 Shuxian Li
Shuxian Li Bryan Musungu
Bryan Musungu David Lightfoot3
David Lightfoot3 Pingsheng Ji
Pingsheng Ji- 1Crop Genetics Research Unit, United States Department of Agriculture, Agricultural Research Service, Stoneville, MS, United States
- 2Department of Plant Biology, Southern Illinois University, Carbondale, IL, United States
- 3Department of Plant, Soil, and Agricultural Systems, Southern Illinois University, Carbondale, IL, United States
- 4Department of Plant Pathology, University of Georgia, Tifton, GA, United States
Phomopsis longicolla T. W. Hobbs (syn. Diaporthe longicolla) is the primary cause of Phomopsis seed decay (PSD) in soybean, Glycine max (L.) Merrill. This disease results in poor seed quality and is one of the most economically important seed diseases in soybean. The objectives of this study were to infer protein–protein interactions (PPI) and to identify conserved global networks and pathogenicity subnetworks in P. longicolla including orthologous pathways for cell signaling and pathogenesis. The interlog method used in the study identified 215,255 unique PPIs among 3,868 proteins. There were 1,414 pathogenicity related genes in P. longicolla identified using the pathogen host interaction (PHI) database. Additionally, 149 plant cell wall degrading enzymes (PCWDE) were detected. The network captured five different classes of carbohydrate degrading enzymes, including the auxiliary activities, carbohydrate esterases, glycoside hydrolases, glycosyl transferases, and carbohydrate binding molecules. From the PPI analysis, novel interacting partners were determined for each of the PCWDE classes. The most predominant class of PCWDE was a group of 60 glycoside hydrolases proteins. The glycoside hydrolase subnetwork was found to be interacting with 1,442 proteins within the network and was among the largest clusters. The orthologous proteins FUS3, HOG, CYP1, SGE1, and the g5566t.1 gene identified in this study could play an important role in pathogenicity. Therefore, the P. longicolla protein interactome (PiPhom) generated in this study can lead to a better understanding of PPIs in soybean pathogens. Furthermore, the PPI may aid in targeting of genes and proteins for further studies of the pathogenicity mechanisms.
Introduction
Proteins in living organisms perform many functions by physically interacting with each other (Bork et al., 2004). Interactomes have been described as the genome-wide roadmaps of inferred protein–protein interactions (PPIs). Investigating the model of PPI can enhance our understanding of the cellular process and biological interactions within an organism. Interactomes of model organisms such as Arabidopsis thaliana (L.) and Saccharomyces cerevisiae (Meyen) were built using high-throughput experimental methodologies (Arabidopsis Interactome Mapping Consortium, 2011). Interactomes have also recently begun to expand to the non-model organisms by predictions based on orthology. Predicted interactomes in agronomically important organisms, such as Citrus sinensis, Oryza sativa (L.), Glycine max (L.) Merrill, and Zea mays (L.), have also provided valuable insight into disease resistance (Afzal et al., 2009; Musungu et al., 2015). In recent years, an abundance of PPI data has been developed through high-throughput technologies, including plant pathogens such as Fusarium graminearum (Zhao et al., 2009) that causes Fusarium head blight of both wheat and barley; Gibberella stalk rot of maize (Goswami and Kistler, 2004); and Magnaporthe grisea (He et al., 2008), cause of rice blast (Talbot, 2003; Dean et al., 2005).
Phomopsis longicolla T. W. Hobbs (syn. Diaporthe longicolla) is a fungal species of Ascomycota in the Diaporthaceae family. It is the primarily causal agent of Phomopsis seed decay (PSD) in soybean, G. max (L.) Merrill (Hobbs et al., 1985; Santos et al., 2011; Li et al., 2015a). This pathogen also causes stem lesion on velvet leaf plants (Li et al., 2001) and can live as an endophyte in the mangrove and Meliaceae plant species (Rhoden et al., 2012). P. longicolla has also been reported to produce a number of cytotoxic and antimicrobial secondary metabolites, such as dicerandrols and phomoxanthones (Isaka et al., 2001; Lin et al., 2010).
Soybean is one of the most important economic crops in the world with global production over 340 million metric tons (ASA, 2017). The soybean PSD disease results in poor seed quality and it is one of the most economically important seed diseases in soybean (Sinclair, 1993; Li, 2011). Management of PSD has been conducted using conventional tillage to reduce pathogen inoculum, rotation with non-host or non-legume crops, and early harvest (once soybeans have matured) to avoid late season wet weather. However, inconsistent reductions of PSD have been reported when those common agronomic practices were used. Fungicide treatments have also been an option to reduce PSD, but they were not always effective in controlling PSD (TeKrony et al., 1985; Wrather et al., 2004). Planting PSD-resistant cultivars is a cost-effective and long-term strategy to manage PSD. In past decades, research has been conducted to identify PSD-resistance sources by screening soybean germplasms, commercial cultivars, and breeding lines (Li et al., 2011, 2015c; Li and Chen, 2013; Li and Smith, 2016), investigating inheritance of resistance and genetic mapping of resistance to PSD (Zimmerman and Minor, 1993; Jackson et al., 2005, 2009; Smith et al., 2008), and breeding for resistant lines and cultivars (Minor et al., 1993; Pathan et al., 2009). However, information about mechanisms underlying the pathogenicity of P. longicolla on soybean is lacking. The prediction of PPI networks in P. longicolla has not been investigated and reported.
Although the genome of the P. longicolla isolate MSPL 10-6 has been sequenced (Li et al., 2015b, 2017), there are still many genes with unknown functions. Therefore, using the computational biology approach to analyze the interactome will help understand the different mechanisms underlying pathogenicity in PPI networks. Hence, the objectives of this study were to perform a genome-wide analysis of the predicted proteins interactome and to identify conserved global networks and pathogenicity subnetworks in P. longicolla causing PSD of soybean. This research will enhance our knowledge of the biology, pathogenicity, and protein interactions of P. longicolla and aid in developing improved disease management strategies for PSD.
Materials and Methods
Constructing a Protein–Protein Interaction Network for Phomopsis longicolla
The interolog method (Yu et al., 2004) was used to predict protein interactions in P. longicolla. The framework for the pipeline involved retrieving the protein sequences from NCBI and using the Inparanoid 4.1 software to infer one-to-one and many-to-many orthology. For one-to-one orthology selection, the proteins pairs with the most significant inparanoid orthology score were considered one-to-one. For the remaining proteins in the cluster, each of the protein pairs was considered to be in the many-to-many ortholog group. Afterwards, in house Python and R scripts were used to combine BioGrid data to allow for development of confidence values (CV).
The genome of a P. longicolla isolate MSPL 10-6 has been sequenced and the amino acid sequences of the P. longicolla proteins predicted to be encoded were retrieved from the National Center for Biotechnology Information (NCBI; Li et al., 2015b, 2017). The protein sequences of thirteen reference species, including eight eukaryotes (Candida albicans, C. elegans, Drosophila melanogaster, Homo sapiens, Mus musculus, Rattus norvegicus, Saccharomyces cerevisiae, S. pombe) and four prokaryotes (Bacillus subtilis, Campylobacter jejuni, Escherichia coli, and Helicobacter pylori) were retrieved from ENSEMBL www.ensembl.org/index.html (access date November 2011; Flicek et al., 2013) and NCBI http://www.ncbi.nlm.nih.gov (Geer et al., 2010).
Previous methodologies described by Geisler-Lee et al. (2007) and Musungu et al. (2015) were used in the Inparanoid 4.1 analysis. Briefly, the following organisms, Bacillus subtilis, C. elegans, C. jejuni, D. melanogaster, E. coli, H. sapiens, M. Musculus, R. norvegicus, S. cerevisiae, and S. pombe, were used as reference organisms in the Inparanoid pipeline (Koonin, 2005; Östlund et al., 2010). Inparanoid works by performing a proteome wide blast comparison between different organisms. It also allows for one-to-one and many-to-many predicted protein interactions to be inferred.
Predicting Phomopsis longicolla Interactions From Conserved Orthologs
The interactome of P. longicolla was constructed from an all-inclusive analysis of physical interactions between proteins of P. longicolla that were predicted based on experimentally determined interactions for the organisms utilized in the study. For determining the protein interacting partners within the PPI network, the previously developed methods (Geisler-Lee et al., 2007; Musungu et al., 2015) were used to infer the unique interactions from a publically available interactome database (BioGRID, version 3.1.841; Stark et al., 2006). The confidence value (CV), gene ontology, and the analysis methods described by Geisler-Lee et al. (2007) were used (Supplementary Tables S1, S2). Additionally, the gene ontology analysis used was the best BLAST hit in F. graminearum because the protein domain information for P. longicolla was not available (Güldener et al., 2006; Carbon et al., 2009). InterproScan analysis was also used for the genome to identify domains in the genome (Zdobnov and Apweiler, 2001). The presence of pathogenicity genes in the P. longicolla interactome was determined using the curated dataset from plant host interactions (Urban et al., 2016).
Modeling Phomopsis longicolla Interactome Using Cytoscape
To visualize the PPI interactions from the network analysis, the P. longicolla protein data (Supplementary File S1 and Supplemental Table S1) was used as the input file in the Cytoscape (version 3.5.1) analysis (Shannon et al., 2003; Cline et al., 2007).
Cross Validation Analysis
Cross validation was performed in R statistical language using the caret package doing K-fold cross validation (Kuhn, 2008). For feature selection we used the PFAM information available for each of the protein sequences after performing InterproScan. For the cross validation, positive interactions were identified using interlog method and the random interlog dataset was created from non-interacting proteins. Due to lack of experimentally determined data for P. longicolla, the PFAM domain information for the proteins was used. Rules for the features listed were:
(1) If the set of domains in Protein A {domain A, domain B, domain C} and Protein B {domain A, domain B, domain C} is true, then the value in matrix was set 1.
(2) If the set of domains in Protein A {domain A, domain B, domain C, domain D} and Protein B {domain A, domain B, domain C} are different, then the value in the matrix was set 2.
(3) If there were no domains or interaction found for the protein, then the interaction was set at 0.
(4) The methodology framework for this study was illustrated in Figure 1.
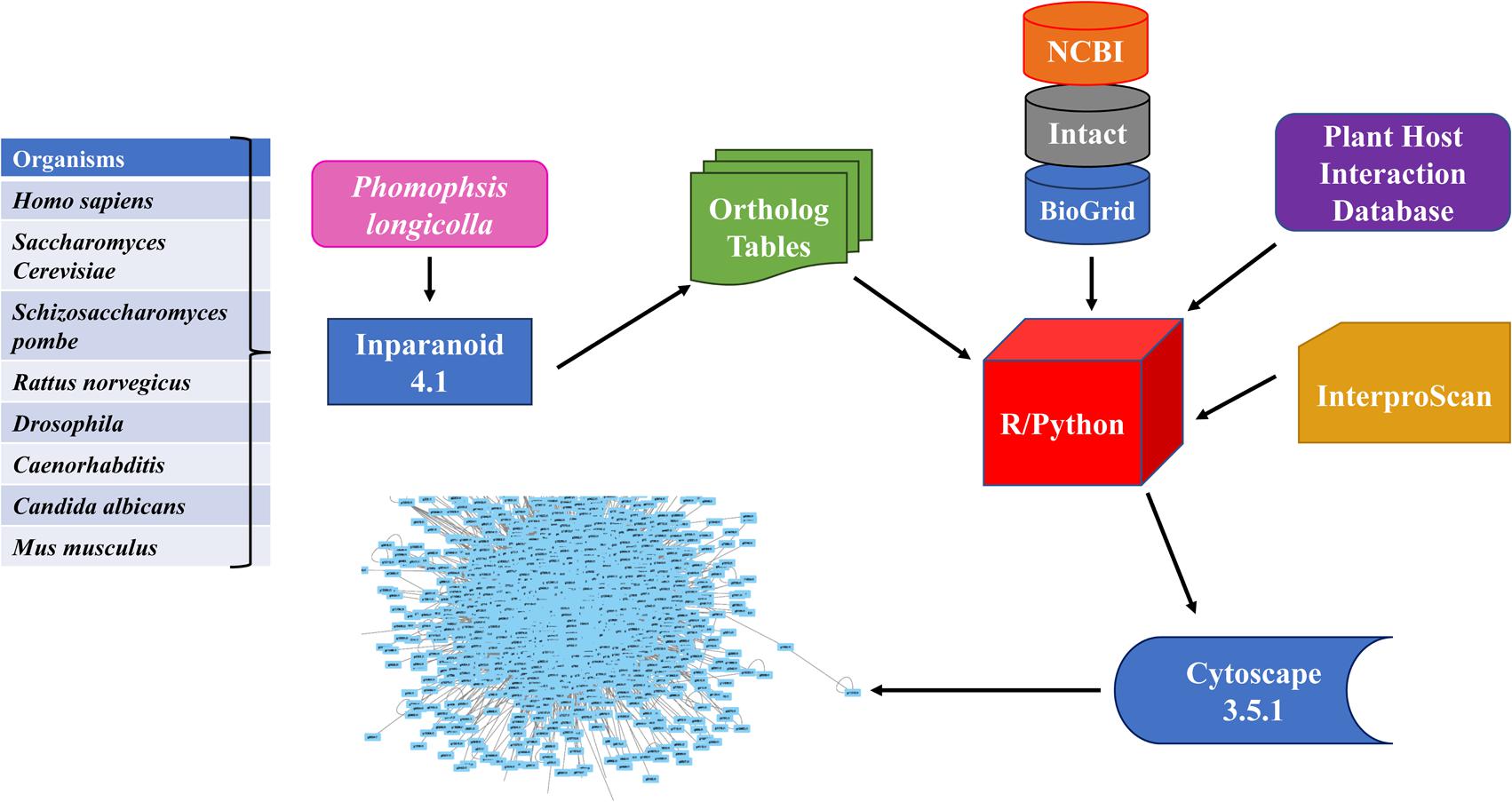
FIGURE 1. Flowchart for developing Phomopsis longicolla interactome. Generation of the interactome was accomplished by using publicly available resources (refer to section “Materials and Methods”) from multiple reference genomes. The ortholog prediction software Inparanoid was used for identification of orthologous proteins between P. longicolla and reference organism. The confidence values were calculated using R statistical language and Python. Additionally, verification was done with cross validation in R statistical language using domain information from EMBL Interproscan, Plant Host Interaction (PHI) data base and NCBI.
Results
General Features of the Phomopsis longicolla Interactome
To investigate predicted physical protein–protein interactions all the predicted proteins encoded by the P. longicolla genome were used. There were 215,255 unique PPIs among 3,868 of 16,595 predicted proteins. The relative contribution of each reference species to the predicted interactions is summarized in Supplementary Table S1. The resulting P. longicolla protein interactome (PiPhom) encompassed just 23% of the total proteome because the paralogous and duplicated genes from the genome were excluded. When duplicated genes were included in the prediction of the interactome, using a many-to-many ortholog matching method that allows the inclusion of paralogs, 50 P. longicolla proteins that were only in the many-to-many set, as well as 189 unique interologs, were added to the uniqueinteractome (Table 1). A premade Cytoscape formatted graphical visualization of P. longicolla interactome for this combined set of proteins was included (Supplementary File S1). In addition, contributions from each organism were highlighted (Supplementary Table S1), where S. cerevisiae had the largest contribution of total interactions including both “one to one” and “many to many” for the PPI data set (78%, Figure 2). H. sapiens contributed the second largest number (13%) of interactions to the PiPhom (Supplementary Table S1).

TABLE 1. Predicted protein–protein interactions in Phomopsis longicolla.
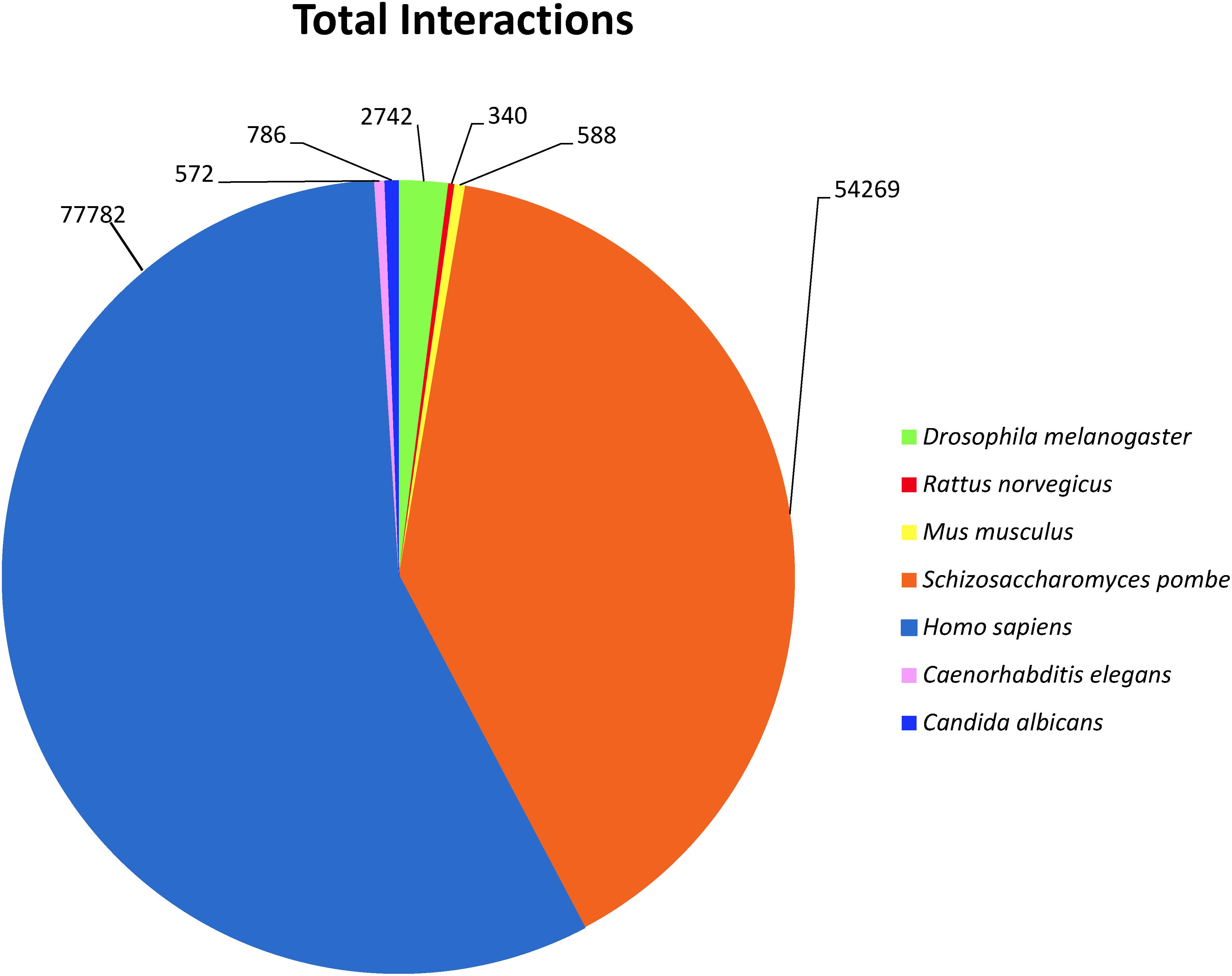
FIGURE 2. Analysis of reference organism proteins in PiPhom interactome. For each of the proteomes the distribution was calculated for their overall contribution for the total (One to One/Many to Many) interactome for the organisms Caenorhabditis elegans, Candida albicans Drosophila melanogaster, Homo sapiens, Mus musculus, Rattus norvegicus, Saccharomyces cerevisiae, and Saccharomyces pombe (S. cerevisiae was analyzed but not included in the figure due to large percentage of orthologous proteins).
Validation and Network Analysis of Predicted Phomopsis longicolla Interactions
In order to determine the significance of the PiPhom interactome, analysis of conserved functional subnetwork models was conducted to determine if conservation of biological pathways in eukaryotes were present within the PiPhom interactome. The analysis of conservation indicated the subnetworks with the strongest confidences values which were similar to previously reported interactomes.
Confidence values (CVs) for each interaction in the PiPhom interactome are listed (Supplementary Table S1) and added to the network visualization (Supplementary File S1) as an edge feature. Interactions with a CV of 1 were ranked as a low confidence data set. They were identified in a single reference source using only one species and one experimental method. P. longicolla had 9,897 such interactions. The next level contained 191,407 interactions at the medium CV score of (2 ≤x ≤ 10). The high confident set contained 13,952 interactions with a CV (x > 10). The frequency profile of CV was similar to previous work where the large portion of unique interactions had medium confidence which is likely due to the high similarity between reference organisms. Due to novelty of the genome, K-fold 10 cross-validation was used with the domain information. From the K-fold, the best model for the data using the domain information inferred a 56% accuracy rating.
In the PiPhom interactome, many homo-interactions were identified. The top 20 conserved interactions in the PiPhom interactome were found to contain protein kinases and cellular machinery such as histone proteins (Table 2). The networks of P. longicolla displayed many of the core regulatory machineries in the cell, such as proteins involved in DNA-repair, zinc finger proteins, and heat shock proteins that were important in multiple eukaryotic systems and organisms (Tables 2, 3). One of the expected pathways mined from the CV analysis of the network was the DNA repair machinery subnetworks (Supplementary File S1). The higher the CV, the greater the likelihood of the conserved interactions detected in PiPhom interactome.
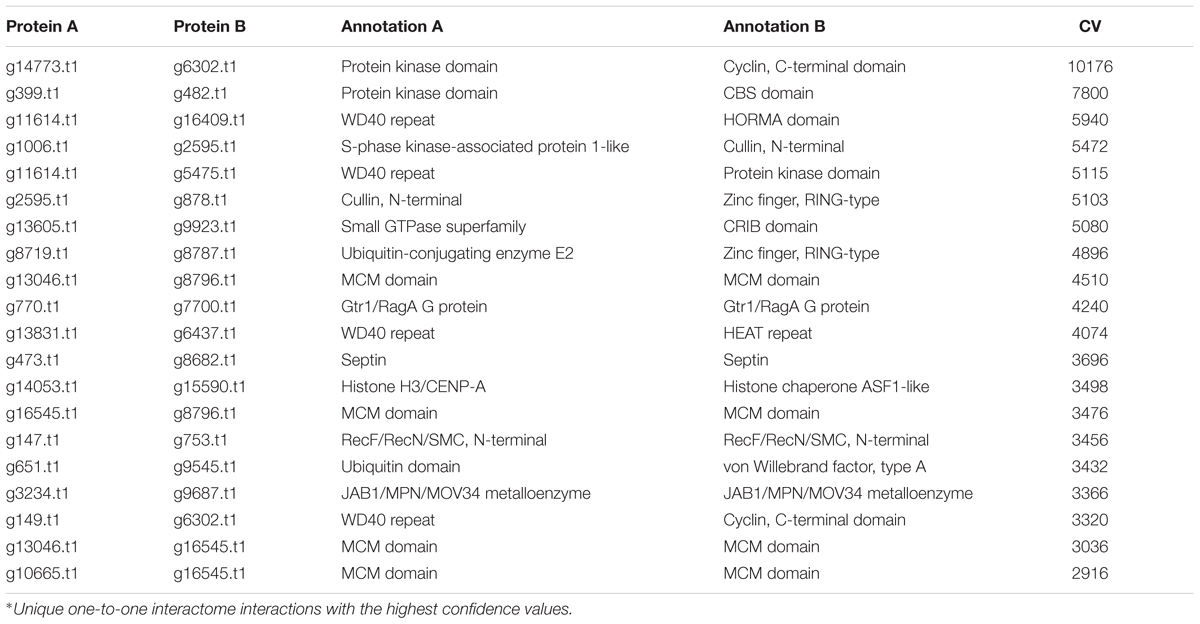
TABLE 2. The highest confidence hetero-interactions in the predicted Phomopsis longicolla interactome∗.
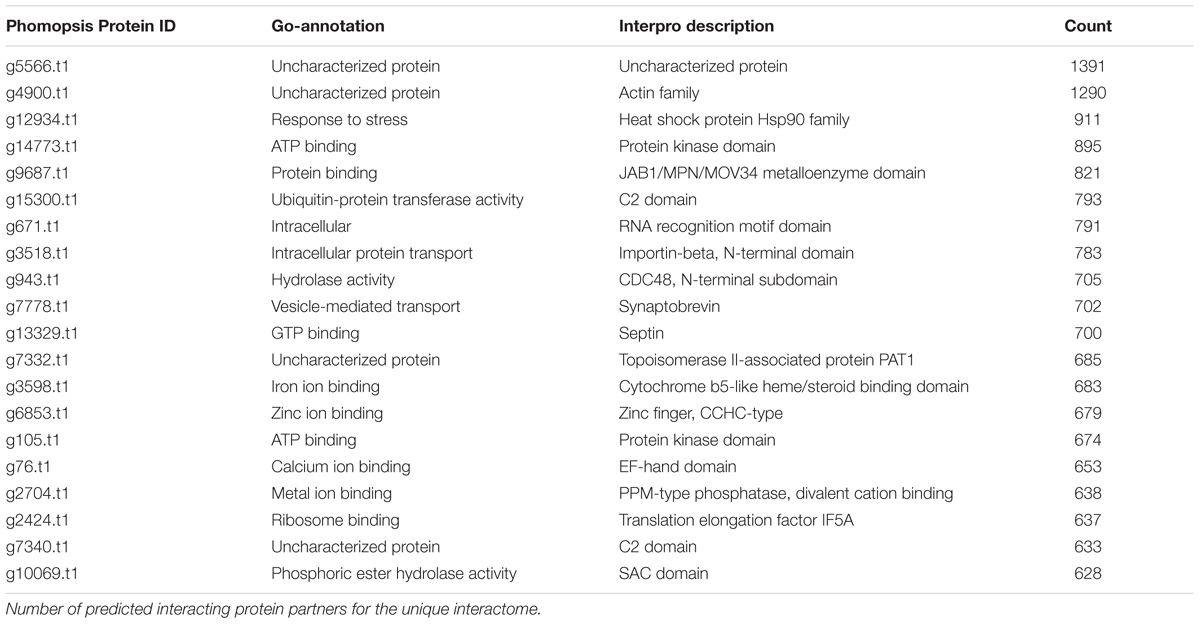
TABLE 3. The most connected proteins in the predicted Phomopsis longicolla interactome.
Proteins in PiPhom interactome with a large number of interacting partners were found to be highly conserved (Table 3). The highly connected proteins were ubiquitous partners and co-factors such as cullins, scaffolding proteins, and proteins involved in degradation pathways. However, the protein with the highest connectivity was uncharacterized (g5566.t1), which had 1,391 different predicted protein partners. Additional analysis of the gene using InterproScan found it to be a cytochrome P450 domain containing gene. BLAST searching against F. graminearum failed, but mining the interactome of yeast inferred g5566.t1 encoded an ortholog of NAB2, a protein involved in RNA transport. Other highly interconnected conserved proteins, such as chaperonins, heat shock proteins, and members of large protein complexes were also identified (Table 3). Among both plants and animals those key protein complexes were conserved within the highly connected hubs. Interestingly, conserved interactions between histones, proteasome components, MutS type DNA repair proteins, and cytochromes were also found in PiPhom interactome. Thus, there is a similar hub pattern in P. longicolla to previously reported interactomes where there was a high degree of confidence. The hubs in the PiPhom interactome were similar to other eukaryotes even when comparing the small proteome to the large genomes with multiple divergent protein copies (Bork et al., 2004).
There was connection between phosphorylation of serine-threonine/tyrosine-protein kinases and transcription factors within the PiPhom interactome. Interestingly, the PiPhom interactome had orthologs to many of these signaling proteins. This may be due to the use of references proteomes from pathogens in the analysis, such as C. albicans, S. cerevisiae and S. pombe. An example could be seen with the (g15658.t1) FUS3, which is a signaling protein in the network with 224 proteins. Another protein was (g2599.t1) PMK1 which had a connectivity of 450 genes. Additional kinase modules included SNF1, STE11, STE12, HOG1, and RAC1 which were found to be hubs in abundant predicted interactions in PiPhom. Each of the connection contained larger than average degree of connectivity for these signaling proteins. When observing transcription factors, many of the conserved transcription factor complexes were present in the interactome, such as TFII complex and cell cycle proteins.
In addition, PiPhom contained pathways of interest to pathogenesis involved in pH, nitrogen metabolism, reactive oxygen species metabolism/catabolism and carbohydrate catabolism/metabolism which are conserved throughout the ascomycetes. These subnetworks represent modules of interest that have not previously been shown in other reported interactomes.
Structural Analyses of the PiPhom Networks
The paths and trees in the PiPhom networks were measured by the structure analysis for mathematical properties, such as shortest paths, connectivity and circuits. The intermediate sized hubs were 10 to 100 interacting partners for the majority of proteins in PiPhom (Figure 3). The unique interactome had an average degree of connectivity of 110.496 neighbors per node.
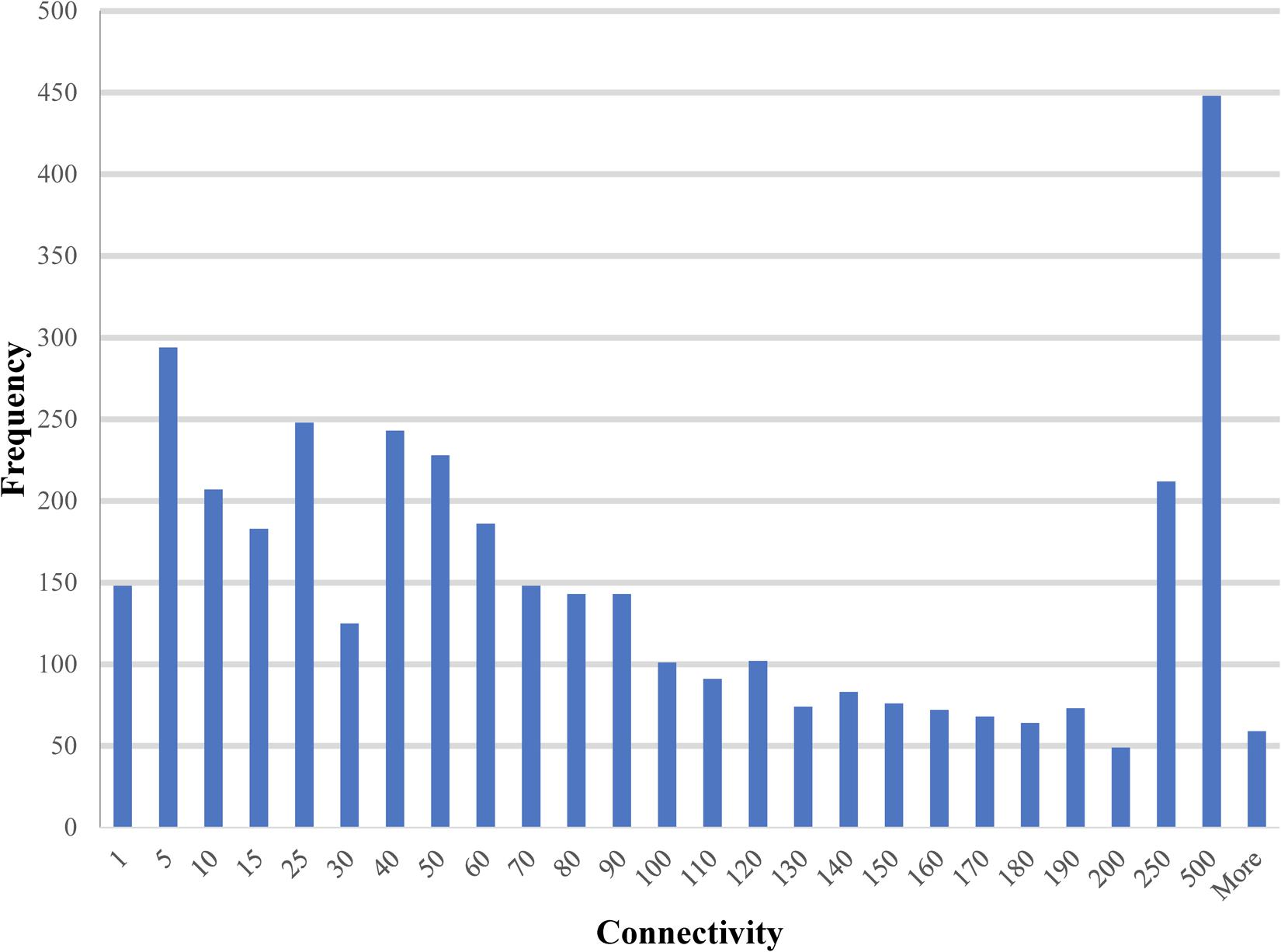
FIGURE 3. Degree of connectivity for unique proteins in the Phomopsis longicolla interactomes. The degree which is a measure of connectivity for vertexes and edges was analyzed for each of the proteins in the P. longicolla interactome.
The path length for PiPhom was defined by analyzing the average distance of Protein A to Protein B which was between 2 and 4 for the PPI network. Further analysis using the network analyzer module in Cytoscape indicated the mean path length to be 3.942 nodes (Supplementary File S1). Topology is a key indicator in inference of PPI networks. PiPhom shared similar network properties to previously reported interactomes and fungal specific structures within the network topologies.
Fungal Gene Ontology Analysis of PiPhom
The interactome of P. longicolla was evaluated for enriched and depleted GO terms using the best E-value BLAST hits against F. graminearum. The PiPhom interactome was enriched significantly in GO: 0016301 (Supplementary Table S2). There was also a significant enrichment in ncRNA metabolic (GO: 0034660) processes within the network.
Conserved Interactions Within the Network
When observing evolutionary conservation by species for the network, the largest subset of enriched interactions contained three or more reference organisms in the network. It has been demonstrated that conserved pathways are likely to be preserved throughout eukaryotes. There were 788 interactions identified in the high confidence set, in which the number or reference was greater than 3 and the CV value was greater than 10. The largest CV interaction seen in the network was (g14773.t1) protein kinase and (g6302.t1) cyclin, C-terminal domain which had a confidence value of 10,176 in the network (Table 2). The high confidence represented a portion of the network that had highly conserved interactions within the network (Figure 4). Within the stringent networks, proteins contained complexes such as the prefoldin, proteasome and vacuolar transport, which are all important mechanisms in eukaryotes.
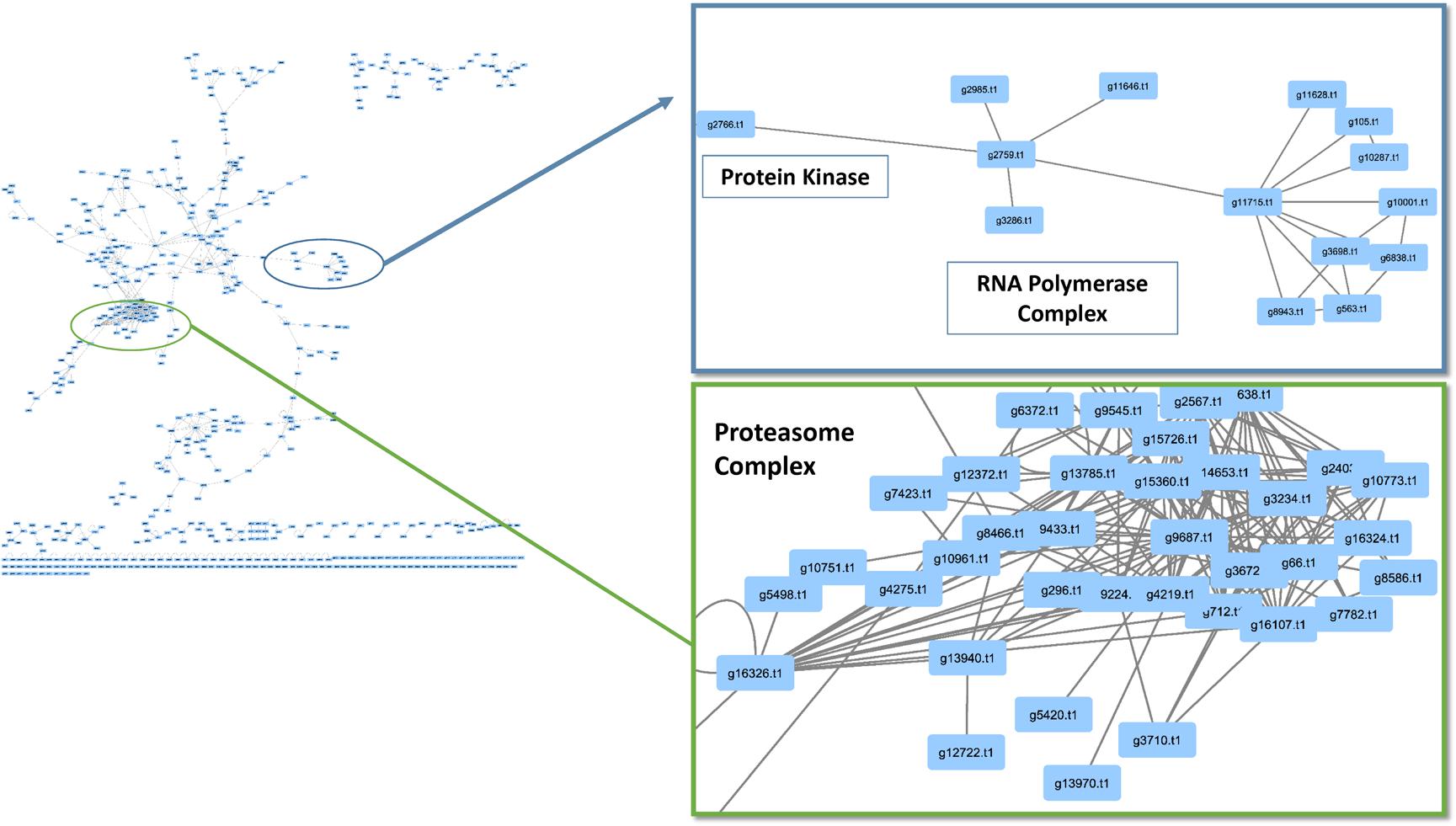
FIGURE 4. Conservation analysis of Phomopsis longicolla interactomes. Confidence value (CV) and at least 3 reference organisms were used to generate a subnetwork of protein and protein interactions of interest. Within the network there were proteasome complex, RNA-polymerase and small subsets of interactions.
Plant Cell Wall Degrading Enzymes
In the PiPhom interactome, there were 149 PCWDEs related proteins with 378 edges (Figure 5). The network captured five different classes of carbohydrate degrading enzymes, such as auxiliary activities (AA), carbohydrate esterases (CE), glycoside hydrolases (GH), glycosyl transferases (GT), and carbohydrate binding molecules (CBM). The most predominant class of PCWDEs was a group of 60 GH proteins which had been implicated in multiple pathogenicity studies (Daguerre et al., 2017). The smallest group of carbohydrate degrading proteins was the PCWDEs, belonging to the CBM family with just two proteins found in PiPhom. The two proteins were (g14970.t1), a CS domain containing 250 protein–protein interacting partners and (g7307.t1) a cysteine-rich secretory protein that had five interacting partners within the network.
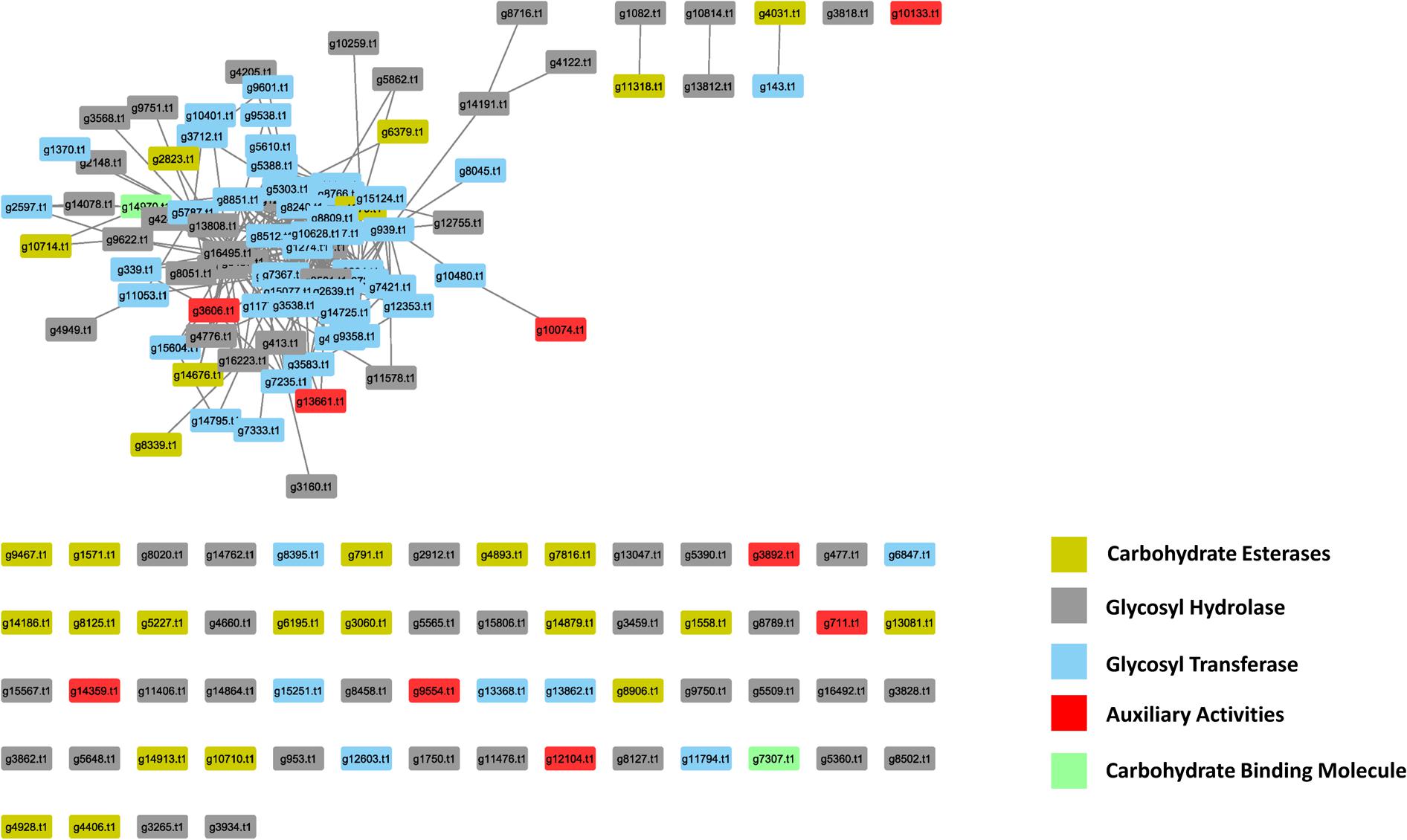
FIGURE 5. Analysis of the abundance of the cell wall degrading enzymes in the Phomopsis longicolla interactome. Five different classes were observed in the interactome: AA, auxiliary activities; CE, carbohydrate esterases; GH, glycoside hydrolases; GT, glycosyl transferase; CBM, carbohydrate binding molecule.
Pathogenicity Genes
There were 1,414 pathogenicity genes that were identified in P. longicolla (Supplementary Table S3). Similarity among orthologous proteins was found for ascomycetes, basidiomycetes and eubacteria. Examples of taxa exhibiting hits within the curated data were A. flavus, A. fumigatus, Alternaria alternata, and F. graminearum. When focusing on proteins that have been inferred to be responsible for pathogenicity, 477 proteins were detected in the interactome (Figure 6). The high confidence data within pathogenicity network was 180 nodes and 257 edges.
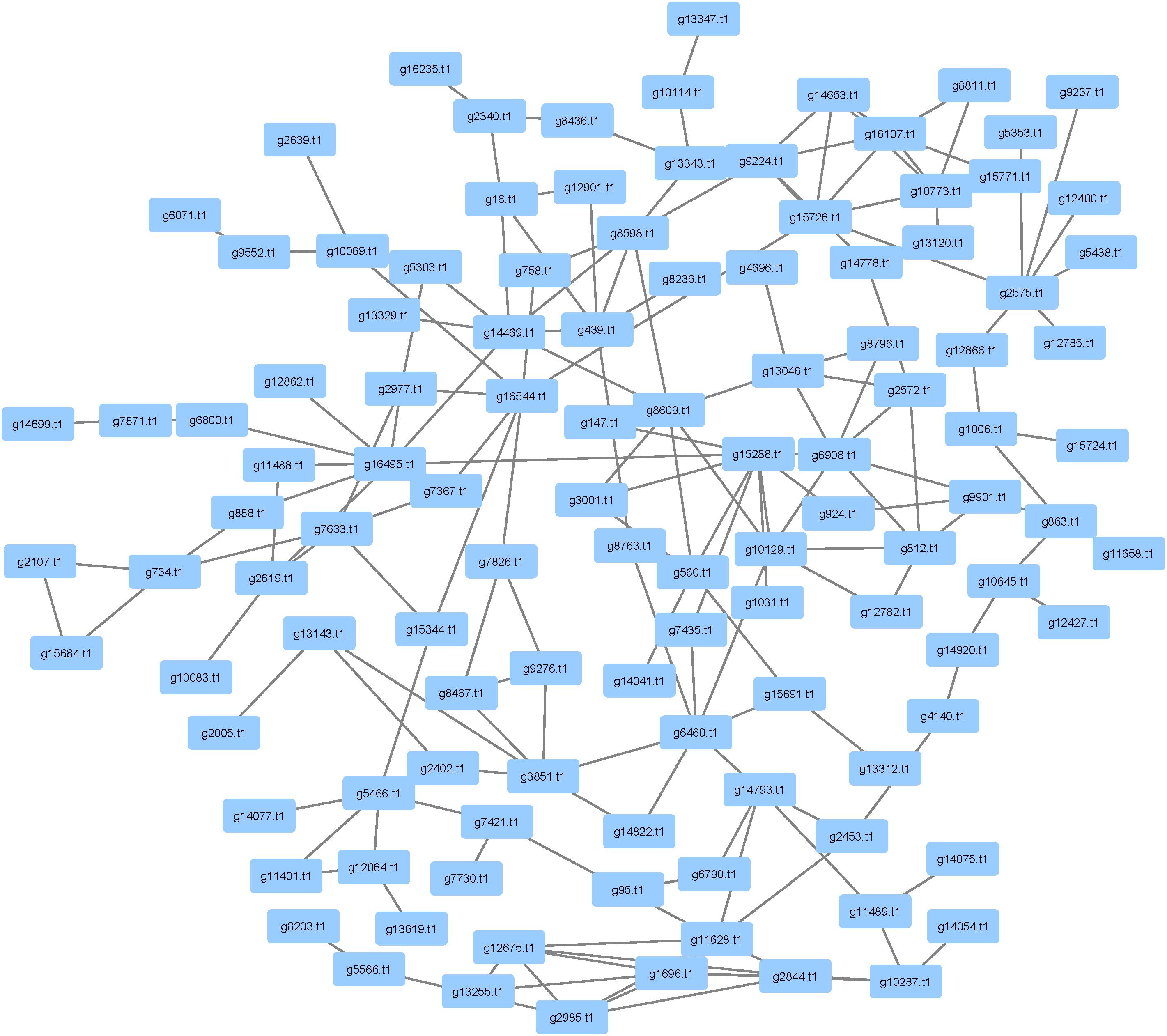
FIGURE 6. Analysis of pathogenicity genes using PHI-base and mined within the Phomopsis longicolla interactome. The network contains 477 proteins that were associated with pathogenicity. The network also contained several cell wall degrading enzymes including glycosyl transferase, auxiliary activities and carbohydrate esterases.
Discussion
Interologs were defined as a conserved interaction between a pair of proteins of a given organism which have interacting homologs in another organism (Yu et al., 2004). This method has been used to study and predict protein interactions successfully in other multiple organisms (Gu et al., 2011; Ho et al., 2012; Weßling et al., 2014), including but not limit to O. sativa, C. elegans, and S. pombe. Bioinformatic algorithms and programs, and corresponding parameters and weights used to produce P. longicolla interactomes in this study were similar to those used in A. thaliana (Geisler-Lee et al., 2007), O. sativa (Ho et al., 2012), and Z. mays (Musungu et al., 2015). In this study, a proteome-wide analysis of a predicted protein interactome using the interlog method was used to predict protein interactions in P. longicolla and developed to create resource for understanding the biology of the ascomycete P. longicolla. The PiPhom interactome can assist plant pathologists interested in possible gene-for-gene interactions and mycologists interested in possible industrial applications in agriculture. Additionally, the PiPhom may lead to a better understanding of this economically important soybean pathogen that causes seed decay. To date, analysis of the P. longicolla genome has been conducted to determine the genome features (Li et al., 2017) and comparative genome study with other soybean ascomycete pathogens (Li and Musungu, unpublished). The primary goal of the study was to predict PPIs and gain a functional understanding of proteins involved in the developmental processes, plant cell wall degrading enzymes (PCWDEs) and pathogenicity proteins which are important components of P. longicolla. In our previous study, PCWDEs encoded within the P. longicolla genome were determined (Li et al., 2017). In this study, a cysteine-rich secretory protein (g7307.t1) that had five interacting partners within the network was identified. The cysteine rich protein was an interesting conserved discovery in the network because many of these proteins have been shown to function as secretory proteins in fungi and oomycetes, such as Phytophthora cactorum, Leptosphaeria maculans, and F. oxysporum (Sperschneider et al., 2015). Using the Systems Biology approaches, networks among families of PCWDEs were identified (Li et al., 2017). Additionally, proteins that represented a graphical significance within the network through degree of connectivity were characterized.
The level of CV can be used as a filter to identify true hypotheses and reduce the false positives when the data is used to build networks. Many previously reported common hetero-interactions were often the most abundant interactions in PPIs (Musungu et al., 2015). However, that was not the case in the PiPhom interactome, in which many homo-interactions were identified. The networks of P. longicolla displayed many of the core regulatory machineries in the cell that were important in multiple eukaryotic systems and organisms. One of the expected pathways mined from the CV analysis of the network was the DNA repair machinery subnetworks, which were conserved throughout eukaryotes (Liu et al., 1999; Ohbayashi et al., 1999) and recovered from similar interactomes, such as S. cerevisiae (Yu et al., 2008), Z. mays (Musungu et al., 2015), and A. thaliana (Geisler-Lee et al., 2007) among others. The higher the CV, the greater the likelihood of the conserved interactions detected in PiPhom interactome.
Connectivity in computational biology has been demonstrated to aid in the generation of hypotheses for targeted pathogenicity analysis. This has been demonstrated in multiple interactome studies when working on systems with minimal biological information. In the PiPhom interactome, the highly connected proteins were ubiquitous partners and co-factors such as cullins, scaffolding proteins, and proteins involved in degradation pathways. This overlapped with the previous interactome studies, such as A. thaliana, Z. mays, and P. patens, which had highly conserved pathways represented by cullins. The highly connected interactions are likely to have a large degree of connectivity since it is evolutionarily conserved (Evlampiev and Isambert, 2008).
The enrichment of the PiPhom protein network led to identification of pathogenesis pathways, such as nitrogen metabolism, carbohydrate degrading enzymes and cell to cell signaling processes. It has already been demonstrated that mitogen activated protein kinases are important in the pathogenicity of multiple pathogens such as F. graminearum, F. solani, and M. grisea (Xu and Hamer, 1996; Di Pietro et al., 2001; Jenczmionka et al., 2003; Ramamoorthy et al., 2007). Moreover, a total of 477 pathogenicity-associated proteins were detected in the PiPhom interactome. The abundance of pathogenicity factors was likely due to yeast data as well as the addition of the pathogen C. albicans in which 80 of 478 proteins were inferred within pathogenicity networks. This is an abundant amount in comparison to previous studies where about 100 pathogenicity proteins were detected for the M. grisea PPI (He et al., 2008). Within the network, there were common pathogenicity genes, which are seen in other ascomycetes. For example, STE11 was associated with pathogenicity in Botrytis cinerea and F. graminearum (Izumitsu et al., 2009; Leroch et al., 2013; Gu et al., 2015). Connection between phosphorylation of serine-threonine/tyrosine-protein kinases and transcription factors was present within the interactome. Those pathogenic proteins have been used as targets for targeted mutagenesis (Caracuel et al., 2003; Shimizu et al., 2003; Ramamoorthy et al., 2007).
Analysis of orthology is a key concept during interactome analysis of protein–protein interactions. This is because interactions are likely to occur if there is conservation through different organisms which can be attributed to fitness of organism or random mutations (Lynch, 2007). These features of the interlog method have been demonstrated in previous interactomes such as A. thaliana, Z. mays, H. sapiens, and S. cerevisiae. Additionally, because our dataset is made up of interactions from S. cerevisiae, multiple developmental pathways in important life cycles of plant pathogens were identified. This was highlighted in the results with proteins such as FUS3 which has been implicated in pathogenesis in fungi such as A. alternata, F. oxysporum, and M. oryzae (Wilson and Talbot, 2009; Lin et al., 2010; Pareek and Rajam, 2017). Moreover, the HOG1 protein was identified, which was initially characterized and conserved in S. cerevisiae to be involved in osmotic signaling. It has also been demonstrated in multiple fungi to be involved in the regulation of pathogenesis. For example, in Zymoseptoria tritici, a hemibiotrophic pathogen, when HOG1 was targeted for knockout it displaced a loss in pathogenicity (Mehrabi et al., 2006). This was additionally seen in pathogens such as Penicillium digitatum, Magnaporthe oryzae and several others (Motoyama et al., 2008; Wang et al., 2014).
The PiPhom interactome was built using preexisting data from the biogrid and resembles other inferred networks built by the same methodology. The connectivity of PiPhom differed from previous interactomes like Zea mays, Physcomitrella patens, D. melanogaster, and O. sativa (Giot et al., 2003; Stark et al., 2006; Gu et al., 2011; Musungu et al., 2015; Schuette et al., 2015). The characteristic path length of PiPhom differed from the previously reported interactomes for organisms, such as A. thaliana (3.4), S. cerevisiae (2.6), Stegodyphus mimosarum (2.5) and H. sapiens (between 1 and 3; Gursoy et al., 2008; Taylor et al., 2009; Chen et al., 2012; Schuette et al., 2015; Wang and Jin, 2017). When contrasting the PiPhom with previous plant, animal, and fungal work, there was an abundance of signaling proteins in the conserved interactions, suggesting a difference in wiring vs. other fungal species. In contrast, the PiPhom conserved subnetwork modules were comprised of many of the pathways previously found in other interactomes, such as ubiquitination, methylation, pheromone signaling, developmental pathways and chromatin remodeling (Mosca et al., 2012). Thus the similarity between PiPhom and previous interactomes produced by similar methodology leads to increase in confidence in the novelties discovered (Geisler-Lee et al., 2007; Gu et al., 2011; Musungu et al., 2015). Furthermore, the other proteins that were found in the network will become targets for in-depth lab studies. Scientists who are interested in a particular PCWDE family would have the ability to mine the network. For example, the g5566t.1 gene that was identified to represent the highest degree of connectivity was initially identified as uncharacterized and containing cytochrome P450 domain information. While multiple genes have been identified to be involved with cytochrome P450 and pathogenicity for other pathogens, the protein sequence would have been initially missed because it was returned as uncharacterized during the initial BLAST analysis against F. graminearum. Utilizing the metadata in PiPhom, the yeast one-to-one ortholog NAB2 was inferred to be the closest ortholog of the g5566t.1 protein, partly because it had the largest degree of connectivity within the network. The protein was essential for cell viability in yeast (Anderson et al., 1993). Its primary function was inferred to be involved in RNA transport and confirmed primarily in yeast. However, mutations in A. oryzae have been able to show that knocking out of the gene altered the ability of the pathogen to grow compared to wild type (Yamada et al., 1999).
In addition, the interactions are informative for study of other biological processes due to the similarity between PhiBase proteins and other pathogens. From the analysis against the pathogen host interaction (PHI) database genes, multiple BLAST hits on virulence and pathogenicity factors were shared among ascomycetes. PiPhom will be used to select for genes of interest. For example, some of the orthologous proteins identified included CYP1, SGE1, PMK1 and many others from fungi and bacteria. The CYP1 and SGE1 have been characterized and previously shown to be involved in pathogenicity (Winnenburg et al., 2006). Another feature of the PiPhom interactome can be used to filter/identify the non-virulence and non-pathogenic hits that are stored within PHI database.
Overall, PiPhom showed several novel interactions specific to P. longicolla. This is likely due to the close ortholog overlap for P. longicolla proteins vs. yeast proteins within the network. The PiPhom interactome appeared different from previous studies of plant interactomes, such as maize (Musungu et al., 2015), in which the amount of interactions generated was limited by plant specific data.
Conclusion
The PiPhom interactome generated from this study provides a valuable resource for understanding the complexity of pathogenicity in P. longicolla. The orthologous proteins, such as FUS3, HOG, CYP1, SGE1, PCWDE, and the g5566t.1 gene identified in this study could play an important role in pathogenicity of P. longicolla. This research enhances our knowledge of the biology, pathogenicity, and protein interactions of P. longicolla and aids in developing improved strategies for managing PSD. Moreover, the PiPhom interactome can also lead to a better understanding of PPIs in soybean pathogens. Furthermore, the PPI may aid in targeting of genes and proteins for further studies of pathogenicity mechanisms.
Author Contributions
SL conceived and led the project, interpreted results and wrote the manuscript. BM analyzed data and wrote the manuscript. DL and PJ provided suggestions for the project and edited the manuscript. All authors reviewed and approved the final version of the manuscript.
Funding
This research was funded by the USDA-ARS Project 6402-21220-012-00D, Crop Genetics Research Unit at Stoneville, MS, United States.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Deepak Bhatnagar at the USDA-ARS, Southern Regional Research Center, New Orleans, LA, United States, for valuable suggestions of the data analysis. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture. USDA is an equal opportunity provider and employer.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00104/full#supplementary-material
FILE S1 | Cytoscape PiPhom. This file contains the PiPhom predicted interactome as a Cytoscape ver 3.5.1 format file. Interactions shown are indicated and filterable by orthology (many-to-many and one-to-one), confidence value (numerical), and connectivity. Each of subnetworks contains cell wall degrading enzymes, parthenogenesis genes and conserved proteins.
TABLE S1 | PiPhom Interactome Data Tables. The Master Interactome sheet contains all interolog predictions for the Phomopsis longicolla interactome using either many to many or one to one orthology. The supplemental data also contains the connectivity numbers for each of the interlog pairs for the interactome calculated from the master interactome. The dataset also includes the raw tables that were used within the publication. A Node Property sheet provides annotation and shows the number of interactions each proteins has in either the unique or combined datasets. The supplemental dataset also contains the unique data sheet for uploading data into Cytoscape for producing Supplemental File S1.
TABLE S2 | gProfiler Results. Gene Ontology analysis was run on the proteins in the Phomopsis longicolla interactome using Fusarium graminearum orthologs. The significant pathways that were enriched had p-value < 0.05 to be considered an enriched pathway.
TABLE S3 |Phomopsis longicolla Blast Results against Phibase. This supplemental table contains the results of Phibase database blast against the P. longicolla interactome. It shows P. longicolla proteins from the interactome that are inferred to be involved with virulence, non-virulence and pathogenicity factors.
Footnotes
References
Afzal, A. J., Natarajan, A., Saini, N., Iqbal, M. J., Geisler, M., Shemy, A. E. I., et al. (2009). The Nematode resistance allele at the rhg1 locus alters the proteome and primary metabolism of soybean roots. Plant Physiol. 151, 1264–1280. doi: 10.1104/pp.109.138149
Anderson, J. T., Wilson, S. M., Datar, K. V., and Swanson, M. S. (1993). NAB2: a yeast nuclear polyadenylated RNA-binding protein essential for cell viability. Mol. Cell. Biol. 13, 2730–2741. doi: 10.1128/mcb.13.5.2730
Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. doi: 10.1126/science.1203877
Bork, P., Jensen, L. J., von Mering, C., Ramani, A. K., Lee, I., and Marcotte, E. M. (2004). Protein interaction networks from yeast to human. Curr. Opin. Struct. Biol. 14, 292–299. doi: 10.1016/j.sbi.2004.05.003
Caracuel, Z., Roncero, M. I. G., Espeso, E. A., González-Verdejo, C. I., García-Maceira, F. I., and Di Pietro, A. (2003). The pH signalling transcription factor PacC controls virulence in the plant pathogen Fusarium oxysporum. Mol. Microbiol. 48, 765–779. doi: 10.1046/j.1365-2958.2003.03465.x
Carbon, S., Ireland, A., Mungall, C. J., Shu, S., Marshall, B., and Lewis, S. (2009). AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289. doi: 10.1093/bioinformatics/btn615
Chen, J., Lalonde, S., Obrdlik, P., Noorani Vatani, A., Parsa, S. A., Vilariño, C., et al. (2012). Uncovering Arabidopsis membrane protein interactome enriched in transporters using mating-based split ubiquitin assays and classification models. Front. Plant Sci. 3:124. doi: 10.3389/fpls.2012.00124
Cline, M. S., Smoot, M., Cerami, E., Kuchinsky, A., Landys, N., Workman, C., et al. (2007). Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382. doi: 10.1038/nprot.2007.324
Daguerre, Y., Edel-Hermann, V., and Steinberg, C. (2017). “Fungal genes and metabolites associated with the biocontrol of soil-borne plant pathogenic fungi,” in Fungal Metabolites, eds J. M. Mérillon and K. G. Ramawat (Cham: Springer International Publishing), 33–104.
Dean, R. A., Talbot, N. J., Ebbole, D. J., Farman, M. L., Mitchell, T. K., Orbach, M. J., et al. (2005). The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434, 980–986. doi: 10.1038/nature03449
Di Pietro, A., García-Maceira, F. I., Meglecz, E., and Roncero, M. I. G. (2001). A MAP kinase of the vascular wilt fungus Fusarium oxysporum is essential for root penetration and pathogenesis. Mol. Microbiol. 39, 1140–1152. doi: 10.1111/j.1365-2958.2001.02307.x
Evlampiev, K., and Isambert, H. (2008). Conservation and topology of protein interaction networks under duplication-divergence evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 9863–9868. doi: 10.1073/pnas.0804119105
Flicek, P., Ahmed, I., Amode, M. R., Barrell, D., Beal, K., Brent, S., et al. (2013). Ensembl 2013. Nucleic Acids Res. 41, D48–D55. doi: 10.1093/nar/gks1236
Geer, L. Y., Marchler-Bauer, A., Geer, R. C., Han, L., He, J., He, S., et al. (2010). The NCBI BioSystems database. Nucleic Acids Res. 38, D492–D496. doi: 10.1093/nar/gkp858
Geisler-Lee, J., O’Toole, N., Ammar, R., Provart, N. J., Millar, A. H., and Geisler, M. (2007). A predicted interactome for Arabidopsis. Plant Physiol. 145, 317–329. doi: 10.1104/pp.107.103465
Giot, L., Bader, J. S., Brouwer, C., Chaudhuri, A., Kuang, B., Li, Y., et al. (2003). A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736. doi: 10.1126/science.1090289
Goswami, R. S., and Kistler, H. C. (2004). Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5, 515–525. doi: 10.1111/j.1364-3703.2004.00252.x
Gu, H., Zhu, P., Jiao, Y., Meng, Y., and Chen, M. (2011). PRIN: a predicted rice interactome network. BMC Bioinformatics 12:161. doi: 10.1186/1471-2105-12-161
Gu, Q., Chen, Y., Liu, Y., Zhang, C., and Ma, Z. (2015). The transmembrane protein FgSho1 regulates fungal development and pathogenicity via the MAPK module Ste50-Ste11-Ste7 in Fusarium graminearum. New Phytol. 206, 315–328. doi: 10.1111/nph.13158
Güldener, U., Mannhaupt, G., Münsterkötter, M., Haase, D., Oesterheld, M., Stümpflen, V., et al. (2006). FGDB: a comprehensive fungal genome resource on the plant pathogen Fusarium graminearum. Nucleic Acids Res. 34(Suppl. 1), D456–D458. doi: 10.1093/nar/gkj026
Gursoy, A., Keskin, O., and Nussinov, R. (2008). Topological properties of protein interaction networks from a structural perspective. Biochem. Soc. Trans. 36(Pt 6), 1398–1403. doi: 10.1042/BST0361398
He, F., Zhang, Y., Chen, H., Zhang, Z., and Peng, Y.-L. (2008). The prediction of protein-protein interaction networks in rice blast fungus. BMC Genomics 9:519. doi: 10.1186/1471-2164-9-519
Ho, C.-L., Wu, Y., Shen, H.-B., Provart, N. J., and Geisler, M. (2012). A predicted protein interactome for rice. Rice 5, 1–14. doi: 10.1186/1939-8433-5-15
Hobbs, T. W., Schmitthenner, A., and Kuter, G. A. (1985). A new Phomopsis species from soybean. Mycologia 77, 535–544. doi: 10.2307/3793352
Isaka, M., Jaturapat, A., Rukseree, K., Danwisetkanjana, K., Tanticharoen, M., and Thebtaranonth, Y. (2001). Phomoxanthones A and B, novel xanthone dimers from the endophytic fungus Phomopsis species. J. Nat. Prod. 64, 1015–1023. doi: 10.1021/np010006h
Izumitsu, K., Yoshimi, A., Kubo, D., Morita, A., Saitoh, Y., and Tanaka, C. (2009). The MAPKK kinase ChSte11 regulates sexual/asexual development, melanization, pathogenicity, and adaptation to oxidative stress in Cochliobolus heterostrophus. Curr. Genet. 55, 439–448. doi: 10.1007/s00294-009-0257-7
Jackson, E. W., Feng, C., Fenn, P., and Chen, P. (2009). Genetic mapping of resistance to Phomopsis seed decay in the soybean breeding line MO/PSD-0259 (PI562694) and plant introduction 80837. J. Hered. 100, 777–783. doi: 10.1093/jhered/esp042
Jackson, E. W., Fenn, P., and Chen, P. (2005). Inheritance of resistance to Phomopsis seed decay in soybean PI 80837 and MO/PSD-0259 (PI 562694). Crop Sci. 45, 2400–2404. doi: 10.2135/cropsci2004.0525
Jenczmionka, N. J., Maier, F. J., Lösch, A. P., and Schäfer, W. (2003). Mating, conidiation and pathogenicity of Fusarium graminearum, the main causal agent of the head-blight disease of wheat, are regulated by the MAP kinase gpmk1. Curr. Genet. 43, 87–95.
Koonin, E. V. (2005). Orthologs, paralogs, and evolutionary genomics 1. Annu. Rev. Genet. 39, 309–338. doi: 10.1146/annurev.genet.39.073003.114725
Kuhn, M. (2008). Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26. doi: 10.18637/jss.v028.i05
Leroch, M., Kleber, A., Silva, E., Coenen, T., Koppenhöfer, D., Shmaryahu, A., et al. (2013). Transcriptome profiling of Botrytis cinerea conidial germination reveals upregulation of infection-related genes during the prepenetration stage. Eukaryot. Cell 12, 614–626. doi: 10.1128/EC.00295-12
Li, S. (2011). “Phomopsis seed decay of soybean,” in Soybean – Molecular Aspects of Breeding, ed. A. Sudaric (Vienna: Intech Publisher), 277–292.
Li, S., Bradley, C. A., Hartman, G. L., and Pedersen, W. L. (2001). First report of Phomopsis longicolla from velvetleaf causing stem lesions on inoculated soybean and velvetleaf plants. Plant Dis. 85:1031. doi: 10.1094/PDIS.2001.85.9.1031A
Li, S., and Chen, P. (2013). Resistance to Phomopsis seed decay in soybean. ISRN Agron. 2013:738379. doi: 10.1155/2013/738379
Li, S., Chen, P., and Hartman, G. (2015a). “Phomopsis seed decay,” in Compendium of Soybean Diseases and Pests, 5th Edn, eds G. L. Hartman, J. C. Rupe, E. J. Sikora, L. L. Domier, J. A. Davis, and K. L. Steffey (St. Paul, MN: APS Press), 47–48.
Li, S., Darwish, O., Alkharouf, N., Matthews, B., Ji, P., Domier, L. L., et al. (2015b). Draft genome sequence of Phomopsis longicolla isolate MSPL 10-6. Genomics Data 3, 55–56. doi: 10.1016/j.gdata.2014.11.007
Li, S., Darwish, O., Alkharouf, N., Musungu, B., and Matthews, B. F. (2017). Analysis of the genome sequence of Phomopsis longicolla: a fungal pathogen causing Phomopsis seed decay in soybean. BMC Genomics 18:688. doi: 10.1186/s12864-017-4075-x
Li, S., Rupe, J., Chen, P., Shannon, G., Wrather, A., and Boykin, D. (2015c). Evaluation of diverse soybean germplasm for resistance to Phomopsis seed decay. Plant Dis. 99, 1517–1525. doi: 10.1094/PDIS-04-14-0429-RE
Li, S., and Smith, J. (2016). Evaluation of soybean breeding lines for resistance to Phomopsis seed decay in Stoneville, Mississippi, 2014. Plant Dis. Manag. Rep. 10:FC045.
Li, S., Smith, J., and Nelson, R. (2011). Resistance to Phomopsis seed decay identified in maturity group V soybean plant introductions. Crop Sci. 51, 2681–2688. doi: 10.2135/cropsci2011.03.0162
Lin, C.-H., Yang, S. L., Wang, N.-Y., and Chung, K.-R. (2010). The FUS3 MAPK signaling pathway of the citrus pathogen Alternaria alternata functions independently or cooperatively with the fungal redox-responsive AP1 regulator for diverse developmental, physiological and pathogenic processes. Fungal Genet. Biol. 47, 381–391. doi: 10.1016/j.fgb.2009.12.009
Liu, L., White, M. J., and Macrae, T. H. (1999). Transcription factors and their genes in higher plants. Eur. J. Biochem. 262, 247–257. doi: 10.1046/j.1432-1327.1999.00349.x
Lynch, M. (2007). The evolution of genetic networks by non-adaptive processes. Nat. Rev. Genet. 8, 803–813. doi: 10.1038/nrg2192
Mehrabi, R., Zwiers, L.-H., De Waard, M. A., and Kema, G. H. J. (2006). MgHog1 regulates dimorphism and pathogenicity in the fungal wheat pathogen Mycosphaerella graminicola. Mol. Plant Microbe Interact. 19, 1262–1269. doi: 10.1094/MPMI-19-1262
Minor, H., Brown, E., Doupnik, B. Jr., Elmore, R., and Zimmerman, M. (1993). Registration of Phomopsis seed decay resistant soybean germplasm MO/PSD-0259. Crop Sci. 33:1105. doi: 10.2135/cropsci1993.0011183X003300050052x
Mosca, R., Pache, R. A., and Aloy, P. (2012). The role of structural disorder in the rewiring of protein interactions through evolution. Mol. Cell. Proteomics 11:M111.014969. doi: 10.1074/mcp.M111.014969
Motoyama, T., Ochiai, N., Morita, M., Iida, Y., Usami, R., and Kudo, T. (2008). Involvement of putative response regulator genes of the rice blast fungus Magnaporthe oryzae in osmotic stress response, fungicide action, and pathogenicity. Curr. Genet. 54, 185–195. doi: 10.1007/s00294-008-0211-0
Musungu, B., Bhatnagar, D., Brown, R. L., Fakhoury, A. M., and Geisler, M. (2015). A predicted protein interactome identifies conserved global networks and disease resistance subnetworks in maize. Front. Genet. 6:201. doi: 10.3389/fgene.2015.00201
Ohbayashi, T., Makino, Y., and Tamura, T.-A. (1999). Identification of a mouse TBP-like protein (TLP) distantly related to the Drosophila TBP-related factor. Nucleic Acids Res. 27, 750–755. doi: 10.1093/nar/27.3.750
Östlund, G., Schmitt, T., Forslund, K., Köstler, T., Messina, D. N., Roopra, S., et al. (2010). InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 38(Suppl. 1), D196–D203. doi: 10.1093/nar/gkp931
Pareek, M., and Rajam, M. V. (2017). RNAi-mediated silencing of MAP kinase signalling genes (Fmk1, Hog1 and Pbs2) in Fusarium oxysporum reduces pathogenesis on tomato plants. Fungal Biol. 121, 775–784. doi: 10.1016/j.funbio.2017.05.005
Pathan, M., Clark, K., Wrather, J., Sciumbato, G., Shannon, J., Nguyen, H., et al. (2009). Registration of soybean germplasm SS93-6012 and SS93-6181 resistant to Phomopsis seed decay. J. Plant Regist. 3, 91–93. doi: 10.3198/jpr2008.01.0002crg
Ramamoorthy, V., Zhao, X., Snyder, A. K., Xu, J. R., and Shah, D. M. (2007). Two mitogen-activated protein kinase signalling cascades mediate basal resistance to antifungal plant defensins in Fusarium graminearum. Cell. Microbiol. 9, 1491–1506. doi: 10.1111/j.1462-5822.2006.00887.x
Rhoden, S. A., Garcia, A., Rubin-filho, C. J., Azevedo, J. L., and Pamphile, J. A. (2012). Phylogenetic diversity of endophytic leaf fungus isolates from the medicinal tree Trichilia elegans (Meliaceae). Genet. Mol. Res. 11, 2513–2522. doi: 10.4238/2012.June.15.8
Santos, J. M., Vrandečić, K., ćosić, J., Duvnjak, T., and Phillips, A. J. L. (2011). Resolving the Diaporthe species occurring on soybean in Croatia. Persoonia 27, 9–19. doi: 10.3767/003158511X603719
Schuette, S., Piatkowski, B., Corley, A., Lang, D., and Geisler, M. (2015). Predicted protein-protein interactions in the moss Physcomitrella patens: a new bioinformatic resource. BMC Bioinformatics 16:89. doi: 10.1186/s12859-015-0524-1
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shimizu, K., Hicks, J. K., Huang, T.-P., and Keller, N. P. (2003). Pka, Ras and RGS protein interactions regulate activity of AflR, a Zn(II)2Cys6 transcription factor in Aspergillus nidulans. Genetics 165, 1095–1104.
Sinclair, J. (1993). Phomopsis seed decay of soybeans: a prototype for studying seed disease. Plant Dis. 77, 329–334. doi: 10.1094/PD-77-0329
Smith, S., Fenn, P., Chen, P., and Jackson, E. (2008). Inheritance of resistance to Phomopsis seed decay in PI360841 soybean. J. Hered. 99, 588–592. doi: 10.1093/jhered/esn037
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Manners, J. M., Singh, K. B., and Taylor, J. M. (2015). Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 11:e1004806. doi: 10.1371/journal.ppat.1004806
Stark, C., Breitkreutz, B.-J., Reguly, T., Boucher, L., Breitkreutz, A., and Tyers, M. (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34(Suppl. 1), D535–D539. doi: 10.1093/nar/gkj109
Talbot, N. J. (2003). Functional genomics of plant–pathogen interactions. New Phytol. 159, 1–4. doi: 10.1046/j.1469-8137.2003.00809.x
Taylor, I. W., Linding, R., Warde-Farley, D., Liu, Y., Pesquita, C., Faria, D., et al. (2009). Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 27, 199–204. doi: 10.1038/nbt.1522
TeKrony, D., Egli, D., Stuckey, R., and Loeffler, T. (1985). Effect of benomyl applications on soybean seedborne fungi, seed germination, and yield. Plant Dis. 69, 763–765. doi: 10.1094/PD-69-763
Urban, M., Cuzick, A., Rutherford, K., Irvine, A., Pedro, H., Pant, R., et al. (2016). PHI-base: a new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 45, D604–D610. doi: 10.1093/nar/gkw1089
Wang, M., Chen, C., Zhu, C., Sun, X., Ruan, R., and Li, H. (2014). Os2 MAP kinase-mediated osmostress tolerance in Penicillium digitatum is associated with its positive regulation on glycerol synthesis and negative regulation on ergosterol synthesis. Microbiol. Res. 169, 511–521. doi: 10.1016/j.micres.2013.12.004
Wang, X., and Jin, Y. (2017). Predicted networks of protein-protein interactions in Stegodyphus mimosarum by cross-species comparisons. BMC Genomics 18:716. doi: 10.1186/s12864-017-4085-8
Weßling, R., Epple, P., Altmann, S., He, Y., Yang, L., Henz, S. R., et al. (2014). Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 16, 364–375. doi: 10.1016/j.chom.2014.08.004
Wilson, R. A., and Talbot, N. J. (2009). Under pressure: investigating the biology of plant infection by Magnaporthe oryzae. Nat. Rev. Microbiol. 7, 185–195. doi: 10.1038/nrmicro2032
Winnenburg, R., Baldwin, T. K., Urban, M., Rawlings, C., Köhler, J., and Hammond-Kosack, K. E. (2006). PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 34, D459–D464. doi: 10.1093/nar/gkj047
Wrather, J., Shannon, J., Stevens, W., Sleper, D., and Arelli, A. (2004). Soybean cultivar and foliar fungicide effects on Phomopsis sp. seed infection. Plant Dis. 88, 721–723. doi: 10.1094/Pdis.2004.88.7.721
Xu, J.-R., and Hamer, J. E. (1996). MAP kinase and cAMP signaling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea. Genes Dev. 10, 2696–2706. doi: 10.1101/gad.10.21.2696
Yamada, O., Lee, B. R., Gomi, K., and Iimura, Y. (1999). Cloning and functional analysis of the Aspergillus oryzae conidiation regulator gene brlA by its disruption and misscheduled expression. J. Biosci. Bioeng. 87, 424–429. doi: 10.1016/S1389-1723(99)80089-9
Yu, H., Braun, P., Yildirim, M. A., Lemmens, I., Venkatesan, K., Sahalie, J., et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–110. doi: 10.1126/science.1158684
Yu, H., Luscombe, N. M., Lu, H. X., Zhu, X., Xia, Y., Han, J. D., et al. (2004). Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 14, 1107–1118. doi: 10.1101/gr.1774904
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhao, X.-M., Zhang, X.-W., Tang, W.-H., and Chen, L. (2009). FPPI: Fusarium graminearum protein-protein interaction database. J. Proteome Res. 8, 4714–4721. doi: 10.1021/pr900415b
Keywords: Phomopsis longicolla, soybean, interactome, network, protein–protein interactions, pathogenicity
Citation: Li S, Musungu B, Lightfoot D and Ji P (2018) The Interactomic Analysis Reveals Pathogenic Protein Networks in Phomopsis longicolla Underlying Seed Decay of Soybean. Front. Genet. 9:104. doi: 10.3389/fgene.2018.00104
Received: 20 September 2017; Accepted: 15 March 2018;
Published: 03 April 2018.
Edited by:
Xiaogang Wu, Institute for Systems Biology, United StatesReviewed by:
Syed Aun Muhammad, Bahauddin Zakariya University, PakistanOksana Sorokina, The University of Edinburgh, United Kingdom
Copyright © 2018 Li, Musungu, Lightfoot and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuxian Li, c2h1eGlhbi5saUBhcnMudXNkYS5nb3Y=
†Present address: Bryan Musungu, Warm Water Aquaculture Unit, United States Department of Agriculture, Agricultural Research Service, Stoneville, MS, United States