 Qian Wang
Qian Wang Qiongshi Lu
Qiongshi Lu Hongyu Zhao
Hongyu Zhao- 1Program of Computational Biology and Bioinformatics, Yale University, New Haven, CT, USA
- 2Department of Biostatistics, Yale School of Public Health, New Haven, CT, USA
- 3Veterans Affairs Cooperative Studies Program Coordinating Center, West Haven, CT, USA
Results from numerous linkage and association studies have greatly deepened scientists’ understanding of the genetic basis of many human diseases, yet some important questions remain unanswered. For example, although a large number of disease-associated loci have been identified from genome-wide association studies in the past 10 years, it is challenging to interpret these results as most disease-associated markers have no clear functional roles in disease etiology, and all the identified genomic factors only explain a small portion of disease heritability. With the help of next-generation sequencing (NGS), diverse types of genomic and epigenetic variations can be detected with high accuracy. More importantly, instead of using linkage disequilibrium to detect association signals based on a set of pre-set probes, NGS allows researchers to directly study all the variants in each individual, therefore promises opportunities for identifying functional variants and a more comprehensive dissection of disease heritability. Although the current scale of NGS studies is still limited due to the high cost, the success of several recent studies suggests the great potential for applying NGS in genomic epidemiology, especially as the cost of sequencing continues to drop. In this review, we discuss several pioneer applications of NGS, summarize scientific discoveries for rare and complex diseases, and compare various study designs including targeted sequencing and whole-genome sequencing using population-based and family-based cohorts. Finally, we highlight recent advancements in statistical methods proposed for sequencing analysis, including group-based association tests, meta-analysis techniques, and annotation tools for variant prioritization.
Introduction
The rapid advancement of biotechnology has brought paradigm shifts in genetic/genomic epidemiology. From linkage studies to genome-wide association studies (GWAS) to the extensive application of next-generation sequencing (NGS), technological developments have improved study designs, deepened our understanding of disease etiology, and led to numerous scientific discoveries (Figure 1). This can be seen in the study of Crohn’s disease, an inflammatory bowel disease with prevalence 0.32% in Europe and North America (Molodecky et al., 2012). Twin-based epidemiological analysis first suggested that there is a genetic component of Crohn’s disease (Molodecky et al., 2012); family-based linkage studies then identified six loci associated to the disease (Hugot et al., 1996; Cardinale et al., 2013); GWAS identified 163 loci at genome-wide significance level, which collectively explain 13.6% of the phenotypic variance (Duerr et al., 2006; Imielinski et al., 2009; Jostins et al., 2012); and re-sequencing GWAS loci identified several causal variants with low minor allele frequencies (Momozawa et al., 2011; Rivas et al., 2011; Cardinale et al., 2013; Ellinghaus et al., 2013; Hunt et al., 2013).
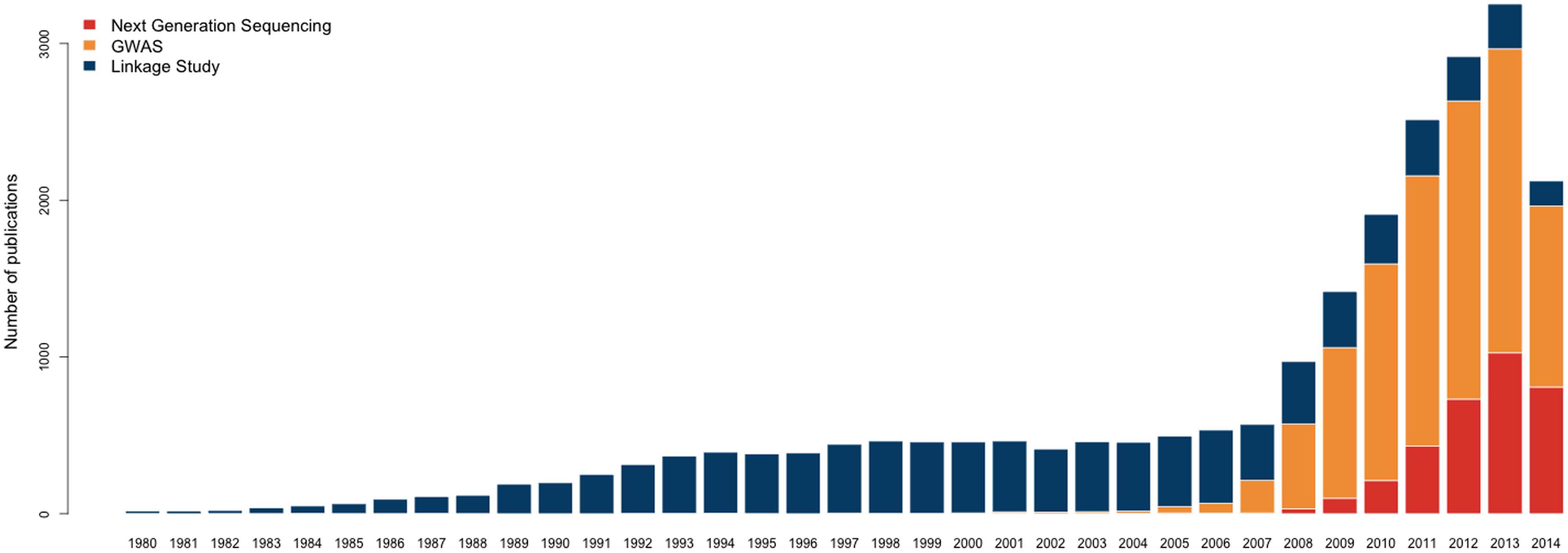
FIGURE 1. Number of publications studying human traits using various techniques (1980–2014). Only the publications in core clinical journals or MEDLINE are included. The vertical axis indicates the number of publications and the horizontal axis indicates different years. The three colors correspond to genetic linkage studies, GWAS, and next generation sequencing, respectively.
Twin-based epidemiological studies can be used to estimate the broad-sense disease heritability, i.e., the amount of phenotypic variance that can be explained by all genetic factors, through comparing phenotype concordance rate in monozygotic and dizygotic twins (Veale, 1960). Linkage studies, which combine information from family pedigrees and sparse genetic markers, can be used to locate disease-associated loci on a very rough scale (Nance et al., 1969; Greer et al., 1989; St George-Hyslop et al., 1990; Dawn Teare and Barrett, 2005). GWAS benefits from the array technology that allows millions of markers to be genotyped at reasonable cost and speed with high accuracy. It has deepened our understanding of disease etiology in multiple directions. First, since the first successful GWAS was published Klein et al. (2005), GWAS has become widely adopted and led to the identifications of a large number of disease-associated genomic loci. As of February 20, 2015, 15,396 single nucleotide polymorphisms (SNPs) from 2,111 publications have been documented in the GWAS Catalog (Welter et al., 2014; Hindorff et al., 2015). Second, the associated loci discovered from GWAS could serve as risk predictors for some diseases, provided large enough GWAS discovery sample size (Wray et al., 2008). Those genetic risk predictors, either used alone or combined with traditional non-genetic risk prediction, have the potential to improve risk-prediction accuracy, which might benefit clinical diagnose and personalized treatment (Jostins and Barrett, 2011). Third, dense genome-wide markers enable a reasonable approximation of narrow-sense heritability (phenotypic variance explained by additive genetic factors) using chip heritability (phenotypic variance explained by genotyped SNPs; Speed et al., 2012; Furlotte et al., 2014). Moreover, valuable insight has been learned on the genetic etiology of many diseases through analyzing the variance contribution of SNPs from certain genomic regions, pathways, as well as variant groups based on minor allele frequencies (Davis et al., 2013; Zaitlen et al., 2014).
As fruitful as GWAS is, it still leaves many intriguing questions unanswered, top on which are the following two problems. The first problem is the difficulty in interpreting GWAS results. This is partly due to our limited understanding of genomic function, especially for non-coding regions, in which considerably many disease-associated loci have been identified. The correlation structure among neighboring variants, often referred to as linkage disequilibrium (LD), also impacts our ability to interpret the results. In fact, it is the haplotype blocks that a GWAS actually identifies, not the real functional variants (Cooper and Shendure, 2011). The second problem is the missing heritability, usually referring to the large gap between the proportion of the variance explained by significant SNPs identified from GWAS and the estimated narrow-sense heritability from twin and pedigree analysis (Manolio et al., 2009; Witte et al., 2014). This has been partly resolved by estimating the chip heritability using linear mixed models and restricted maximum-likelihood estimation in genome-wide complex trait analysis (Yang et al., 2010, 2011). However, a large part of narrow-sense heritability still remains missing. One explanation is the imperfect LD between tagged SNPs and causal variants. Other potential contributors to missing heritability include small insertions and deletions, large structural variants (SVs; Frazer et al., 2009; Mefford and Eichler, 2009), epigenetic factors, gene-by-gene and gene-by-environment interactions (Frazer et al., 2009), and phantom heritability (Zuk et al., 2012). All these genetic and non-genetic factors may have substantial contributions to the etiology for some diseases and disorders.
The rapidly developing NGS technology promises many opportunities to answer some of these questions. Ultimately, it has the potential to provide further biological insight into disease etiology, which may lead to important clinical applications including disease prevention, diagnosis, and treatment. In this review, we discuss why and how, and to which extent NGS techniques can address the issues mentioned above, i.e., zooming in to identify more disease-associated variants or even the real causal ones, and zooming out to recover the missing heritability. We also summarize study designs, statistical methods for analyzing sequencing data, current findings, and challenges.
Zooming In and Out: Identification of Causal Variants and Dissection of Disease Heritability
Next-generation sequencing can be used to identify not only single nucleotide variants (SNVs), but also SVs and epigenetic variations. SNVs are the easiest to call from sequencing data compared to other variant types. Many methods have been proposed to call SNVs with high accuracy from NGS data [e.g., GATK (McKenna et al., 2010), cortex (Iqbal et al., 2012), and DISCOVAR (Weisenfeld et al., 2014)]. SVs in the human genome include copy number variants, copy-number neutral (balanced) translocations, and inversions of various sizes. Traditionally, large SVs are studied using cytogenetics (Langer-Safer et al., 1982; Schrock et al., 1996), while small SVs require finer technologies such as array comparative genomic hybridization (CGH; de Ravel et al., 2007). A number of methods have also been developed to call SVs from sequencing data, including re-sequencing and de novo assembly methods such as MultiBreak-SV (Ritz et al., 2014), GASV (Sindi et al., 2009), LUMPY (Layer et al., 2014), DELLY (Rausch et al., 2012), cn.MOPS (Klambauer et al., 2012), as well as methods reviewed in Medvedev et al. (2009), Tan et al. (2014). Epigenetic variations are the modifications on DNA or chromatin without altering the DNA sequence. Before NGS, detection of epigenetic variations relies on PCR assays (Herman et al., 1996), DNA methylation profiling arrays (Bibikova et al., 2006; Schumacher et al., 2006), and ChIP-chip (Buck and Lieb, 2004). Now, with the help of NGS, epigenetic variations can be detected with ultra high resolution. Moreover, it has become possible to detect allele-specific epigenetic variations through ChIP-seq and bisulfite sequencing (Meissner et al., 2005; Kerkel et al., 2008; Park, 2009; Schalkwyk et al., 2010). Since many types of genomic and epigenetic variations can be detected with improved coverage and accuracy using sequencing data, NGS has the potential to partly recover the missing heritability. Therefore, a more comprehensive view of the decomposition of phenotypic variance is expected from applications of NGS in genetic epidemiology. However, although the variant-calling accuracy of structural and epigenetic variations has been significantly improved, it is still relatively low compared to that of SNVs, especially for earlier sequencing technology with shorter reads. It is also particularly challenging to call SVs using whole-exome sequencing (WES) compared with whole-genome sequencing (WGS). In this review, we focus on the detection of causal SNVs using NGS technology. Issues related to SVs and epigenetic variations can be found elsewhere (Medvedev et al., 2009; Meaburn and Schulz, 2012).
By its study design, GWAS works ideally under the common-disease common-variant (CDCV) hypothesis (Visscher et al., 2012). However, the CDCV hypothesis may not hold for many common diseases as recent studies have suggested a substantial contribution of rare variants to many diseases and traits, as reviewed in Gibson (2012). GWAS will likely fail to detect signals from rare variants in these cases unless the effect size is very large or the causal variants are in strong LD with genotyped markers. With data generated from NGS, researchers can identify more signals in two situations. First, if GWAS does detect the signal (likely to be weak) due to LD structures, the implicated genomic regions can be re-sequenced to uncover the candidates of causal variants. Second, if the disease risk is driven by rare variants independent from the genotyped SNVs, then GWAS will completely miss the signal. In this case, WES or WGS could be used to scan the exome or the genome and detect rare-variant associations in a hypothesis-free manner.
Instead of using LD to detect signals near the probed SNVs, NGS allows researchers to study all the SNVs in each individual directly. However, it will also reveal an overwhelmingly large number of rare variants, most of which have no functional relevance. Therefore, it is non-trivial to identify the causal variants even after accurate variant calling. Moreover, due to the relatively high cost of sequencing compared to other technological options today, most sequencing studies do not have a very large sample size to have adequate statistical power to detect signals through traditional univariate statistical tests. In order to tackle this problem, various strategies in terms of study designs and statistical models have been proposed and implemented. The first strategy is targeted sequencing, including WES and candidate-gene studies. This type of studies usually significantly reduces the cost of sequencing so that larger samples may be analyzed. The second strategy is to focus on a certain type of variants that are more likely to be causal by properly choosing the control group. One example is to use family-trio data to identify de novo mutations that only exist in affected children but not in healthy parents (Xu et al., 2011, 2012; Gauglerl et al., 2014; Muona et al., 2014). Finally, statistically sound methods have also been proposed, including group-based association tests (Lee et al., 2014), meta-analysis techniques for sequencing data (Evangelou and Ioannidis, 2013; Lee et al., 2014), and bioinformatics tools for genome annotation (Cooper and Shendure, 2011; Ward and Kellis, 2012b; Hou and Zhao, 2013), to extract most information from the data. These statistical methods are crucial when applying NGS in population-based association studies. All these strategies are discussed below.
Application of NGS Under Various Study Designs
Next-generation sequencing has brought great success to many different types of studies. First, in terms of cohort type, some studies use family-based data (Vissers et al., 2010; Fromer et al., 2014; Iossifov et al., 2014) while others collect data from unrelated individuals (Hunt et al., 2013; Morrison et al., 2013). Sample sizes are also highly variable in different studies, with extreme cases as small as one individual (Lupski et al., 2010) or as large as tens of thousands subjects (Hunt et al., 2013; De Rubeis et al., 2014). Finally, in terms of sequencing target, WGS, WES, and candidate-gene sequencing all have been applied. Although cost and budget are important factors in choosing a study design, it is crucial to choose the most appropriate study design according to the underlying genetic etiology in order to make meaningful scientific discoveries. For example, tissue-specific diseases may indicate a possible contribution from somatic mutations (Stratton, 2009); diseases that are highly detrimental yet have high prevalence, e.g., autism disorder, may be implicated in causal de novo mutations; and common diseases tend to have more complicated genetic etiology than monogenic diseases, usually involving a large number of genetic factors with small effect individually. Prior knowledge of diseases can guide researchers to make the best use of NGS when designing studies. In this section, we review some pioneer research with different study designs.
Targeted Sequencing and Whole-Genome Sequencing
Due to the still high cost of WGS, targeted sequencing is more commonly used as a cost-effective study design. Popular options include WES using exome-capturing technologies and candidate-gene studies where only a set of preselected genes (e.g., genes close to significant loci identified in GWAS) is sequenced. Although WES misses the entire non-coding genome and sometimes part of the coding regions, numerous scientific discoveries have been made using WES (Majewski et al., 2011). The synthetic association hypothesis (Dickson et al., 2010) provides the theoretical support for re-sequencing GWAS candidate genes in search of causal rare variants. The greater interpretability for variants in the coding regions, the increased statistical power due to less severe multiple testing and the larger sample size due to the much lower cost altogether make WES and candidate-gene studies popular choices.
Re-sequencing GWAS loci of autoimmune diseases have yielded rich positive results (Hunt et al., 2013). By re-sequencing the GWAS significant loci of Crohn’s disease, deleterious or protective variants with low frequencies have been identified in multiple genes, e.g., IL23R and NOD2 (Momozawa et al., 2011; Rivas et al., 2011). By re-sequencing 55 ulcerative colitis GWAS loci in 200 cases and 150 controls, variants with low frequencies were detected in CARD9, IL23R, and RNF186 (Beaudoin et al., 2013). However, re-sequencing 25 GWAS loci in a very large sample (24,892 cases for six autoimmune disease phenotypes and 17,019 controls) did not yield a supportive result for the synthetic association (Hunt et al., 2013). The contribution of rare variants in coding regions is negligible compared with that of common variants. Notably, although this study has a very large sample size, only the exons of 25 GWAS loci were re-sequenced. Therefore, the contribution of rare variants to Crohn’s disease still awaits further assessment with genome-wide screening.
Relatively fewer studies use population-based WGS for complex disorders or traits due to the high cost. However, several pioneer studies have demonstrated the potential of WGS in understanding the genetic architecture of complex diseases, especially for discovering the contribution of rare variants and variants in non-coding regions. For example, the CHARGE consortium (Psaty et al., 2009) performed WGS on 962 individuals to study the levels of high-density lipoprotein cholesterol (HDL-C) and it was found that common (MAF > 0.01) and rare variants (MAF < 0.01) explain about 61.8 and 7.8% of HDL-C level variance, respectively (Morrison et al., 2013). As the sequencing cost continues to drop (now less than $1000 per genome at 30x coverage with the announcement of Illumina Hiseq X Ten system), we can expect more population-based WGS studies in the future. Results from those studies may greatly deepen our understanding of disease architecture, due to their unparalleled coverage of the human genome.
Rare Diseases and Common Complex Diseases
Next-generation sequencing can be applied to study both rare diseases and common diseases. For rare monogenic diseases, causal variants may be identified even with a small sample size. However, it remains challenging to identify causal alleles for most common diseases as well as some rare diseases with genetic heterogeneity (McClellan and King, 2010), which typically require a larger sample size.
Whole-exome sequencing and WGS allow a revisit to monogenic diseases that are traditionally studied using linkage analysis, and bring the opportunities of finding genetic causes for intractable patients with no previously known causes. Many novel causal variants have been identified, as reviewed in Bamshad et al. (2011), Ku et al. (2011), Dewey et al. (2012). For example, gene SH3TC2 was found to contain causative alleles for Charcot-Marie-Tooth disease using WGS on one patient (Lupski et al., 2010); WES of four patients identified a causal gene OHODH for Miller syndrome (Ng et al., 2010). These results demonstrate the power of NGS in identifying causal variants for monogenic diseases even with very small sample size. There are certain complications, though, caused by potential unnoticed environmental risk factors (Weatherall, 2001) and existence of functionally redundant paralogs of disease genes (Chen et al., 2013).
Not all rare diseases have such a simple genetic structure as monogenic diseases do. Rare diseases are usually diagnosed or defined by symptoms, whereas the same symptom can be induced by different mechanisms. In fact, some rare diseases are a group of diseases manifesting similar symptoms. The identification of causal variants for those diseases generally requires larger sample sizes than monogenic diseases. Muona et al. (2014) recently found a causal de novo mutation in gene KCNC1 for progressive myoclonus epilepsy (PME), a group of rare disorders, through WES on 110 unrelated patients (Muona et al., 2014). By doing WGS on 50 patients, Gilissen et al. (2014) discovered major genetic causes for severe and genetically heterogeneous intellectual disability that affects 0.5% of newborns.
Genetic heterogeneity can have many faces for common diseases. Individuals with the same disease may have different causal variants from the same gene or different genes in the disease pathway(s). These variants can be common or rare, coding or non-coding. On the other hand, individuals with the same causal genetic factors may not manifest the same phenotype due to incomplete penetrance, interaction with other genetic, epigenetic, or environmental factors. All of these scenarios may exist simultaneously among patients of a complex disease, making it difficult to characterize the genetic etiology. GWAS can only identify common variants with reasonable effect sizes, which are usually non-causal. NGS may help in this situation as a tool to screen rare variants. One successful application is for neurodevelopmental disorders.
Whole-exome sequencing has achieved great success for de novo mutation detection in neurodevelopmental disorders such as autism spectrum disorder (O’Roak et al., 2011, 2012; Iossifov et al., 2012, 2014; Neale et al., 2012; Sanders et al., 2012; Hamilton et al., 2013; De Rubeis et al., 2014; Robinson et al., 2014), mental retardation (Vissers et al., 2010; Gilissen et al., 2014; Wen et al., 2014), and schizophrenia (Awadalla et al., 2010; Girard et al., 2011; Xu et al., 2011, 2012; Fromer et al., 2014; McCarthy et al., 2014; Purcell et al., 2014). Causal de novo mutations for these neurodevelopmental disorders are not randomly distributed in the genome, as converging evidence has pointed to their enrichment in synaptic, transcriptional and chromatin remodeling genes (De Rubeis et al., 2014; Fromer et al., 2014; McCarthy et al., 2014; Wen et al., 2014). These studies not only identified de novo causal rare variants, but also demonstrated how large their contributions are to neurodevelopmental disorders, which brings new insight into the genetic etiology. For example, according to twin studies, autism disorder has an estimated broad-sense heritability of over 0.9 (for the narrow phenotype of autism), while GWAS loci can only explain a small part of the heritability (Freitag, 2007). A recent WES study on more than 2500 simplex families showed that 12% of autism diagnoses can be explained by 13% of de novo missense mutations, and 9% of autism diagnoses can be explained by 43% of de novo likely gene-disruption mutations (Iossifov et al., 2014). Another study, using a Swedish sample, confirmed the substantial contribution of de novo mutations to individual autism liability, but also pointed out that population-wise, their contribution to autism liability is only 2.6%, accounting for a very modest proportion of the estimated narrow-sense heritability 52.4%, which is mostly contributed by common variation (Gauglerl et al., 2014). Notably, although contribution from de novo mutation to population-level phenotypic variation is small compared with common variants, de novo mutations are very important for individual phenotype, and thus detecting those causal de novo mutations is important and may lead to improvements in disease risk prediction and personalized treatment. Moreover, the fact that the detected de novo mutations tend to come from certain pathways further reveals the pathological mechanisms of those disorders, which may lead to novel treatment strategies.
Statistical Methods to Detect Rare Variant Association
Effects of rare variants vary across different diseases. Even if there is a substantial contribution from rare variants, it remains challenging to detect rare variant associations due to low statistical power. Many statistical methods have been proposed to increase the signal or reduce the noise in testing variant-disease association using sequencing data. We group these methods into three general categories: group-based association test, meta-analysis, and functional annotation. However, despite using very different techniques, these three categories are closely related to each other and are often used in combination.
Group-Based Association Tests
The major strategy used in GWAS analysis is to evaluate each SNP individually with a univariate statistic. However, standard individual variant tests are underpowered to detect rare variant effects due to the low minor allele frequency (MAF) unless effect sizes or sample sizes are very large. Moreover, rare variant association studies usually involve extreme multiple testing due to the large number of rare variants in each individual. Pelak et al. (2010) reported about 3.5 million SNVs per genome using WGS on 20 samples. This further reduces the power when type-I error is controlled. Therefore, many group-based association tests that assess the cumulative effects of multiple variants have been proposed for sequencing studies. For simplicity, we describe these strategies for the analysis of a single genomic region, e.g., a gene.
The earliest collapsing methods, also known as burden tests (e.g., CAST Morgenthaler and Thilly, 2007), collapse all rare variants in a genomic region into a single variable. This can be done either through an indicator of whether an individual has any rare variants, or through summing up the total number of rare alleles (Morris and Zeggini, 2010). Both schemes completely ignore the effect of common variants and weight all rare variants equally, independent of their allele frequency. Several weighted sum tests (WSTs) generalize these ideas and suggest weighting variants according to their frequencies (Madsen and Browning, 2009; Price et al., 2010). In this way, contributions from both common and rare variants are incorporated. Different WST approaches use different weighting schemes, but in general, they all down-weight common variants and up-weight rare ones. The variable threshold (VT) approach generalizes burden tests in another direction (Price et al., 2010). Instead of using a pre-fixed threshold for rarity, the VT approach computes the test statistics over a series of reasonable thresholds τ, and adaptively chooses the τ that maximizes the test statistic.
None of the methods discussed so far allow variants to influence the phenotype in different directions. Variants with similar MAF are also assumed to have similar effect sizes. The adaptive summation (aSUM) approach is the first method that distinguishes protective variants from deleterious ones (Han and Pan, 2010). The flexibility is further improved by kernel-based methods, e.g., sequence kernel association test (SKAT; Wu et al., 2011). Similar to the WSTs, SKAT also incorporates a weighting scheme, but both the weight and the kernel can be modified based on the prior knowledge of disease etiology. No matter what kernel or weighting scheme is used, the score test guarantees the type-I error being well-controlled. Appropriate choices would simply increase the power. Other than that, both magnitudes and directionality of the associations are estimated from data instead of pre-fixed, which again introduces great flexibility.
Burden tests and the kernel-based variance-component tests have very different model assumptions. However, Lin and Tang (2011) developed a general regression framework for rare variant association testing that unifies existing methods including WST, VT, and SKAT. Lee et al. (2012) also generalized the variance-component testing framework used in SKAT by incorporating correlation structure into the random effect so that the burden test and the original SKAT both become special cases of this general framework.
Finally, although rare variants are more likely to be causal because of selection pressure, common variants could still have substantial effects in some diseases. Therefore, it would be wise to combine the effects of common and rare variants using a statistically justified framework. Right after CAST came out, the combined multivariate and collapsing (CMC) method (Li and Leal, 2008) improved CAST by collapsing variants into subgroups based on their allele frequencies, and then applying a multivariate test. More recently, Ionita-Laza et al. (2013) used the similar idea to generalize SKAT. Different weights and kernels are chosen for common and rare variants. Then, several combination approaches can be implemented to test for the combined effect (Ionita-Laza et al., 2013).
After years of exploratory research, scientists have acquired a rich collection of methods to test for group-based association. However, each method has its unique assumptions and limitations. For example, if a large proportion of rare variants are causal at the same direction, burden tests will be the most powerful; if a genomic region consists of a mixture of deleterious and protective variants, SKAT should become the superior choice. Although general frameworks have been proposed, those models often include more parameters and use more degrees of freedom. Currently, large-scale sequencing studies are still costly, so the sample size is often not very large. Whether the general and flexible frameworks could work well in such circumstances remains to be thoroughly investigated using empirical data (Liu et al., 2014). In practice, researchers should choose the statistical method tailored to the most reasonable assumptions according to the prior knowledge of disease etiology.
Meta-Analysis
Meta-analysis is a statistical method for pooling results from multiple independent studies. It essentially increases the sample size by incorporating summary statistics rather than relying on individual-level data from different studies, which is an important feature since individual-level data usually cannot be shared due to policies and ethical concerns. While its basic idea originated back to the 17th century (Plackett, 1970), meta-analysis is still a popular approach in biomedical research, especially in genomic studies where limited sample size is often a key limiting factor for significant discoveries. Numerous meta-analysis methods and software have been developed for GWAS [e.g., METAL (Willer et al., 2010) and GWAMA (Mägi and Morris, 2010)]. These methods have enjoyed a great success, with hundreds of GWAS meta-analyses being published (Panagiotou et al., 2013). A comprehensive comparison of these meta-analysis methods is reviewed elsewhere (Evangelou and Ioannidis, 2013).
There are three major meta-analysis strategies for individual variants: approaches based on p-values or Z scores, fixed-effects models, and random-effects models. It would be natural to extend these approaches for group-based tests in sequencing studies (Table 1). In fact, approaches based on p-values or Z scores can be applied to group-based association tests directly using Fisher’s or Stouffer’s methods (Fisher, 1934; Stouffer et al., 1949). However, these methods are unable to deal with the heterogeneity among studies or to estimate the overall effect size. Moreover, they have been shown to be less powerful than fixed-effects models in both simulation and real data analysis (Liu et al., 2013, 2014). In 2013, several groups independently developed score-based fixed-effects models that incorporate diverse types of group-based association tests (Lee et al., 2013; Tang and Lin, 2013; Liu et al., 2014). Traditional meta-analysis approaches for single variant associations usually involve the estimation of single-variant effect that is not stable for rare variants. Methods based on score statistics avoid this issue because it only requires fitting the null model. Another advantage of score-based procedure is that it does not require different studies to have the same set of variants. This is crucial for sequencing analysis because very rare variants are not guaranteed to exist in all the cohorts being studied. Moreover, these meta-analysis approaches have been shown to be numerically equivalent to the mega-analysis using individual-level data. Finally, Hu et al. (2013) further extended the fixed-effects models based on a key observation that the multivariate score statistic as well as the corresponding information matrix can be recovered from test statistics for single variants. This adds more flexibility into the meta-analysis framework because only statistics for individual variants need to be shared. However, this simplification is valid only under the assumption of additive mode of inheritance.

TABLE 1. A list of meta-analysis software for group-based association tests.
Fixed-effects models assume the genetic effects to be the same in different studies. In contrast, random-effects models test for the heterogeneous genetic associations by allowing the genetic effects to vary across studies. Traditional random-effects meta-analysis models test for the mean effect of genetic variables (DerSimonian and Laird, 1986). Although it reflects the heterogeneous nature, this type of methods tend to be less powerful than fixed-effects models (Evangelou and Ioannidis, 2013). Han and Eskin (2011) improved the discovery power of random-effects models by testing for the joint null hypothesis of the absence of any genetic effects and between-study variance. Tang and Lin (2014) extended the same idea to random-effects group-based meta-analysis while using the score-based framework. Lee et al. (2013) also proposed a random-effects model. It demonstrated comparable discovery power in simulations compared to the method developed by Tang and Lin (2014).
Functional Annotation
Genomic functional annotation is crucial for prioritizing variants and interpreting results in association studies. This is especially helpful for predicting causal variants among a group of SNVs with strong LD. With the help of appropriate annotation tools, both random and systematic noise in the data can be greatly reduced. There are some techniques that use special study design or simple filtering rather than statistical models to incorporate functional annotations. For example, in terms of study design, we have discussed that WES is sometimes more preferred than WGS. One consideration is that variants in the protein-coding regions are more likely to be functional. In terms of variant-filtering procedures, various pipelines have been developed to focus on a certain type of variants such as non-synonymous SNPs or frame-shifting insertions and deletions (Vissers et al., 2010; Xu et al., 2011; Kim et al., 2012; Reumers et al., 2012). These variant filters suffer from a high chance of missing real causal candidates by enforcing variants to satisfy every screening condition. In contrast, well-justified statistical methods allow incorporating diverse types of information to collectively evaluate the functional potential of variants. Here, we focus on the statistical tools that predict functional genomic variants.
Methods for predicting deleterious variants in the protein-coding regions are the richest of the available approaches. Numerous tools have been developed to serve this purpose, including SIFT (Ng and Henikoff, 2001), PolyPhen (Ramensky et al., 2002; Adzhubei et al., 2010), MutationTaster (Schwarz et al., 2010), SAPRED (Ye et al., 2007), and SNPs3D (Yue et al., 2006), among others. Most of these methods are statistical classifiers using both evolutionary and biochemical information of proteins as annotation features (Cooper and Shendure, 2011). The major differences among these tools are the choices of training data, covariates, and classification methods. Compared with the “mysterious” non-coding regions in the human genome, researchers have gained a much deeper understanding of the protein-coding regions through tracing the functional mechanisms in transcription and translation. Therefore, it is not surprising that some covariates (e.g., amino acid properties and protein structural information) are informative for predicting deleteriousness of variants in coding regions. The positive training data are usually collected from large databases for pathogenic variants [e.g., OMIM (Hamosh et al., 2005) and ClinVar (Landrum et al., 2014)], while some matched benign variants are used as the negative training set. Finally, statistical classification methods (e.g., naïve Bayes classifier and support vector machine) are trained on the training data using collected covariates. The informative covariates, the gold-standard training data, and the statistically justified classification frameworks altogether guarantee the predictive ability of these tools.
Compared to the well-understood coding regions, the non-coding regions in the human genome are much less explored. However, it has been established that ∼98% of the human genome is non-coding DNA (Elgar and Vavouri, 2008). About 95% of known variants within sequenced genomes and nearly 90% of the significant variants from GWAS lie outside of protein-coding regions (Hindorff et al., 2009). All these pieces of evidence, as well as the expected wide applications of WGS in the near future, suggest that the scope of the annotation tools should be extended to the whole-genome. Several tools have been developed, including HaploReg (Ward and Kellis, 2012a), RegulomeDB (Boyle et al., 2012), CADD (Kircher et al., 2014), and GWAVA (Ritchie et al., 2014; Table 2). Among these tools, HaploReg and RegulomeDB are more of databases than prediction tools. They both offer well-designed user interfaces that present many useful annotation data collected from different sources such as the ENCODE project (Dunham et al., 2012). The users need to judge the functional potential of variant candidates based on these annotations by themselves. CADD and GWAVA are similar to the deleteriousness prediction tools developed for coding regions. CADD is based on support vector machine, while GWAVA uses the random forest algorithm. Large consortia such as the ENCODE project have generated a vast amount of regulatory information for the human genome (Dunham et al., 2012). Among those, information of the transcriptional binding sites, histone modification, DNase I hypersensitivity, DNA methylation, and many others all have the potential to serve as predictive covariates in non-coding functional annotation tools. However, current supervised-learning-based methods still suffer from potentially biased training data due to our limited knowledge of non-coding functional mechanism. Therefore, methods based on unsupervised learning may be advantageous at this early stage, but no such method has been proposed yet.
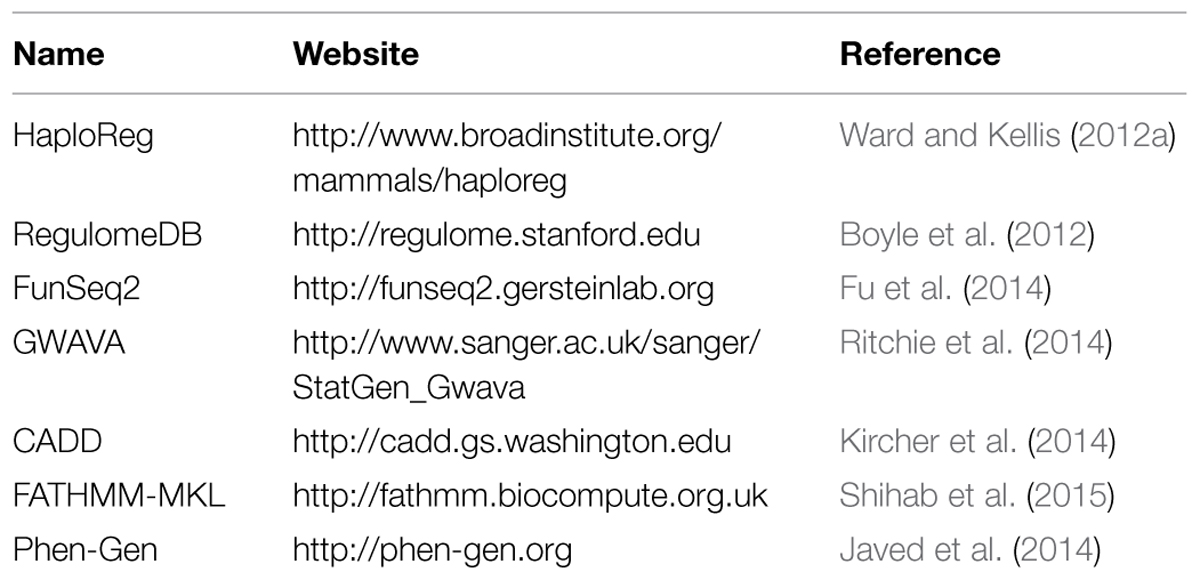
TABLE 2. A list of tools for annotating variants in non-coding regions.
Integrating functional annotations in causal variant detection is a very active research field with many challenging open questions. While this review was in preparation, a new non-coding variant functional prediction method based on multiple kernel learning was published (Shihab et al., 2015). Here we only introduced some of the existing tools that are closely related to sequencing study. It is worth noting that researchers should choose the most appropriate annotation tool based on the scientific hypothesis. For example, in cancer studies, methods that predict regulatory somatic mutations will probably be favored (Khurana et al., 2013; Fu et al., 2014). When the phenotypic information is available, methods that integrate phenotype-specific gene prioritization may be advantageous (Sifrim et al., 2013; Javed et al., 2014; Singleton et al., 2014).
Discussion
In the last 10 years, GWAS has transformed genetic epidemiology to genomic epidemiology. More than 2,000 GWAS have been done for almost all known complex diseases, leading to the identification of a vast number of disease-associated genomic loci. Despite these discoveries, more and more people have realized the limitations of this experiment design. In this review, we discussed some of the well-known issues in GWAS analysis, including missing heritability and lack of interpretability. Recent advances of the next generation sequencing technology have made sequencing faster, more affordable and more accurate. These technological advances as well as the success of pioneer sequencing studies strongly suggest that NGS has the potential to lead genomic epidemiology into a new era. It allows systematic assessment of rare SNVs as well as many other diverse types of genomic and epigenetic variations using hypothesis-free whole-genome scans. Large consortia and programs have also been formed, e.g., the 1000 Genomes Project Consortium (Genomes Project et al., 2012) and the NHGRI Genome Sequencing Program (GSP), in this mission of decoding the variations of human genome using NGS technologies. All these advances would bring biological insight and benefit scientific researches.
Besides its benefits to the basic science, NGS also has a bright future in clinical applications. For example, WES identified a missense mutation in a 15-months child patient with symptoms similar to Crohn’s disease. Based on this, the child received proper diagnosis and treatment, which would otherwise be intractable (Worthey et al., 2011). Notably, clinical cases like this also benefit scientific research as novel causes of disorders are revealed in the process. Programs that use NGS to aid diagnosis have been launched, e.g., “3Gb-testing” project (Boccia et al., 2014). As sequencing technologies become more mature and affordable, we expect the potential of NGS to be fully realized as a bridge between clinical applications and research progresses.
Although the future of NGS is very promising, many challenges still remain. First, although the cost of generating sequencing data continues to drop, it is still substantially greater than the cost of more traditional technologies. Currently, sequencing cost ranges from 500 (70-fold WES) to 1000 dollars (30-fold WGS) per sample, which is nearly 10 times greater than using high-quality microarrays. Apart from the cost of data generation, the demanding requirement of sample recruitment, data storage, and downstream processing all act as barriers to sample size and statistical power (Sboner et al., 2011). Moreover, some issues such as the optimal combination of sequencing depth and sample size can only be answered using empirical data, and are still far from being fully understood. When designing NGS-based studies, researchers should take all these factors into consideration in order to choose the most appropriate study design. Finally, finding causal variants from the overwhelmingly large number of background mutations is a great challenge. In large population-based WGS studies, the number of SNVs that appear at least once can easily go beyond 10 million. This leads to extreme multiple testing problems for which any traditional statistical procedure is likely to be underpowered. We have discussed several categories of novel statistical methods designed for sequencing analysis, including group-based association tests, meta-analysis approaches, and annotation tools for variant prioritization. But these do not cover all aspects of statistical methods that can be used in sequencing studies. With the popularization of next generation sequencing, we expect to see a boom in novel and powerful statistical approaches, amazing scientific discoveries, as well as clinical breakthroughs.
Author Contributions
QW and QL conceived and wrote the paper. HZ advised on statistical and genetic issues.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the National Institutes of Health (R01 GM59507 and U01 HG005718), the VA Cooperative Studies Program of the Department of Veterans Affairs, Office of Research and Development, and the Yale World Scholars Program from the China Scholarship Council.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P.,et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Awadalla, P., Gauthier, J., Myers, R. A., Casals, F., Hamdan, F. F., Griffing, A. R.,et al. (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet. 87, 316–324. doi: 10.1016/J.Ajhg.2010.07.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bamshad, M. J., Ng, S. B., Bigham, A. W., Tabor, H. K., Emond, M. J., Nickerson, D. A.,et al. (2011). Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755. doi: 10.1038/Nrg3031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beaudoin, M., Goyette, P., Boucher, G., Lo, K. S., Rivas, M. A., Stevens, C.,et al. (2013). Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 9:e1003723. doi: 10.1371/journal.pgen.1003723
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bibikova, M., Lin, Z. W., Zhou, L. X., Chudin, E., Garcia, E. W., Wu, B.,et al. (2006). High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 16, 383–393. doi: 10.1101/Gr.4410706
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boccia, S., Mc Kee, M., Adany, R., Boffetta, P., Burton, H., Cambon-Thomsen, A.,et al. (2014). Beyond public health genomics: proposals from an international working group. Eur. J. Public Health 24, 876–878. doi: 10.1093/eurpub/cku142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boyle, A. P., Hong, E. L., Hariharan, M., Cheng, Y., Schaub, M. A., Kasowski, M.,et al. (2012). Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797. doi: 10.1101/gr.137323.112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buck, M. J., and Lieb, J. D. (2004). ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83, 349–360. doi: 10.1016/J.Ygeno.2003.11.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cardinale, C. J., Kelsen, J. R., Baldassano, R. N., and Hakonarson, H. (2013). Impact of exome sequencing in inflammatory bowel disease. World J. Gastroenterol. 19, 6721–6729. doi: 10.3748/Wjg.V19.I40.6721
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, W. H., Zhao, X. M., Van Noort, V., and Bork, P. (2013). Human monogenic disease genes have frequently functionally redundant paralogs. PLoS Comput. Biol. 9:e1003073. doi: 10.1371/journal.pcbi.1003073
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cooper, G. M., and Shendure, J. (2011). Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 12, 628–640. doi: 10.1038/nrg3046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, L. K., Yu, D., Keenan, C. L., Gamazon, E. R., Konkashbaev, A. I., Derks, E. M.,et al. (2013). Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet. 9:e1003864. doi: 10.1371/journal.pgen.1003864
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dawn Teare, M., and Barrett, J. H. (2005). Genetic linkage studies. Lancet 366, 1036–1044. doi: 10.1016/S0140-6736(05)67382-5
de Ravel, T. J., Devriendt, K., Fryns, J. P., and Vermeesch, J. R. (2007). What’s new in karyotyping? The move towards array comparative genomic hybridisation (CGH). Eur. J. Pediatr. 166, 637–643. doi: 10.1007/s00431-007-0463-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Ercument Cicek, A.,et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DerSimonian, R., and Laird, N. (1986). Meta-analysis in clinical trials. Control Clin. Trials 7, 177–188. doi: 10.1016/0197-2456(86)90046-2
Dewey, F. E., Pan, S., Wheeler, M. T., Quake, S. R., and Ashley, E. A. (2012). DNA Sequencing clinical applications of new DNA sequencing technologies. Circulation 125, 931–944. doi: 10.1161/Circulationaha.110.972828
Dickson, S. P., Wang, K., Krantz, I., Hakonarson, H., and Goldstein, D. B. (2010). Rare variants create synthetic genome-wide associations. PLoS Biol. 8:e1000294. doi: 10.1371/journal.pbio.1000294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duerr, R. H., Taylor, K. D., Brant, S. R., Rioux, J. D., Silverberg, M. S., Daly, M. J.,et al. (2006). A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463. doi: 10.1126/Science.1135245
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dunham, I., Kundaje, A., Aldred, S. F., Collins, P. J., Davis, C., Doyle, F.,et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/Nature11247
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elgar, G., and Vavouri, T. (2008). Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends Genet. 24, 344–352. doi: 10.1016/j.tig.2008.04.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ellinghaus, D., Zhang, H., Zeissig, S., Lipinski, S., Till, A., Jiang, T.,et al. (2013). Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 145, 339–347. doi: 10.1053/J.Gastro.2013.04.040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Evangelou, E., and Ioannidis, J. P. (2013). Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 14, 379–389. doi: 10.1038/nrg3472
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Feng, S., Liu, D., Zhan, X., Wing, M. K., and Abecasis, G. R. (2014). RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics 30, 2828–2829. doi: 10.1093/bioinformatics/btu367
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frazer, K. A., Murray, S. S., Schork, N. J., and Topol, E. J. (2009). Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251. doi: 10.1038/Nrg2554
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Freitag, C. M. (2007). The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol. Psychiatry 12, 2–22. doi: 10.1038/sj.mp.4001896
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P.,et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/Nature12929
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fu, Y., Liu, Z., Lou, S., Bedford, J., Mu, X., Yip, K. Y.,et al. (2014). FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 15, 480. doi: 10.1186/s13059-014-0480-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Furlotte, N. A., Heckerman, D., and Lippert, C. (2014). Quantifying the uncertainty in heritability. J. Hum. Genet. 59, 269–275. doi: 10.1038/Jhg.2014.15
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gauglerl, T., Klei, L., Sanders, S. J., Bodea, C. A., Goldberg, A. P., Lee, A. B.,et al. (2014). Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885. doi: 10.1038/Ng.3039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Genomes Project, C., Abecasis, G. R., Auton, A., Brooks, L. D., Depristo, M. A., Durbin, R. M.,et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gibson, G. (2012). Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145. doi: 10.1038/Nrg3118
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gilissen, C., Hehir-Kwa, J. Y., Thung, D. T., Van De Vorst, M., Van Bon, B. W. M., Willemsen, M. H.,et al. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347. doi: 10.1038/Nature13394
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Girard, S. L., Gauthier, J., Noreau, A., Xiong, L., Zhou, S. R., Jouan, L.,et al. (2011). Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863. doi: 10.1038/Ng.886
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Greer, W. L., Mahtani, M. M., Kwong, P. C., Rubin, L. A., Peacocke, M., Willard, H. F.,et al. (1989). Linkage studies of the Wiskott-Aldrich syndrome: polymorphisms at TIMP and the X chromosome centromere are informative markers for genetic prediction. Hum. Genet. 83, 227–230. doi: 10.1007/BF00285161
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hamilton, P. J., Campbell, N. G., Sharma, S., Erreger, K., Herborg Hansen, F., Saunders, C.,et al. (2013). De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 18, 1315–1323. doi: 10.1038/mp.2013.102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A., and Mckusick, V. A. (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517. doi: 10.1093/nar/gki033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, B., and Eskin, E. (2011). Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598. doi: 10.1016/j.ajhg.2011.04.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, F., and Pan, W. (2010). A data-adaptive sum test for disease association with multiple common or rare variants. Hum. Hered. 70, 42–54. doi: 10.1159/000288704
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herman, J. G., Graff, J. R., Myohanen, S., Nelkin, B. D., and Baylin, S. B. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826. doi: 10.1073/Pnas.93.18.9821
Hindorff, L. A., Macarthur, J., Morales, J., Junkins, H. A., Hall, P. N., Klemm, A. K.,et al. (2015). A Catalog of Published Genome-Wide Association Studies. Available at: http://www.genome.gov/gwastudies.
Hindorff, L. A., Sethupathy, P., Junkins, H. A., Ramos, E. M., Mehta, J. P., Collins, F. S.,et al. (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 106, 9362–9367. doi: 10.1073/pnas.0903103106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hou, L., and Zhao, H. (2013). A review of post-GWAS prioritization approaches. Front. Genet. 4:280. doi: 10.3389/fgene.2013.00280
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hu, Y. J., Berndt, S. I., Gustafsson, S., Ganna, A., Hirschhorn, J., North, K. E.,et al. (2013). Meta-analysis of gene-level associations for rare variants based on single-variant statistics. Am. J. Hum. Genet. 93, 236–248. doi: 10.1016/j.ajhg.2013.06.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hugot, J. P., Laurent-Puig, P., Gower-Rousseau, C., Olson, J. M., Lee, J. C., Beaugerie, L.,et al. (1996). Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature 379, 821–823. doi: 10.1038/379821a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hunt, K. A., Mistry, V., Bockett, N. A., Ahmad, T., Ban, M., Barker, J. N.,et al. (2013). Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 498, 232–235. doi: 10.1038/Nature12170
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imielinski, M., Baldassano, R. N., Griffiths, A., Russell, R. K., Annese, V., Dubinsky, M.,et al. (2009). Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 41, 1335–1340. doi: 10.1038/Ng.489
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J. D., and Lin, X. (2013). Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet. 92, 841–853. doi: 10.1016/j.ajhg.2013.04.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D.,et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iossifov, I., Ronemus, M., Levy, D., Wang, Z. H., Hakker, I., Rosenbaum, J.,et al. (2012). De Novo Gene Disruptions in children on the Autistic Spectrum. Neuron 74, 285–299. doi: 10.1016/J.Neuron.2012.04.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iqbal, Z., Caccamo, M., Turner, I., Flicek, P., and Mcvean, G. (2012). De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 44, 226–232. doi: 10.1038/Ng.1028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Javed, A., Agrawal, S., and Ng, P. C. (2014). Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat. Methods 11, 935–937. doi: 10.1038/nmeth.3046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jostins, L., and Barrett, J. C. (2011). Genetic risk prediction in complex disease. Hum. Mol. Genet. 20, R182–R188. doi: 10.1093/Hmg/Ddr378
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jostins, L., Ripke, S., Weersma, R. K., Duerr, R. H., Mcgovern, D. P., Hui, K. Y.,et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. doi: 10.1038/nature11582
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kerkel, K., Spadola, A., Yuan, E., Kosek, J., Jiang, L., Hod, E.,et al. (2008). Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 40, 904–908. doi: 10.1038/Ng.174
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khurana, E., Fu, Y., Colonna, V., Mu, X. J., Kang, H. M., Lappalainen, T.,et al. (2013). Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342, 1235587. doi: 10.1126/science.1235587
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. J., Park, Y. M., Baik, K. H., Choi, H. Y., Yang, G. S., Koh, I.,et al. (2012). Exome sequencing and subsequent association studies identify five amino acid-altering variants influencing human height. Hum. Genet. 131, 471–478. doi: 10.1007/S00439-011-1096-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kircher, M., Witten, D. M., Jain, P., O’roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klambauer, G., Schwarzbauer, K., Mayr, A., Clevert, D. A., Mitterecker, A., Bodenhofer, U.,et al. (2012). cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40:e69. doi: 10.1093/nar/gks003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klein, R. J., Zeiss, C., Chew, E. Y., Tsai, J. Y., Sackler, R. S., Haynes, C.,et al. (2005). Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389. doi: 10.1126/science.1109557
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ku, C. S., Naidoo, N., and Pawitan, Y. (2011). Revisiting Mendelian disorders through exome sequencing. Hum. Genet. 129, 351–370. doi: 10.1007/S00439-011-0964-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Landrum, M. J., Lee, J. M., Riley, G. R., Jang, W., Rubinstein, W. S., Church, D. M.,et al. (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985. doi: 10.1093/nar/gkt1113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langer-Safer, P. R., Levine, M., and Ward, D. C. (1982). Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. U.S.A. 79, 4381–4385. doi: 10.1073/pnas.79.14.4381
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Layer, R. M., Chiang, C., Quinlan, A. R., and Hall, I. M. (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84. doi: 10.1186/gb-2014-15-6-r84
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, S., Abecasis, G. R., Boehnke, M., and Lin, X. (2014). Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet. 95, 5–23. doi: 10.1016/j.ajhg.2014.06.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, S., Teslovich, T. M., Boehnke, M., and Lin, X. (2013). General framework for meta-analysis of rare variants in sequencing association studies. Am. J. Hum. Genet. 93, 42–53. doi: 10.1016/j.ajhg.2013.05.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, S., Wu, M. C., and Lin, X. (2012). Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775. doi: 10.1093/biostatistics/kxs014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, B., and Leal, S. M. (2008). Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321. doi: 10.1016/j.ajhg.2008.06.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, D. Y., and Tang, Z. Z. (2011). A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 89, 354–367. doi: 10.1016/j.ajhg.2011.07.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, D. J., Peloso, G. M., Zhan, X., Holmen, O. L., Zawistowski, M., Feng, S.,et al. (2014). Meta-analysis of gene-level tests for rare variant association. Nat. Genet. 46, 200–204. doi: 10.1038/ng.2852
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, L., Sabo, A., Neale, B. M., Nagaswamy, U., Stevens, C., Lim, E.,et al. (2013). Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 9:e1003443. doi: 10.1371/journal.pgen.1003443
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lupski, J. R., Reid, J. G., Gonzaga-Jauregui, C., Rio Deiros, D., Chen, D. C., Nazareth, L.,et al. (2010). Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 362, 1181–1191. doi: 10.1056/NEJMoa0908094
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Madsen, B. E., and Browning, S. R. (2009). A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 5:e1000384. doi: 10.1371/journal.pgen.1000384
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mägi, R., and Morris, A. P. (2010). GWAMA: software for genome-wide association meta-analysis. BMC Bioinform. 11:288. doi: 10.1186/1471-2105-11-288
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Majewski, J., Schwartzentruber, J., Lalonde, E., Montpetit, A., and Jabado, N. (2011). What can exome sequencing do for you? J. Med. Genet. 48, 580–589. doi: 10.1136/Jmedgenet-2011-100223
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J.,et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/Nature08494
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McCarthy, S. E., Gillis, J., Kramer, M., Lihm, J., Yoon, S., Berstein, Y.,et al. (2014). De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658. doi: 10.1038/Mp.2014.29
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McClellan, J., and King, M. C. (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. doi: 10.1016/j.cell.2010.03.032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A.,et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/Gr.107524.110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meaburn, E., and Schulz, R. (2012). Next generation sequencing in epigenetics: insights and challenges. Semin. Cell Dev. Biol. 23, 192–199. doi: 10.1016/J.Semcdb.2011.10.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Medvedev, P., Stanciu, M., and Brudno, M. (2009). Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods 6, S13–S20. doi: 10.1038/nmeth.1374
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mefford, H. C., and Eichler, E. E. (2009). Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev. 19, 196–204. doi: 10.1016/J.Gde.2009.04.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., and Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877. doi: 10.1093/nar/gki901
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Molodecky, N. A., Soon, I. S., Rabi, D. M., Ghali, W. A., Ferris, M., Chernoff, G.,et al. (2012). Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46.e42–54.e42. doi: 10.1053/j.gastro.2011.10.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Momozawa, Y., Mni, M., Nakamura, K., Coppieters, W., Almer, S., Amininejad, L.,et al. (2011). Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat. Genet. 43, 43–47. doi: 10.1038/ng.733
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morgenthaler, S., and Thilly, W. G. (2007). A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutat. Res. 615, 28–56. doi: 10.1016/j.mrfmmm.2006.09.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morris, A. P., and Zeggini, E. (2010). An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 34, 188–193. doi: 10.1002/gepi.20450
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morrison, A. C., Voorman, A., Johnson, A. D., Liu, X., Yu, J., Li, A.,et al. (2013). Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat. Genet. 45, 899–901. doi: 10.1038/ng.2671
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muona, M., Berkovic, S. F., Dibbens, L. M., Oliver, K. L., Maljevic, S., Bayly, M. A.,et al. (2014). A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat. Genet. 47, 39–46. doi: 10.1038/ng.3144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nance, W. E., Hara, S., Hansen, A., Elliott, J., Lewis, M., and Chown, B. (1969). Genetic linkage studies in a Negro kindred with Norrie’s disease. Am. J. Hum. Genet. 21, 423–429.
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A.,et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/Nature11011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ng, P. C., and Henikoff, S. (2001). Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. doi: 10.1101/gr.176601
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M.,et al. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–34. doi: 10.1038/Ng.499
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S.,et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/Ng.835
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P.,et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Panagiotou, O. A., Willer, C. J., Hirschhorn, J. N., and Ioannidis, J. P. (2013). The power of meta-analysis in genome Wide Association Studies. Annu. Rev. Genomics Hum. Genet. 14, 441. doi: 10.1146/annurev-genom-091212-153520
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680. doi: 10.1038/nrg2641
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pelak, K., Shianna, K. V., Ge, D., Maia, J. M., Zhu, M., Smith, J. P.,et al. (2010). The characterization of twenty sequenced human genomes. PLoS Genet. 6:e1001111. doi: 10.1371/journal.pgen.1001111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Price, A. L., Kryukov, G. V., De Bakker, P. I., Purcell, S. M., Staples, J., Wei, L.-J.,et al. (2010). Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 86, 832–838. doi: 10.1016/j.ajhg.2010.04.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Psaty, B. M., O’Donnell, C. J., Gudnason, V., Lunetta, K. L., Folsom, A. R., Rotter, J. I.,et al. (2009). Cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2, 73–80. doi: 10.1161/CIRCGENETICS.108.829747
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P.,et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/Nature12975
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramensky, V., Bork, P., and Sunyaev, S. (2002). Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900. doi: 10.1093/nar/gkf493
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rausch, T., Zichner, T., Schlattl, A., Stutz, A. M., Benes, V., and Korbel, J. O. (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. doi: 10.1093/bioinformatics/bts378
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reumers, J., De Rijk, P., Zhao, H., Liekens, A., Smeets, D., Cleary, J.,et al. (2012). Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 30, 61–68. doi: 10.1038/Nbt.2053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ritchie, G. R., Dunham, I., Zeggini, E., and Flicek, P. (2014). Functional annotation of noncoding sequence variants. Nat. Methods 11, 294–296. doi: 10.1038/nmeth.2832
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ritz, A., Bashir, A., Sindi, S., Hsu, D., Hajirasouliha, I., and Raphael, B. J. (2014). Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics. 30, 3458–3466. doi: 10.1093/bioinformatics/btu714
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rivas, M. A., Beaudoin, M., Gardet, A., Stevens, C., Sharma, Y., Zhang, C. K.,et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073. doi: 10.1038/ng.952
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Robinson, E. B., Samocha, K. E., Kosmicki, J. A., Mcgrath, L., Neale, B. M., Perlis, R. H.,et al. (2014). Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl. Acad. Sci. U.S.A. 111, 15161–15165. doi: 10.1073/Pnas.1409204111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J.,et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/Nature10945
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sboner, A., Mu, X. J., Greenbaum, D., Auerbach, R. K., and Gerstein, M. B. (2011). The real cost of sequencing: higher than you think! Genome Biol. 12, 125. doi: 10.1186/gb-2011-12-8-125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schalkwyk, L. C., Meaburn, E. L., Smith, R., Dempster, E. L., Jeffries, A. R., Davies, M. N.,et al. (2010). Allelic Skewing of DNA Methylation Is Widespread across the Genome. Am. J. Hum. Genet. 86, 196–212. doi: 10.1016/J.Ajhg.2010.01.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schrock, E., Du Manoir, S., Veldman, T., Schoell, B., Wienberg, J., Ferguson-Smith, M. A.,et al. (1996). Multicolor spectral karyotyping of human chromosomes. Science 273, 494–497. doi: 10.1126/science.273.5274.494
Schumacher, A., Kapranov, P., Kaminsky, Z., Flanagan, J., Assadzadeh, A., Yau, P.,et al. (2006). Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res. 34, 528–542. doi: 10.1093/Nar/Gkj461
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shihab, H. A., Rogers, M. F., Gough, J., Mort, M., Cooper, D. N., Day, I. N.,et al. (2015). An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. doi: 10.1093/bioinformatics/btv009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sifrim, A., Popovic, D., Tranchevent, L. C., Ardeshirdavani, A., Sakai, R., Konings, P.,et al. (2013). eXtasy: variant prioritization by genomic data fusion. Nat. Methods 10, 1083–1084. doi: 10.1038/nmeth.2656
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sindi, S., Helman, E., Bashir, A., and Raphael, B. J. (2009). A geometric approach for classification and comparison of structural variants. Bioinformatics 25, i222–i230. doi: 10.1093/bioinformatics/btp208
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singleton, M. V., Guthery, S. L., Voelkerding, K. V., Chen, K., Kennedy, B., Margraf, R. L.,et al. (2014). Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am. J. Hum. Genet. 94, 599–610. doi: 10.1016/j.ajhg.2014.03.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Speed, D., Hemani, G., Johnson, M. R., and Balding, D. J. (2012). Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 91, 1011–1021. doi: 10.1016/J.Ajhg.2012.10.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
St George-Hyslop, P. H., Haines, J. L., Farrer, L. A., Polinsky, R., Van Broeckhoven, C., Goate, A.,et al. (1990). Genetic linkage studies suggest that Alzheimer’s disease is not a single homogeneous disorder. Nature 347, 194–197. doi: 10.1038/347194a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stouffer, S. A., Suchman, E. A., Devinney, L. C., Star, S. A., and Williams, R. M. Jr. (1949). The American Soldier: Adjustment During Army Life (Studies in Social Psychology in World War II, Vol. 1). Princeton: Princeton University Press.
Stratton, M. (2009). Patterns of somatic mutation in human cancer genomes. Chromosome Res. 17, 16–16.
Tan, R., Wang, Y., Kleinstein, S. E., Liu, Y., Zhu, X., Guo, H.,et al. (2014). An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum. Mutat. 35, 899–907. doi: 10.1002/humu.22537
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, Z. Z., and Lin, D. Y. (2013). MASS: meta-analysis of score statistics for sequencing studies. Bioinformatics 29, 1803–1805. doi: 10.1093/bioinformatics/btt280
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, Z. Z., and Lin, D. Y. (2014). Meta-analysis of sequencing studies with heterogeneous genetic associations. Genet. Epidemiol. 38, 389–401. doi: 10.1002/gepi.21798
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Veale, A. M. O. (1960). Introduction to quantitative genetics - falconer,Ds. R. Statist. Soc. Ser. C Appl. Stat. 9, 202–203. doi: 10.2307/2985722
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Visscher, P. M., Brown, M. A., Mccarthy, M. I., and Yang, J. (2012). Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24. doi: 10.1016/j.ajhg.2011.11.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vissers, L. E. L. M., De Ligt, J., Gilissen, C., Janssen, I., Steehouwer, M., De Vries, P.,et al. (2010). A de novo paradigm for mental retardation. Nat. Genet. 42, 1109–1112. doi: 10.1038/Ng.712
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ward, L. D., and Kellis, M. (2012a). HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934. doi: 10.1093/nar/gkr917
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ward, L. D., and Kellis, M. (2012b). Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 30, 1095–1106. doi: 10.1038/nbt.2422
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weatherall, D. J. (2001). Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat. Rev. Genet. 2, 245–255. doi: 10.1038/35066048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weisenfeld, N. I., Yin, S., Sharpe, T., Lau, B., Hegarty, R., Holmes, L.,et al. (2014). Comprehensive variation discovery in single human genomes. Nat. Genet. 46, 1350–1355. doi: 10.1038/ng.3121
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Welter, D., Macarthur, J., Morales, J., Burdett, T., Hall, P., Junkins, H.,et al. (2014). The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006. doi: 10.1093/nar/gkt1229
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wen, Z., Nguyen, H. N., Guo, Z., Lalli, M. A., Wang, X., Su, Y.,et al. (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418. doi: 10.1038/nature13716
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Willer, C. J., Li, Y., and Abecasis, G. R. (2010). METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. doi: 10.1093/bioinformatics/btq340
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Witte, J. S., Visscher, P. M., and Wray, N. R. (2014). The contribution of genetic variants to disease depends on the ruler. Nat. Rev. Genet. 15, 765–776. doi: 10.1038/Nrg3786
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Worthey, E. A., Mayer, A. N., Syverson, G. D., Helbling, D., Bonacci, B. B., Decker, B.,et al. (2011). Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet. Med. 13, 255–262. doi: 10.1097/GIM.0b013e3182088158
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wray, N. R., Goddard, M. E., and Visscher, P. M. (2008). Prediction of individual genetic risk of complex disease. Curr. Opin. Genet. Dev. 18, 257–263. doi: 10.1016/J.Gde.2008.07.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M., and Lin, X. (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93. doi: 10.1016/j.ajhg.2011.05.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, B., Ionita-Laza, J., Roos, J. L., Boone, B., Woodrick, S., Sun, Y.,et al. (2012). De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 44, 1365–1369. doi: 10.1038/Ng.2446
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, B., Roos, J. L., Dexheimer, P., Boone, B., Plummer, B., Levy, S.,et al. (2011). Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43, 864–868. doi: 10.1038/Ng.902
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, J. A., Benyamin, B., Mcevoy, B. P., Gordon, S., Henders, A. K., Nyholt, D. R.,et al. (2010). Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569. doi: 10.1038/Ng.608
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ye, Z. Q., Zhao, S. Q., Gao, G., Liu, X. Q., Langlois, R. E., Lu, H.,et al. (2007). Finding new structural and sequence attributes to predict possible disease association of single amino acid polymorphism (SAP). Bioinformatics 23, 1444–1450. doi: 10.1093/bioinformatics/btm119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yue, P., Melamud, E., and Moult, J. (2006). SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinform. 7:166. doi: 10.1186/1471-2105-7-166
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zaitlen, N., Pasaniuc, B., Sankararaman, S., Bhatia, G., Zhang, J., Gusev, A.,et al. (2014). Leveraging population admixture to characterize the heritability of complex traits. Nat. Genet. 46, 1356–1362. doi: 10.1038/ng.3139
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zuk, O., Hechter, E., Sunyaev, S. R., and Lander, E. S. (2012). The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. U.S.A. 109, 1193–1198. doi: 10.1073/Pnas.1119675109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: next-generation sequencing, genomic epidemiology, study design, statistical methods, genetic etiology
Received: 25 January 2015; Paper pending published: 07 March 2015;
Accepted: 30 March 2015; Published online: 20 April 2015
Citation: Wang Q, Lu Q and Zhao H (2015) A review of study designs and statistical methods for genomic epidemiology studies using next generation sequencing. Front. Genet. 6:149. doi: 10.3389/fgene.2015.00149
Edited by:
Yiran Guo, Children’s Hospital of Philadelphia, USAReviewed by:
Kui Zhang, University of Alabama at Birmingham, USADajiang Liu, University of Michigan, USA
Copyright © 2015 Wang, Lu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyu Zhao, Department of Biostatistics, Yale School of Public Health, 60 College Street, New Haven, CT 06510, USAaG9uZ3l1LnpoYW9AeWFsZS5lZHU=
†These authors have contributed equally to this work.