 Himjyot Jaiswal
Himjyot Jaiswal- Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden
The DNA damage response (DDR) has two main goals, to repair the damaged DNA and to communicate the presence of damaged DNA. This communication allows the adaptation of cellular behavior to minimize the risk associated with DNA damage. In particular, cell cycle progression must be adapted after a DNA-damaging insult, and cells either pause or terminally exit the cell cycle during a DDR. As cells can accumulate mutations after a DDR due to error-prone DNA repair, terminal cell cycle exit may prevent malignant transformation. The tumor suppressor p53 plays a key role in promoting terminal cell cycle exit. Interestingly, p53 has been implicated in communication of a stress response to surrounding cells, known as the bystander response. Recently, surrounding cells have also been shown to affect the damaged cell, suggesting the presence of intercellular feedback loops. How such feedback may affect terminal cell cycle exit remains unclear, but its presence calls for caution in evaluating cellular outcome without controlling the cellular surrounding. In addition, such feedback may contribute to how the cellular environment affects malignant transformation after DNA damage.
Introduction
Changes in the genome can be a potential threat to the cell and to organism survival. However, the genome is continuously exposed to a variety of genotoxic stresses. These are endogenous insults such as the production of reactive oxygen species (ROS) or various metabolite byproducts, or exogenous insults such as UV radiation, heavy metals, air pollutants, bacterial toxins, and inflammatory responses. All of these agents cause structural damage and can hinder or abolish cellular processes as transcription or DNA replication. Of the various DNA lesions, DNA double strand breaks (DSBs) are considered most deleterious, because if unrepaired they can lead to chromosomal aberrations such as deletions, translocations, and amplifications. These chromosomal aberrations may result in deregulation of gene expression and altered cellular function, which may eventually cause cell death or tumor initiation and progression (Lord and Ashworth, 2012).
To minimize the risk to genome integrity, cells have evolved the DNA damage response (DDR)—a highly regulated signaling network that responds to the presence of DNA lesions (Bartek and Lukas, 2007; Jackson and Bartek, 2009). A prime function of the DDR is to ensure that lesions in DNA are recognized and repaired. Simultaneously, the repair needs to be coordinated with other cellular processes, in particular cell cycle progression. Therefore, the DDR can be divided into two major pathways, one that assembles and repairs the lesions and one that amplifies and conveys the signal away from the break site to modify cellular behavior. In all eukaryotes these two processes are initiated by sensor proteins such as the Mre11-Rad50-Nbs1 (MRN) complex or the Ku70/Ku80 dimer, that detect the presence of DSBs. The binding of sensor proteins to damaged DNA recruits the phosphoinositide 3-kinase related kinases ATM, ATR, or DNA-PK leading to activation of these kinases (Falck et al., 2005). Once activated these kinases initiate cascades that enforce local and global rearrangement of chromatin, involving recruitment of multiple proteins and posttranslational modifications as phosphorylation, ubiquitylation, sumoylation, and methylation (Lukas et al., 2011). For example, phosphorylation of histone 2A variant (H2AX) at C-termini near a break site by ATM serves as a platform for the protein MDC1, who in turn can function as a recruitment platform for the ubiquitin ligase RNF8. RNF8-mediated ubiquitylation recruits RNF168, whose ubiquitylation of chromatin proteins attract BRCA1 and 53BP1, proteins that affect how the DNA break will be repaired (Kolas et al., 2007; Bekker-Jensen and Mailand, 2011).
DSB Repair is not Always Perfect
A majority of DSBs are repaired by three pathways—homologous recombination (HR), non-homologous end joining (NHEJ), and microhomology-mediated end joining (MMEJ). NHEJ is a fast repair process using template independent ligation of two ends of DNA and is functional throughout the cell cycle; in contrast HR is a slow repair process that depends upon the use of a sister chromatid as template and is functional only in late S- and G2 phase of the cell cycle (Chapman et al., 2012). Whereas NHEJ only requires minor modifications of the DNA to allow for ligation, HR requires resection to create stretches of single-stranded DNA that can be used for base-pairing with the sister chromatid (Hartlerode and Scully, 2009). The amount of resection at a DSB is influenced by proteins as BRCA1 and 53BP1, and is considered as a determinant for which repair mechanism will be utilized (Chapman et al., 2012). Importantly, during NHEJ the DNA ends are frequently modified to allow efficient ligation, resulting in a change in the genetic information (Lieber, 2010). Similarly, MMEJ, an alternative version of end-joining contains even larger modifications of DNA ends, giving rise to deletions (Decottignies, 2013). In contrast, due to the use of a template for sequence information, HR is largely considered as an error-free repair process, although its accuracy is debated. Two different processes are described to support that HR may be an error-prone process—first the unequal sister chromatid exchange (SCE) which has been observed in highly repetitive sequences, and second the involvement of translesion synthesis polymerases in synthesizing the DNA (Guirouilh-Barbat et al., 2014). Thus, cells that have repaired DSBs are likely to contain changes in the genetic information.
Checkpoint Maintenance is not Always Perfect
The repair of damaged DNA needs to be coordinated with various other cellular processes, in particular cell cycle progression. Therefore, in addition to stimulating repair, the DDR enforces a cell-cycle arrest, referred to as a DNA damage checkpoint. At the heart of the checkpoint are the ATM and ATR kinases, which initiate a signaling cascade by phosphorylating the effector kinases Chk2 and Chk1. Chk1/Chk2 in return phosphorylate cell cycle regulators as Cdc25 phosphatases. Phosphorylation of Cdc25s leads to their functional inactivation and subsequent inhibition of Cdk activity, causing rapid inhibition of cell cycle progression (Peng et al., 1997; Mailand et al., 2000; Karlsson-Rosenthal and Millar, 2006). In addition, inhibition of indirect regulators of Cdk activity as Plk1 and Aurora A support a rapid cell cycle arrest (Smits et al., 2000; Krystyniak et al., 2006). Checkpoint signaling also maintains the arrest by stabilizing p53 that transcriptionally regulates a large number of genes involved in DDR and other stress pathways (Allen et al., 2014). In addition, p38-dependent pathways contribute to regulate protein expression to maintain a checkpoint over time (Reinhardt et al., 2010). However, although the DDR is a very tightly regulated process, evidence of H2AX phosphorylation, chromosomal rearrangements and breakage during the transition from G2 to mitosis suggest that checkpoint signaling is not always stringent (Syljuasen et al., 2006; Deckbar et al., 2007; Lobrich and Jeggo, 2007). Thus, cells that have initiated a DDR and a checkpoint arrest may resume proliferation before all damaged DNA is repaired.
Cell Cycle Exit
As an alternative to a temporal cell cycle arrest, cells may permanently leave the cell cycle and become senescent. The duration from infliction of DNA damage to cell cycle exit depends directly on the cell cycle state. Whereas an untransformed G2 cell exits the cell cycle if damage is not repaired within a couple of hours, an S-phase cell first finishes DNA replication and only leaves the cell cycle in G2 (Baus et al., 2003; Krenning et al., 2014; Müllers et al., 2014). Cell cycle exit in G2 phase depends on activation of the ubiquitin ligase APC/C-Cdh1, which efficiently targets a large amount of cell cycle regulators for proteasome-mediated degradation (Wiebusch and Hagemeier, 2010). How APC/C-Cdh1 is activated after DNA damage remains unclear, but the process depends on expression of p53 and its transcriptional target p21, and at least in the case of Cyclin B1, nuclear translocation of the protein to be degraded (Wiebusch and Hagemeier, 2010; Johmura et al., 2014; Krenning et al., 2014; Müllers et al., 2014). Thus, the regulation of p53 is a key determinant for whether cell cycle exit or resumed proliferation occurs after initiation of a DDR.
p53 and Cell Fate
The level and activity of p53 is upregulated in response to various stresses and has been shown to play a role in different pathways including DDR, hypoxia, apoptosis, metabolism and senescence (Gonfloni et al., 2014; Pflaum et al., 2014). Functioning as a complex signaling node, the p53 protein contains a large amount of post-translational modifications, which together with differential affinity for transcriptional elements and expression of regulatory proteins impact on cell fate decisions (Kruse and Gu, 2009; Carvajal and Manfredi, 2013). Interestingly, although p53 levels are similarly induced, different stimuli can elicit different responses on p53-transcription targets such as p21 (Espinosa et al., 2003; Donner et al., 2007), highlighting that p53 function may be modulated by the integration of a wide variety of signaling pathways (Sullivan et al., 2012). One factor that can affect p53 function is its temporal dynamics in cells. Rather than accumulating at a certain level, cellular p53 can oscillate after induction of DSBs (Lahav et al., 2004). In contrast to sustained p53 induction that stimulates cell cycle exit, the oscillatory pulses of p53 favor eventual resumption of proliferation after damage (Purvis et al., 2012). However, exactly how integration of signals determines p53 behavior remains unclear, in particular in the context of a population of cells.
The Bystander Response
During the past few decades the DDR pathway has been studied extensively in cells that have experienced damage directly. However, cells experiencing a DDR can communicate this to surrounding cells (Klammer et al., 2013). The first evidence of propagation of the DDR came from experiments performed in Chinese hamster ovary cell lines, in which 1% of nuclei hit by α-particles resulted in more than 30% of the cell population showing increased incidence of SCE (Nagasawa and Little, 1992). Supported by other observations, this phenomenon was later termed the radiation-induced bystander effect (RIBE), which is defined as physiological changes in unirradiated cells manifested by cells exposed to radiation (Sokolov et al., 2005, 2007; Klammer et al., 2013). Apart from SCE, various biological consequences of RIBE have been observed in different studies such as genomic instability, micronuclei formation, apoptosis, micro RNA (miRNA) regulation, and differentiation (Lorimore et al., 1998; Belyakov et al., 2002; Kovalchuk et al., 2010; Vinnikov et al., 2012). A common feature of RIBE seems to be induction of DNA damage. Indeed, Ku70, Ku80, or DNA-PKcs knockout bystander cells that are repair deficient are sensitive to the induction of mutations and chromosomal aberrations (Little et al., 2003; Nagasawa et al., 2003). However, the number of DSBs generated in directly irradiated and bystander cells differ, and point mutations are predominant in bystander cells as compared to partial or total gene deletion in directly irradiated cells (Little et al., 1997; Huo et al., 2001; Sedelnikova et al., 2007). Mechanistically, deregulation of redox homeostasis may be a major cause of DNA damage in bystander cells (Azzam et al., 2002; Sokolov et al., 2007). Indeed, addition of Vitamin C or E to cell culture reduces the frequency of micronuclei formation, suggesting that ROS contributes to DNA damage formation (Narayanan et al., 1997; Konopacka and Rzeszowska-Wolny, 2006). The occurrence of DSBs in bystander cells is more frequent during DNA replication or active transcription, indicating that energy-dependent processes may underlie some of the damage (Burdak-Rothkamm et al., 2007; Dickey et al., 2012). In addition, these processes involve opening up double-stranded DNA, suggesting a mechanism for how ROS-induced single-stranded breaks can be transformed to DSBs, and indicating that the bystander effect may be particularly efficient during late cell cycle stages where replication and transcription is high.
The bystander effects appear to be cell and genotype specific and also depend upon the type of radiation (Baskar, 2010). Most of the RIBE studies have been performed in cell and tissue culture models where non-irradiated cells were co-cultured with either irradiated cells or with the conditioned medium from irradiated cells. Using mice models, Koturbash et al. (2008) showed that the bystander effect occurs in vivo as cranial irradiation led to DNA damage in protected spleen tissues. The RIBE also led to a profound epigenetic change in different bystander parts of the animal and, interestingly, the bystander response could differ between male and female (Besplug et al., 2005; Koturbash et al., 2006, 2007).
The above observations suggest that paracrine or endocrine signaling molecules from irradiated cells are responsible for the bystander effect. However, in addition to secretion of extracellular factors, transmission through gap junctions has also been implicated in RIBE, suggesting that multiple factors may propagate a bystander effect (Azzam et al., 2001; Hubackova et al., 2012; Klammer et al., 2013; He et al., 2014). Some of the factors implicated in transmitting the bystander response are interleukins, transforming growth factor beta (TGFβ), and nitric oxide (NO) (Iyer et al., 2000; Shao et al., 2002; Dieriks et al., 2010). As a consequence of RIBE, a DNA damage-response pathway is initiated in bystander cells. Apart from the p53 pathway, the DDR also initiates stress signaling through JNK and p38 MAPK signaling cascades including NF-kB, a major regulator of cell survival, inflammation, autophagy, and differentiation (Azzam et al., 1998; Piret et al., 1999). Activation of such a signaling network reprograms a cell to react to external danger and may coordinate a response in a complex tissue environment.
Reciprocal Bystander Effect
Proper tissue homeostasis is dependent on bidirectional rather than unidirectional communication between cells. It is therefore reasonable to expect that an exchange of signaling molecules between non-irradiated and irradiated cells occurs (Goldberg and Lehnert, 2002; Chen et al., 2011; Widel et al., 2012; He et al., 2014). Indeed, the first observation of bidirectional communication between cells was seen by Mackonis et al. (2007), who reported an increased rate of survival of cells receiving a high radiation dose when their nearby cells received a low radiation dose. This interesting observation was termed a type III effect. Later on Chen et al. (2011) showed that there is a decrease in micronuclei formation and apoptosis in irradiated cells when co-cultured with non-irradiated cells. However, although the mechanisms of a reciprocal bystander effect are not yet clear, recently He et al. (2014) used co-culture of irradiated macrophages and non-irradiated hepatocytes to postulate that cAMP released from bystander hepatocytes could lead to a decreased micronuclei formation in irradiated macrophages. These studies suggest that reciprocal communication is important to react to external damage in an efficient and flexible manner. Interestingly, incorporation of both bystander and reciprocal bystander responses suggests the presence of intercellular feedback loops that may augment responses in both damaged and non-damaged cells.
p53 in the Bystander Response
One of the promising candidates that can function as a connecting link between intrinsic and extrinsic signals is the p53 protein. Apart from cell autonomous responses, such as activation in response to DSBs in bystander cells, p53 also plays a role in transmission of the bystander response (Lorimore et al., 2013). In particular, cytochrome C release from damaged cells has been shown to be involved in RIBE in a p53-dependent manner, suggesting that p53 can both transmit and respond to RIBE (He et al., 2011). The oscillatory behavior of p53 over time has attracted the attention of modeling efforts to predict the potential outcome on cell fate (Lev Bar-Or et al., 2000; Geva-Zatorsky et al., 2006; Wee et al., 2009). A recent study based on mathematical modeling proposed that cytochrome C could couple the p53 oscillatory behavior in damaged and non-damaged cells to enhance the robustness and sustainability of p53 pulses (Kim and Jackson, 2013). Although this model needs further validation in an experimental setup, a reciprocal bystander effect imposed by cytochrome C on p53 pulses may impact on cell fate decisions, as p53 oscillations favor resumed proliferation rather than cell cycle exit (Purvis et al., 2012).
Conclusion
As both DNA repair and cell cycle checkpoint maintenance is not perfect, the occurrence of DNA damage to a cell constitutes a risk for establishment and propagation of genomic changes. By forcing a cell to permanently withdraw from the cell cycle, the risk associated with such changes can be reduced. Indeed, a permanent cell cycle exit is suggested to function as a tumor barrier after oncogene-induced DNA damage in S phase (Bartkova et al., 2006; Di Micco et al., 2006), a phase that may be particularly susceptible for RIBE (Burdak-Rothkamm et al., 2007). However, the determinants for when cells exit the cell cycle are not clear. Interestingly, p53, the key regulator of cell cycle exit may both modulate and respond to bystander communication. This opens up for the possibility that feedback within a population impacts on whether cell cycle exit occurs (Figure 1).
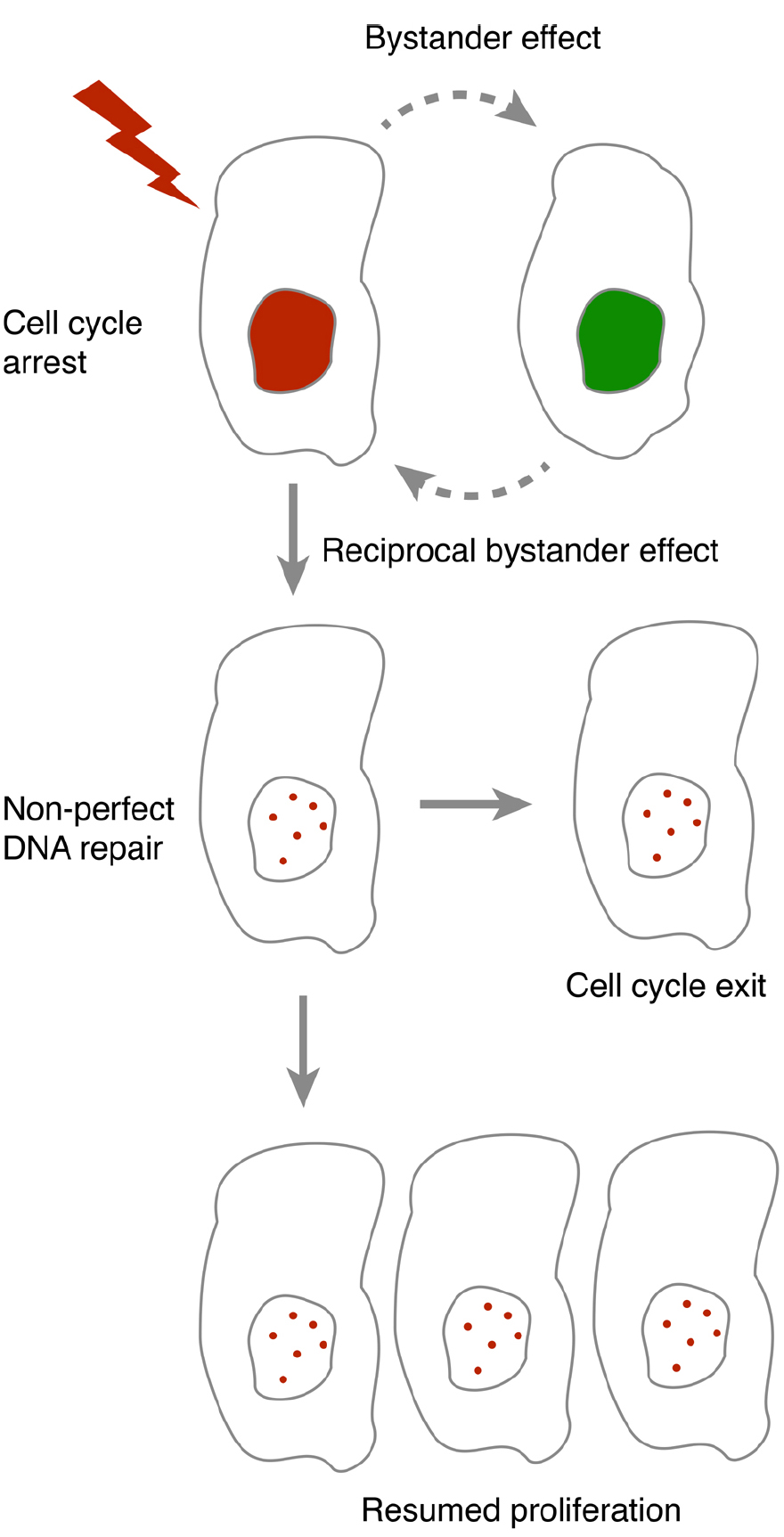
Figure 1. In the presence of DNA damage, cells either pause or terminally exit the cell cycle. As DNA repair and cell cycle checkpoint maintenance are not always accurate, resumption of proliferation after a cell cycle pause may lead to propagation of mutations. Bystander communication forms an intercellular feedback that may contribute to whether resumption of proliferation may occur.
The original definition of a checkpoint is a mechanism that is checking to see that the prerequisites (for a process as cell cycle progression) have been properly satisfied (Hartwell and Weinert, 1989). A growing body of evidence suggests that upon damage a cell changes its microenvironment and spreads a signal to neighboring cells to communicate that damage is inflicted. Whether the spread of a signal from a damaged cell is a call for help or a warning is still not clear. The spread is likely to contribute to an effective population response and to assist to eliminate severely damaged cells. However, how cell intrinsic and cell extrinsic pathways interact to determine the fate of a damaged cell remains unclear. Nonetheless, the existence of cell–cell communication affecting DDR pathways calls for caution in evaluating experiments without controlling the local environment, as factors as cell confluence may impact on experimental outcome.
Outlook
The bystander response, as a cause of genome instability, is implicated in induction of mutations leading to secondary cancers (Coates et al., 2008; Lorimore et al., 2008; Mancuso et al., 2008). In contrast to partial or total gene deletion in directly irradiated cells, bystander cells show primarily point mutations (Huo et al., 2001). Thus, surrounding cells may receive a more subtle genomic change that may promote survival. Early tumor development is accompanied by DNA damage also in the absence of treatment, where activation of a DDR can precede p53 mutations and defects in DNA damage signaling (Bartkova et al., 2005; Gorgoulis et al., 2005). Whether a bystander effect may contribute to increase malignant transformation during tumorigenesis remains to be studied. However, it is possible that a group of early tumor cells may not only collectively enhance the amount of DNA damage per cell, but may also impact on whether proliferation will be resumed. Due to the non-perfect DNA repair and checkpoint maintenance, such resumed proliferation may increase the risk for malignant transformation.
p53 and its associated pathways are altered in more than half of all human cancers, likely reflecting the importance of p53 for cellular fate. It is tempting to speculate that alteration in the p53 pathway can give flexibility to a cell to respond to different extrinsic signals and to better adapt to the environment. Understanding how the bystander effect couples to cell fate decision may impact on risk assessment and indicate novel targets to increase the efficiency of chemo- and radiation therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen, M. A., Andrysik, Z., Dengler, V. L., Mellert, H. S., Guarnieri, A., Freeman, J. A., et al. (2014). Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 3, e02200. doi: 10.7554/eLife.02200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azzam, E. I., De Toledo, S. M., Gooding, T., and Little, J. B. (1998). Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat. Res. 150, 497–504. doi: 10.2307/3579865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azzam, E. I., De Toledo, S. M., and Little, J. B. (2001). Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from α-particle irradiated to nonirradiated cells. Proc. Natl. Acad. Sci. U.S.A. 98, 473–478. doi: 10.1073/pnas.011417098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azzam, E. I., De Toledo, S. M., Spitz, D. R., and Little, J. B. (2002). Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from α-particle-irradiated normal human fibroblast cultures. Cancer Res. 62, 5436–5442.
Bartek, J., and Lukas, J. (2007). DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 19, 238–245. doi: 10.1016/j.ceb.2007.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. doi: 10.1038/nature03482
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. doi: 10.1038/nature05268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baskar, R. (2010). Emerging role of radiation induced bystander effects: cell communications and carcinogenesis. Genome Integr. 1, 13. doi: 10.1186/2041-9414-1-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baus, F., Gire, V., Fisher, D., Piette, J., and Dulic, V. (2003). Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 22, 3992–4002. doi: 10.1093/emboj/cdg387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bekker-Jensen, S., and Mailand, N. (2011). The ubiquitin- and SUMO-dependent signaling response to DNA double-strand breaks. FEBS Lett. 585, 2914–2919. doi: 10.1016/j.febslet.2011.05.056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belyakov, O. V., Folkard, M., Mothersill, C., Prise, K. M., and Michael, B. D. (2002). Bystander-induced apoptosis and premature differentiation in primary urothelial explants after charged particle microbeam irradiation. Radiat. Prot. Dosimetry 99, 249–251. doi: 10.1093/oxfordjournals.rpd.a006775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Besplug, J., Burke, P., Ponton, A., Filkowski, J., Titov, V., Kovalchuk, I., et al. (2005). Sex and tissue-specific differences in low-dose radiation-induced oncogenic signaling. Int. J. Radiat. Biol. 81, 157–168. doi: 10.1080/09553000500103512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burdak-Rothkamm, S., Short, S. C., Folkard, M., Rothkamm, K., and Prise, K. M. (2007). ATR-dependent radiation-induced gamma H2AX foci in bystander primary human astrocytes and glioma cells. Oncogene 26, 993–1002. doi: 10.1038/sj.onc.1209863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carvajal, L. A., and Manfredi, J. J. (2013). Another fork in the road—life or death decisions by the tumour suppressor p53. EMBO Rep. 14, 414–421. doi: 10.1038/embor.2013.25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chapman, J. R., Taylor, M. R., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510. doi: 10.1016/j.molcel.2012.07.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S., Zhao, Y., Han, W., Chiu, S. K., Zhu, L., Wu, L., et al. (2011). Rescue effects in radiobiology: unirradiated bystander cells assist irradiated cells through intercellular signal feedback. Mutat. Res. 706, 59–64. doi: 10.1016/j.mrfmmm.2010.10.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coates, P. J., Rundle, J. K., Lorimore, S. A., and Wright, E. G. (2008). Indirect macrophage responses to ionizing radiation: implications for genotype-dependent bystander signaling. Cancer Res. 68, 450–456. doi: 10.1158/0008-5472.CAN-07-3050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deckbar, D., Birraux, J., Krempler, A., Tchouandong, L., Beucher, A., Walker, S., et al. (2007). Chromosome breakage after G2 checkpoint release. J. Cell Biol. 176, 749–755. doi: 10.1083/jcb.200612047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Decottignies, A. (2013). Alternative end-joining mechanisms: a historical perspective. Front. Genet. 4:48. doi: 10.3389/fgene.2013.00048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dickey, J. S., Baird, B. J., Redon, C. E., Avdoshina, V., Palchik, G., Wu, J., et al. (2012). Susceptibility to bystander DNA damage is influenced by replication and transcriptional activity. Nucleic Acids Res. 40, 10274–10286. doi: 10.1093/nar/gks795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dieriks, B., De Vos, W. H., Derradji, H., Baatout, S., and Van Oostveldt, P. (2010). Medium-mediated DNA repair response after ionizing radiation is correlated with the increase of specific cytokines in human fibroblasts. Mutat. Res. 687, 40–48. doi: 10.1016/j.mrfmmm.2010.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642. doi: 10.1038/nature05327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Donner, A. J., Szostek, S., Hoover, J. M., and Espinosa, J. M. (2007). CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 27, 121–133. doi: 10.1016/j.molcel.2007.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Espinosa, J. M., Verdun, R. E., and Emerson, B. M. (2003). p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol. Cell 12, 1015–1027. doi: 10.1016/S1097-2765(03)00359-9
Falck, J., Coates, J., and Jackson, S. P. (2005). Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611. doi: 10.1038/nature03442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geva-Zatorsky, N., Rosenfeld, N., Itzkovitz, S., Milo, R., Sigal, A., Dekel, E., et al. (2006). Oscillations and variability in the p53 system. Mol. Syst. Biol. 2, 2006.0033. doi: 10.1038/msb4100068
Goldberg, Z., and Lehnert, B. E. (2002). Radiation-induced effects in unirradiated cells: a review and implications in cancer. Int. J. Oncol. 21, 337–349. doi: 10.3892/ijo.21.2.337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonfloni, S., Iannizzotto, V., Maiani, E., Bellusci, G., Ciccone, S., and Diederich, M. (2014). P53 and Sirt1: routes of metabolism and genome stability. Biochem. Pharmacol. 92, 149–156. doi: 10.1016/j.bcp.2014.08.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913. doi: 10.1038/nature03485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guirouilh-Barbat, J., Lambert, S., Bertrand, P., and Lopez, B. S. (2014). Is homologous recombination really an error-free process? Front. Genet. 5:175. doi: 10.3389/fgene.2014.00175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hartlerode, A. J., and Scully, R. (2009). Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 423, 157–168. doi: 10.1042/BJ20090942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hartwell, L. H., and Weinert, T. A. (1989). Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629–634. doi: 10.1126/science.2683079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, M., Dong, C., Xie, Y., Li, J., Yuan, D., Bai, Y., et al. (2014). Reciprocal bystander effect between α-irradiated macrophage and hepatocyte is mediated by cAMP through a membrane signaling pathway. Mutat. Res. Fundam. Mol. Mech. Mutagen. 763–764, 1–9. doi: 10.1016/j.mrfmmm.2014.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, M., Zhao, M., Shen, B., Prise, K. M., and Shao, C. (2011). Radiation-induced intercellular signaling mediated by cytochrome-c via a p53-dependent pathway in hepatoma cells. Oncogene 30, 1947–1955. doi: 10.1038/onc.2010.567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hubackova, S., Krejcikova, K., Bartek, J., and Hodny, Z. (2012). IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence.’ Aging (Albany NY) 4, 932–951.
Huo, L., Nagasawa, H., and Little, J. B. (2001). HPRT mutants induced in bystander cells by very low fluences of α particles result primarily from point mutations. Radiat. Res. 156, 521–525. doi: 10.1667/0033-7587(2001)156[0521:HMIIBC]2.0.CO;2
Iyer, R., Lehnert, B. E., and Svensson, R. (2000). Factors underlying the cell growth-related bystander responses to α particles. Cancer Res. 60, 1290–1298.
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johmura, Y., Shimada, M., Misaki, T., Naiki-Ito, A., Miyoshi, H., Motoyama, N., et al. (2014). Necessary and sufficient role for a mitosis skip in senescence induction. Mol. Cell 55, 73–84. doi: 10.1016/j.molcel.2014.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karlsson-Rosenthal, C., and Millar, J. B. (2006). Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 16, 285–292. doi: 10.1016/j.tcb.2006.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J. K., and Jackson, T. L. (2013). Mechanisms that enhance sustainability of p53 pulses. PLoS ONE 8:e65242. doi: 10.1371/journal.pone.0065242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klammer, H., Mladenov, E., Li, F., and Iliakis, G. (2013). Bystander effects as manifestation of intercellular communication of DNA damage and of the cellular oxidative status. Cancer Lett. doi: 10.1016/j.canlet.2013.12.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640. doi: 10.1126/science.1150034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konopacka, M., and Rzeszowska-Wolny, J. (2006). The bystander effect-induced formation of micronucleated cells is inhibited by antioxidants, but the parallel induction of apoptosis and loss of viability are not affected. Mutat. Res. 593, 32–38. doi: 10.1016/j.mrfmmm.2005.06.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koturbash, I., Boyko, A., Rodriguez-Juarez, R., Mcdonald, R. J., Tryndyak, V. P., Kovalchuk, I., et al. (2007). Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis 28, 1831–1838. doi: 10.1093/carcin/bgm053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koturbash, I., Loree, J., Kutanzi, K., Koganow, C., Pogribny, I., and Kovalchuk, O. (2008). In vivo bystander effect: cranial X-irradiation leads to elevated DNA damage, altered cellular proliferation and apoptosis, and increased p53 levels in shielded spleen. Int. J. Radiat. Oncol. Biol. Phys. 70, 554–562. doi: 10.1016/j.ijrobp.2007.09.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koturbash, I., Rugo, R. E., Hendricks, C. A., Loree, J., Thibault, B., Kutanzi, K., et al. (2006). Irradiation induces DNA damage and modulates epigenetic effectors in distant bystander tissue in vivo. Oncogene 25, 4267–4275. doi: 10.1038/sj.onc.1209467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kovalchuk, O., Zemp, F. J., Filkowski, J. N., Altamirano, A. M., Dickey, J. S., Jenkins-Baker, G., et al. (2010). microRNAome changes in bystander three-dimensional human tissue models suggest priming of apoptotic pathways. Carcinogenesis 31, 1882–1888. doi: 10.1093/carcin/bgq119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krenning, L., Feringa, F. M., Shaltiel, I. A., Van Den Berg, J., and Medema, R. H. (2014). Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 55, 59–72. doi: 10.1016/j.molcel.2014.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kruse, J. P., and Gu, W. (2009). Modes of p53 regulation. Cell 137, 609–622. doi: 10.1016/j.cell.2009.04.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krystyniak, A., Garcia-Echeverria, C., Prigent, C., and Ferrari, S. (2006). Inhibition of Aurora A in response to DNA damage. Oncogene 25, 338–348. doi: 10.1038/sj.onc.1209056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lahav, G., Rosenfeld, N., Sigal, A., Geva-Zatorsky, N., Levine, A. J., Elowitz, M. B., et al. (2004). Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36, 147–150. doi: 10.1038/ng1293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lev Bar-Or, R., Maya, R., Segel, L. A., Alon, U., Levine, A. J., and Oren, M. (2000). Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc. Natl. Acad. Sci. U.S.A. 97, 11250–11255. doi: 10.1073/pnas.210171597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi: 10.1146/annurev.biochem.052308.093131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Little, J. B., Nagasawa, H., Li, G. C., and Chen, D. J. (2003). Involvement of the nonhomologous end joining DNA repair pathway in the bystander effect for chromosomal aberrations. Radiat. Res. 159, 262–267. doi: 10.1667/0033-7587(2003)159[0262:IOTNEJ]2.0.CO;2
Little, J. B., Nagasawa, H., Pfenning, T., and Vetrovs, H. (1997). Radiation-induced genomic instability: delayed mutagenic and cytogenetic effects of X rays and α particles. Radiat. Res. 148, 299–307. doi: 10.2307/3579514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lobrich, M., and Jeggo, P. A. (2007). The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 7, 861–869. doi: 10.1038/nrc2248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lord, C. J., and Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature 481, 287–294. doi: 10.1038/nature10760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lorimore, S. A., Chrystal, J. A., Robinson, J. I., Coates, P. J., and Wright, E. G. (2008). Chromosomal instability in unirradiated hemaopoietic cells induced by macrophages exposed in vivo to ionizing radiation. Cancer Res. 68, 8122–8126. doi: 10.1158/0008-5472.CAN-08-0698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lorimore, S. A., Kadhim, M. A., Pocock, D. A., Papworth, D., Stevens, D. L., Goodhead, D. T., et al. (1998). Chromosomal instability in the descendants of unirradiated surviving cells after α-particle irradiation. Proc. Natl. Acad. Sci. U.S.A. 95, 5730–5733. doi: 10.1073/pnas.95.10.5730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lorimore, S. A., Rastogi, S., Mukherjee, D., Coates, P. J., and Wright, E. G. (2013). The influence of p53 functions on radiation-induced inflammatory bystander-type signaling in murine bone marrow. Radiat. Res. 179, 406–415. doi: 10.1667/RR3158.2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lukas, J., Lukas, C., and Bartek, J. (2011). More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 13, 1161–1169. doi: 10.1038/ncb2344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mackonis, E. C., Suchowerska, N., Zhang, M., Ebert, M., Mckenzie, D. R., and Jackson, M. (2007). Cellular response to modulated radiation fields. Phys. Med. Biol. 52, 5469–5482. doi: 10.1088/0031-9155/52/18/001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mailand, N., Falck, J., Lukas, C., Syljuasen, R. G., Welcker, M., Bartek, J., et al. (2000). Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425–1429. doi: 10.1126/science.288.5470.1425
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mancuso, M., Pasquali, E., Leonardi, S., Tanori, M., Rebessi, S., Di Majo, V., et al. (2008). Oncogenic bystander radiation effects in Patched heterozygous mouse cerebellum. Proc. Natl. Acad. Sci. U.S.A. 105, 12445–12450. doi: 10.1073/pnas.0804186105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Müllers, E., Cascales, H. S., Jaiswal, H., Saurin, A. T., and Lindqvist, A. (2014). Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 13, 2733–2743. doi: 10.4161/15384101.2015.945831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagasawa, H., Huo, L., and Little, J. B. (2003). Increased bystander mutagenic effect in DNA double-strand break repair-deficient mammalian cells. Int. J. Radiat. Biol. 79, 35–41. doi: 10.1080/713864984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagasawa, H., and Little, J. B. (1992). Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res. 52, 6394–6396.
Narayanan, P. K., Goodwin, E. H., and Lehnert, B. E. (1997). Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 57, 3963–3971.
Peng, C. Y., Graves, P. R., Thoma, R. S., Wu, Z., Shaw, A. S., and Piwnica-Worms, H. (1997). Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277, 1501–1505. doi: 10.1126/science.277.5331.1501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pflaum, J., Schlosser, S., and Muller, M. (2014). p53 family and cellular stress responses in cancer. Front. Oncol. 4:285. doi: 10.3389/fonc.2014.00285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piret, B., Schoonbroodt, S., and Piette, J. (1999). The ATM protein is required for sustained activation of NF-κB following DNA damage. Oncogene 18, 2261–2271. doi: 10.1038/sj.onc.1202541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purvis, J. E., Karhohs, K. W., Mock, C., Batchelor, E., Loewer, A., and Lahav, G. (2012). p53 dynamics control cell fate. Science 336, 1440–1444. doi: 10.1126/science.1218351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reinhardt, H. C., Hasskamp, P., Schmedding, I., Morandell, S., Van Vugt, M. A., Wang, X., et al. (2010). DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 40, 34–49. doi: 10.1016/j.molcel.2010.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sedelnikova, O. A., Nakamura, A., Kovalchuk, O., Koturbash, I., Mitchell, S. A., Marino, S. A., et al. (2007). DNA double-strand breaks form in bystander cells after microbeam irradiation of three-dimensional human tissue models. Cancer Res. 67, 4295–4302. doi: 10.1158/0008-5472.CAN-06-4442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shao, C., Furusawa, Y., Aoki, M., Matsumoto, H., and Ando, K. (2002). Nitric oxide-mediated bystander effect induced by heavy-ions in human salivary gland tumour cells. Int. J. Radiat. Biol. 78, 837–844. doi: 10.1080/09553000210149786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smits, V. A., Klompmaker, R., Arnaud, L., Rijksen, G., Nigg, E. A., and Medema, R. H. (2000). Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2, 672–676. doi: 10.1038/35023629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sokolov, M. V., Dickey, J. S., Bonner, W. M., and Sedelnikova, O. A. (2007). gamma-H2AX in bystander cells: not just a radiation-triggered event, a cellular response to stress mediated by intercellular communication. Cell Cycle 6, 2210–2212. doi: 10.4161/cc.6.18.4682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sokolov, M. V., Smilenov, L. B., Hall, E. J., Panyutin, I. G., Bonner, W. M., and Sedelnikova, O. A. (2005). Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene 24, 7257–7265. doi: 10.1038/sj.onc.1208886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sullivan, K. D., Gallant-Behm, C. L., Henry, R. E., Fraikin, J. L., and Espinosa, J. M. (2012). The p53 circuit board. Biochim. Biophys. Acta 1825, 229–244. doi: 10.1016/j.bbcan.2012.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Syljuasen, R. G., Jensen, S., Bartek, J., and Lukas, J. (2006). Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res. 66, 10253–10257. doi: 10.1158/0008-5472.CAN-06-2144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinnikov, V., Lloyd, D., and Finnon, P. (2012). Bystander apoptosis in human cells mediated by irradiated blood plasma. Mutat. Res. 731, 107–116. doi: 10.1016/j.mrfmmm.2011.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wee, K. B., Surana, U., and Aguda, B. D. (2009). Oscillations of the p53-Akt network: implications on cell survival and death. PLoS ONE 4:e4407. doi: 10.1371/journal.pone.0004407
Widel, M., Przybyszewski, W. M., Cieslar-Pobuda, A., Saenko, Y. V., and Rzeszowska-Wolny, J. (2012). Bystander normal human fibroblasts reduce damage response in radiation targeted cancer cells through intercellular ROS level modulation. Mutat. Res. 731, 117–124. doi: 10.1016/j.mrfmmm.2011.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiebusch, L., and Hagemeier, C. (2010). p53- and p21-dependent premature APC/C-Cdh1 activation in G2 is part of the long-term response to genotoxic stress. Oncogene 29, 3477–3489. doi: 10.1038/onc.2010.99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: bystander effect, p53, cell cycle, senescence, DNA damage, cell cycle exit, G2
Citation: Jaiswal H and Lindqvist A (2015) Bystander communication and cell cycle decisions after DNA damage. Front. Genet. 6:63. doi: 10.3389/fgene.2015.00063
Received: 01 December 2014; Accepted: 08 February 2015;
Published online: 27 February 2015.
Edited by:
Marcel Van Vugt, University of Groningen, NetherlandsReviewed by:
Philipp Kaldis, Agency for Science, Technology and Research, SingaporeAnabelle Decottignies, Catholic University of Louvain, Belgium
Copyright © 2015 Jaiswal and Lindqvist. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arne Lindqvist, Department of Cell and Molecular Biology, Karolinska Institutet, von Eulers väg 3, SE-171 77 Stockholm, Sweden e-mail:YXJuZS5saW5kcXZpc3RAa2kuc2U=