induced significantly, with the pregnane X-receptor acting as a key mediator of regulation. Currently available data indicates that DNA methylation plays a role in the regulation of CYP3A4 – treatment with 5′-aza-deoxycytidine increased the expression of CYP3A4 in a variety of cell lines, with induction in some cell systems by a factor of more than 100-fold (Dannenberg and Edenberg, 2006; Habano et al., 2011). Due to the sensitivity of CYP3A4 to PXR, these effects are likely due to differential methylation at the promoter of PXR rather than a direct effect on CYP3A4. In studies on the mouse ortholog Cyp3a11, Li et al. (2009) demonstrated that increased expression of Cyp3a11 was associated with increased histone H3 lysine-9 dimethylation. In the brain, which does not express Cyp3a11, there was extensive histone-H3 lysine-9 dimethylation and lysine-27 trimethylation at the Cyp3a locus (Mikkelsen et al., 2007). At a mechanistic level, it has been shown that ligand binding to PXR triggers recruitment of protein arginine N-methyltransferase-1 (PRMT1) to the CYP3A4 promoter which triggers increased histone acetylation and methylation (Xie et al., 2009).
Oxysterol 7α-hydroxylase (CYP7B1) is responsible for the 7α-hydroxylation of intermediates in bile acid synthesis such as 27-OHC and 25-OHC but not 24S-OHC (cf below). In similarity with 27-OHC, CYP7B1 is considered to be an ubiquitous enzyme although its expression is greater in males than in females (Leuenberger et al., 2009; Stiles et al., 2009). It is known that the CYP7B1 promoter is methylated and treatment of the prostate cancer cell line LNCaP with DNMT inhibitors leads to a reduction in the mRNA levels (Olsson et al., 2007). The results following HDACi treatment are more variable and appear to be dependent on the cell type – HDACi treatment leads to a reduction in SH-SY5Y cells but an induction in HepG2 cells (Chittur et al., 2008; Shafaati et al., 2009). This latter induction was shown to be dependent on HDAC1 and HDAC3 dependent processes. Methylation has a key role on the sexual dimorphic expression of CYP7B1 – sumoylation of the nuclear hormone receptor peroxisome-proliferator activated receptor triggers enhanced interaction with GA-binding protein (GABP) and the recruitment of various HDAC and DNMT. The net effect is the methylation of an important SP1 binding site in the CYP7B1 promoter, preventing its binding to the promoter and so the expression is reduced (Leuenberger et al., 2009).
24S-hydroxycholesterol 7α-hydroxylase (CYP39A1) is responsible for the 7α-hydroxylation of the 24S-OHC. It has limited expression throughout the body, with liver, eye and brain expression reported (Li-Hawkins et al., 2000; Steckelbroeck et al., 2002; Ikeda et al., 2003; Shafaati et al., 2009). In similarity with CYP7B1, CYP39A1 is expressed in a sexually dimorphic manner, with higher levels in females compared to males (Li-Hawkins et al., 2000). Data on the epigenetic regulation of CYP39A1 are very limited – HDACi treatment in mice results in an increase in Cyp39a1 in the brain while at the same time decreasing hepatic expression (Shafaati et al., 2009). Huang et al. (2009) have described that CYP39A1 may be hypermethylated in ovarian cancer, but did not report effects on mRNA levels. Using a hypomorphic DNMT1 mouse line, Kutay et al. (2012) recently reported that the expression change in the liver of Cyp39a1 in response to alcohol challenge was blunted by more than half in the DNMT1 deficient lineage.
Lipoproteins
To date, there have been few reports on the epigenetic regulation of plasma lipoproteins or the proteins involved in their turnover and there do not appear to be any regulation of the structural apolipoproteins APOA1 and APOB. However, there is some recent evidence that APOE, which is present on both APOB and APOA1 containing lipoproteins in the circulation, is epigenetically regulated. In an early study on hyperhomocystenemia Yi-Deng et al. (2007) reported a decrease in Apoe mRNA and protein following homocysteine treatment, a decrease which was reversed with folate treatment. These changes were matched by an concomitant increase promoter methylation, linking the change to differential methylation of the DNA. Very recently, Yu et al. (2013) reported that exon 4 of the APOE gene, which contains the important e2/e3/e4 polymorphic region, contained a CpG island. This region was highly methylated in numerous brain regions and also in peripheral blood lymphocytes and the degree of methylation appeared to be sensitive to APOE allele present in exon 4. Moreover, there were functional consequences – the differentially methylated alleles had independent enhancer/silencer effects (Yu et al., 2013). Thus, although the direct data is limited, it appears that methylation at least plays a key role in the expression of APOE.
Apolipoprotein J (CLU, initially described as clusterin) is found in APOA1 (i.e., HDL-type) containing-lipoproteins. Salminen’s group has studied the epigenetic regulation of CLU in various cellular systems. Treatment with different HDACi increased the mRNA expression of CLU in normal human astrocytes, C6 glioma cells and both SK-N-AS and HN10 neuroblastoma cells (Nuutinen et al., 2005). The expression in SK-N-AS cells was further enhanced by a combination of HDACi and DNMT inhibition. Intracellular and secreted CLU was also significantly increased by HDACi treatment (Suuronen et al., 2007). Taken together these results indicate that epigenetic mechanisms play an important role in the regulation of CLU.
In HepG2 cells, HDACi treatment appears to reduce the expression of the LDL-receptor (Chittur et al., 2008; Nunes et al., 2012). In addition, the proprotein convertase subtilisin/kexin type 9 protein (PCSK9), which controls the degradation of the LDL-receptor and is a critical regulator of the amount of LDLR expressed on the cell surface, is epigenetically regulated. SIRT6 is recruited to the promoter by FOXO3 and promotes the deacetylation of histone H3 proteins at lysines 9 and 56 (Tao et al., 2013). This has the effect of reducing PCSK9 expression, preserving LDL-R exposure and function and thus lowering plasma low density lipoprotein cholesterol levels. In the Sirt6 knockout mouse, Pcsk9 levels are elevated with a concomitant reduction in the expression of the Ldlr and an increases the level of cholesterol in the blood (ibid).
Intriguingly, some very recent data indicates that lipoproteins may trigger epigenetic changes in genes involved in disease. Kumar et al. (2013) demonstrated that DNMT1 expression and activity were induced in endothelial cells following treatment with LDL cells. Binding of MECP2 was also enhanced, with a net result of a decrease in endothelial Kruppel-like factor 2 (KLF2) expression. The overall effect was an enhanced procoagulant status of the endothelium a change which may have functional significance (ibid).
Accumulating evidence indicates that microRNAs (miRNA) are involved in the regulation of lipoprotein homeostasis, adding an additional level of complexity and control to the process. Studies by Esau et al. (2006) identified that miR-122 was highly enriched in the liver and was able to regulate plasma cholesterol in the mouse and in non-human primates. Ablation of miR-122 using antisense oligonucleotides resulted in a decrease in total cholesterol by 25–30%, with the effect in non-human primates due to a reduction in LDL cholesterol. However, the mechanism for these changes was not obvious, despite the use of genome wide expression profiling approaches. miR-370 has also been shown to indirectly influence plasma cholesterol levels, via modulation of miR-122 levels (Iliopoulos et al., 2010). On the other hand, several miRNAs – including miR-33a/b, miR-128, and miR-144 – have been reported to modulate the expression of components of the pathway for HDL-dependent cholesterol efflux. This regulatory mechanism appears to be of functional importance, as silencing of these miRNAs in vivo leads to an increase in plasma HDL (Rayner et al., 2011a; Adlakha et al., 2013; Ramírez et al., 2013). This may represent a regulatory mechanism which converges on the flux-governing steps of sterol efflux.
Salerno et al. recently described two miRNAs which appear to have a role in regulation of the LDLR – miR-520d and miR-224 both target genes involved in determining how much LDLR is expressed on the cell surface, namely IDOL and PCSK9 (Salerno et al., 2013). In vitro overexpression of miR-520d decreased the expression of both genes by 30–50%, while similar experiments with miR-224 resulted in a decrease in the order of 75%. The net effect of these decreases was an increase in the cell-surface expression of the LDLR and greater LDL binding.
Cholesterol Storage
Excess cholesterol in the cell can be converted to choelsteryl esters by sterol O-acyl transferases 1 and 2 (SOAT1 and SOAT2, respectively, also known as ACAT1 and ACAT2). This provides a mechanism whereby cells can store cholesterol and remove it from the metabolically active pool present in the cell. Data on the regulation of SOAT1 and SOAT2 are very limited and is found only in studies examining other aspects of lipid metabolism. Tessema and Belinsky (2008) identified that the SOAT2 promoter was methylated to varying degrees among a panel of lung cancer adenocarcinoma cells. Liang et al. (2013) reported a modest decrease in SOAT1 promoter methylation following homocysteine treatment, which was contemporaneous with an increase in SOAT1 mRNA and cholesteryl ester concentration. Finally, Devlin et al. (2010) reported that hepatic expression of Soat2 in hyperhomocystenemic mice was reduced in the context of elevated DNA methylation. Thus, although the available data is limited, there are indications that sterol esterification is likely to be under epigenetic regulation.
Potential of Epigenetic Therapies in Dyslipidemias
There is relatively limited direct data on the potential for HDACi and related epigenetic therapies to influence lipid levels and the data that is available is typically in connection with consumption of juices and other supplements. In an early study, Murashima et al. (2004) examined the effects of 1-week intake of broccoli sprouts – which rich in the phytochemical HDACi sulforaphane – on various biochemical parameters in man and reported positive effects on HDL-cholesterol. Subsequent studies of hypercholesterolemic men treated with kale juice (also rich in sulforaphane) in demonstrated similar beneficial increases in HDL-cholesterol and a significant and clinically relevant decrease in the atherogenic index (Kim et al., 2008). In a randomized double-blind placebo-controlled study on type 2 diabetic patients, using broccoli sprout powder (BSP) Bahadoran et al. (2012) reported a significantly higher HDL-cholesterol in the group receiving the highest dose of BSP and a reduction in the atherogenic index similar to that described by Kim et al. (2008). Thus, although the data is both limited and confounded by the well known anti-oxidant effects of sulforaphane, it does appear that targeted dietary epigenetic therapies may be viable options for treatment of dyslipidemia.
The key role for miR-33 in regulation of cholesterol efflux has prompted several investigators to explore the possibility that antagonism of miR-33 may be a viable HDL-raising strategy. Initial studies from Moore’s group demonstrated that treatment of atherprone mice with anti-miR-33 lead to an increase in plasma HDL levels with no obvious hepatotoxicity and moderate regression of atherosclerosis (Rayner et al., 2011b). Subsequent studies where non-human primates were treated for a total of 12 weeks demonstrated an elevation in Abca1 expression and increase in HDL levels (to 1.5-fold of the mismatch control), again with no apparent hepatotoxicity (Rayner et al., 2011a). Similar changes were also reported by Rottiers et al. (2013). Inhibition of the activity of miR-33 thus appears to be a potential mechanism to enhance reverse cholesterol transport in vivo. However, very recently Goedeke et al. (2014) reported that long-term inhibition of miR-33 in the setting of a high-fat diet may lead to hepatic steatosis. Although these studies were carried out in mice, they do indicate that anti-miR-33 therapies may have adverse effects when used for long term treatment.
Due to its hydrophobic nature, the movement of cholesterol within the cells relies on a wide variety of membrane bound and soluble proteins. The Niemann Pick Disease Type C proteins 1 and 2 (NPC1 and NPC2, respectively) work in a coordinated manner to ferry cholesterol from the endolysosomal system to the endoplasmic reticulum (Yu et al., 2014). There is compelling evidence that both of these genes are regulated by histone acetylation status and treatment of cells with a variety of different HDACi leads to a significant increase in the expression of these genes (Gévry et al., 2003; Kim et al., 2007; Pipalia et al., 2011; Helquist et al., 2013; Maceyka et al., 2013). The increase in the expression of the mutated NPC proteins, which have some by not sufficient activity, resulted in a quantitative increase in activity which was sufficient to correct the cellular defect (Munkacsi et al., 2011; Pipalia et al., 2011; Yang et al., 2014). Due the success of the HDACi treatment in pre-clinical models, HDACi therapy is currently being trialed in a small group of NPC patients. This underscores the potential for therapies based on epigenetic approaches to address deficits in cholesterol homeostasis which are currently intractable by other approaches.
Concluding Remarks
Epigenetic regulation of cholesterol and sterol homeostatic genes is a burgeoning field which is already showing great potential to explain the behavior of genes involved in sterol balance. There are numerous questions outstanding in the field, but there are excellent investigators working intensively to address them. The goal will be to translate basic research findings into transcriptional therapies for non-cancer human diseases, in a similar manner to NPC disease as outline above. The availability of pharmaceutical compounds developed for other diseases (e.g., vorinostat) as well as the ever increasing amount of dietary epigenetic modulators provides a good foundation for translation into the clinical setting as possible therapies for cardiovascular and neurodegenerative diseases. The efforts of dedicated investigators who continue to shed light on the importance of epigenetic mechanisms will contribute to this development in the coming years.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^It should be noted that cholesterol itself is not a direct precursor for vitamin D; rather the sterol 7-dehydrocholesterol, itself an intermediate in cholesterol biosynthesis, fulfils this role.
References
Adlakha, Y. K., Khanna, S., Singh, R., Singh, V. P., Agrawal, A., and Saini, N. (2013). Pro-apoptotic miRNA-128-2 modulates ABCA1, ABCG1 and RXRα expression and cholesterol homeostasis. Cell Death Dis. 4:e780. doi: 10.1038/cddis.2013.301
Bahadoran, Z., Mirmiran, P., Hosseinpanah, F., Rajab, A., Asghari, G., and Azizi, F. (2012). Broccoli sprouts powder could improve serum triglyceride and oxidized LDL/LDL-cholesterol ratio in type 2 diabetic patients: a randomized double-blind placebo-controlled clinical trial. Diabetes Res. Clin. Pract. 96, 348–354. doi: 10.1016/j.diabres.2012.01.009
Ballas, N., Grunseich, C., Lu, D. D., Speh, J. C., and Mandel, G. (2005). REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657. doi: 10.1016/j.cell.2005.03.013
Bauman, D. R., Bitmansour, A. D., McDonald, J. G., Thompson, B. M., Liang, G., and Russell, D. W. (2009). 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. U.S.A. 106, 16764–16769. doi: 10.1073/pnas.0909142106
Björkhem, I. (2013). Five decades with oxysterols. Biochimie 95, 448–454. doi: 10.1016/j.biochi.2012.02.029
Björkhem, I., and Meaney, S. (2004). Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24, 806–815. doi: 10.1161/01.ATV.0000120374.59826.1b
Blanc, M., Hsieh, W. Y., Robertson, K. A., Kropp, K. A., Forster, T., Shui, G.,et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. doi: 10.1016/j.immuni.2012.11.004
Bobrowska, A., Donmez, G., Weiss, A., Guarente, L., and Bates, G. (2012). SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of Huntington’s disease phenotypes in vivo. PLoS ONE 7:e34805. 10.1371/journal.pone.0034805
Buchovecky, C. M., Turley, S. D., Brown, H. M., Kyle, S. M., McDonald, J. G., Liu, B.,et al. (2013). A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 45, 1013–1020. doi: 10.1038/ng.2714
Chanda, D., Xie, Y. B., and Choi, H. S. (2010). Transcriptional corepressor SHP recruits SIRT1 histone deacetylase to inhibit LRH-1 transactivation. Nucleic Acids Res. 38, 4607–4619. doi: 10.1093/nar/gkq227
Chiang, J. Y. L. (2013). Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212. doi: 10.1002/cphy.c120023
Chittur, S. V., Sangster-Guity, N., and McCormick, P. J. (2008). Histone deacetylase inhibitors: a new mode for inhibition of cholesterol metabolism. BMC Genomics 9:507. doi: 10.1186/1471-2164-9-507
Dannenberg, L. O., and Edenberg, H. J. (2006). Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics 7:181. doi: 10.1186/1471-2164-7-181
Davis, H. R., and Altmann, S. W. (2009). Niemann-Pick C1 Like 1 (NPC1L1) an intestinal sterol transporter. Biochim. Biophys. Acta 1791, 679–683. doi: 10.1016/j.bbalip.2009.01.002
Devlin, A. M., Singh, R., Bottiglieri, T., Innis, S. M., and Green, T. J. (2010). Hepatic acyl-coenzyme a: cholesterol acyltransferase-2 expression is decreased in mice with hyperhomocysteinemia. J. Nutr. 140, 231–237. doi: 10.3945/jn.109.112920
Diczfalusy, U. (2013). On the formation and possible biological role of 25-hydroxycholesterol. Biochimie 95, 455–460. doi: 10.1016/j.biochi.2012.06.016
Dietschy, J. M. (2009). Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 390, 287–293. doi: 10.1515/BC.2009.035
Dietschy, J. M., and Turley, S. D. (2002). Control of cholesterol turnover in the mouse. J. Biol. Chem. 277, 3801–3804. doi: 10.1074/jbc.R100057200
Drzewinska, J., Walczak-Drzewiecka, A., and Ratajewski, M. (2011). Identification and analysis of the promoter region of the human DHCR24 gene: involvement of DNA methylation and histone acetylation. Mol. Biol. Rep. 38, 1091–1101. doi: 10.1007/s11033-010-0206-z
Elhanati, S., Kanfi, Y., Varvak, A., Roichman, A., Carmel-Gross, I., Barth, S.,et al. (2013). Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 4, 905–912. doi: 10.1016/j.celrep.2013.08.006
Esau, C., Davis, S., Murray, S. F., Yu, X. X., Pandey, S. K., Pear, M.,et al. (2006). miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 3, 87–98. doi: 10.1016/j.cmet.2006.01.005
Escher, G., Hoang, A., Georges, S., Tchoua, U., El-Osta, A., Krozowski, Z.,et al. (2005). Demethylation using the epigenetic modifier, 5-azacytidine, increases the efficiency of transient transfection of macrophages. J. Lipid Res. 46, 356–365. doi: 10.1194/jlr.D400014-JLR200
Giandomenico, V., Simonsson, M., Grönroos, E., and Ericsson, J. (2003). Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol. Cell Biol. 23, 2587–2599. doi: 10.1128/MCB.23.7.2587-2599.2003
Gill, S., Stevenson, J., Kristiana, I., and Brown, A. J. (2011). Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 13, 260–273. doi: 10.1016/j.cmet.2011.01.015
Goedeke, L., Salerno, A., Ramírez, C. M., Guo, L., Allen, R. M., Yin, X.,et al. (2014). Long-term therapeutic silencing of miR-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol. Med. 6, 1133–1141. doi: 10.15252/emmm.201404046
Gold, E. S., Ramsey, S. A., Sartain, M. J., Selinummi, J., Podolsky, I., Rodriguez, D. J.,et al. (2012). ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J. Exp. Med. 209, 807–817. doi: 10.1084/jem.20111202
Gévry, N. Y., Lalli, E., Sassone-Corsi, P., and Murphy, B. D. (2003). Regulation of niemann-pick c1 gene expression by the 3′5′-cyclic adenosine monophosphate pathway in steroidogenic cells. Mol. Endocrinol. 17, 704–715. doi: 10.1210/me.2002-0093
Habano, W., Gamo, T., Terashima, J., Sugai, T., Otsuka, K., Wakabayashi, G.,et al. (2011). Involvement of promoter methylation in the regulation of Pregnane X receptor in colon cancer cells. BMC Cancer 11:81. doi: 10.1186/1471-2407-11-81
Helquist, P., Maxfield, F. R., Wiech, N. L., and Wiest, O. (2013). Treatment of Niemann–pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 10, 688–697. doi: 10.1007/s13311-013-0217-2
Honda, A., Miyazaki, T., Ikegami, T., Iwamoto, J., Maeda, T., Hirayama, T.,et al. (2011). Cholesterol 25-hydroxylation activity of CYP3A. J. Lipid Res. 52, 1509–1516. doi: 10.1194/jlr.M014084
Huang, Y. W., Jansen, R. A., Fabbri, E., Potter, D., Liyanarachchi, S., Chan, M. W.,et al. (2009). Identification of candidate epigenetic biomarkers for ovarian cancer detection. Oncol. Rep. 22, 853–861.
Ikeda, H., Ueda, M., Ikeda, M., Kobayashi, H., and Honda, Y. (2003). Oxysterol 7alpha-hydroxylase (CYP39A1) in the ciliary nonpigmented epithelium of bovine eye. Lab. Invest. 83, 349–355. doi: 10.1097/01.LAB.0000059933.35157.40
Iliopoulos, D., Drosatos, K., Hiyama, Y., Goldberg, I. J., and Zannis, V. I. (2010). MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J. Lipid Res. 51, 1513–1523. doi: 10.1194/jlr.M004812
Imai, S., Kikuchi, R., Kusuhara, H., Yagi, S., Shiota, K., and Sugiyama, Y. (2009). Analysis of DNA methylation and histone modification profiles of liver-specific transporters. Mol. Pharmacol. 75, 568–576. doi: 10.1124/mol.108.052589
Jeon, T. I., and Osborne, T. F. (2012). SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 23, 65–72. doi: 10.1016/j.tem.2011.10.004
Kim, S. J., Lee, B. H., Lee, Y. S., and Kang, K. S. (2007). Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 360, 593–599. doi: 10.1016/j.bbrc.2007.06.116
Kim, S. Y., Yoon, S., Kwon, S. M., Park, K. S., and Lee-Kim, Y. C. (2008). Kale juice improves coronary artery disease risk factors in hypercholesterolemic men. Biomed. Environ. Sci. 21, 91–97. doi: 10.1016/S0895-3988(08)60012-4
Kingwell, B. A., Chapman, M. J., Kontush, A., and Miller, N. E. (2014). HDL-targeted therapies: progress, failures and future. Nat. Rev. Drug Discov. 13, 445–464. doi: 10.1038/nrd4279
Knutson, S. K., Chyla, B. J., Amann, J. M., Bhaskara, S., Huppert, S. S., and Hiebert, S. W. (2008). Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 27, 1017–1028. doi: 10.1038/emboj.2008.51
Kumar, A., Kumar, S., Vikram, A., Hoffman, T. A., Naqvi, A., Lewarchik, C. M.,et al. (2013). Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler. Thromb. Vasc. Biol. 33, 1936–1942. doi: 10.1161/ATVBAHA.113.301765
Kutay, H., Klepper, C., Wang, B., Hsu, S. H., Datta, J., Yu, L.,et al. (2012). Reduced susceptibility of DNA methyltransferase 1 hypomorphic (Dnmt1N/+) mice to hepatic steatosis upon feeding liquid alcohol diet. PLoS ONE 7:e41949. doi: 10.1371/journal.pone.0041949
Leuenberger, N., Pradervand, S., and Wahli, W. (2009). Sumoylated PPARalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J. Clin. Invest. 119, 3138–3148. doi: 10.1172/JCI39019
Liang, Y., Yang, X., Ma, L., Cai, X., Wang, L., Yang, C.,et al. (2013). Homocysteine-mediated cholesterol efflux via ABCA1 and ACAT1 DNA methylation in THP-1 monocyte-derived foam cells. Acta Biochim. Biophys. Sin. (Shanghai), 45, 220–228. doi: 10.1093/abbs/gms119
Li-Hawkins, J., Lund, E. G., Bronson, A. D., and Russell, D. W. (2000). Expression cloning of an oxysterol 7alpha-hydroxylase selective for 24-hydroxycholesterol. J. Biol. Chem. 275, 16543–16549. doi: 10.1074/jbc.M001810200
Liu, S. Y., Aliyari, R., Chikere, K., Li, G., Marsden, M. D., Smith, J. K.,et al. (2013). Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. doi: 10.1016/j.immuni.2012.11.005
Li, Y., Cui, Y., Hart, S. N., Klaassen, C. D., and Zhong, X. B. (2009). Dynamic patterns of histone methylation are associated with ontogenic expression of the Cyp3a genes during mouse liver maturation. Mol. Pharmacol. 75, 1171–1179. doi: 10.1124/mol.108.052993
Luthi-Carter, R., Taylor, D. M., Pallos, J., Lambert, E., Amore, A., Parker, A.,et al. (2010). SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 7927–7932. doi: 10.1073/pnas.1002924107
Maceyka, M., Milstien, S., and Spiegel, S. (2013). The potential of histone deacetylase inhibitors in Niemann-Pick type C disease. FEBS J. 280, 6367–6372. doi: 10.1111/febs.12505
Malhotra, P., Soni, V., Anbazhagan, A. N., Kumar, A., Dudeja, A., Saksena, S.,et al. (2014). Epigenetic modulation of intestinal cholesterol transporter Niemann-Pick C1-Like 1 (NPC1L1) gene expression by DNA methylation. J. Biol. Chem. 289, 23132–23140. doi: 10.1074/jbc.M113.546283
Marquart, T. J., Allen, R. M., Ory, D. S., and Baldán, A. (2010). miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. U.S.A. 107, 12228–12232. doi: 10.1073/pnas.1005191107
McDonald, J. G., and Russell, D. W. (2010). Editorial: 25-Hydroxycholesterol: a new life in immunology. J. Leukoc. Biol. 88, 1071–1072. doi: 10.1189/jlb.0710418
Meaney, S. (2013). Epigenetic regulation of oxysterol formation. Biochimie 95, 531–537. doi: 10.1016/j.biochi.2012.08.020
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G.,et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. doi: 10.1038/nature06008
Milagre, I., Nunes, M. J., Castro-Caldas, M., Moutinho, M., Gama, M. J., and Rodrigues, E. (2012). Neuronal differentiation alters the ratio of Sp transcription factors recruited to the CYP46A1 promoter. J. Neurochem. 120, 220–229. doi: 10.1111/j.1471-4159.2011.07577.x
Milagre, I., Nunes, M. J., Gama, M. J., Silva, R. F., Pascussi, J. M., Lechner, M. C.,et al. (2008). Transcriptional regulation of the human CYP46A1 brain-specific expression by Sp transcription factors. J. Neurochem. 106, 835–849. doi: 10.1111/j.1471-4159.2008.05442.x
Milagre, I., Nunes, M. J., Moutinho, M., Rivera, I., Fuso, A., Scarpa, S.,et al. (2010). Chromatin-modifying agents increase transcription of CYP46A1, a key player in brain cholesterol elimination. J. Alzheimers Dis. 22, 1209–1221. doi: 10.3233/JAD-2010-100651
Munkacsi, A. B., Chen, F. W., Brinkman, M. A., Higaki, K., Gutiérrez, G. D., Chaudhari, J.,et al. (2011). An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 286, 23842–23851. doi: 10.1074/jbc.M111.227645
Murashima, M., Watanabe, S., Zhuo, X. G., Uehara, M., and Kurashige, A. (2004). Phase 1 study of multiple biomarkers for metabolism and oxidative stress after one-week intake of broccoli sprouts. Biofactors 22, 271–275. doi: 10.1002/biof.5520220154
Nunes, M. J., Moutinho, M., Milagre, I., Gama, M. J., and Rodrigues, E. (2012). Okadaic acid inhibits the trichostatin A-mediated increase of human CYP46A1 neuronal expression in a ERK1/2-Sp3-dependent pathway. J. Lipid Res. 53, 1910–1919. doi: 10.1194/jlr.M027680
Nuutinen, T., Suuronen, T., Kyrylenko, S., Huuskonen, J., and Salminen, A. (2005). Induction of clusterin/apoJ expression by histone deacetylase inhibitors in neural cells. Neurochem. Int. 47, 528–538. doi: 10.1016/j.neuint.2005.07.007
Ohyama, Y., Meaney, S., Heverin, M., Ekström, L., Brafman, A., Shafir, M.,et al. (2006). Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J. Biol. Chem. 281, 3810–3820. doi: 10.1074/jbc.M505179200
Olsson, M., Gustafsson, O., Skogastierna, C., Tolf, A., Rietz, B. D., Morfin, R.,et al. (2007). Regulation and expression of human CYP7B1 in prostate: overexpression of CYP7B1 during progression of prostatic adenocarcinoma. Prostate 67, 1439–1446. doi: 10.1002/pros.20630
Pipalia, N. H., Cosner, C. C., Huang, A., Chatterjee, A., Bourbon, P., Farley, N.,et al. (2011). Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 108, 5620–5625. doi: 10.1073/pnas.1014890108
Poirier, S., Samami, S., Mamarbachi, M., Demers, A., Chang, T. Y., Vance, D. E.,et al. (2014). The epigenetic drug 5-azacytidine interferes with cholesterol and lipid metabolism. J. Biol. Chem. 289, 18736–18751. doi: 10.1074/jbc.M114.563650
Porter, F. D. (2003). Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr. Opin. Pediatr. 15, 607–613. doi: 10.1097/00008480-200312000-00011
Ramírez, C. M., Rotllan, N., Vlassov, A. V., Dávalos, A., Li, M., Goedeke, L.,et al. (2013). Control of cholesterol metabolism and plasma high-density lipoprotein levels by microRNA-144. Circ. Res. 112, 1592–1601. doi: 10.1161/CIRCRESAHA.112.300626
Rawson, R. B., DeBose-Boyd, R., Goldstein, J. L., and Brown, M. S. (1999). Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J. Biol. Chem. 274, 28549–28556. doi: 10.1074/jbc.274.40.28549
Rayner, K. J., Esau, C. C., Hussain, F. N., McDaniel, A. L., Marshall, S. M., van Gils, J. M.,et al. (2011a). Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478, 404–407. doi: 10.1038/nature10486
Rayner, K. J., Sheedy, F. J., Esau, C. C., Hussain, F. N., Temel, R. E., Parathath, S.,et al. (2011b). Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Invest. 121, 2921–2931. doi: 10.1172/JCI57275
Rottiers, V., Obad, S., Petri, A., McGarrah, R., Lindholm, M. W., Black, J. C.,et al. (2013). Pharmacological inhibition of a microRNA family in nonhuman primates by a seed-targeting 8-mer antimiR. Sci. Transl. Med. 5, 212ra162. doi: 10.1126/scitranslmed.3006840
Salerno, A., Rayner, K., Wanschel, A., Oldebeken, S., Scotti, E., Tontonoz, P.,et al. (2013). Post-transcriptional control of the LDLR pathway by cholesterol-responsive microRNAs. Athersc. Thromb. Vasc. Biol. 33, A63.
Shafaati, M., O’Driscoll, R., Björkhem, I., and Meaney, S. (2009). Transcriptional regulation of cholesterol 24-hydroxylase by histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 378, 689–694. doi: 10.1016/j.bbrc.2008.11.103
Sharpe, L. J., and Brown, A. J. (2013). Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J. Biol. Chem. 288, 18707–18715. doi: 10.1074/jbc.R113.479808
Smith, Z., Ryerson, D., and Kemper, J. K. (2013). Epigenomic regulation of bile acid metabolism: emerging role of transcriptional cofactors. Mol. Cell Endocrinol. 368, 59–70. doi: 10.1016/j.mce.2012.04.008
Song, K. H., Li, T., Owsley, E., and Chiang, J. Y. L. (2010). A putative role of micro RNA in regulation of cholesterol 7alpha-hydroxylase expression in human hepatocytes. J. Lipid Res. 51, 2223–2233. doi: 10.1194/jlr.M004531
Steck, T. L., and Lange, Y. (2010). Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol. 20, 680–687. doi: 10.1016/j.tcb.2010.08.007
Steckelbroeck, S., Watzka, M., Lütjohann, D., Makiola, P., Nassen, A., Hans, V. H.,et al. (2002). Characterization of the dehydroepiandrosterone (DHEA) metabolism via oxysterol 7alpha-hydroxylase and 17-ketosteroid reductase activity in the human brain. J. Neurochem. 83, 713–726. doi: 10.1046/j.1471-4159.2002.01187.x
Stiles, A. R., McDonald, J. G., Bauman, D. R., and Russell, D. W. (2009). CYP7B1: one cytochrome P450, two human genetic diseases, and multiple physiological functions. J. Biol. Chem. 284, 28485–28489. doi: 10.1074/jbc.R109.042168
Sundqvist, A., Bengoechea-Alonso, M. T., Ye, X., Lukiyanchuk, V., Jin, J., Harper, J. W.,et al. (2005). Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 1, 379–391. doi: 10.1016/j.cmet.2005.04.010
Suuronen, T., Nuutinen, T., Ryhänen, T., Kaarniranta, K., and Salminen, A. (2007). Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 357, 397–401. doi: 10.1016/j.bbrc.2007.03.135
Tao, R., Xiong, X., DePinho, R. A., Deng, C. X., and Dong, X. C. (2013). FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 288, 29252–29259. doi: 10.1074/jbc.M113.481473
Tessema, M., and Belinsky, S. A. (2008). Mining the epigenome for methylated genes in lung cancer. Proc. Am. Thorac. Soc. 5, 806–810. doi: 10.1513/pats.200805-045TH
van der Wulp, M. Y., Verkade, H. J., and Groen, A. K. (2013). Regulation of cholesterol homeostasis. Mol. Cell Endocrinol. 368, 1–16. doi: 10.1016/j.mce.2012.06.007
Vance, D. E., and Vance, J. E. (2002). Biochemistry of Lipids, Lipoproteins, and Membranes, New Comprehensive Biochemistry. 4th ed. Amsterdam: Elsevier.
Vance, D., and Vance, J. E. (2008). Biochemistry of Lipids, Lipoproteins and Membranes. 5th ed. Amsterdam: Elsevier.
Villagra, A., Ulloa, N., Zhang, X., Yuan, Z., Sotomayor, E., and Seto, E. (2007). Histone deacetylase 3 down-regulates cholesterol synthesis through repression of lanosterol synthase gene expression. J. Biol. Chem. 282, 35457–35470. doi: 10.1074/jbc.M701719200
Walker, A. K., Yang, F., Jiang, K., Ji, J. Y., Watts, J. L., Purushotham, A.,et al. (2010). Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 24, 1403–1417. doi: 10.1101/gad.1901210
Wang, D. Q. (2007). Regulation of intestinal cholesterol absorption. Annu. Rev. Physiol. 69, 221–248. doi: 10.1146/annurev.physiol.69.031905.160725
Xie, Y., Ke, S., Ouyang, N., He, J., Xie, W., Bedford, M. T.,et al. (2009). Epigenetic regulation of transcriptional activity of pregnane X receptor by protein arginine methyltransferase 1. J. Biol. Chem. 284, 9199–9205. doi: 10.1074/jbc.M806193200
Yang, M., Liu, W., Pellicane, C., Sahyoun, C., Joseph, B. K., Gallo-Ebert, C.,et al. (2014). Identification of miR-185 as a regulator of de novo cholesterol biosynthesis and low density lipoprotein uptake. J. Lipid Res. 55, 226–238. doi: 10.1194/jlr.M041335
Ye, J., and DeBose-Boyd, R. A. (2011). Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 3, a004754. doi: 10.1101/cshperspect.a004754
Yi-Deng, J., Tao, S., Hui-Ping, Z., Jian-Tuan, X., Jun, C., Gui-Zhong, L.,et al. (2007). Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 26, 737–744. doi: 10.1089/dna.2007.0619
Yu, C. E., Cudaback, E., Foraker, J., Thomson, Z., Leong, L., Lutz, F.,et al. (2013). Epigenetic signature and enhancer activity of the human APOE gene. Hum. Mol. Genet. 22, 5036–5047. doi: 10.1093/hmg/ddt354
Keywords: cholesterol, oxysterol, HMGCR, PCSK9, LDLR, APOE, APOJ, CYP46A1
Citation: Meaney S (2014) Epigenetic regulation of cholesterol homeostasis. Front. Genet. 5:311. doi: 10.3389/fgene.2014.00311
Received: 25 July 2014; Accepted: 20 August 2014;
Published online: 24 September 2014.
Edited by:
Steven G. Gray, Trinity College Dublin, IrelandReviewed by:
Steven G. Gray, Trinity College Dublin, IrelandAnne-Marie Baird, Queensland University of Technology, Australia
Copyright © 2014 Meaney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steve Meaney, School of Biological Sciences, College of Sciences and Health, and Environmental Sustainability and Health Institute, Dublin Institute of Technology, Kevin Street, Dublin 8, Ireland e-mail:c3RldmUubWVhbmV5QGRpdC5pZQ==