 Gero Knittel
Gero Knittel Paul Liedgens
Paul Liedgens Hans C. Reinhardt
Hans C. Reinhardt- 1Department of Internal Medicine, University Hospital of Cologne, Cologne, Germany
- 2Cologne Excellence Cluster on Cellular Stress Response in Aging-Associated Diseases, University of Cologne, Cologne, Germany
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia in the Western world and accounts for approximately 30% of adult leukemias and 25% of non-Hodgkin lymphomas. The median age at diagnosis is 72 years. During recent years numerous genetic aberrations have been identified that are associated with an aggressive course of the disease and resistance against genotoxic chemotherapies. The DNA damage-responsive proapoptotic ATM-CHK2-p53 signaling pathway is frequently mutationally inactivated in CLL either through large deletions on chromosome 11q (ATM) or 17p (TP53), or through protein-damaging mutations. Here, we focus on the role of ATM signaling for the immediate DNA damage response, DNA repair and leukemogenesis. We further discuss novel therapeutic concepts for the targeted treatment of ATM-defective CLLs. We specifically highlight the potential use of PARP1 and DNA-PKcs inhibitors for the treatment of ATM-mutant CLL clones. Lastly, we briefly discuss the current state of genetically engineered mouse models of the disease and emphasize the use of these preclinical tools as a common platform for the development and validation of novel therapeutic agents.
Background
Genome maintenance is a major challenge for all life on earth. In mammals, genomic integrity is preserved through mechanisms that ensure the faithful transmission of fully replicated and undamaged DNA during each cell division (Hoeijmakers, 2001, 2009). For this purpose, eukaryotic organisms evolved a complex DNA surveillance program: Prior to mitosis, cells progress through G1/S-, intra-S and G2/M cell cycle checkpoints (Bartek and Lukas, 2007; Reinhardt and Yaffe, 2013). These checkpoints are activated in response to incomplete DNA replication (e.g., due to stalled replication forks), as well as genotoxic damage induced by internal and external sources, such as UV radiation, reactive oxygen species, ionizing radiation (IR) or DNA-damaging chemotherapeutic agents (Weinert, 1998; Zhou and Elledge, 2000; Abraham, 2001; Kastan and Bartek, 2004; Lukas et al., 2004; Bartek and Lukas, 2007). Active checkpoints halt cell cycle progression and thus provide the time necessary to resolve genomic damage (Reinhardt and Yaffe, 2013). If the genotoxic insult exceeds repair capacity, additional signaling cascades, leading to programmed cell death, are activated (Reinhardt and Yaffe, 2013). Thus, DNA damage checkpoints serve as an effective mechanism to provide and maintain genomic stability (Zhou and Elledge, 2000; Kastan and Bartek, 2004; Reinhardt and Yaffe, 2013).
Coherent with a prominent role of the DNA damage response (DDR) in genome maintenance, many DDR-associated genes have been found to be altered in the germline of patients suffering from cancer-prone inherited syndromes, such as Li-Fraumeni (TP53), Ataxia telangiectasia (ATM), Seckel syndrome (ATR), Nijmegen breakage syndrome (NBS1), A-T-like disease (MRE11), Xeroderma pigmentosum (XP complementation groups) or familial breast and ovarian cancer (BRCA1, BRCA2, RAD51C) (Frebourg and Friend, 1992; Lavin and Shiloh, 1997; Lehmann, 2003; O'Driscoll et al., 2003; Shiloh, 2003; Taylor et al., 2004; Nevanlinna and Bartek, 2006; Fackenthal and Olopade, 2007; Meindl et al., 2010). Disabling mutations within DDR genes have been proposed to result in a so-called “mutator phenotype,” which is thought to drive the runaway proliferation of incipient cancer cells through the accumulation of additional cancer-driving or resistance-causing genomic aberrations (Loeb et al., 2003, 2008; Jiricny, 2006; Jackson and Bartek, 2009; Lord and Ashworth, 2012). While defects in DDR genes appear to facilitate malignant transformation, exploiting these genome-destabilizing alterations for targeted anti-cancer therapy offers a promising therapeutic avenue. In this review, we will focus on cancer-associated defects in ATM-mediated DNA double-strand (DSB) repair and their potential targeting. We will further pinpoint the lack of suitable genetically engineered mouse models of CLL as a critical bottleneck for the rapid preclinical evaluation of novel targeted therapies.
DNA Double Strand Break Repair
DSBs can be inflicted by different agents, such as IR and topoisomerase II inhibitors (e.g., etoposide) (Reinhardt and Yaffe, 2009). Mammalian cells use two major DSB repair mechanisms (Figures 1A,B). The error-prone non-homologous end joining (NHEJ) pathway, which does not depend on an intact DNA replication product as a template for repair, can be employed throughout all cell cycle phases (Figure 1B) (Dietlein and Reinhardt, 2014; Dietlein et al., 2014a). NHEJ is primarily used throughout G1-phase, when no intact sister chromatid is available as a template for repair. NHEJ-mediated DSB repair relies on the catalytic activity of the protein kinase DNA-PKcs, which is recruited to the break site through physical interactions with the non-catalytic subunits Ku70 and Ku80 (Lees-Miller and Meek, 2003). DNA-PKcs activity mediates the assembly of additional NHEJ factors, such as XRCC4- and Lig4, which facilitate re-ligation of the DSB ends during NHEJ (Lees-Miller and Meek, 2003). Homologous recombination (HR)-mediated DSB repair is the second DSB repair pathway employed by mammalian cells (Figure 1A). HR is an error-free DSB repair mechanism that requires the presence of an intact DNA replication product, which is used as a template. This template dependence leads to a restriction of HR use to late S- and G2-phase (Chapman et al., 2012; Dietlein and Reinhardt, 2014; Dietlein et al., 2014a). One of the earliest steps of the HR process is resection of the DSB to create a single-stranded 3′-DNA overhang, which is engaged and coated by the single-stranded DNA (ssDNA)-binding protein RPA (Cimprich and Cortez, 2008; Lyndaker and Alani, 2009). RPA is subsequently replaced by RAD51 in an ATM/CHK2/BRCA1/BRCA2/PALB2-dependent process (Sung and Klein, 2006; San Filippo et al., 2008; Heyer et al., 2010; Krejci et al., 2012). This ssDNA overhang then serves to invade the intact sister chromatid as an intact copy for DNA repair (Sung and Klein, 2006; San Filippo et al., 2008; Krejci et al., 2012). During the HR process, RAD51 fulfills a key role by mediating homology search, strand exchange, and Holliday junction formation (Chapman et al., 2012).
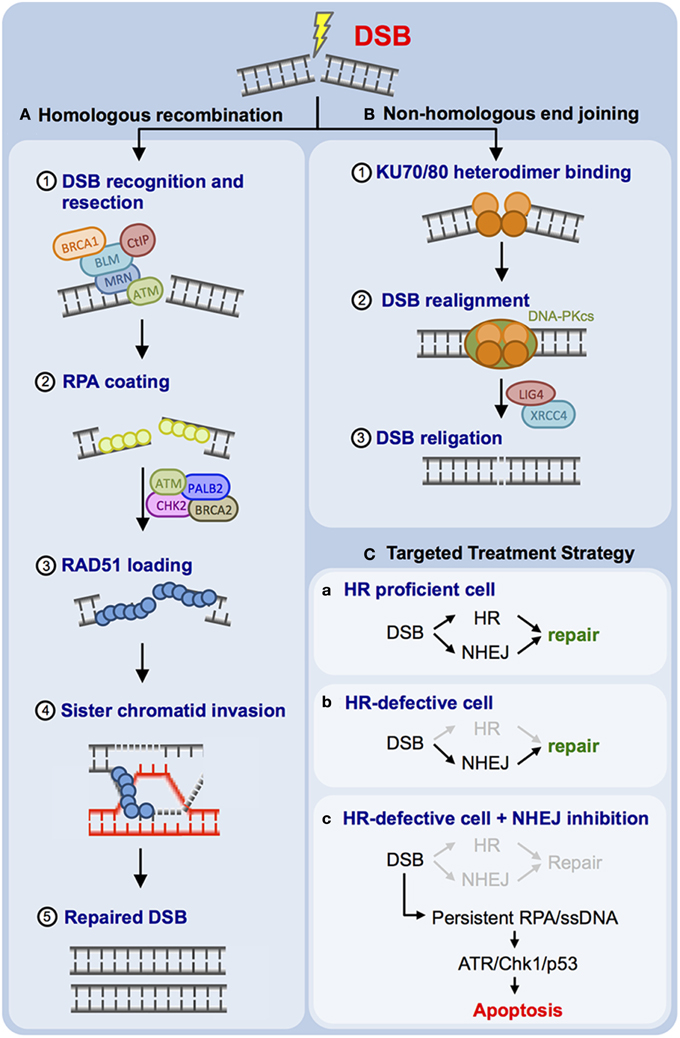
Figure 1. Mammalian cells employ two principal DNA double-strand break (DSB) repair pathways. (A) Schematic representation of the error-free homologous recombination (HR) pathway. DSB resection (1), RPA coating (2), RAD51 coating (3), strand invasion (4), and DSB repair are illustrated (5). (B) Schematic representation of non-homologous end joining (NHEJ). Ku70/Ku80 binding (1), DNA-PK holo-enzyme assembly and recruitment of additional NHEJ factors, such as LIG4 and XRCC4 (2), as well as DSB religation (3) are illustrated. (C) Proposed targeting of HR-defective human cancer through DNA-PKcs inhibition is outlined (for details please refer to the main text).
ATM Signaling and the DNA Damage Response
The proximal DDR kinase ATM, which is mutated in the human cancer-prone disorder Ataxia telangiectasia (A-T), is a key regulator of the cellular DDR and essentially controls three different functional outcomes of DDR signaling: cell cycle checkpoints, DNA repair and apoptosis (Reinhardt and Yaffe, 2009; Shiloh and Ziv, 2013). Immediately following the occurrence of a DSB, the trimeric MRN complex, consisting of MRE11, RAD50 and NBS1, is recruited to the site of the lesion (Chapman and Jackson, 2008; Reinhardt and Yaffe, 2013). In parallel, ATM is activated and tethered to the site of the DSB via a physical interaction with the C-terminus of NBS1 (Falck et al., 2005). ATM subsequently phosphorylates histone H2AX on Ser-139. The resulting phospho-H2AX is commonly referred to as γ-H2AX (Rogakou et al., 1998, 1999; Bartek and Lukas, 2007; Reinhardt and Yaffe, 2009). The phosphorylated Ser-139 residue in the C-terminal region of γ-H2AX subsequently binds with high affinity to the phosphopeptide-recognizing BRCT domains of the mediator protein MDC1 (Lee et al., 2005; Stucki et al., 2005; Lou et al., 2006), which in turn is phosphorylated by ATM at multiple residues (Matsuoka et al., 2007). In addition, MDC1 is phosphorylated by the constitutively active Ser/Thr kinase CK2 (Spycher et al., 2008). The resulting phospho-motif is recognized through the phosphopeptide-binding FHA and/or BRCT domains of NBS1 (Chapman and Jackson, 2008; Spycher et al., 2008; Lloyd et al., 2009). This CK2-dependent NBS1 recruitment retains the MRN complex and NBS1-bound ATM at the DSB site (Melander et al., 2008; Spycher et al., 2008). Thus, MDC1, through ATM- and CK2-directed phosphorylation, tethers both the MRN complex and active ATM at the break site, essentially forming an ATM auto-amplification loop.
Coherent with its role in checkpoint signaling and genome maintenance, ATM is frequently mutated in various human cancer entities, ranging from solid tumors to lymphomas and leukemias (Haidar et al., 2000; Ripolles et al., 2006; Ding et al., 2008; Waddell et al., 2015). Moreover, bi-allelic loss of ATM was shown to be associated with resistance against genotoxic chemotherapy and reduced patient survival (Ripolles et al., 2006; Austen et al., 2007; Skowronska et al., 2012). Recent in vitro experiments suggest that ATM is required for the execution of chemotherapy-induced p53-mediated apoptosis (Jiang et al., 2009). Together these data might rationalize why disabling ATM alterations are a selected genomic aberration in human neoplastic disease.
Intriguingly, ATM is not only a critical mediator of DNA damage-induced apoptosis, but has also been shown to play a major role in DNA repair, specifically HR-mediated DSB repair, with a less well-characterized role in NHEJ (Luo et al., 1996; Dar et al., 1997; Chen et al., 1999; Morrison et al., 2000; Yuan et al., 2003; Kuhne et al., 2004; Riballo et al., 2004; Xie et al., 2004; Bredemeyer et al., 2006; Shrivastav et al., 2009). Experiments performed with ATM-deficient DT40 cells, as well as A-T cells derived from patients have shown that these cells display a mild, but distinct HR defect as the result of impaired assembly and functioning of RAD51-associated protein complexes (Morrison et al., 2000; Shiloh, 2003; Yuan et al., 2003). Specifically, a decreased and delayed formation of RAD51 foci was observed in A-T cells following IR (Shiloh, 2003; Morrison et al., 2000; Yuan et al., 2003). As detailed above, RAD51 recruitment requires an RPA-coated 3′-single-stranded overhang and thus prior DSB resection. This DSB resection process was shown to be ATM-dependent (Adams et al., 2006; Jazayeri et al., 2006; Myers and Cortez, 2006). Further investigation revealed that ATM is specifically involved in HR-mediated DSB repair during the G2-phase of the cell cycle. For instance, it was recently shown that IR-induced sister chromatid exchanges in G2 are ATM-dependent (Beucher et al., 2009; Conrad et al., 2011; Jeggo et al., 2011). Furthermore, CtBP-interacting protein (CtIP), which promotes efficient DSB resection during the HR process, recently emerged as an ATM substrate (Shibata et al., 2011). The rather mild DNA repair defect that is observed in ATM-deficient cells might be explained by the recent observation that ATM appears to control HR-mediated DSB repair specifically in heterochromatin (HC) regions of the genome (Goodarzi et al., 2008, 2010; Jeggo et al., 2011). These experiments revealed that approximately 85% of IR-induced DSBs are rapidly repaired through a largely ATM-independent process. Approximately 15% of IR-induced DSBs are repaired via a slow-acting repair process that depends on ATM (Goodarzi et al., 2010). Intriguingly, DSBs that undergo delayed repair are mainly restricted to areas of the genome that consist of HC (Goodarzi et al., 2010). It was further shown that ATM directly phosphorylates the HC-building factor KAP-1. This KAP-1 phosphorylation allows HR-mediated DSB repair within HC regions. Furthermore, KAP-1-depletion was demonstrated to rescue the DSB repair defect induced by ATM deficiency (Goodarzi et al., 2008, 2010; Jeggo et al., 2011). Altogether these data strongly suggest that the apoptosis-evading effect of ATM-deficiency, which likely stems from insufficient p53 activation, is associated with a potentially druggable HR defect.
Defective ATM-dependent DSB Repair as a Potential Therapeutic Target in CLL
Chronic lymphocytic leukemia (CLL) is a lymphoproliferative disorder that accounts for approximately 30% of adult leukemias and 25% of non-Hodgkin lymphomas (NHL) (Hallek and Pflug, 2010). It is the most common form of leukemia in the western world with an incidence rate of 4-5/100.000 (Hallek and Pflug, 2010). CLL is a disease of the elderly with <10% of the patients being <40 years of age and a median age at diagnosis of 72 years (Hallek and Pflug, 2010). CLL is extraordinarily heterogeneous in its clinical manifestation, treatment response and course. Some patients live for decades and do not require any therapeutic intervention, while others suffer from rapidly progressive and refractory disease (Cramer and Hallek, 2011). It is this extraordinary heterogeneity, which makes treatment of CLL especially challenging. To date, no curative therapy exists besides allogeneic stem cell transplantation, for which most patients do not qualify due to age or reduced performance status. However, it is important to note that we are witnessing a paradigm shift in the treatment of CLL with new, targeted agents recently approved (e.g., ibrutinib, idelalisib), or being evaluated in advanced approval trials (ABT-199). These novel agents interfere directly with B cell receptor signaling (ibrutinib—BTK Inhibitor, idelalisib—PI3Kδ Inhibitor), or relieve repression of the pro-apoptotic proteins BAX and BAK through BCL2 blockade (ABT-199) (for an excellent review, please refer to Thompson et al., 2015).
A hallmark feature of CLL cells is an extraordinarily high frequency of genomic aberrations, which can be documented in more than 80% of CLL patients (Dohner et al., 2000; Di Bernardo et al., 2008; Crowther-Swanepoel et al., 2010; Ouillette et al., 2010). Moreover, the failure of all conventional chemotherapies to induce long-lasting remissions strongly suggests that the apoptosis-mediating DDR is crippled in CLL. The genomic instability of CLL cells is reflected by a number of cytogenetic abnormalities that occur recurrently in CLL. For instance, deletions of the short arm of chromosome 17 (del(17p)) are found in 5–8% of chemotherapy-naïve patients. These deletions almost always include band 17p13, where the prominent tumor suppressor gene TP53 is located. CLL patients carrying a del(17p) clone show marked resistance against genotoxic chemotherapies that cannot be overcome by the addition of anti-CD20 antibodies in the context of state of the art chemo-immunotherapy (Hallek et al., 2010). Among cases with confirmed del(17p), the majority show mutations in the remaining TP53 allele (>80%) (Seiffert et al., 2012). Disabling TP53 mutations are enriched in chemotherapy-treated patients, suggesting that an inactivation of the pro-apoptotic ATM-CHK2-p53 signaling cascade is selected for in CLL (Puente et al., 2011; Quesada et al., 2011).
Deletions of the long arm of chromosome 11 (del(11q)) can be found in approximately 25% of chemotherapy-naïve patients with advanced disease stages and 10% of patients with early stage disease (Zenz et al., 2010; Puente et al., 2011; Quesada et al., 2011). These deletions frequently encompass band 11q23 harboring the ATM gene. A subset of approximately 40% of patients carrying a del(11q) clone display inactivating mutations of the second ATM allele and these cases show a poor chemotherapy response, reminiscent of what has been described for TP53-defective CLLs (Austen et al., 2007). In addition, patients carrying a del(11q) clone typically show rapid progression, and reduced overall survival (Seiffert et al., 2012). As for TP53, disabling ATM mutations are enriched in chemotherapy-treated patients, again suggesting that an inactivation of the pro-apoptotic DDR is selected for in CLL (Puente et al., 2011; Quesada et al., 2011). It remains to be seen whether the novel agents, including ibrutinib, idelalisib, ABT-199, obinotuzumab or lenalidomide might overcome the reduced prognosis of del(17p)/TP53 and del(11q)/ATM altered cases.
Recently, two novel potential therapeutic approaches to specifically treat ATM-deficient neoplastic disease have emerged from in vitro and in vivo experiments performed in different laboratories.
As ATM is involved in HR-mediated DSB repair (Figure 1A), it was proposed that repression of NHEJ, the second prominent DSB repair pathway employed by mammalian cells, might display selective toxicity against ATM-defective cells while sparing healthy cells (Figure 1C) (Gurley and Kemp, 2001; Jiang et al., 2009; Reinhardt et al., 2009; Riabinska et al., 2013; Dietlein and Reinhardt, 2014; Dietlein et al., 2014a,b). Early experiments performed with ATM−/− and PRKDC−/− (encoding DNA-PKcs) mice revealed that double knockout animals undergo early embryonic lethality (E7.5), while single knockout animals were born alive (Xu et al., 1996; Gao et al., 1998; Gurley and Kemp, 2001). These data revealed a robust synthetic lethal interaction between ATM and PRKDC and suggest that pharmacological interception of DNA-PKcs signaling might be detrimental to ATM-defective del(11q) CLLs. Consistent with this hypothesis, combined depletion of Atm and Prkdc in Myc-driven transplanted murine lymphomas led to a massive sensitization of these lymphomas against the anthracycline doxorubicine (Figure 1C) (Jiang et al., 2009; Reinhardt et al., 2009). Pharmacological DNA-PKcs inhibition has recently been evaluated in preclinical systems (Figure 1C). DNA-PKcs repression with the ATP-competitive small molecule inhibitor KU-0060648 resulted in robust induction of apoptosis of ATM-defective cells in vitro (Riabinska et al., 2013). Furthermore, KU-0060648 displayed substantial cytotoxicity against Atm-depleted Myc-driven murine lymphomas, while Atm-proficient lymphomas were entirely resistant (Riabinska et al., 2013). The authors next extended their observations to freshly isolated CLL cells. While KU-0060648 displayed marked single agent activity against del(11q) CLL cells, cytogenetically normal cells did not show any apoptosis following drug exposure (Riabinska et al., 2013). Further analyses revealed that DNA-PKcs inhibition in ATM-defective cells prevents effective DSB repair (Riabinska et al., 2013). On a molecular level, the authors showed that KU-0060648-exposed ATM-defective cells initiate DSB resection and accumulate RPA-coated ssDNA intermediates. These structures ultimately trigger apoptotic cell death through activation of the RPA/ATRIP/ATR/CHK1/p53/Puma apoptotic signaling cascade (Riabinska et al., 2013). Further experiments showed that not only ATM-deficiency, but also other HR-impairing genetic aberrations, such as BRCA1-, BRCA2-, FANCD2- or RAD50 mutations were associated with DNA-PKcs dependence (Dietlein et al., 2014b). Together these data suggest that DNA-PKcs inhibitors either as single agents or in combination with DSB-inducing chemotherapeutics might be a viable treatment option for del(11q) CLLs. Intriguingly, Celgene has developed CC-115, a small molecule compound that is currently being evaluated in phase I/II clinical trials as a combined DNA-PKcs/mTOR inhibitor for the treatment of both solid tumors and hematological malignancies, including CLL (ClinicalTrials.gov identifier: NCT01353625).
A second potential therapeutic approach for ATM-defective human neoplastic disease has recently emerged from preclinical model systems. Different groups have shown that PARP1 inhibitors display selective toxicity against ATM-defective cells (Williamson et al., 2012; Gilardini Montani et al., 2013; Kubota et al., 2014) (Figures 2A–C). PARP1 inhibitors have recently gained the attention of the biomedical community, as they have been demonstrated to selectively eradicate BRCA1- or BRCA2-deficient cells and tumors (Figure 2C) (Bryant et al., 2005; Farmer et al., 2005). PARP1 inhibitor treatment was shown to induce DNA damage in BRCA1 or BRCA2-proficient and -deficient cells (Farmer et al., 2005). However, only BRCA1 or BRCA2-defective cells were sensitive to PARP1 inhibition, while BRCA1/2 wildtype cells were PARP1 inhibitor-resistant (Farmer et al., 2005). Subsequent experiments revealed that additional DNA repair-disabling cancer-associated mutations in genes such as RAD51, RAD54, DSS1, RPA1, NBS1, ATR, ATM, CHK1, CHK2, FANCD2, FANCA, or FANCC were also associated with PARP1 inhibitor sensitivity (McCabe et al., 2006). These results motivated additional experiments that tested the hypothesis that ATM deficiency could be an actionable genetic alteration that might be susceptible to PARP1 inhibition. In this regard, four pieces of data have recently been published. First, RNA interference-mediated ATM repression was shown to sensitize MCF-7 and ZR-75-1 breast cancer cells (ER-positive, HER2-negative, BRCA1/2 wildtype, TP53 wildtype) to the PARP1 inhibitor olaparib (Gilardini Montani et al., 2013). Second, a focused gastric cancer cell line screen revealed that low ATM protein expression significantly correlated with olaparib sensitivity (Kubota et al., 2014). A further characterization revealed that pharmacological- or RNA-interference-mediated repression of ATM kinase activity enhanced olaparib sensitivity in gastric cancer cell lines with parallel depletion or inactivation of p53 (Kubota et al., 2014). In addition to these solid tumor entities, PARP inhibitors have also been evaluated in hematological malignancies. In mantle cell lymphoma xenograft transplants it was recently shown that animals carrying lymphomas lacking both ATM and TP53 (UPN2) displayed significant olaparib sensitivity. Similarly, in mice transplanted with lymphomas lacking ATM and one copy of TP53, olaparib induced a significant survival gain. In contrast, mice transplanted with ATM- and p53-proficient lymphomas (JVM-2), or lymphomas with isolated p53 inactivation (HBL-2), did not derive a survival benefit from olaparib (Williamson et al., 2010, 2012). Lastly, proliferating primary ATM-deficient CLL cells were shown to display increased olaparib sensitivity, compared to ATM-proficient counterparts (Weston et al., 2010). Both genetic and pharmacological experiments validated that this effect was ATM-dependent (Weston et al., 2010). Furthermore, the authors employed a murine xenograft model of an ATM-mutant mantle cell lymphoma cell line to demonstrate a significantly reduced lymphoma burden and an increased survival of animals following olaparib treatment in vivo (Weston et al., 2010). Altogether, these data suggest that PARP1 inhibition might be a useful strategy for the treatment of refractory ATM-defective CLLs (Figure 2C).
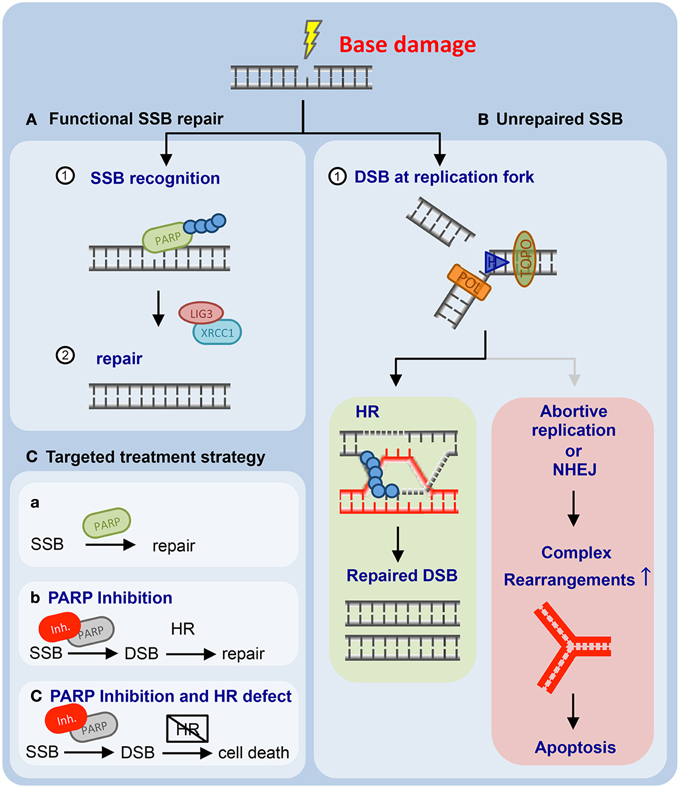
Figure 2. Mammalian cells employ base excision repair to resolve single-strand breaks (SSBs) and non-helix-distorting base modifications. (A) Unperturbed base excision repair (BER) requires PARP1 and LIG3 and XRCC1. (B) PARP1 inhibition leads to the accumulation of genotoxic lesions that are subsequently repaired through homologous recombination (HR)-mediated DNA repair (left panel). If HR-mediated DNA repair is unavailable, PARP1 inhibitor-induced genotoxic damage accumulates and ultimately results in apoptotic cells death (right panel). (C) Proposed targeting of HR-defective human cancer through PARP1 inhibition is outlined (for details please refer to the main text).
Perspectives
One of the biggest hurdles in preclinical CLL research and preclinical development of targeted CLL therapeutics is the lack of mouse models that faithfully mimic the genetic events leading to human CLL development. Although several models exist (for an excellent review, please refer to Simonetti et al., 2014), none of these models truly recapitulates the multistep leukemogenesis typically observed in CLL patients. Specifically the high-risk aberrations, such as Tp53- or Atm deletion/mutation are thus far not sufficiently recapitulated. Although Tp53−/− mice have been crossed with Eμ-Tcl1 transgenic animals, the resulting compound-mutant Eμ-Tcl1;Tp53−/− mice carried a homozygous germline deletion of Tp53, which limits their use as a preclinical model to mirror somatic del(17p) or TP53-mutation in CLL (Liu et al., 2014). Of note, Eμ-Tcl1;Tp53−/− mice develop B-CLL substantially earlier than Eμ-Tcl1 mice with an early appearance of CD5+/IgM+ B cells in the spleen (Liu et al., 2014). These animals display an aggressive course of disease development, as well as a drug resistance phenotype reminiscent of human del(17p) CLL (Liu et al., 2014). These data suggest that a B cell-specific conditional Tp53 deletion, for instance through the use of Cd19-CreERT2 deleter mice on the Eμ-Tcl1 background, might be a useful experimental strategy to faithfully mimic clonal evolution of p53-defective CLL. In addition, B cell-specific conditional Atm deletion using the recently published Atmfl allele (Zha et al., 2008) should be performed with Cd19-CreERT2 deleter mice in the Eμ-Tcl1 background. Furthermore, it is desirable to translate recent large scale CLL genome sequencing data into preclinical platforms. For instance, generation of mice carrying a B cell-specific Myd88L265P mutation, which has recently been described as a potential early driver lesion in CLL (Landau et al., 2013), should be pursued (Figure S1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the members of the Reinhardt lab for helpful discussions. We apologize to our colleagues for the omission of important contributions and their references, due to space limitations. This work was supported by the Volkswagenstiftung (HCR), the Deutsche Forschungsgemeinschaft (KFO-286, RE2246/2-1, HCR), the Helmholtz-Gemeinschaft (HCR), the Deutsche Jose Carreras Stiftung (HCR), the Deutsche Krebshilfe (HCR), the Else Kröner Fresenius Stiftung (HCR), and the Boehringer Ingelheim Stiftung (HCR).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fgene.2015.00207/abstract
Figure S1. Schematic proposal of the use and early integration of genetically engineered mouse models for the development of novel CLL therapeutics. Recent large-scale chronic lymphocytic leukemia (CLL) genome sequencing efforts have unraveled the identity of numerous potential driver mutations in CLL. With this genomic information in hand, we propose the generation of novel genetically-engineered mouse models of CLL to serve as a preclinical platform for the identification and validation of novel CLL therapeutics.
References
Abraham, R. T. (2001). Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196. doi: 10.1101/gad.914401
Adams, K. E., Medhurst, A. L., Dart, D. A., and Lakin, N. D. (2006). Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 25, 3894–3904. doi: 10.1038/sj.onc.1209426
Austen, B., Skowronska, A., Baker, C., Powell, J. E., Gardiner, A., Oscier, D., et al. (2007). Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J. Clin. Oncol. 25, 5448–5457. doi: 10.1200/JCO.2007.11.2649
Bartek, J., and Lukas, J. (2007). DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 19, 238–245. doi: 10.1016/j.ceb.2007.02.009
Beucher, A., Birraux, J., Tchouandong, L., Barton, O., Shibata, A., Conrad, S., et al. (2009). ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 28, 3413–3427. doi: 10.1038/emboj.2009.276
Bredemeyer, A. L., Sharma, G. G., Huang, C. Y., Helmink, B. A., Walker, L. M., Khor, K. C., et al. (2006). ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442, 466–470. doi: 10.1038/nature04866
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. doi: 10.1038/nature03443
Chapman, J. R., and Jackson, S. P. (2008). Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 9, 795–801. doi: 10.1038/embor.2008.103
Chapman, J. R., Taylor, M. R., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510. doi: 10.1016/j.molcel.2012.07.029
Chen, G., Yuan, S. S., Liu, W., Xu, Y., Trujillo, K., Song, B., et al. (1999). Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 274 12748–12752. doi: 10.1074/jbc.274.18.12748
Cimprich, K. A., and Cortez, D. (2008). ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627. doi: 10.1038/nrm2450
Conrad, S., Kunzel, J., and Lobrich, M. (2011). Sister chromatid exchanges occur in G2-irradiated cells. Cell Cycle 10, 222–228. doi: 10.4161/cc.10.2.14639
Cramer, P., and Hallek, M. (2011). Prognostic factors in chronic lymphocytic leukemia-what do we need to know? Nat. Rev. Clin. Oncol. 8, 38–47. doi: 10.1038/nrclinonc.2010.167
Crowther-Swanepoel, D., Broderick, P., Di Bernardo, M. C., Dobbins, S. E., Torres, M., Mansouri, M., et al. (2010). Common variants at 2q37.3, 8q24.21, 15q21.3 and 16q24.1 influence chronic lymphocytic leukemia risk. Nat. Genet. 42, 132–136. doi: 10.1038/ng.510
Dar, M. E., Winters, T. A., and Jorgensen, T. J. (1997). Identification of defective illegitimate recombinational repair of oxidatively-induced DNA double-strand breaks in ataxia-telangiectasia cells. Mutat. Res. 384, 169–179. doi: 10.1016/S0921-8777(97)00021-9
Di Bernardo, M. C., Crowther-Swanepoel, D., Broderick, P., Webb, E., Sellick, G., Wild, R., et al. (2008). A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 40, 1204–1210. doi: 10.1038/ng.219
Dietlein, F., and Reinhardt, H. C. (2014). Molecular pathways: exploiting tumor-specific molecular defects in DNA repair pathways for precision cancer therapy. Clin. Cancer Res. 20, 5882–5887. doi: 10.1158/1078-0432.CCR-14-1165
Dietlein, F., Thelen, L., Jokic, M., Jachimowicz, R. D., Ivan, L., Knittel, G., et al. (2014b). A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 4, 592–605. doi: 10.1158/2159-8290.CD-13-0907
Dietlein, F., Thelen, L., and Reinhardt, H. C. (2014a). Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 30, 326–339. doi: 10.1016/j.tig.2014.06.003
Ding, L., Getz, G., Wheeler, D. A., Mardis, E. R., McLellan, M. D., Cibulskis, K., et al. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. doi: 10.1038/nature07423
Dohner, H., Stilgenbauer, S., Benner, A., Leupolt, E., Krober, A., Bullinger, L., et al. (2000). Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 343, 1910–1916. doi: 10.1056/nejm200012283432602
Fackenthal, J. D., and Olopade, O. I. (2007). Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer 7, 937–948. doi: 10.1038/nrc2054
Falck, J., Coates, J., and Jackson, S. P. (2005). Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611. doi: 10.1038/nature03442
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. doi: 10.1038/nature03445
Frebourg, T., and Friend, S. H. (1992). Cancer risks from germline p53 mutations. J. Clin. Invest. 90, 1637–1641. doi: 10.1172/JCI116034
Gao, Y., Chaudhuri, J., Zhu, C., Davidson, L., Weaver, D. T., and Alt, F. W. (1998). A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity 9, 367–376. doi: 10.1016/S1074-7613(00)80619-6
Gilardini Montani, M. S., Prodosmo, A., Stagni, V., Merli, D., Monteonofrio, L., Gatti, V., et al. (2013). ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J. Exp. Clin. Cancer Res. 32:95. doi: 10.1186/1756-9966-32-95
Goodarzi, A. A., Jeggo, P., and Lobrich, M. (2010). The influence of heterochromatin on DNA double strand break repair: getting the strong, silent type to relax. DNA Repair (Amst). 9, 1273–1282. doi: 10.1016/j.dnarep.2010.09.013
Goodarzi, A. A., Noon, A. T., Deckbar, D., Ziv, Y., Shiloh, Y., Lobrich, M., et al. (2008). ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177. doi: 10.1016/j.molcel.2008.05.017
Gurley, K. E., and Kemp, C. J. (2001). Synthetic lethality between mutation in Atm and DNA-PK(cs) during murine embryogenesis. Curr. Biol. 11, 191–194. doi: 10.1016/S0960-9822(01)00048-3
Haidar, M. A., Kantarjian, H., Manshouri, T., Chang, C. Y., O'Brien, S., Freireich, E., et al. (2000). ATM gene deletion in patients with adult acute lymphoblastic leukemia. Cancer 88, 1057–1062. doi: 10.1002/(SICI)1097-0142(20000301)88:5<1057::AID-CNCR16>3.0.CO;2-6
Hallek, M., Fischer, K., Fingerle-Rowson, G., Fink, A. M., Busch, R., Mayer, J., et al. (2010). Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 376, 1164–1174. doi: 10.1016/s0140-6736(10)61381-5
Hallek, M., and Pflug, N. (2010). Chronic lymphocytic leukemia. Ann. Oncol. 21(Suppl. 7), vii154–vii164. doi: 10.1093/annonc/mdq373
Heyer, W. D., Ehmsen, K. T., and Liu, J. (2010). Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139. doi: 10.1146/annurev-genet-051710-150955
Hoeijmakers, J. H. (2001). Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. doi: 10.1038/35077232
Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485. doi: 10.1056/nejmra0804615
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jazayeri, A., Falck, J., Lukas, C., Bartek, J., Smith, G. C., Lukas, J., et al. (2006). ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 8, 37–45. doi: 10.1038/ncb1337
Jeggo, P. A., Geuting, V., and Lobrich, M. (2011). The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 101, 7–12. doi: 10.1016/j.radonc.2011.06.019
Jiang, H., Reinhardt, H. C., Bartkova, J., Tommiska, J., Blomqvist, C., Nevanlinna, H., et al. (2009). The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 23, 1895–1909. doi: 10.1101/gad.1815309
Jiricny, J. (2006). The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 7, 335–346. doi: 10.1038/nrm1907
Kastan, M. B., and Bartek, J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. doi: 10.1038/nature03097
Krejci, L., Altmannova, V., Spirek, M., and Zhao, X. (2012). Homologous recombination and its regulation. Nucleic Acids Res. 40, 5795–5818. doi: 10.1093/nar/gks270
Kubota, E., Williamson, C. T., Ye, R., Elegbede, A., Peterson, L., Lees-Miller, S. P., et al. (2014). Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 13, 2129–2137. doi: 10.4161/cc.29212
Kuhne, M., Riballo, E., Rief, N., Rothkamm, K., Jeggo, P. A., and Lobrich, M. (2004). A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 64, 500–508. doi: 10.1158/0008-5472.CAN-03-2384
Landau, D. A., Carter, S. L., Stojanov, P., McKenna, A., Stevenson, K., Lawrence, M. S., et al. (2013). Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–26. doi: 10.1016/j.cell.2013.01.019
Lavin, M. F., and Shiloh, Y. (1997). The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol. 15, 177–202. doi: 10.1146/annurev.immunol.15.1.177
Lee, M. S., Edwards, R. A., Thede, G. L., and Glover, J. N. (2005). Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J. Biol. Chem. 280, 32053–32056. doi: 10.1074/jbc.C500273200
Lees-Miller, S. P., and Meek, K. (2003). Repair of DNA double strand breaks by non-homologous end joining. Biochimie 85, 1161–1173. doi: 10.1016/j.biochi.2003.10.011
Lehmann, A. R. (2003). DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85, 1101–1111. doi: 10.1016/j.biochi.2003.09.010
Liu, J., Chen, G., Feng, L., Zhang, W., Pelicano, H., Wang, F., et al. (2014). Loss of p53 and altered miR15-a/16-1short right arrowMCL-1 pathway in CLL: insights from TCL1-Tg:p53(-/-) mouse model and primary human leukemia cells. Leukemia 28, 118–128. doi: 10.1038/leu.2013.125
Lloyd, J., Chapman, J. R., Clapperton, J. A., Haire, L. F., Hartsuiker, E., Li, J., et al. (2009). A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 139, 100–111. doi: 10.1016/j.cell.2009.07.043
Loeb, L. A., Bielas, J. H., and Beckman, R. A. (2008). Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 68, 3551–3557; discussion 3557. doi: 10.1158/0008-5472.can-07-5835
Loeb, L. A., Loeb, K. R., and Anderson, J. P. (2003). Multiple mutations and cancer. Proc. Natl. Acad. Sci. U.S.A. 100, 776–781. doi: 10.1073/pnas.0334858100
Lord, C. J., and Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature 481, 287–294. doi: 10.1038/nature10760
Lou, Z., Minter-Dykhouse, K., Franco, S., Gostissa, M., Rivera, M. A., Celeste, A., et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200. doi: 10.1016/j.molcel.2005.11.025
Lukas, J., Lukas, C., and Bartek, J. (2004). Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst). 3, 997–1007. doi: 10.1016/j.dnarep.2004.03.006
Luo, C. M., Tang, W., Mekeel, K. L., DeFrank, J. S., Anne, P. R., and Powell, S. N. (1996). High frequency and error-prone DNA recombination in ataxia telangiectasia cell lines. J. Biol. Chem. 271, 4497–4503. doi: 10.1074/jbc.271.8.4497
Lyndaker, A. M., and Alani, E. (2009). A tale of tails: insights into the coordination of 3' end processing during homologous recombination. Bioessays 31, 315–321. doi: 10.1002/bies.200800195
Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R. III., Hurov, K. E., Luo, J., et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. doi: 10.1126/science.1140321
McCabe, N., Turner, N. C., Lord, C. J., Kluzek, K., Bialkowska, A., Swift, S., et al. (2006). Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 66, 8109–8115. doi: 10.1158/0008-5472.CAN-06-0140
Meindl, A., Hellebrand, H., Wiek, C., Erven, V., Wappenschmidt, B., Niederacher, D., et al. (2010). Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 42, 410–414. doi: 10.1038/ng.569
Melander, F., Bekker-Jensen, S., Falck, J., Bartek, J., Mailand, N., and Lukas, J. (2008). Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 181, 213–226. doi: 10.1083/jcb.200708210
Morrison, C., Sonoda, E., Takao, N., Shinohara, A., Yamamoto, K., and Takeda, S. (2000). The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 19, 463–471. doi: 10.1093/emboj/19.3.463
Myers, J. S., and Cortez, D. (2006). Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 281, 9346–9350. doi: 10.1074/jbc.m513265200
Nevanlinna, H., and Bartek, J. (2006). The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 25, 5912–5919. doi: 10.1038/sj.onc.1209877
O'Driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A., and Goodship, J. A. (2003). A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501. doi: 10.1038/ng1129
Ouillette, P., Fossum, S., Parkin, B., Ding, L., Bockenstedt, P., Al-Zoubi, A., et al. (2010). Aggressive chronic lymphocytic leukemia with elevated genomic complexity is associated with multiple gene defects in the response to DNA double-strand breaks. Clin. Cancer Res. 16, 835–847. doi: 10.1158/1078-0432.CCR-09-2534
Puente, X. S., Pinyol, M., Quesada, V., Conde, L., Ordóñez, G. R., Villamor, N., et al. (2011). Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475, 101–105. doi: 10.1038/nature10113
Quesada, V., Conde, L., Villamor, N., Ordonez, G. R., Jares, P., Bassaganyas, L., et al. (2011). Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 44, 47–52. doi: 10.1038/ng.1032
Reinhardt, H. C., Jiang, H., Hemann, M. T., and Yaffe, M. B. (2009). Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle 8, 3112–3119. doi: 10.4161/cc.8.19.9626
Reinhardt, H. C., and Yaffe, M. B. (2009). Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255. doi: 10.1016/j.ceb.2009.01.018
Reinhardt, H. C., and Yaffe, M. B. (2013). Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 14, 563–580. doi: 10.1038/nrm3640
Riabinska, A., Daheim, M., Herter-Sprie, G. S., Winkler, J., Fritz, C., Hallek, M., et al. (2013). Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 5, 189ra78. doi: 10.1126/scitranslmed.3005814
Riballo, E., Kuhne, M., Rief, N., Doherty, A., Smith, G. C., Recio, M. J., et al. (2004). A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 16, 715–724. doi: 10.1016/j.molcel.2004.10.029
Ripolles, L., Ortega, M., Ortuno, F., Gonzalez, A., Losada, J., Ojanguren, J., et al. (2006). Genetic abnormalities and clinical outcome in chronic lymphocytic leukemia. Cancer Genet. Cytogenet. 171, 57–64. doi: 10.1016/j.cancergencyto.2006.07.006
Rogakou, E. P., Boon, C., Redon, C., and Bonner, W. M. (1999). Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905–916. doi: 10.1083/jcb.146.5.905
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., and Bonner, W. M. (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868. doi: 10.1074/jbc.273.10.5858
San Filippo, J., Sung, P., and Klein, H. (2008). Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257. doi: 10.1146/annurev.biochem.77.061306.125255
Seiffert, M., Dietrich, S., Jethwa, A., Glimm, H., Lichter, P., and Zenz, T. (2012). Exploiting biological diversity and genomic aberrations in chronic lymphocytic leukemia. Leuk. Lymphoma 53, 1023–1031. doi: 10.3109/10428194.2011.631638
Shibata, A., Conrad, S., Birraux, J., Geuting, V., Barton, O., Ismail, A., et al. (2011). Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 30, 1079–1092. doi: 10.1038/emboj.2011.27
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155–168. doi: 10.1038/nrc1011
Shiloh, Y., and Ziv, Y. (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210. doi: 10.1038/nrm3546
Shrivastav, M., Miller, C. A., De Haro, L. P., Durant, S. T., Chen, B. P., Chen, D. J., et al. (2009). DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst). 8, 920–929. doi: 10.1016/j.dnarep.2009.05.006
Simonetti, G., Bertilaccio, M. T., Ghia, P., and Klein, U. (2014). Mouse models in the study of chronic lymphocytic leukemia pathogenesis and therapy. Blood 124, 1010–1019. doi: 10.1182/blood-2014-05-577122
Skowronska, A., Parker, A., Ahmed, G., Oldreive, C., Davis, Z., Richards, S., et al. (2012). Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J. Clin. Oncol. 30, 4524–4532. doi: 10.1200/JCO.2011.41.0852
Spycher, C., Miller, E. S., Townsend, K., Pavic, L., Morrice, N. A., Janscak, P., et al. (2008). Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J. Cell Biol. 181, 227–240. doi: 10.1083/jcb.200709008
Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J., and Jackson, S. P. (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226. doi: 10.1016/j.cell.2005.09.038
Sung, P., and Klein, H. (2006). Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 7, 739–750. doi: 10.1038/nrm2008
Taylor, A. M., Groom, A., and Byrd, P. J. (2004). Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst). 3, 1219–1225. doi: 10.1016/j.dnarep.2004.04.009
Thompson, P. A., Shpall, E. J., and Keating, M. J. (2015). Shifting paradigms in the treatment of chronic lymphocytic leukemia. Future Oncol. 11, 641–657. doi: 10.2217/fon.14.288
Waddell, N., Pajic, M., Patch, A.-M., Chang, D. K., Kassahn, K. S., et al. (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501. doi: 10.1038/nature14169
Weinert, T. (1998). DNA damage checkpoints update: getting molecular. Curr. Opin. Genet. Dev. 8, 185–193. doi: 10.1016/S0959-437X(98)80140-8
Weston, V. J., Oldreive, C. E., Skowronska, A., Oscier, D. G., Pratt, G., Dyer, M. J., et al. (2010). The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 116, 4578–4587. doi: 10.1182/blood-2010-01-265769
Williamson, C. T., Kubota, E., Hamill, J. D., Klimowicz, A., Ye, R., Muzik, H., et al. (2012). Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 4, 515–527. doi: 10.1002/emmm.201200229
Williamson, C. T., Muzik, H., Turhan, A. G., Zamo, A., O'Connor, M. J., Bebb, D. G., et al. (2010). ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 9, 347–357. doi: 10.1158/1535-7163.MCT-09-0872
Xie, A., Puget, N., Shim, I., Odate, S., Jarzyna, I., Bassing, C. H., et al. (2004). Control of sister chromatid recombination by histone H2AX. Mol. Cell 16, 1017–1025. doi: 10.1016/j.molcel.2004.12.007
Xu, Y., Ashley, T., Brainerd, E. E., Bronson, R. T., Meyn, M. S., and Baltimore, D. (1996). Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 10, 2411–2422. doi: 10.1101/gad.10.19.2411
Yuan, S. S., Chang, H. L., and Lee, E. Y. (2003). Ionizing radiation-induced Rad51 nuclear focus formation is cell cycle-regulated and defective in both ATM(-/-) and c-Abl(-/-) cells. Mutat. Res. 525, 85–92. doi: 10.1016/S0027-5107(03)00009-5
Zenz, T., Mertens, D., Kuppers, R., Dohner, H., and Stilgenbauer, S. (2010). From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat. Rev. Cancer 10, 37–50. doi: 10.1038/nrc2764
Zha, S., Sekiguchi, J., Brush, J. W., Bassing, C. H., and Alt, F. W. (2008). Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc. Natl. Acad. Sci. U.S.A. 105, 9302–9306. doi: 10.1073/pnas.0803520105
Keywords: chronic lymphocytic leukemia, DNA damage response, PARP inhibitor, DNA-PKcs inhibitor, precision medicine
Citation: Knittel G, Liedgens P and Reinhardt HC (2015) Targeting ATM-deficient CLL through interference with DNA repair pathways. Front. Genet. 6:207. doi: 10.3389/fgene.2015.00207
Received: 12 March 2015; Accepted: 28 May 2015;
Published: 10 June 2015.
Edited by:
Ashani Weeraratna, The Wistar Institute, USAReviewed by:
Alessandra Montecucco, Consiglio Nazionale delle Ricerche, ItalyReeti Behera, The Wistar Institute, USA
Copyright © 2015 Knittel, Liedgens and Reinhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans C. Reinhardt, Department of Internal Medicine I, University Hospital of Cologne, Weyertal 115b, Bldg. 73/CCG, 50931 Cologne, Germany,Y2hyaXN0aWFuLnJlaW5oYXJkdEB1ay1rb2Vsbi5kZQ==
†These authors have contributed equally to this work.