 Giuseppe Andolfo1†
Giuseppe Andolfo1† Antimo Di Donato1†
Antimo Di Donato1† Reza Darrudi1,2
Reza Darrudi1,2 Angela Errico1
Angela Errico1 Riccardo Aiese Cigliano3
Riccardo Aiese Cigliano3 Maria R. Ercolano1*
Maria R. Ercolano1*- 1Department of Agriculture Sciences, University of Naples ‘Federico II’, Naples, Italy
- 2Department of Horticulture, College of Agriculture and Natural Resources, University of Tehran, Karaj, Iran
- 3Sequentia Biotech Eureka, Barcelona, Spain
Introduction
The Cucurbitaceae family is the second most large horticultural family in terms of economic importance after Solanaceae. It includes several important crops, such as melon (Cucumis melo), watermelon (Citrullus lanatus), cucumber (Cucumis sativus) and many Cucurbita species with edible fruits (Jeffrey, 1980). The genus Cucurbita (2x = 2n = 40), originated in the Americas, encompasses three economically important crop species such as Cucurbita pepo, Cucurbita moschata, and Cucurbita maxima, cultivated throughout temperate, sub-tropical, and tropical regions (Wang et al., 2011). Cucurbita pepo includes a wide assortment of varieties and cultivars, known for their unique fruit shape and color and appreciated for their culinary properties. Among different species of this genus, Cucurbita pepo have the greatest monetary value (Paris, 2008). Botanical classification based on allozyme variation recognized three subspecies in this species including: pepo, ovifera (syn. texana), and fraterna. Paris (1986) classified edible-fruited C. pepo into eight cultivar-groups: Acorn, Crookneck, Scallop, and Straightneck that belong to subsp. ovifera and Pumpkin, Zucchini, Cocozelle, and Vegetable Marrow that belong to subsp. pepo (Paris, 2010). The genome size of Cucurbita spp. is approximately 500 Mb (Arumuganathan and Earle, 1991). Recently, a high-quality draft of C. pepo (subsp. pepo cultivar-group Zucchini) genome with a sequences length of about 265 million base pairs (Mbp) was made available on CucurbiGene database as well as several C. pepo transcriptomes have been explored (Blanca et al., 2011; Wyatt et al., 2015; Vitiello et al., 2016; Xanthopoulou et al., 2016, 2017; Montero-Pau et al., 2017). However, still little is known about the genetic diversity of this noteworthy crop and even less has been done to explore its proteome. High-throughput sequencing of transcriptomes has opened the way to study the genetic and functional information stored within any organism at an unprecedented scale and speed.
Transcriptome generation through RNA sequencing (RNA-seq) is a technology that can be used in the high resolution and broad dynamic range gene expression studies and in the simultaneous understanding of the genes function (Wang et al., 2009). Basically, the protein-coding genes function is inferred by the analysis of structure, function and evolution of the proteins they encode (Guo, 2013). For the characterization of unannotated proteins, can result particularly useful to undertake orthology analysis. Proteome data are important resources for having an overall genome vision but at the same time achieving a high level of accuracy in comparative studies (Andolfo et al., 2014a). To this end, we sequenced and assembled the first transcriptome of zucchini cultivar “True French,” founder of important pathogen resistant commercial varieties and to harness the full potential of such data we performed also an high-quality proteome annotation. A total of 33,966 protein sequences were predicted, functionally annotated and compared to cucumber, melon, watermelon and Arabidopsis proteomes. In addition, disease resistance (R) gene family was finely characterized and several specie-specific R-genes expansion was detected in C. pepo.
Value of the Data
• The transcriptome obtained can be used as reference for gene expression analysis. Genetic and breeding studies will be enhanced by tools and insights developed from this resource.
• The transcriptome sequence data were assembled and annotated to create a C. pepo reference proteome for future genomic works in this species.
• Zucchini is an important crop that lack of molecular genetics information. The transcriptome and proteome released will drive new discovery to understand complex agronomic traits and to identify novel resistance gene loci.
• The predicted proteome and comparative dataset provided will facilitate the understanding of evolutionary mechanisms of expansion/contraction of important gene families, such as resistance genes, in Cucurbita spp.
Experimental Design, Materials and Methods
Plant Material, Total RNA Extraction and Quality Control, Library Preparation and RNA-Seq
Plants of Cucurbita pepo subsp. pepo cultivar-group Zucchini, variety True French, were grown in greenhouse facility at Department of Agricultural Science of University of Naples “Federico II” using standard horticultural practices. C. pepo cv. True French tissue samples were collected from young plants of about 10 cm high. Total RNA was isolated from ground, frozen leaf tissues using the SpectrumTM Plant Total RNA Kit (Sigma-Aldirch). A complete removal of traces of DNA was performed using On-Column DNase I Digest Set (Sigma-Aldirch). Quantity and integrity of the extracted total RNA were determined using NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific Inc., USA), on a denaturing formaldehyde gel and Agilent 2100 bioanalyzer (Agilent Technologies, USA) respectively, to be RIN > 8. Library preparation and sequencing were performed by the Genomix4Life S.r.l., spin-off of Salerno University. The sequencing library was prepared using the TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, CA, USA) and paired-end reads of 100 bp were sequenced from the three independent samples on one lane of an illumine HiSeq 2000.
Preprocessing and Transcriptome Assembly
The quality control checks on raw sequence data (75,22 millions of paired reads totalling 15 e12 bp) from all the three data sets was performed using FastQC (Andrews, 2010). Raw reads were filtered to remove the adapter sequences and the poorer quality regions with sequence pre-processing tool, Trimmomatic (Bolger et al., 2014). Paired-end read duplicates from the PCR amplification step in the sequencing process were removed and only those reads with a mapping score ≥ 30 were kept in the alignments. The high quality reads were aligned against the C. pepo reference genome sequence version 3.2 (https://cucurbigene.upv.es/) with STAR aligner (version 2.4.0j). The resulting alignment was used as input to Cufflinks (version 2.2.1) for transcript assembly. PASA pipeline (version 2.0.2) was used to combine Cufflinks results with the public transcriptome version 3.0 (https://cucurbigene.upv.es/).
Proteome Annotation and Characterization of R-Genes
The proteome functional annotation was performed through a match search against four database (TAIR10, SWISS-PROT, TrEMBL and GenBank-NR) using DIAMOND in sensitive mode with a cut-off e-value of 1 e−5 (Buchfink et al., 2015). To add information about protein function to our proteome, a Blast2GO (Conesa et al., 2005) annotation, using default parameters, were conducted. Finally, the zucchini proteome was scanned with InterProScan v.5.13 (Jones et al., 2014) against the InterPro protein signature databases to identify and finely characterize plant resistance proteins.
Orthology Analysis
To identify orthologous gene groups among C. pepo, C. melo, C. sativus, C. lanatus and A. thaliana we used OrthoMCL software with default settings. The association between reference R-genes (http://prgdb.crg.eu/) and relative orthologous group (OG) was detected using Best BLAST Hit method (BlastP, E < 1 e−5) and the output was filtered for a query coverage and identity percentage, both >50%.
Results and Discussion
Transcriptome Sequencing, Assembly, and Annotation
The sequencing produced a total of 69,5 millions of clean paired reads, obtaining 13,9 e12 bp of RNA-Seq data for C. pepo (Supplementary Table 1). Transcriptome assembly yielded 68,720 transcripts, with mean length of 1,534 bp. The transcripts were translated and a high-quality proteome of 33,966 primary protein sequences, with mean length of 316 AA, were obtained. DIAMOND similarity-based searches were performed against the publically available databases (SWISS-PROT, TrEMBL, TAIR10, and GenBank-NR) to annotate C. pepo proteome (Supplementary Table 2). About 85% of proteins encoded by genes had homology with four principal databases and over 75% were functionally annotated (Table 1). In addition, a GO-annotation using Blast2GO were effected and a total of 256,138 GO-terms were assigned to about 65% (21480) of the predicted proteins (Supplementary Table 3).
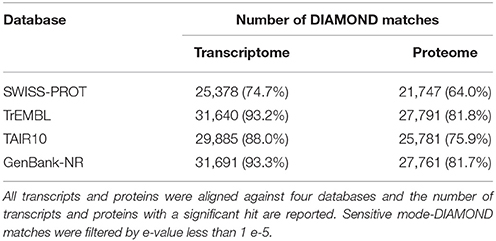
Table 1. Annotation of C. pepo leaf tissues transcriptome based on homology.
R-Gene Annotation
A fine characterization of genes encoding domains similar to plant resistance (R) proteins, in C. pepo proteome was conducted. R-proteins can be categorized according to the presence and organization of protein domains, such as Toll/Interleukin-1 receptor (TIR), coiled coil (CC), the nucleotide-binding site (NBS), leucine-rich repeats (LRRs). A total of 64 R-proteins (also called NLR, NB-LRR, NBS-LRR, or NB-ARC-LRR proteins) were identified (Supplementary Table 4). The CNL (Coiled coil, Nucleotide-binding site, Leucine-rich repeats) class was divided into sub-classes based on sequence similarity with the canonical CNLs that contain an EDVID amino-acid motif, and the RPW8-like proteins (Andolfo et al., 2014b). Interestingly, an expansion of RPW8-NLR genes (11 out of 64) in C. pepo was discovered. Diversely, C. melo, C. sativus, and C. lanatus presented only three RPW8-NLRs for each species (Figure 1A). It is now well-known that RPW8-NLRs can function as helper NLRs for well-defined NLR–mediated resistance responses. Thus, they may enhance the C. pepo defense system to offset its reduced number of NRL receptors available (Sanseverino and Ercolano, 2012). In addition, C. pepo RPW8-NLRs showed a very high homology to ADR1 (activated disease resistance 1), R-gene that confer resistance again Erysiphe cichoracearumi, the causal agent of Powdery Mildew (PM) in A. thaliana (Micali et al., 2008). PM disease, caused by Podosphaera xanthii (syn. Sphaerotheca fuliginea) has an important economic impact on C. pepo varieties. ADR1-like proteins expansion, identified in C. pepo, could suggest an adaptive diversification induced by specie-specific pathogen pressure (Andolfo and Ercolano, 2015).
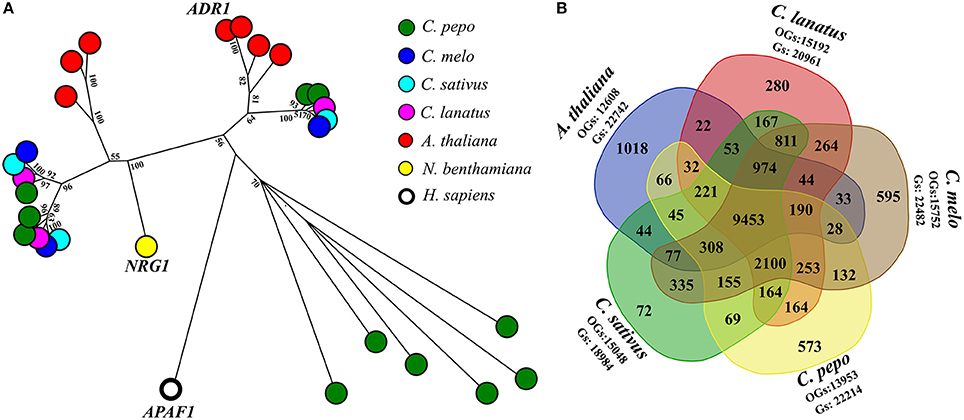
Figure 1. RPW8-NLR genes expansion and comparative analysis. (A) Evolutionary history of RPW8-NLR proteins annotated in Cucurbita pepo. Full NB domains (PF00931) of 26 RPW8-NLR proteins were used together with 3 well characterized reference genes (short gene name: ADR1, NRG1, and APAF1) to do a maximum likelihood analysis based on the Jones et al. w/freq. model. Model with the lowest BIC score (Bayesian Information Criterion) was considered to describe the substitution pattern the best. Sequence similarities were determined performing a ClustalW (default settings) multiple alignment. Evolutionary analyses were conducted in MEGA7. Labels show the bootstrap values higher than 50 (out of 100), are indicated above the branches. The tree is drawn to scale, with branch lengths proportional to the number of substitutions per site. Species to which belong sequences are indicated by colored spots. (B) Venn diagram of genes (Gs) clustered into orthogroups (OGs). Five species (Cucurbita pepo, Citrullus lanatus, Cucumis melo, Cucumis sativus, and A. thaliana) were used to generate the Venn diagram. In the graph were reported the number of specie-specific and common OGs.
Orthologous Groups
A comparative analysis among C. pepo, C. melo, C. sativus, C. lanatus, and A. thaliana were performed to obtain functional information on our proteome. A total of 18,742 orthologous groups (OGs), which included 107,386 sequences, were identified (Supplementary Table 5). 9,453 OGs enclosed 69,982 sequences and were highly conserved in all analyzed genomes (Figure 1B). A core Cucurbitaceae proteins (9,465 sequences) clustered in 2099 OGs were detected. About 65% (22,214) of C. pepo predicted proteins were grouped in 13,953 OGs (Figure 1B). Several C. pepo gene family expansions associated to transmembrane R-genes were discovered. One hundred zucchini proteins, annotated as Receptor-like Kinase (RLK) and Receptor-like Protein (RLP) were clustered in five OGs (OG_00004, OG_00027, OG_00038, OG_00053, OG_01889, and OG_00579) and associated a well-characterized R-genes (Supplementary Table 5). Probably the expansion of cell surface receptors (RLKs and RLPs) and relative strengthening of first defence line represent adaptive dynamics to balance the limited number of cytoplasmic receptors (NRLs) (Andolfo and Ercolano, 2015). The C. pepo gene family expansions could be associated to the cucurbit-common tetraploidization recently identified by Wang et al. (2017). Furthermore, we identified a number of gene families related to important agronomical traits. Fourteen OGs related to OVATE gene family grouped 19 zucchini genes. OVATE is an important locus for fruit shape determination and plant development (Rodríguez et al., 2011). We identified three zucchini ortholog genes to PSY1 and PSY3 melon genes that putatively involved in carotenoid metabolism and fruit ripening (Garcia-Mas et al., 2012). The Cup000085g037789 is the ortholog gene of OR, a cloned gene that governs the fruit flesh colur in melon and in other important crops (Tzuri et al., 2015). Comparative analysis of C. pepo proteome can be used to identify orthologous genes for functional study. Our dataset represented a very important resource to reduce the plant breeding work for the identification of candidates for important agronomical traits (Supplementary Table 5).
Direct Link to Deposited Data and Information to Users
The dataset submitted to NCBI include the raw read sequences of three biological replicates of Cucurbita pepo subsp. pepo cultivar-group Zucchini, variety True French, in FASTQ format. The raw reads of C. pepo can be accessed at NCBI with the following BioSample accession number: SAMN07426850 (www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP114337). The C. pepo transcriptome annotation, in GTF format, and primary protein sequences in FASTA format can be accessed at FIGSHARE with the following link (https://figshare.com/s/8a083f60df238acdbc19). The Supplementary Material (Supplementary Tables 1–5) for this article can be found online at: (https://figshare.com/s/8a083f60df238acdbc19). Users can download and use the data freely for research purpose only with acknowledgment to us and quoting this paper as reference to the data.
Author Contributions
GA was chiefly involved in data analysis, results interpretation and manuscript writing. AD was mainly involved in data analysis, results interpretation and manuscript writing. RD drafted the manuscript. AE provided a critical reading of the manuscript. RA assembled the transcriptome. ME coordinated the project and contributed to data analysis and results interpretation. All of the authors read and approved the final manuscript.
Funding
This work was supported by the Ministry of University and Research (GenHORT project).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Cucurbigene team for giving us the permission to use the C. pepo genome v3.2. Plants of Cucurbita pepo subsp. pepo cultivar-group Zucchini, variety True French, were kindly provided by the Semiorto Sementi Seed Company (Sarno, Italy).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2017.00181/full#supplementary-material
References
Andolfo, G., and Ercolano, M. R. (2015). Plant innate immunity multicomponent model. Front. Plant Sci. 6:987. doi: 10.3389/fpls.2015.00987
Andolfo, G., Ferriello, F., Tardella, L., Ferrarini, A., Sigillo, L., Frusciante, L., et al. (2014a). Tomato genome-wide transcriptional responses to Fusarium wilt, and tomato Mosaic virus. PLoS ONE 9:e94963. doi: 10.1371/journal.pone.0094963
Andolfo, G., Jupe, F., Witek, K., Etherington, G. J., Ercolano, M. R., and Jones, J. D. G. (2014b). Defining the full tomato NB-LRR resistance gene repertoire using genomic and cDNA RenSeq. BMC Plant Biol. 14:120. doi: 10.1186/1471-2229-14-120
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Arumuganathan, K., and Earle, E. D. (1991). Nuclear DNA content of some important plant species nuclear DNA content material and methods. Plant Mol. Biol. Rep. 9, 208–218. doi: 10.1007/BF02672069
Blanca, J., Cañizares, J., Roig, C., Ziarsolo, P., Nuez, F., and Picó, B. (2011). Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics 12:104. doi: 10.1186/1471-2164-12-104
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Garcia-Mas, J., Benjak, A., Sanseverino, W., Bourgeois, M., Mir, G., González, V. M., et al. (2012). The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. U.S.A. 109, 11872–11877. doi: 10.1073/pnas.1205415109
Guo, Y. L. (2013). Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 73, 941–951. doi: 10.1111/tpj.12089
Jeffrey, C. (1980). A review of the Cucurbitaceae. Bot. J. Linn. Soc. 81, 233–247. doi: 10.1111/j.1095-8339.1980.tb01676.x
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Micali, C., Göllner, K., Humphry, M., Consonni, C., and Panstruga, R. (2008). The powdery mildew disease of arabidopsis: a paradigm for the interaction between plants and biotrophic fungi. Arabidopsis Book 6:e0115. doi: 10.1199/tab.0115
Montero-Pau, J., Blanca, J., Bombarely, A., Ziarsolo, P., Esteras, C., Martí-Gómez, C., et al. (2017). De-novo assembly of zucchini genome reveals a whole genome duplication associated with the origin of the Cucurbita genus. bioRxiv. doi: 10.1101/147702
Paris, H. S. (1986). A proposed subspecific classification for Cucurbita pepo. Phytologia 61, 133–138.
Paris, H. S. (2008). “Summer squash,” in Vegetables I. Handbook of Plant Breeding, Vol. 1, eds J. Prohens and F. Nuez (New York, NY: Springer).
Paris, H. S. (2010). History of the Cultivar-Groups of Cucurbita pepo. Horticult. Rev. 25, 71–170. doi: 10.1002/9780470650783.ch2
Rodríguez, G. R., Munos, S., Anderson, C., Sim, S.-C., Michel, A., Causse, M., et al. (2011). Distribution of SUN, OVATE, LC, and FAS in the tomato germplasm and the relationship to fruit shape diversity. Plant Physiol. 156, 275–285. doi: 10.1104/pp.110.167577
Sanseverino, W., and Ercolano, M. R. (2012). In silico approach to predict candidate R proteins and to define their domain architecture. BMC Res. Notes 5:678. doi: 10.1186/1756-0500-5-678
Tzuri, G., Zhou, X., Chayut, N., Yuan, H., Portnoy, V., Meir, A., et al. (2015). A “golden” SNP in CmOr governs the fruit flesh color of melon (Cucumis melo). Plant J. 82, 267–279. doi: 10.1111/tpj.12814
Vitiello, A., Scarano, D., D'Agostino, N., Digilio, M. C., Pennacchio, F., Corrado, G., et al. (2016). Unraveling zucchini transcriptome response to aphids (No. e1635v1). PeerJ.
Wang, J., Sun, P., Li, Y., Liu, Y., Yang, N., Yu, J., et al. (2017). An overlooked paleo-tetraploidization in Cucurbitaceae. Mol. Biol. Evol. doi: 10.1093/molbev/msx242. [Epub ahead of print].
Wang, Y. H., Behera, T. K., and Kole, C. (eds.). (2011). Genetics, Genomics and Breeding of Cucurbits. CRC Press.
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wyatt, L. E., Strickler, S. R., Mueller, L. A., and Mazourek, M. (2015). An acorn squash (Cucurbita pepo ssp. ovifera) fruit and seed transcriptome as a resource for the study of fruit traits in Cucurbita. Hortic. Res. 2:14070. doi: 10.1038/hortres.2014.70
Xanthopoulou, A., Ganopoulos, I., Psomopoulos, F., Manioudaki, M., Moysiadis, T., Kapazoglou, A., et al. (2017). De novo comparative transcriptome analysis of genes involved in fruit morphology of pumpkin cultivars with extreme size difference and development of EST-SSR markers. Gene 622, 50–66. doi: 10.1016/j.gene.2017.04.035
Keywords: RNA-seq, proteome, Cucurbita pepo, R-genes, orthology
Citation: Andolfo G, Di Donato A, Darrudi R, Errico A, Aiese Cigliano R and Ercolano MR (2017) Draft of Zucchini (Cucurbita pepo L.) Proteome: A Resource for Genetic and Genomic Studies. Front. Genet. 8:181. doi: 10.3389/fgene.2017.00181
Received: 04 September 2017; Accepted: 06 November 2017;
Published: 21 November 2017.
Edited by:
Youri I. Pavlov, University of Nebraska Medical Center, United StatesReviewed by:
Alexander V. Rodionov, Botanical Institute VL Komarova (RAS), RussiaXiyin Wang, North China University of Science and Technology, China
Copyright © 2017 Andolfo, Di Donato, Darrudi, Errico, Aiese Cigliano and Ercolano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria R. Ercolano, ZXJjb2xhbm9AdW5pbmEuaXQ=
†These authors have contributed equally to this work.