 Yuan Wang1†
Yuan Wang1† Xiangdong Ding1†Zhen Tan1
Xiangdong Ding1†Zhen Tan1 Chao Ning1Kai Xing1
Chao Ning1Kai Xing1 Ting Yang1Yongjie Pan2Dongxiao Sun1*Chuduan Wang1*
Ting Yang1Yongjie Pan2Dongxiao Sun1*Chuduan Wang1*- 1Key Laboratory of Animal Genetics and Breeding of Ministry of Agriculture, National Engineering Laboratory of Animal Breeding, College of Animal Science and Technology, China Agricultural University, Beijing, China
- 2Beijing Shunxin Agriculture Co., Ltd., Beijing, China
Piglet uniformity (PU) and farrowing interval (FI) are important reproductive traits related to production and economic profits in the pig industry. However, the genetic architecture of the longitudinal trends of reproductive traits still remains elusive. Herein, we performed a genome-wide association study (GWAS) to detect potential genetic variation and candidate genes underlying the phenotypic records at different parities for PU and FI in a population of 884 Large White pigs. In total, 12 significant SNPs were detected on SSC1, 3, 4, 9, and 14, which collectively explained 1–1.79% of the phenotypic variance for PU from parity 1 to 4, and 2.58–4.11% for FI at different stages. Of these, seven SNPs were located within 16 QTL regions related to swine reproductive traits. One QTL region was associated with birth body weight (related to PU) and contained the peak SNP MARC0040730, and another was associated with plasma FSH concentration (related to FI) and contained the SNP MARC0031325. Finally, some positional candidate genes for PU and FI were identified because of their roles in prenatal skeletal muscle development, fetal energy substrate, pre-implantation, and the expression of mammary gland epithelium. Identification of novel variants and candidate genes will greatly advance our understanding of the genetic mechanisms of PU and FI, and suggest a specific opportunity for improving marker assisted selection or genomic selection in pigs.
Introduction
In the pig production industry, reproductive traits such as litter size, litter birth weight, and litter mortality play an important role in the development of production and economic profits. In the past few decades, litter size at birth has been treated as an important criterion to evaluate sow productivity (Southwood and Kennedy, 1991; Blasco et al., 1998). However, piglet survival after birth is greatly negatively affected by increasing litter size. Previous genetic studies have shown that piglet uniformity (PU, the within-litter birth weight variability) is positively correlated with piglet mortality (Milligan et al., 2002; Damgaard et al., 2003), and that low birth weight piglets may experience morbidity and mortality. In addition, the farrowing interval (FI, the number of days between two adjacent litters) in a sow's productive life is also an important index in evaluating reproductive ability (Serenius et al., 2003). FI can be considered a comprehensive trait that contains lactation length, weaning to estrus mating interval, and gestation length. Therefore, more attention was focused on PU and FI (Wolf et al., 2005; Cavalcante Neto et al., 2009) than other traits. Both PU and FI are low-heritability traits, where the estimated heritabilities range from 0.05 to 0.12 (Wittenburg et al., 2008; Cavalcante Neto et al., 2009; Zhang et al., 2016). Although we have already made some progress in traditional genetic improvement of reproductive traits, it is still a challenge to understand the biological mechanisms of complex traits (Andersson et al., 2009), which can be an effective alternative basis for breeding programs.
A total of 16,516 QTL associated with 626 different traits in pigs have been reported in previous studies (http://www.animalgenome.org/cgi-bin/QTLdb/SS/index). Among them, 1,416 QTL are associated with reproductive traits. Fifty QTL for PU have been detected across the majority of chromosomes, but there are still no QTL for FI. Although numerous QTL have been detected in domestic animals (King et al., 2003; Holl et al., 2004), these findings are still insufficient because of the low power of linkage analyses and poor resolution in most QTL (Tabor et al., 2002). In recent years, genome-wide association studies (GWAS) have become a powerful strategy for the detection of variation in different traits based on high throughput SNP platforms. It has been widely used in humans (Hindorff et al., 2009; Lauc et al., 2010; Hoffmann et al., 2017) and domestic animals (Petersen et al., 2013; Kominakis et al., 2017). In pigs, many GWAS have been performed on various economically important traits, including immune traits (Luo et al., 2012; Ponsuksili et al., 2016), meat quality traits (Casiró et al., 2017; Verardo et al., 2017), and structural soundness traits (Fan et al., 2011). However, few GWAS have been conducted to assess the genetic architecture of PU and FI. In addition, it is notable that many complex traits undergo dynamic alterations as animals age (Van de Pol and Verhulst, 2006), so the phenotypes of animals at different stages should be used in GWAS to detect genetic variants and increase the statistical power.
In our study, we performed a GWAS on the PU and FI at 4 different stages from first to fourth parity, based on farrowing records using a PorcineSNP80 BeadChip in a Large White pig population. The purpose of this study was to identify the genomic variants and candidate genes that contribute to the phenotypic variability of PU and FI, and promote the improvement of pig breeding programs.
Materials and Methods
Animals and Phenotypes
A total of 884 Large White pigs from the nucleus pig breeding farm of Beijing Shunxin Agriculture Co., Ltd (http://www.000860.com/sxkg/, Beijing, China) were used in our study. We collected blood samples from the jugular vein using the standard procedure of the breeding program, which was approved by the Animal Welfare Committee of China Agricultural University (GB/T 17236–2008).
All pigs had the same genetic background. Farrowing records were collected from parity 1 to 4 during the years 2010 to 2016. Piglet uniformity was defined as the within-litter birth weight variability, usually represented by the coefficient of variation of birth weights within one litter. Farrowing interval was defined as the number of days between two adjacent litters. The rank-based inverse normal transformation of phenotypic values was performed by the function rntransform in the GenABEL package in R (Aulchenko et al., 2007).
Genotyping and Quality Control
Genomic DNA was extracted from 1-mL blood samples using TIANamp Genomic DNA kits (Tiangen Biotech, Beijing, China). The quality and quantity of the DNA samples were measured with a NanoDrop™ 2000 (Thermo Fisher Scientific, Waltham, MA, USA). All DNA samples were eligible for genotyping with a ratio of light absorption (A260/280) between 1.8 and 2.0, a concentration >50 ng/μL, and total volume <50 μL. Genotyping was conducted using the Porcine SNP80 BeadChip (GeneSeek, Lincoln, Nebraska, USA) which contained 68,528 SNPs across 18 autosomes and two sex chromosomes. Quality control of the genotype data was carried out using Plink software (Purcell et al., 2007). DNA samples with genotyping of <90% of the markers were removed. The SNPs with call rates < 90%, minor allele frequencies <0.03, Hardy–Weinberg equilibrium (HWE) P < 1 × 10−6, and the SNPs with no position information and located on the sex chromosomes were also excluded from the dataset. After quality control, the missing genotypes were imputed using Beagle software (Browning and Browning, 2009) based on the remaining SNP genotypes, SNPs with the highest linkage disequilibrium r2-value larger than 0.3 were retained for further analysis (Yuan et al., 2015).
Genome-Wide Association Studies
GWAS were implemented independently for PU and FI. From parity 1 to 4, most individuals had more than 1 farrowing record which could be treated as “longitudinal data.” A single-SNP GWAS was performed in ASReml software (Gilmour et al., 2009), and testing was done using a Wald F statistic. The repeatability model was as follows:
where Y is the vector of phenotype values; μ is the overall mean; b is the vector of fixed effects including herd, farrowing season, parity, and number of piglets born alive. The fixed effect of number of piglets born alive was only applied in the analysis of PU, where it was classified into four groups: ≤5, 6–11, 12–13, 14–15, and ≥16 (Quesnel et al., 2008); m is the incidence vector of SNP genotype scores with values 0, 1, or 2 corresponding to the three genotypes (11, 12, and 22) of the SNP (where two denotes the allele with a minor frequency); f is the regression coefficient of phenotypes on SNP genotypes; u is the vector of residual polygenetic effects with , where G is the genomic relationship matrix that was constructed through SNP markers (VanRaden, 2008) and is the polygenetic additive variance, compared with pedigree-based relationships, genetic relationship matrices can better remove population structure and control false positives (Kang et al., 2010); pe is the vector of random permanent environmental effects with , where is the permanent environmental variance; e is the vector of residual errors with , where is the residual errors variance; X, Z, and W are the incidence matrices for b, u, and pe, respectively. For most genetic association studies, the effect of any given locus on the trait is very small (Manolio et al., 2009), so we only need to estimate the variance parameters once for each data set, and then apply them to each marker (Kang et al., 2010). We used ASReml software to estimate , , and using average information restricted maximum likelihood.
Since Bonferroni correction is overly conservative and may produce false negative results (Johnson et al., 2010), we used the false discovery rate (FDR) (Benjamini and Hochberg, 1995; Weller et al., 1998) to determine the threshold values. FDR was set as 0.01, and the threshold P-value was calculated as follows:
where n is the number of P < 0.01 in the results, and m is the total number of SNPs.
The Manhattan plots and quantile-quantile (QQ) plots were drawn by R packages (http://cran.r-project.org/web/packages/gap/index.html). Genomic inflation factor λ was calculated to judge the extent of false positive signals with the estlambda function in the GenABEL packages in R.
GCTA software (Yang et al., 2011) was used to calculate phenotypic variances contributed by significant SNPs for each parity. The genetic relationship matrix (GRM) was created based on genotyped SNPs on chromosomes, and fixed effects (herd, farrowing season, and number of piglets born alive) were treated as covariates to account for potential population structure. The liner mixed model as follows:
where Y is an n× 1 vector of phenotypic values for n individuals, β is a vector of fixed effects with its incidence matrix X, gG1 is a vector of aggregate effects of the selected significant SNPs with its incidence matrix Z1, and where VG1 represents the selected SNPs-derived GRM with its additive genetic variance , gG2 is a vector of aggregate effects of the other SNPs except the selected significant SNPs and Z2 is an incidence matrix for gG2, and where VG2 represents the other SNPs-derived GRM with its additive genetic variance , e is the vector of residual errors with , where is the residual errors variance and I is an identity matrix. In this study, the phenotypic variance explained by selected SNPs is defined as where is the phenotypic variance.
Haplotype Block Analysis
To further detect candidate regions associated with the two traits, we conducted linkage disequilibrium analysis for the chromosomal regions with multiple significant SNPs using Haploview v4.2 (Barrett et al., 2005). A block was defined using the solid spin algorithm by the criteria of Gabriel et al. (2002).
Gene Search and Functional Annotation
The functional genes containing or near (within 1 Mb) the identified significant SNPs were selected based on the swine genome assembly 10.2 (https://www.ensembl.org/Sus_scrofa/Info/Index). The annotations of genes were carried out using the NCBI database (https://www.ncbi.nlm.nih.gov/) based on the description of gene function and related literatures, simultaneously, gene ontology (GO) analysis was conducted using the DAVID Bioinformatics Resources (https://david.ncifcrf.gov) for PU and FI, respectively. The Fisher's exact test was used to assess the significance of the enriched terms (Dennis et al., 2003; Rivals et al., 2007), and the enriched GO terms with the P < 0.05 were selected to explore the genes involved in biological processes (Wang et al., 2015; Xing et al., 2016).
Results
Phenotype and SNP Data Statistics
Descriptive statistics of phenotypes of PU and FI from parity 1 to 4 are shown in Table 1. Neither trait had approximately normal distributions, so the rntransform function in the GenABEL package was used to convert these phenotypes for further analysis.
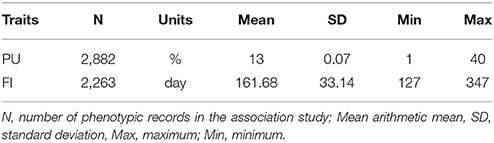
Table 1. Descriptive statistics of PU and FI in a large white population.
After quality control, 880 individuals with a genotyping call rates >90% were retained, and 51,727 SNPs were available for the GWAS. The average distance between the neighboring SNPs on each chromosome was calculated (Table S1). The number of SNPs on each chromosome ranged from 1,424 (SSC18) to 5,041 (SSC1), and the adjacent physical distances between them were 30.9 kb (SSC12) to 62.6 kb (SSC1).
Genome-Wide Association Study
In this study, the threshold P-values were 1.01 × 10−4 and 1.05 × 10−4 for PU and FI, respectively. A total of 12 significant SNPs were detected, located on SSC1, 3, 4, 9, and 14 (Figure 1, Table 2). For PU, five significant SNPs were detected, of which three were located on SSC3. For FI, seven significant SNPs were discovered, of which five were located on SSC14. The Manhattan and QQ plots for PU and FI are shown in Figure 1. The genomic inflation factor λ, calculated to judge the extent of false positive signals, was 1.04 for PU and 1.03 for FI.
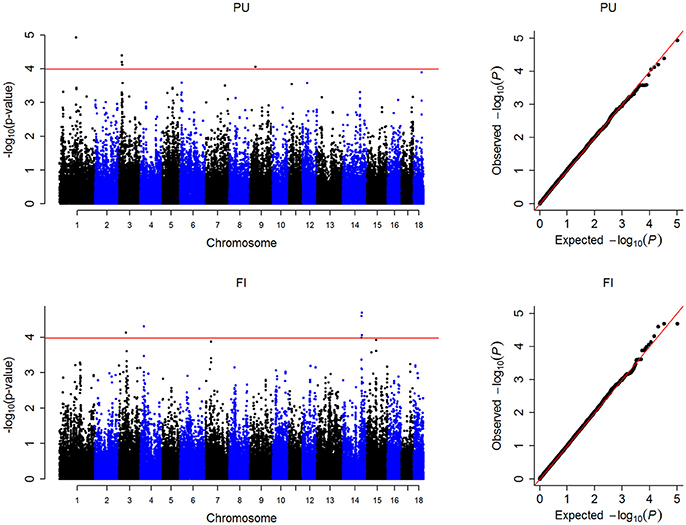
Figure 1. Manhattan plots and Q-Q plots of the observed P-values for PU and FI. The Manhattan plots indicate −log10 (P-values) for genome-wide SNPs (y-axis) plotted against their respective positions on each chromosome (x-axis), the horizontal red lines indicate the thresholds for PU (1.01 × 10−4) and FI (1.05 × 10−4). The Q-Q plots show the observed −log10-transformed P-values (y-axis) and the expected −log10-transformed P-values (x-axis).
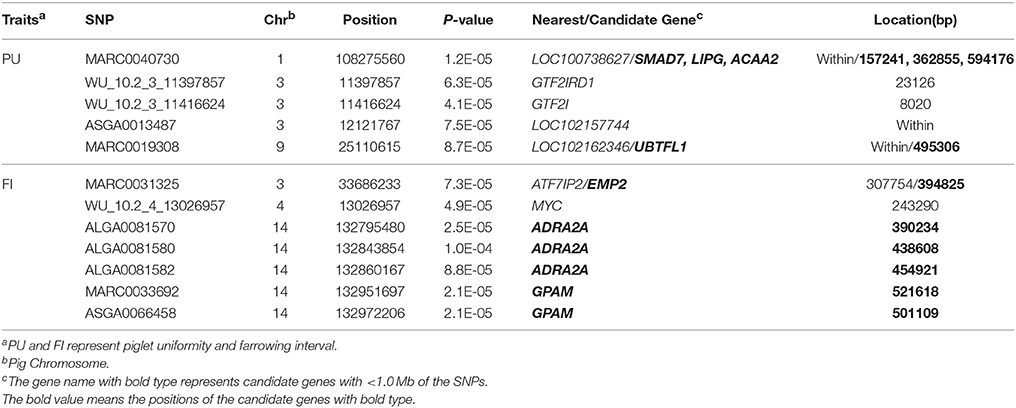
Table 2. All significant SNPs for piglet uniformity and farrowing interval.
SNP Effects
Using GCTA software, the phenotypic variances explained by significant SNPs were estimated for each parity. For PU, five significant SNPs accounted for 1, 1.59, 1.79, and 1.29% of the phenotypic variance from parity 1 to 4, respectively, and the most significant SNP, MARC0040730, located on SSC1, accounted for 0.87–0.95% of the phenotypic variance. Notably, the effect alleles at MARC0040730 were associated with PU at different parities, and the phenotypic differences among the three genotypes at this locus are presented in Figure 2. Along with different parities, each genotype showed different coefficients of variation for birth weights, except for the genotypes AA and GG in the fourth parity. For FI, seven significant SNPs explained 3.17, 4.11, and 2.58% of the phenotypic variance at different parities, respectively. Of note, among them, the peak SNP MARC0033692, located on SSC14, accounted for 1.13–1.18% of the phenotypic variance, and the three genotypes revealed consistent trends for FI at different parities, i.e., the individuals with genotype GG had a longer FI than the other genotypes (Figure 2).
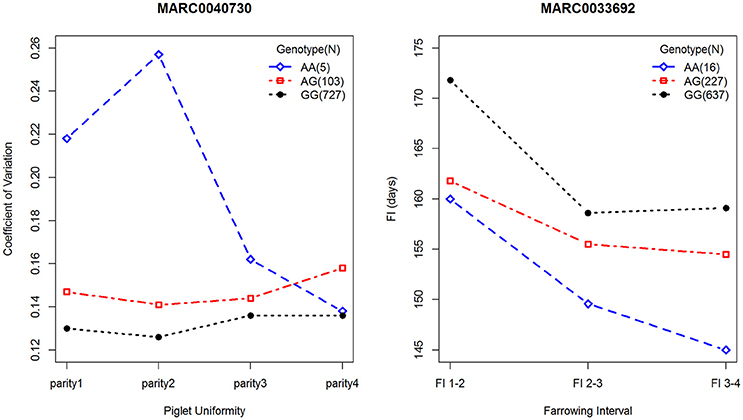
Figure 2. Phenotypic differences contributed by loci of MARC0040730 and MARC0033692. The left plot describes the phenotypes of PU among three genotypes at MARC0040730. The right plot describes the phenotypes of FI among three genotypes at MARC0033692. Blue diamond, red square and black circle represent minor-allele homozygotes, heterozygotes, and major-allele homozygotes, respectively. Number of samples for each genotype is indicated in the top right.
Haplotype Block Analysis
In our study of FI, we found five significant SNPs located on SSC14, and detected a haplotype block (132.8–132.97 Mb) (Figure 3). The LOC106506128 gene was located in this region. In particular, the nearest annotated genes of significant SNPs, including the GPAM and ADRA2A genes, were located downstream (501 Kb) and upstream (390 Kb) of this region, respectively. As for PU, however, relatively few significant SNPs generated haplotype blocks. Two significant SNPs were situated in an 18 Kb block on SSC3 (Figure 3), the GTF2IRD1 gene located downstream (23.1 Kb) of the region, and GTF2I gene located upstream (8 Kb) of the region.
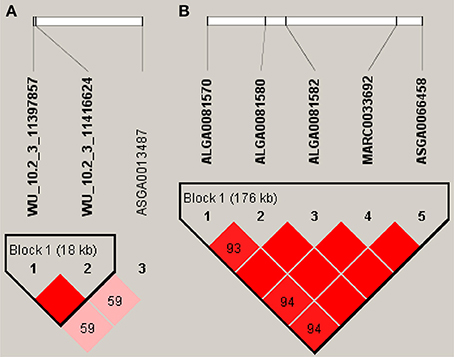
Figure 3. Haplotype blocks for significant SNPs. (A) Indicate a haplotype block composed of significant SNPs located on SSC3 for PU. (B) Indicate a haplotype block composed of significant SNPs located on SSC14 for FI. The black lines mark the identified blocks.
Candidate Genes and Function Analysis
A total of 61 functional genes that contained or were near (within 1 Mb) the identified significant SNPs were obtained based on the swine genome assembly 10.2 (Table S2). These genes were used to perform GO analysis. Thirteen significant GO terms were identified (Table S3). The most significant GO terms for PU and FI were related to transitions between slow and fast fibers, and cellular iron ion homeostasis, respectively. Considering the genes involved in biological process in DAVID, and functional annotations in the NCBI database and literatures, seven genes (SMAD7, LIPG, ACAA2, UBTFL1, GPAM, ADAR2A, and EMP2) with biological functions such as prenatal skeletal muscle development, fetal energy substrate, pre-implantation, and the expression of mammary gland epithelium were selected as promising candidates for swine reproductive traits (Table 2). The majority of these were the nearest functional genes to significant SNPs.
Discussion
Both PU and FI are important indices that can be used to evaluate pig reproductive ability and play crucial roles in production efficiency and economic profits. Thus, it is essential to understand the genetic mechanisms of reproductive performance for future pig breeding programs. GWAS provides an efficient way to detect potential genetic variation and candidate genes in domestic animals (Zhang et al., 2013), especially for some economically valuable traits (Schopen et al., 2011; Sell-Kubiak et al., 2015). In the present study, we conducted a GWAS in a Large White pig population that contained 884 individuals. Our population is larger than that in the PU study of Wang et al. (2016), which consisted of 82 sows. We also made full use of the phenotypic data at four different parities through a repeatability model approach, while some other studies only used the phenotypes of a single parity (Onteru et al., 2012; Schneider et al., 2012). Moreover, this study can detect the genetic variation influencing phenotypic variability over time, and capture parity-independent variation (Smith et al., 2010). Several consistent variants, such as MARC0040730 (located on SSC1) and MARC0033692 (located on SSC14), were found to be associated with PU and FI, respectively, at all parities.
Population stratification is a major factor in false positives in GWAS. Here, through a repeatability model approach, both fixed and random effects were used to adjust the population stratification. In general, a genomic inflation factor λ of <1.05 indicates no population stratification (Price et al., 2010), our values were 1.04 for PU and 1.03 for FI. QQ plots also indicated that we have controlled population stratification, but only a few genomic variants were detected. As we known, quantitative traits are controlled by polygenes (Falconer and Mackay, 1981), and the effect of most given locus on the trait is small. In addition, both PU and FI belong to low-heritability traits, the influence of genetic factor is limited, and the number of samples in our study is still not large enough. Therefore, it's essential to perform studies with a large population and efficient methods for PU and FI in the future.
For PU, considering that the number of piglets born and birth weight were the major influencing factors (Canario et al., 2010), we selected four functional genes near the significant SNPs as important candidates. The SMAD7 gene, near to the peak SNP MARC0040730, belongs to the SAMD gene family, which has been shown to be a key regulator of transforming growth factor β (TGF-β) (Hayashi et al., 1997). Recent studies have shown that it promotes skeletal muscle cell differentiation (Miyake et al., 2010; Cohen et al., 2015) and follicular development in mice (Gao et al., 2013). Hua et al. (2016) demonstrated that the SMAD7 gene plays an important role in prenatal skeletal muscle development and promotes weaning weight in pigs. For better fetal growth, the mother must provide sufficient nutrients such as amino acids, glucose, and lipids through the placenta (Brett et al., 2014). Thus, genes associated with energy metabolism may affect fetal growth and piglet birth weight variation. Both LIPG and ACAA2 genes are involved in lipid metabolism, and lipids such as triglycerides and cholesterol play critical roles in fetal growth (Woollett, 2011). The LIPG gene encodes a member of the triglyceride lipase family of proteins, which participates in glycerolipid metabolism and transportation of lipids. The ACAA2 gene encodes the protein that catalyzes the last step in fatty acid β-oxidation (Eaton et al., 1996), and is involved in the fatty acid β-oxidation pathway that can provide fetal energy substrate. Furthermore, fetal lipid deposition is mainly increased in the gestational period (Haggarty, 2002). During the course of reproduction, pre-implantation stages are important for reproductive and stem cell biology (Riaz et al., 2011). The UBTFL1 gene, also known as Hmgpi, is a preimplantation-specific gene and is involved in early development and implantation. Yamada et al. (Yamada et al., 2010) indicated that Hmgpi plays a critical role in the earliest stages of mammalian embryonic development.
For FI, which contains lactation length, weaning to estrus mating interval, and gestation length, three functional genes related to the three stages and located near the significant SNPs were selected as potential candidates. The GPAM gene encodes a mitochondrial enzyme and catalyzes the initial step in triacylglycerol and phospholipid biosynthesis (Wendel et al., 2009). Knockout studies suggest that the GPAM isoform plays an important role in the development of lactation (Gimeno and Cao, 2008), which is expressed in mammary gland epithelium and is upregulated during lactation. The ADAR2A gene encodes a member of the G protein-coupled receptor superfamily, which plays a critical role in the central nervous system. Philipp et al. (2002) showed that α2-adrenoceptors are important regulators of placental structure and function that can help to establish the circulatory system of the placenta between mother and embryo and thus maintain pregnancy. The EMP2 gene encodes a tetraspan protein that plays an important role in the endometrium and is differentially expressed in the different phases of the estrous cycle (Wadehra et al., 2008). During implantation, EMP2 is involved in the molecular interactions between the blastocyst-stage embryo and maternal endometrium (Wadehra et al., 2006). Thus, based on functional studies of these genes, we hypothesize that GPAM, ADAR2A, and EMP2 may influence different stages of FI.
Five significant SNPs associated with PU are located on eight QTL regions for reproductive traits, including teat number (Ding et al., 2009), corpus luteum number (Sato et al., 2011), and nonfunctional nipples (Jonas et al., 2008). In particular, the peak SNP MARC0040730, located on a QTL region associated with birth body weight (SSC1, 16.1 to 289.6 Mb) (Guo et al., 2008), and the SMAD7, LIPG, and ACAA2 genes are contained within this region. For FI, we found two significant SNPs, WU_10.2_4_13026957 and MARC0031325, were included within eight reproductive QTLs. One of these QTL regions is related to plasma FSH concentration (SSC3, 21.9 to 38.2 Mb) (Rohrer et al., 2001) and contains both the SNP MARC0031325 and the EMP2 gene. To summarize, our findings confirmed the importance of these two QTL.
Conclusion
Our findings provide knowledge on genomic variation and candidate genes that are involved in the genetic mechanisms of PU and FI. The SNPs that are associated with additive genetic variability at different parities can be used as fundamental information in marker assisted selection or genomic selection, which will be helpful in improving pig breeding programs.
Data Accessibility
The genotype and phenotype data of the samples used in the present study are available from the FigShare Repository: https://figshare.com/articles/Genome-wide_Association_Study_of_Piglet_Uniformity_and_Farrowing_Interval/5594230.
Author Contributions
CW and DS designed and supervised the study. YW contributed to genomic DNA extraction and conducted GWAS analysis, XD contributed to population construction and statistical analysis with help from CN, and ZT. KX, ZT, TY, and YP contributed to genomic DNA extraction, phenotypes collection, and samples collection. YW drafted the manuscript, which was critically a remarks by XD, DS, and CW. All authors read and approved the final manuscript.
Funding
This work was supported by the Beijing Innovation Consortium of Agriculture Research System (BAIC02-2016), the China Agriculture Research System (CARS-35), the National Natural Science Foundation of China (31671327), the Beijing City Committee of Science and Technology Key Project (D151100004615004), and the Program for Changjiang Scholar and Innovation Research Team in University (grant number IRT_15R621).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the colleagues in the molecular quantitative genetics team at the National Engineering Laboratory for Animal Breeding of China Agricultural University, for their technical assistance and helpful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2017.00194/full#supplementary-material
References
Andersson, L., Weir, B. S., Hill, W. G., Zhu, J., and Zeng, Z. B. (2009). Genome-wide association analysis in domestic animals: a powerful approach for genetic dissection of trait loci. Genetica 136, 341–349. doi: 10.1007/s10709-008-9312-4
Aulchenko, Y. S., Ripke, S., Isaacs, A., and van Duijn, C. M. (2007). GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296. doi: 10.1093/bioinformatics/btm108
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300.
Blasco, A., Sorensen, D., and Bidanel, J. P. (1998). Bayesian inference of genetic parameters and selection response for litter size components in pigs. Genetics 149, 301–306.
Brett, K. E., Ferraro, Z. M., Yockell-Lelievre, J., Gruslin, A., and Adamo, K. B. (2014). Maternal–fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int. J. Mol. Sci. 15, 16153–16185. doi: 10.3390/ijms150916153
Browning, B. L., and Browning, S. R. (2009). A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 84, 210. doi: 10.1016/j.ajhg.2009.01.005
Canario, L., Lundgren, H., Haandlykken, M., and Rydhmer, L. (2010). Genetics of growth in piglets and the association with homogeneity of body weight within litters. J. Anim. Sci. 88, 1240. doi: 10.2527/jas.2009-2056
Casiró, S., Velez-Irizarry, D., Ernst, C. W., Raney, N. E., Bates, R. O., Charles, M. G., et al. (2017). Genome-wide association study in an F2 Duroc x Pietrain resource population for economically important meat quality and carcass traits. J. Anim. Sci. 95, 545–558. doi: 10.2527/jas2016.1003
Cavalcante Neto, A., Lui, J. F., Sarmento, J. L. R., Ribeiro, M. N., Monteiro, J. M. C., Fonseca, C., et al. (2009). Estimation models of variance components for farrowing interval in swine. Braz. Arch. Biol. Technol. 52, 69–76. doi: 10.1590/S1516-89132009000100009
Cohen, T. V., Kollias, H. D., Liu, N., Ward, C. W., and Wagner, K. R. (2015). Genetic disruption of Smad7 impairs skeletal muscle growth and regeneration. J. Physiol. 593, 2479–2497. doi: 10.1113/JP270201
Damgaard, L. H., Rydhmer, L., Løvendahl, P., and Grandinson, K. (2003). Genetic parameters for within-litter variation in piglet birth weight and change in within-litter variation during suckling. J. Anim. Sci. 81, 604–610. doi: 10.2527/2003.813604x
Dennis, G. Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C., et al. (2003). DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4:R60. doi: 10.1186/gb-2003-4-9-r60
Ding, N., Guo, Y., Knorr, C., Ma, J., Mao, H., Lan, L., et al. (2009). Genome-wide QTL mapping for three traits related to teat number in a White Duroc x Erhualian pig resource population. BMC Genet. 10:6. doi: 10.1186/1471-2156-10-6
Eaton, S., Bartlett, K., and Pourfarzam, M. (1996). Mammalian mitochondrial beta-oxidation. Biochem. J. 320(Pt 2), 345.
Falconer, D. S., and Mackay, T. F. C. (1981). Introduction to quantitative genetics. Q. Rev. Biol. 49, 223–226.
Fan, B., Onteru, S. K., Du, Z.Q., Garrick, D. J., Stalder, K. J., and Rothschild, M. F. (2011). Genome-wide association study identifies loci for body composition and structural soundness traits in pigs. PLoS ONE 6:e14726. doi: 10.1371/journal.pone.0014726
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., et al. (2002). The structure of haplotype blocks in the human genome. Science 296, 2225–2229. doi: 10.1126/science.1069424
Gao, Y., Wen, H., Wang, C., and Li, Q. (2013). SMAD7 antagonizes key TGFβ superfamily signaling in mouse granulosa cells in vitro. Reproduction 146, 1–11. doi: 10.1530/REP-13-0093
Gilmour, A. R., Gogel, B., Cullis, B., Thompson, R., and Butler, D. (2009). ASReml User Guide Release 3.0. Hemel Hempstead: VSN International Ltd.
Gimeno, R. E., and Cao, J. (2008). Mammalian glycerol-3-phosphate acyltransferases: new genes for an old activity. J. Lipid Res. 49, 2079–2088. doi: 10.1194/jlr.R800013-JLR200
Guo, Y. M., Lee, G. J., Archibald, A. L., and Haley, C. S. (2008). Quantitative trait loci for production traits in pigs: a combined analysis of two Meishan x Large white populations. Anim. Genet. 39, 486–495. doi: 10.1111/j.1365-2052.2008.01756.x
Haggarty, P. (2002). Placental regulation of fatty acid delivery and its effect on fetal growth–a review. Placenta 23(Suppl A), S28. doi: 10.1053/plac.2002.0791
Hayashi, H., Abdollah, S., Qiu, Y., Cai, J., Xu, Y. Y., Grinnell, B. W., et al. (1997). The MAD-related protein smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 89, 1165. doi: 10.1016/S0092-8674(00)80303-7
Hindorff, L. A., Sethupathy, P., Junkins, H. A., Ramos, E. M., Mehta, J. P., Collins, F. S., et al. (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 106, 9362–9367. doi: 10.1073/pnas.0903103106
Hoffmann, T. J., Passarelli, M. N., Graff, R. E., Emami, N. C., Sakoda, L. C., Jorgenson, E., et al. (2017). Genome-wide association study of prostate-specific antigen levels identifies novel loci independent of prostate cancer. Nat. Commun. 8:14248. doi: 10.1038/ncomms14248
Holl, J. W., Cassady, J. P., Pomp, D., and Johnson, R. K. (2004). A genome scan for quantitative trait loci and imprinted regions affecting reproduction in pigs. J. Anim. Sci. 82, 3421–3429. doi: 10.2527/2004.82123421x
Hua, C., Wang, Z., Zhang, J., Penx, X., Hou, X., Yang, Y., et al. (2016). SMAD7, an antagonist of TGF-beta signaling, is a candidate of prenatal skeletal muscle development and weaning weight in pigs. Mol. Biol. Rep. 43, 241–251. doi: 10.1007/s11033-016-3960-8
Johnson, R. C., Nelson, G. W., Troyer, J. L., Lautenberger, J. A., Kessing, B. D., Winkler, C. A., et al. (2010). Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genomics 11:724. doi: 10.1186/1471-2164-11-724
Jonas, E., Schreinemachers, H. J., Kleinwächter, T., Un, C., Oltmanns, I., Tetzlaff, S., et al. (2008). QTL for the heritable inverted teat defect in pigs. Mamm. Genome 19, 127–138. doi: 10.1007/s00335-007-9086-5
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348. doi: 10.1038/ng.548
King, A. H., Jiang, Z., Gibson, J. P., Haley, C. S., and Archibald, A. L. (2003). Mapping quantitative trait loci affecting female reproductive traits on porcine chromosome 8. Biol. Reprod. 68, 2172–2179. doi: 10.1095/biolreprod.102.012955
Kominakis, A., Hager-Theodorides, A. L., Zoidis, E., Saridaki, A., Antonakos, G., and Tsiamis, G. (2017). Combined GWAS and 'guilt by association'-based prioritization analysis identifies functional candidate genes for body size in sheep. Genet. Sel. Evol. 49, 41. doi: 10.1186/s12711-017-0316-3
Lauc, G., Essafi, A., Huffman, J. E., Hayward, C., KneŽević, A., Kattla, J. J., et al. (2010). Genomics meets glycomics-the first GWAS study of human N-Glycome identifies HNF1α as a master regulator of plasma protein fucosylation. PLoS Genet. 6:e1001256. doi: 10.1371/journal.pgen.1001256
Luo, W., Cheng, D., Chen, S., Wang, L., Li, Y., Ma, X., et al. (2012). Genome-wide association analysis of meat quality traits in a porcine large White x Minzhu intercross population. Int. J. Biol. Sci. 8, 580–595. doi: 10.7150/ijbs.3614
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Milligan, B. N., Fraser, D., and Kramer, D. L. (2002). Within-litter birth weight variation in domestic pig and its relation to pre-weaning survival, weight gain, and variation in weaning weights. Livestock Prod. Sci. 76, 181–191. doi: 10.1016/S0301-6226(02)00012-X
Miyake, T., Alli, N. S., and McDermott, J. C. (2010). Nuclear function of Smad7 promotes myogenesis. Mol. Cell. Biol. 30, 722. doi: 10.1128/MCB.01005-09
Onteru, S. K., Fan, B., Du, Z. Q., Garrick, D. J., Stalder, K. J., and Rothschild, M. F. (2012). A whole-genome association study for pig reproductive traits. Anim. Genet. 43, 18–26. doi: 10.1111/j.1365-2052.2011.02213.x
Petersen, J. L., Mickelson, J. R., Rendahl, A. K., Valberg, S. J., Andersson, L. S., Axelsson, J., et al. (2013). Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 9:e1003211. doi: 10.1371/journal.pgen.1003211
Philipp, M., Brede, M. E., Hadamek, K., Gessler, M., Lohse, M. J., and Hein, L. (2002). Placental α2-adrenoceptors control vascular development at the interface between mother and embryo. Nat. Genet. 31, 311–315. doi: 10.1038/ng919
Price, A. L., Zaitlen, N. A., Reich, D., and Patterson, N. (2010). New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459. doi: 10.1038/nrg2813
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Quesnel, H., Brossard, L., Valancogne, A., and Quiniou, N. (2008). Influence of some sow characteristics on within-litter variation of piglet birth weight. Animal 2, 1842–1849. doi: 10.1017/S175173110800308X
Riaz, A., Zhao, X., Dai, X., Li, W., Liu, L., Wan, H., et al. (2011). Mouse cloning and somatic cell reprogramming using electrofused blastomeres. Cell Res. 21, 770. doi: 10.1038/cr.2010.180
Rivals, I., Personnaz, L., Taing, L., and Potier, M. C. (2007). Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23, 401–407. doi: 10.1093/bioinformatics/btl633
Rohrer, G. A., Wise, T. H., Lunstra, D. D., and Ford, J. J. (2001). Identification of genomic regions controlling plasma FSH concentrations in Meishan-white composite boars. Physiol. Genomics 6, 145–151. Available online at: http://physiolgenomics.physiology.org/content/6/3/145.full.pdf+html
Sato, S., Hayashi, T., and Kobayashi, E. (2011). Fine mapping the number of corpora lutea quantitative trait loci on SSC3: analysis of the porcine follicle-stimulating hormone receptor gene. Anim. Sci. J. 82, 633–641. doi: 10.1111/j.1740-0929.2011.00899.x
Schneider, J. F., Rempel, L. A., Snelling, W. M., Wiedmann, R. T., Nonneman, D. J., and Rohrer, G. A. (2012). Genome-wide association study of swine farrowing traits. Part II: bayesian analysis of marker data. J. Anim. Sci. 90, 3360–3367. doi: 10.2527/jas.2011-4759
Schopen, G. C., Visker, M. H., Koks, P. D., Mullaart, E., van Arendonk, J. A., and Bovenhuis, H. (2011). Whole-genome association study for milk protein composition in dairy cattle. J. Dairy Sci. 94, 3148–3158. doi: 10.3168/jds.2010-4030
Sell-Kubiak, E., Duijvesteijn, N., Lopes, M. S., Janss, L. L., Knol, E. F., Bijma, P., et al. (2015). Genome-wide association study reveals novel loci for litter size and its variability in a large white pig population. BMC Genomics 16:1049. doi: 10.1186/s12864-015-2273-y
Serenius, T., Sevón-Aimonen, M. L., and Mäntysaari, E. A. (2003). Effect of service sire and validity of repeatability model in litter size and farrowing interval of finnish landrace and large white populations. Livestock Prod. Sci. 81, 213–222. doi: 10.1016/S0301-6226(02)00300-7
Ponsuksili, S., Reyer, H., Trakooljul, N., Murani, E., and Wimmers, K. (2016). Single- and Bayesian multi-marker genome-wide association for haematological parameters in pigs. PLoS ONE 11:e0159212. doi: 10.1371/journal.pone.0159212
Smith, E. N., Chen, W., Kähönen, M., Kettunen, J., Lehtimäki, T., Peltonen, L., et al. (2010). Longitudinal genome-wide association of cardiovascular disease risk factors in the Bogalusa heart study. PLoS Genet. 6:e1001094. doi: 10.1371/journal.pgen.1001094
Southwood, O. I., and Kennedy, B. W. (1991). Genetic and environmental trends for litter size in swine. J. Anim. Sci. 69, 3177–3182. doi: 10.2527/1991.6983177x
Tabor, H. K., Risch, N. J., and Myers, R. M. (2002). Opinion: candidate-gene approaches for studying complex genetic traits: practical considerations. Nat. Rev. Genet. 3, 391–397. doi: 10.1038/nrg796
Van de Pol, M., and Verhulst, S. (2006). Age-dependent traits: a new statistical model to separate within- and between-individual effects. Am. Nat. 167, 766. doi: 10.1086/503331
VanRaden, P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423. doi: 10.3168/jds.2007-0980
Verardo, L. L., Sevón-Aimonen, M. L., Serenius, T., Hietakangas, V., and Uimari, P. (2017). Whole-genome association analysis of pork meat pH revealed three significant regions and several potential genes in Finnish Yorkshire pigs. BMC Genet. 18:13. doi: 10.1186/s12863-017-0482-x
Wadehra, M., Dayal, M., Mainigi, M., Ord, T., Iyer, R., Braun, J., et al. (2006). Knockdown of the tetraspan protein epithelial membrane protein-2 inhibits implantation in the mouse. Dev. Biol. 292, 430–441. doi: 10.1016/j.ydbio.2006.01.015
Wadehra, M., Mainigi, M., Morales, S. A., Rao, R. G., Gordon, L. K., Williams, C. J., et al. (2008). Steroid hormone regulation of EMP2 expression and localization in the endometrium. Reprod. Biol. Endocrinol. 6:15. doi: 10.1186/1477-7827-6-15
Wang, K., Liu, D., Hernandez-Sanchez, J., Chen, J., Liu, C., Wu, Z., et al. (2015). Genome wide association analysis reveals new production trait genes in a male duroc population. PLoS ONE 10:e0139207. doi: 10.1371/journal.pone.0139207
Wang, X., Liu, X., Deng, D., Yu, M., and Li, X. (2016). Genetic determinants of pig birth weight variability. BMC Genet. 17(Suppl. 1):15. doi: 10.1186/s12863-015-0309-6
Weller, J. I., Song, J. Z., Heyen, D. W., Lewin, H. A., and Ron, M. (1998). A new approach to the problem of multiple comparisons in the genetic dissection of complex traits. Genetics 150, 1699–1706.
Wendel, A. A., Lewin, T. M., and Coleman, R. A. (2009). Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochim. Biophys. Acta 1791, 501–506. doi: 10.1016/j.bbalip.2008.10.010
Wittenburg, D., Guiard, V., Teuscher, F., and Reinsch, N. (2008). Comparison of statistical models to analyse the genetic effect on within-litter variance in pigs. Animal 2, 1559–1568. doi: 10.1017/S1751731108002851
Wolf, J., Žáková, E., and Groeneveld, E. (2005). Genetic parameters for a joint genetic evaluation of production and reproduction traits in pigs. Czech J. Anim. Sci. 50, 96–103. Available online at: http://www.agriculturejournals.cz/publicFiles/52522.pdf
Woollett, L. A. (2011). Review: transport of maternal cholesterol to the fetal circulation. Placenta 32(Suppl. 2), 218–221. doi: 10.1016/j.placenta.2011.01.011
Xing, K., Zhu, F., Zhai, L., Chen, S., Tan, Z., Sun, Y., et al. (2016). Identification of genes for controlling swine adipose deposition by integrating transcriptome, whole-genome resequencing, and quantitative trait loci data. Sci. Rep. 6:23219. doi: 10.1038/srep23219
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yamada, M., Hamatani, T., Akutsu, H., Chikazawa, N., Kuji, N., Yoshimura, Y., et al. (2010). Involvement of a novel preimplantation-specific gene encoding the high mobility group box proteinHmgpiin early embryonic development. Hum. Mol. Genet. 19, 480–493. doi: 10.1093/hmg/ddp512
Yuan, J., Wang, K., Yi, G., Ma, M., Dou, T., Sun, C., et al. (2015). Genome-wide association studies for feed intake and efficiency in two laying periods of chickens. Genet. Sel. Evol. 47, 82. doi: 10.1186/s12711-015-0161-1
Zhang, H., Wang, Z., Wang, S., and Li, H. (2013). Progress of genome wide association study in domestic animals. J. Anim. Sci. Biotechnol. 4, 1–1. doi: 10.1186/2049-1891-4-3
Keywords: piglet uniformity, farrowing interval, genome-wide association study, pigs, candidates
Citation: Wang Y, Ding X, Tan Z, Ning C, Xing K, Yang T, Pan Y, Sun D and Wang C (2017) Genome-Wide Association Study of Piglet Uniformity and Farrowing Interval. Front. Genet. 8:194. doi: 10.3389/fgene.2017.00194
Received: 03 September 2017; Accepted: 15 November 2017;
Published: 28 November 2017.
Edited by:
Luis Varona, University of Zaragoza, SpainReviewed by:
Dirk-Jan De Koning, Swedish University of Agricultural Sciences, SwedenJoanna Szyda, Wroclaw University of Environmental and Life Sciences, Poland
Copyright © 2017 Wang, Ding, Tan, Ning, Xing, Yang, Pan, Sun and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongxiao Sun, c3VuZHhAY2F1LmVkdS5jbg==
Chuduan Wang, Y2R3YW5nQGNhdS5lZHUuY24=
†These authors have contributed equally to this work.