 B. G. Welderufael
B. G. Welderufael Peter Løvendahl
Peter Løvendahl Dirk-Jan de Koning
Dirk-Jan de Koning Lucas L. G. Janss
Lucas L. G. Janss W. F. Fikse
W. F. Fikse- 1Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, Uppsala, Sweden
- 2Center for Quantitative Genetics and Genomics, Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark
Because mastitis is very frequent and unavoidable, adding recovery information into the analysis for genetic evaluation of mastitis is of great interest from economical and animal welfare point of view. Here we have performed genome-wide association studies (GWAS) to identify associated single nucleotide polymorphisms (SNPs) and investigate the genetic background not only for susceptibility to – but also for recoverability from mastitis. Somatic cell count records from 993 Danish Holstein cows genotyped for a total of 39378 autosomal SNP markers were used for the association analysis. Single SNP regression analysis was performed using the statistical software package DMU. Substitution effect of each SNP was tested with a t-test and a genome-wide significance level of P-value < 10-4 was used to declare significant SNP-trait association. A number of significant SNP variants were identified for both traits. Many of the SNP variants associated either with susceptibility to – or recoverability from mastitis were located in or very near to genes that have been reported for their role in the immune system. Genes involved in lymphocyte developments (e.g., MAST3 and STAB2) and genes involved in macrophage recruitment and regulation of inflammations (PDGFD and PTX3) were suggested as possible causal genes for susceptibility to – and recoverability from mastitis, respectively. However, this is the first GWAS study for recoverability from mastitis and our results need to be validated. The findings in the current study are, therefore, a starting point for further investigations in identifying causal genetic variants or chromosomal regions for both susceptibility to – and recoverability from mastitis.
Introduction
Because of considerable improvements of production traits and unfavorable genetic correlations, mainly with milk production, mastitis continues to be one of the most frequent dairy cattle diseases with significant economic implications (Halasa et al., 2007; Hogeveen et al., 2011). Breeding can be instrumental in improving resistance to mastitis (Rupp and Boichard, 2003). However, genetic response to traditional selection is small because of the low heritability (h2 ≈ 0.03) (Heringstad et al., 2000; Carlén et al., 2004) of the trait. Moreover, mastitis is not easy to measure directly and often the phenotype data are binary (absent or present) at the observable scale. In genetic evaluations, this all-or-none trait definition may not fully utilize all information available in the data, for instance the duration and levels of infection (Carlén et al., 2005; Vazquez et al., 2009). Genome-wide association studies (GWAS) present options for direct selection of such traits through genetic markers. The results from GWAS can then be used in designing breeding schemes that increase the frequency of favorable alleles (Tiezzi et al., 2015).
Genome-wide association studies utilize information on genetic markers like single nucleotide polymorphisms (SNPs) to determine association with a trait of interest assuming that a marker is in linkage disequilibrium (LD) with, or close to, a causative mutation (Hirschhorn and Daly, 2005; Goddard and Hayes, 2009). In cattle, GWAS have been performed to evaluate marker or SNP association with clinical mastitis (CM) and with milk somatic cell count (SCC) as indicator trait, and many quantitative trait loci (QTLs) have been reported. QTLs for SCC have been reported on: Bos taurus autosomes (BTA) 6, 13, 14, and 20 in Nordic Holstein cattle (Sahana et al., 2013); BTA 6, 10, 15, and 20 in Irish Holstein-Friesian cattle (Meredith et al., 2012); 24 chromosomes (a total of 171 significant SNPs) in Valdostana Red Pied cattle breed (Strillacci et al., 2014); and on BTA 6, 13, 19 and X in German Holstein cows (Abdel-Shafy et al., 2014). A review (Sender et al., 2013) indicated that QTLs for mastitis resistance measured via either SCC or less frequently using CM records have been found on almost all chromosomes. The cattle QTL database1 also showed that QTLs for the keyword ‘mastitis,’ i.e., QTLs for mastitis related traits including CM, SCC or other indicator traits are more or less evenly spread over all chromosomes with the largest number of detected associations on BTA6 (see Supplementary Figure S1). BTA6 is known for harboring QTLs that affect both milk quality traits, e.g., the casein gene cluster (Nilsen et al., 2009; Pirola et al., 2013) and CM (Nilsen et al., 2009; Sodeland et al., 2011).
These studies have been useful for identifying genetic variants and genes associated with CM and indicator traits. However, they lack consistency in reporting SNPs or genes implicated in mastitis, and to our knowledge, none of these studies have reported any genetic variant or chromosomal regions associated with recoverability from mastitis. We, therefore, performed GWAS using single SNP regression analysis for two main objectives: (1) to identify new or confirm previously identified regions of the genome for their association with mastitis susceptibility, and (2) identify variants and regions of the genome associated with recoverability from mastitis in Danish Holstein cows.
Materials and Methods
The raw phenotype data, analyzed in the current study, were edited and used by Welderufael et al. (2017) to estimate genetic parameters for susceptibility to – and recoverability from mastitis in Danish Holstein cows using bi-variate threshold models. The data contained a total of 89,232 weekly transition records for a total of 1,791 Danish Holstein cows in parities 1–3 distributed over one research herd (Danish Cattle Research Center, Tjele, Denmark) and six commercial dairy herds. Welderufael et al. (2017) derived the transition records from SCC data recorded at every milking using voluntary milking system (VMS, DeLaval International AB, Tumba, Sweden) and stored in an online database. Mastitis could be caused by different pathogens to which the cows may respond differently. However, such pathogen-specific data were not available in the current study. Welderufael et al. (2017) used the concept of transition model to define the traits of interest (susceptibility to – and recoverability from mastitis). The transition model helps to capture the variations an individual cow may exhibit in the entire disease course by capturing susceptibility as well as recovery. During a specified period of time, a cow is assumed to move between or within two states – healthy (H) and diseased (D). A cow may return to the same state more than once, meaning repeated disease cases are acknowledged by the model (Franzén et al., 2012). For a healthy cow, there is a risk of becoming infected and for a mastitic cow there is a possibility of recovering, which is conceptualized into probabilities of mastitis and recovery (Franzén et al., 2012). For each cow, several transitions from H to D, denoted as HD, and from D to H, denoted as DH, can occur within a lactation. Franzén et al. (2012) depicted such occurrences in a transition probability matrix, Ti, for cow i, as follows:
where = Probability of remaining in the H state for cow i.
= Probability of moving from H to D state for cow i.
= Probability of moving from D to H state for cow i.
= Probability of remaining in the D state for cow i.
In the above transition probability matrix, the first row consists of the probabilities of being in either of both states at time t+1for cow i that is healthy at time t, and the second row consists of the probabilities of being in either of both states at t+1 for cow i that is diseased at time t. In practice, the transition matrix is desired to have high values of (probability of remaining in the H state for cow i) and (probability of moving from D to H state, i.e., fast recovery if cow i had moved from H to D state), and consequently low values of (probability of moving from H to D state) and (probability of remaining in D state) (Franzén et al., 2012). Note that the probabilities sum to 1 within rows, and only two probabilities (two traits) are needed to model the transitions of animals between the H and D states.
Thus, for each cow and lactation the sequence of H’s and D’s, indicating whether or not a cow had mastitis on subsequent test weeks, was converted into a new sequence of weekly transitions indicators: 0 if a cow remains in the same state and 1 if the cow changes state. This resulted into two series of transition records: one for healthy to diseased (HD, to define susceptibility to mastitis) and the other for diseased to healthy (DH, to define recoverability from mastitis). A more detailed description of the data is given by Welderufael et al. (2017).
Phenotypic Statistical Analyses
Phenotypic statistical analyses were performed to obtain adjusted phenotypes. The raw phenotypes (transition records) were adjusted for different fixed and random effects before using them in the association analysis. Welderufael et al. (2017) modeled the phenotype data as a linear combination of systematic effects and as a function of time. A lactation curve [f(DIM)] was modeled by a combination of Legendre polynomials and a Wilmink term (exp-0.05×DIM) (Wilmink, 1987), to reflect that susceptibility and recovery is not constant during the lactation. A time variable was used to indicate the duration of an episode. Changes of risk during each episode [f(time)] were modeled with Legendre polynomials. The model for the phenotypic analyses included parity (1–3) and herd (1–7) as fixed effects, regression coefficients of a second order polynomial on DIM plus a Wilmink term and regression coefficients of a third order polynomial on time as covariates, and random effects of herd-test-week, cow-parity interaction, and cow effects. The observed transitions were linked to an underlying continues scale called liability (Gianola and Foulley, 1983). This relationship between the observed binary response (y), and the unobservable liability (λ) is formally expressed as:
where τ is a fixed threshold and usually set to an arbitrary value (here 0), such that y = 1 if λ > 0 and 0 otherwise. The mean on the liability scale models the average probability for y = 1. Accordingly, the observed transitions and liabilities were modeled with a threshold model as follows:
where λ was vector of the underlying liabilities linked to the transition scores (y); b was a vector of all fixed effects (herd and parity) and covariates (regression coefficients of a second order polynomial on DIM plus a Wilmink term and regression coefficients of a third order polynomial on time); h, p, and c were vectors of random effects of herd-test-week, cow-parity interaction, and cow effects, respectively; X, Z1, Z2, and Z3 were respective incidence matrices, e was a vector of the residual effects. The vectors of random effects (h, p, c, and e) were assumed to be normally distributed, i.e., and , where , , , and were, respectively, variances for herd-test-week, cow-parity interaction, cow, and residual effects, and I were the identity matrices. The residual variance () is not identifiable, and it was fixed to 1. More detailed definitions and descriptions of the model and traits are provided by Welderufael et al. (2017).
The phenotypic analyses were performed using the statistical software package MCMCglmm (Hadfield, 2010) in R (R Core Team, 2016). Estimation of the random cow plus cow∗parity effects were based on 5,000 samples from a single chain of 250,000 iterations with a burn-in of 50,000 and samples stored at every 50th round. In some association analyses, in particular the so called GRAMMAR analysis (Aulchenko et al., 2007), residuals from a polygenic model are taken as phenotypes. This procedure aims to pre-correct the data for polygenic effects. However, our association analyses include a random genetic (cow) effects in the model and the aim of the phenotypic analysis is to extract meaningful phenotypes for the GWAS. The posterior means and standard deviations of the random cow plus cow∗parity effects were obtained from the sum, mean and standard deviation of the estimates for each animal and trait. The calculated posterior means were used as adjusted phenotype data in the association analyses. The weights used in the association analyses were calculated as weight = 1/(PSD)2, where PSD is the posterior standard deviation of the cow plus cow∗parity effects. Association analyses were carried out on four datasets (each parity separately and all parities together) to investigate parity specific associations. The number of records, means and SD of the adjusted phenotype used in the association analyses are in Table 1.
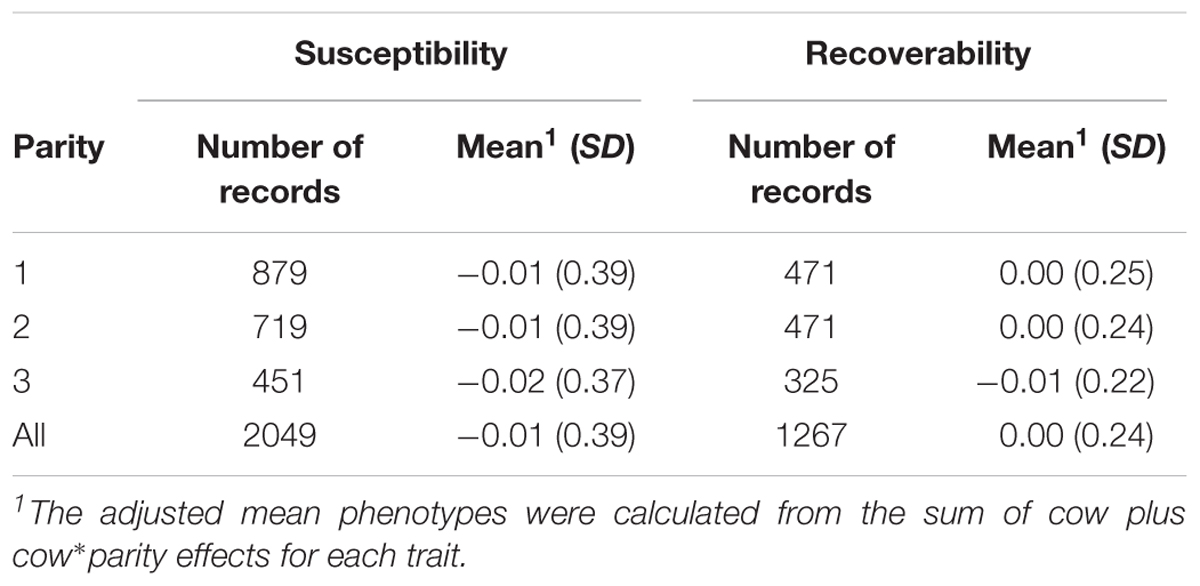
TABLE 1. Number of records and mean (SD) used as phenotype in the association analyses for susceptibility to – and recoverability from mastitis.
Further bivariate and trivariate analyses were performed to study correlations between traits as well as between parities but within traits. The bivariate model was similar to equation (2) and except the residual, all between traits random effects were assumed correlated. The trivariate model used to calculate correlations between parities was also similar to the model in equation (2) but did not include parity and cow-parity interaction effects. In this trivariate model only the cow effects across parities were correlated. Herd-test-week effects were included and assumed uncorrelated.
Genotype Data
The raw data contained 1957 cows genotyped with the Illumina BovineSNP50_v2 (Illumina Inc., San Diego, CA, United States) for a total of 46931 autosomal SNP markers. However, we use phenotypes records from our previous study (Welderufael et al., 2017) and the animals with phonotypes records in our previous study were not all genotyped. In our previous study (Welderufael et al., 2017), we had phenotypes records for a total of 1791 cows. After matching the phenotype to the genotype data, only 997 cows with phenotypes remained in the genotype data. Quality control (QC) for markers was done using PLINK (Purcell et al., 2007). SNPs with more than 10% missing genotypes and SNPs with minor allele frequency (<1%) were excluded. One cow was also removed from next analyses for having less than 90% genotype call rate. All the 39378 SNP markers used for the final association analyses were distributed over the cattle genome as shown in Supplementary Figure S2.
Detection of Population Stratification
Multidimensional scaling (MDS) of SNP genotypes was performed for detection of population stratification for the 996 animals that passed genotyping quality control checks. The aim was to assess potential genetic clustering of animals by herd. The cows came from seven herds: one research herd and six commercial herds. In order to visualize possible population stratifications, MDS plots of an identity-by-state (IBS) matrix was generated using PLINK (Purcell et al., 2007).
As shown in Figures 1A and 1C, the first five MDS components explained 14.3, 6.2, 5.8, 5.4, and 3.9%, respectively, of the total genomic variance, and therefore, we reasoned that fitting these first five MDS components as clustering factor into our model could improve the GWAS results because it corrects for confounding effect of the population structure (Price et al., 2006). The MDS plots of the IBS matrix further revealed that three animals were genetic outliers and appeared to form their own distinct cluster (Figure 1B). After removing the three genetic outlier animals, the remaining animals formed no distinct cluster (Figure 1D).
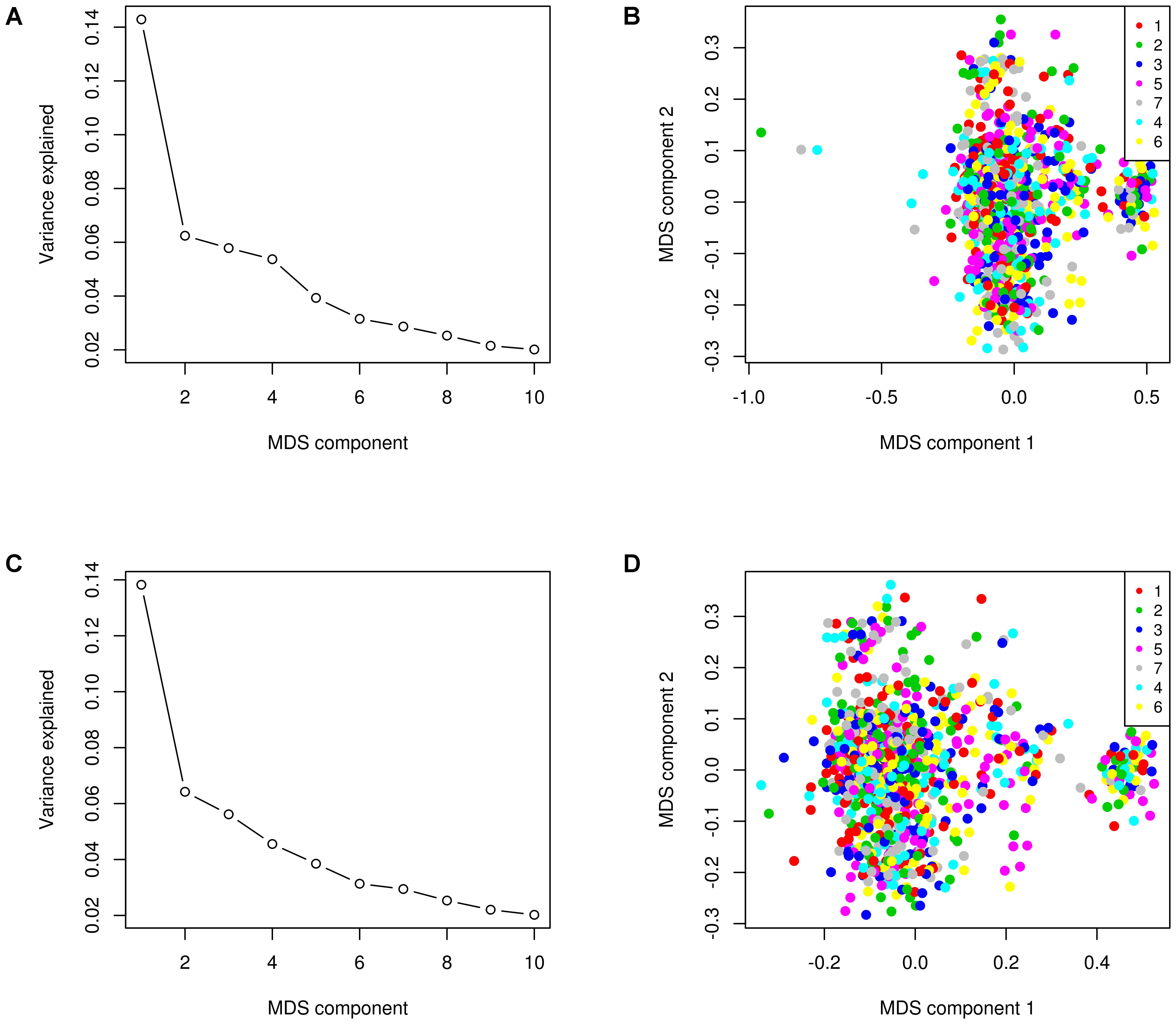
FIGURE 1. Multidimensional scaling (MDS) plot of identity-by-state (IBS) matrix. Genomic variance explained by the first 10 MDS components before (A) and after (C) removing genetic outlier animals. The MDS plot before (B) and after (D) removing the three genetic outlier animals shows the population structure within herds (distinguished by color) identified by the first two dimensions.
After QC and removal of four cows (one for having less than 90% genotype data and three other cows for being genetic outliers), 993 cows and 39378 SNP markers remained and were used for final association analyses.
Association Analysis and Significance Test
Single SNP association analysis was performed with a linear mixed model in DMU (Madsen and Jensen, 2013). Each SNP was fitted as a covariate. To account for shared genetic effects of related individuals, pedigree-based polygenic effect was included by fitting individual animal as a random effect in the model. To account for the confounding effect of population structure observed in the MDS plots, the top five MDS components were included as covariates in the association analysis. The single SNP regression analysis was performed using the following statistical model:
where y was a vector of posterior mean of summed cow and cow∗parity effects obtained from equation (2); 1 was a vector of ones; μ was the general mean; X was a matrix containing the top five MDS components fitted as covariates, and b was a vector of associated effects; S was vector of SNP genotypes coded as 0, 1, or 2 for genotype copies of one of the alleles at each locus; α was the allele substitution effect; a was a vector of random additive polygenic effects, which was assumed to follow normal distribution, where A is the pedigree-based additive genetic relationship matrix; Z was an incidence matrix relating elements of the vector of additive polygenic values a to individual phenotypes; e was a vector of random residuals and the residuals were assumed to follow a normal distribution, , where W was a diagonal matrix with diagonal element ; and were the additive polygenic and residual variances, respectively. The weight wi is used to account for the different residual variances due to parities. The pedigree for the animals (993) in the genotype data was extended to a maximum depth of 10.9 generations and an average of 4.4 known generations. The extended pedigree contained a total of 12779 animals and were offspring of 2659 sires and 9536 dams.
The association analysis was conducted using the statistical software package DMU (Madsen and Jensen, 2013). The null hypothesis H0: b = 0 was tested with a t-test. A genome-wide Bonferroni corrected significance threshold can be obtained by dividing the nominal significance threshold (0.05) by the number of independent tests (total number of SNPs, 39378) in the analyses. The P-value after Bonferroni multiple testing corrections was 1.26974E-06, equivalent to -log10 (P-value) = 5.90. However, in the current study no P-value exceeded the Bonferroni threshold and hence a liberal threshold of P-value < 10-4 was used to declare significant SNP-trait associations.
Candidate Gene Identification
Candidate genes were identified by checking whether significantly associated SNPs were located within a gene, or within 1 Mb up- or downstream from a gene using the Ensembl2 gene annotation system (Aken et al., 2016). We also made use of the human gene database- GeneCards3 for functional annotations of those identified genes. Throughout the text, genomic positions of genetic variants and genes were based on the Bos taurus genome UMD3.1 assembly (Zimin et al., 2009).
Results
Descriptive Statistics of the Phenotypic Analysis
Herd-test-week variation was lower for the HD trait than for the DH trait (Table 2), which translates to less variation in susceptibility to- than in recoverability from mastitis within year across time. On the other hand, animal related variation was higher for susceptibility to mastitis (HD) than for recoverability from mastitis (DH) (Table 2). The between traits posterior mean of cow effect correction [rc = -0.90 (0.07)] was much higher than the cow-parity interaction correction [rcp = -0.29 (0.11)].
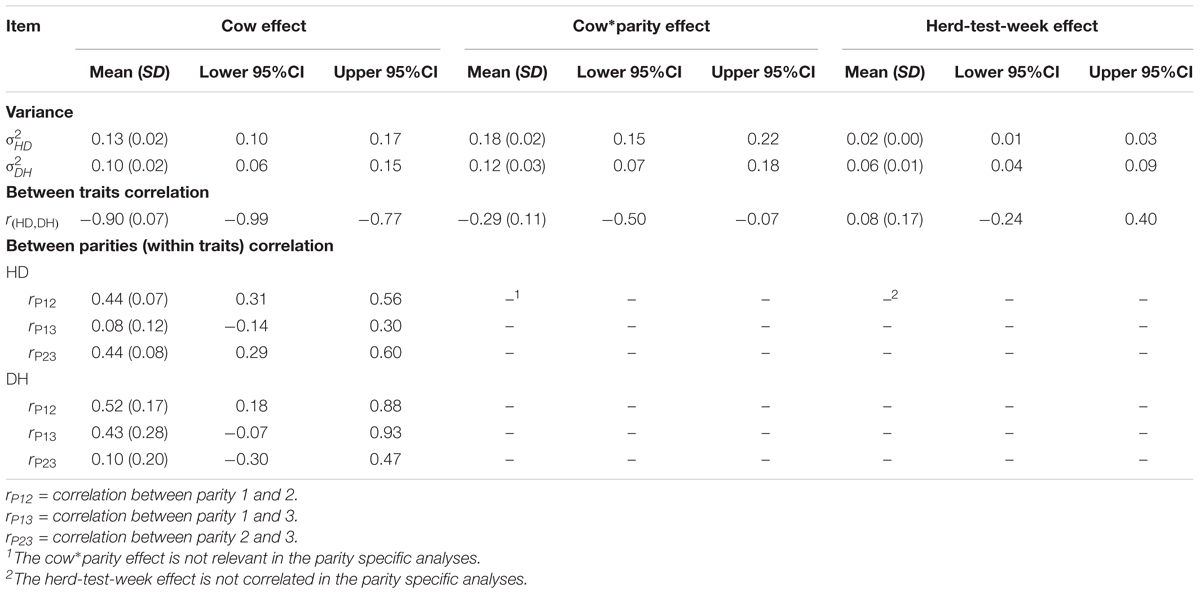
TABLE 2. Posterior mean (SD) of variances and correlations between susceptibility to – (HD) and recoverability (DH) from mastitis, and between parities but within traits.
The variance components presented in Table 2 also showed large cow-parity interaction effects, which indicates these traits to be very parity specific. To show this even clearer we added new parity specific analyses for the phenotype input variable, which confirmed low to medium correlations (0.08–0.52) across parities. As expected, for the HD trait, the smallest between parities cow effect correlation was observed between the non-consecutive parities 1 and 3 [rp13 = 0.08 (0.12)], whereas for the DH trait, the smallest between parities cow effect correlation was observed between the consecutive parities 2 and 3 [rp23 = 0.10 (0.20)] (Table 2).
SNP Variants Associated With Susceptibility to – and Recoverability From Mastitis
The nominal significance level of P-value < 10-4 was considered to determine genome-wide significant SNP-trait association. For the full data (all parities analyzed together), significant SNP-trait associations were detected on BTA3 and 7 for mastitis susceptibility and on BTA7 and 15 for recoverability from mastitis (Table 3). Parity specific significant SNP-trait association were also detected on the same chromosomes but no overlapping association signals were observed. Significant association signals in parity 1 and in the other parities (parity 2 or 3) detected on the same chromosome did not overlap, for either of the traits. For susceptibility to mastitis, the highest number of significant associations were observed on BTA7 and BTA13. For recoverability from mastitis, the highest number of significant associations were observed on BTA12 and BTA13. In total, 29 significant SNPs (14 for susceptibility to mastitis and 15 for recoverability) were detected.
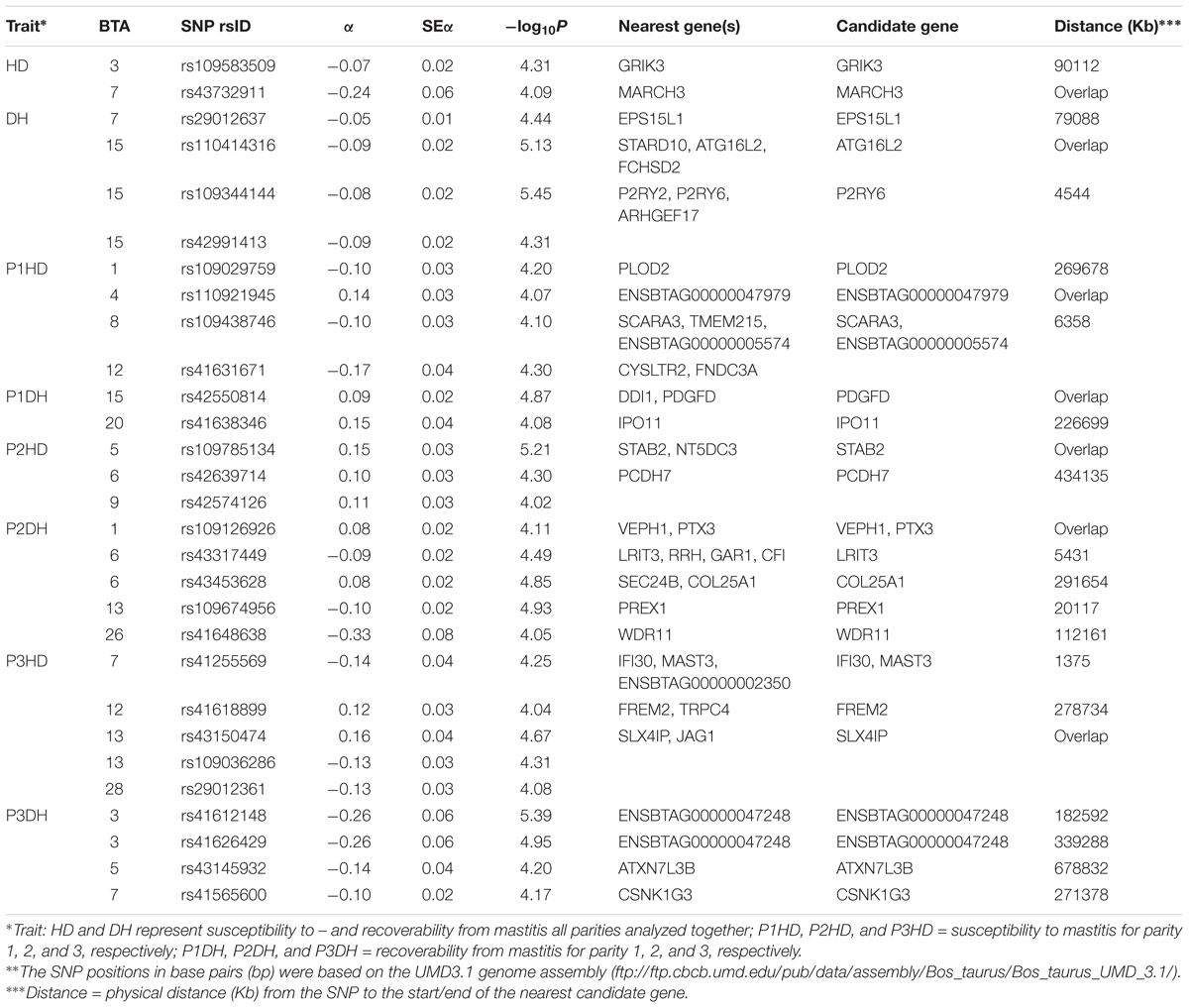
TABLE 3. List of SNPs in the Bos taurus autosomes (BTA) showing significant [-log10(P-value) > 4] associations with susceptibility to – (HD) and recoverability (DH) from mastitis.
As shown in the quantile–quantile (QQ) plot (Figure 2) and Manhattan plot (Figure 3), only 6 SNPs (2 for susceptibility and 4 for recoverability) reached the defined genome-wide significance threshold when all parities were analyzed together. The QQ plots and Manhattan plots for the parity specific associations are provided in the Supplementary Material (Supplementary Figures S3–S8). The clear deviation from the expected P-value only in the tail area of the QQ plots (Figure 2 and QQ plots in the Supplementary Material) further demonstrated that the GWAS results were not substantially influenced by the observed population stratification. Influence of population stratification was adequately controlled by including the MDS components in the model and this was quantified by a calculated genomic inflation factor ≤ 1.05 (on average λ = 1.02, results not shown).
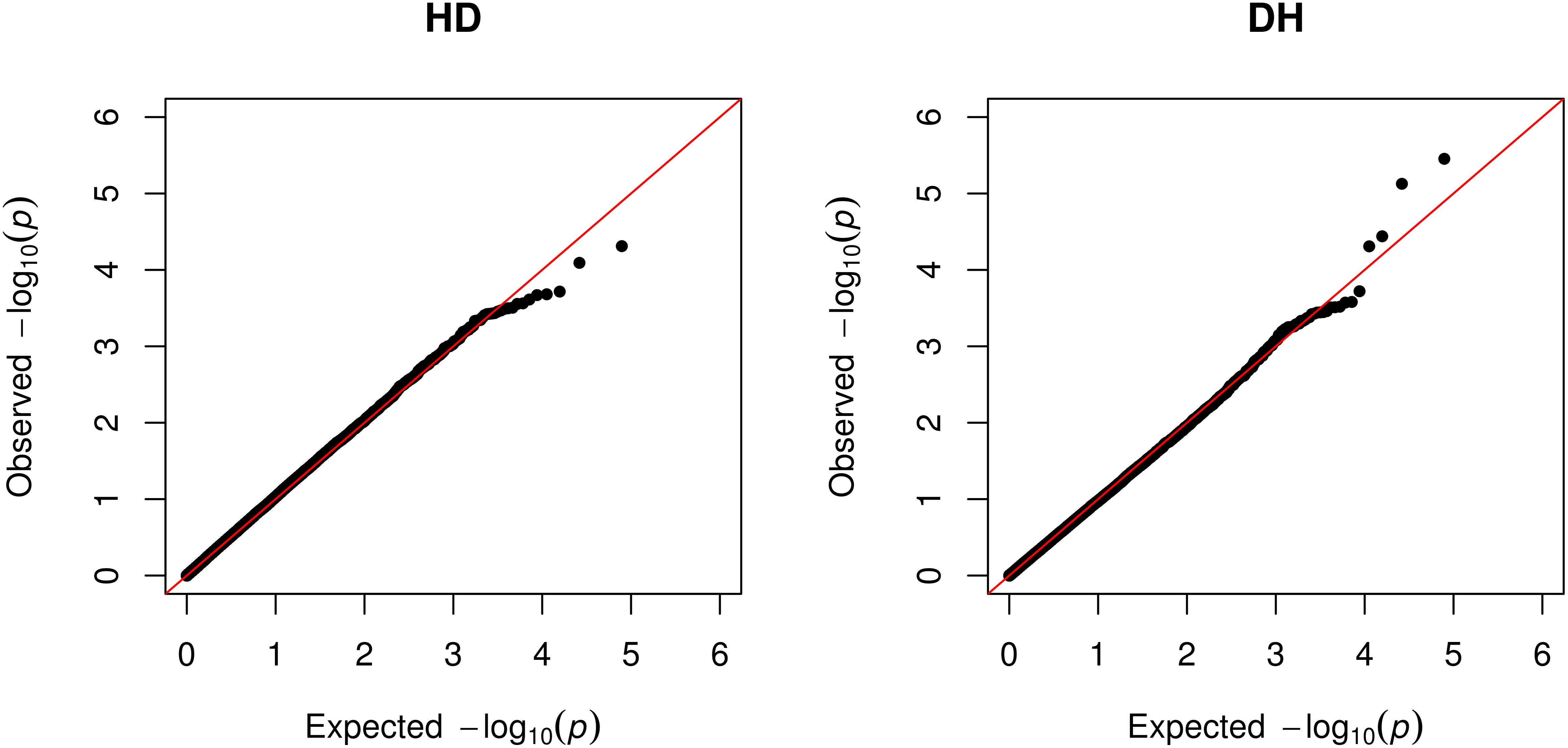
FIGURE 2. QQ-plot of the expected null distribution of the P-values vs. the observed null distribution of the P-values for susceptibility to – (HD) and recoverability (DH) from mastitis for all parities analyzed together.
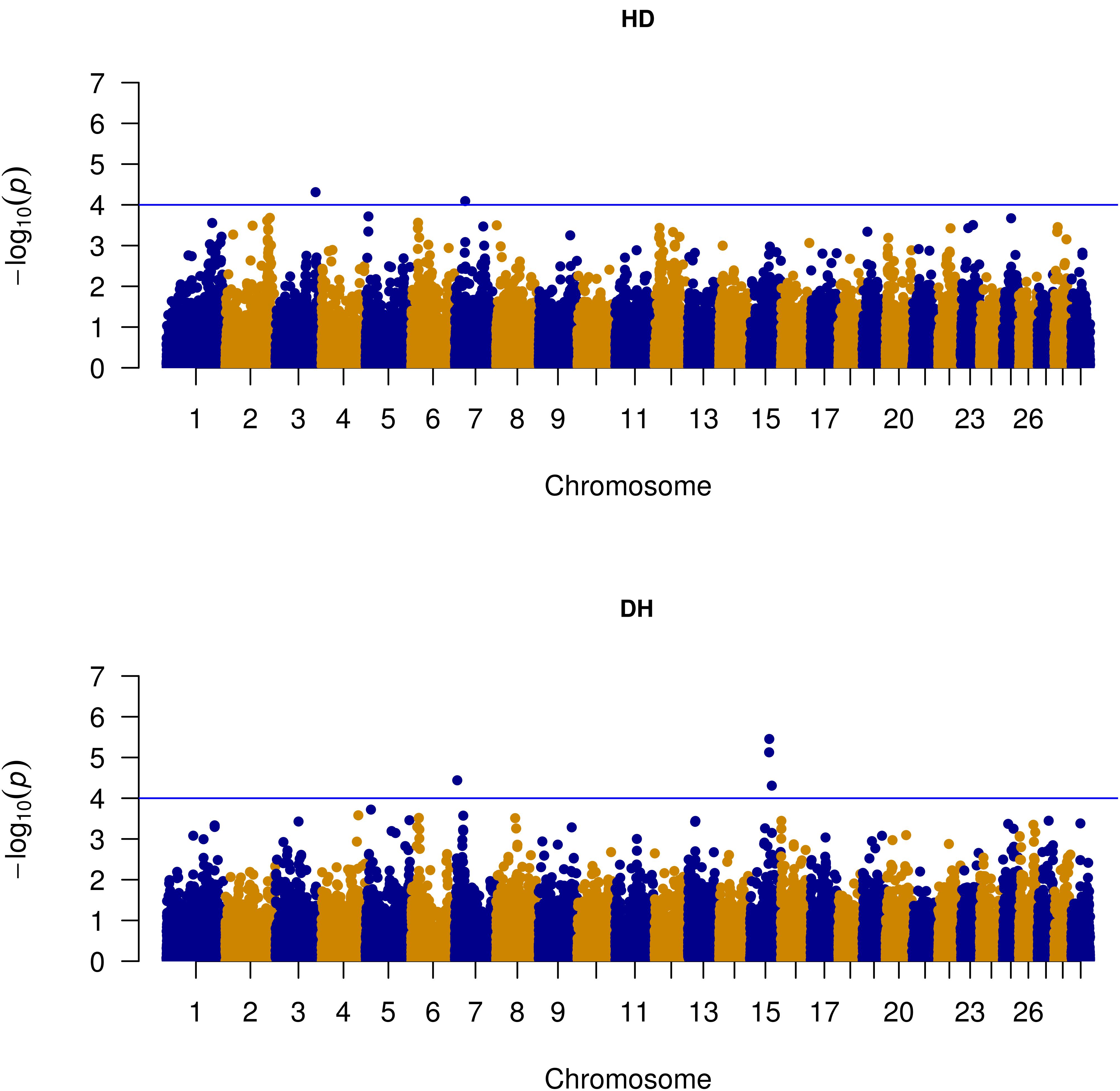
FIGURE 3. Manhattan plot of genome-wide associations with susceptibility to – (HD) and recoverability from mastitis (DH) for all parities. The blue line represents suggestive significance level [–log10(P-value) = 4].
Discussion
Because mastitis is very frequent and unavoidable, adding recovery information to the analysis is of great interest. In this study we have performed GWAS using single SNP regression analysis and identified SNP variants in and near by possible causative genes not only for susceptibility to – but also for recoverability from mastitis. To our knowledge, this is the first study to report associated SNP variants and identify possible causative genes for recoverability from mastitis in Danish Holstein cows.
Variances and Correlations From the Phenotypic Analyses
The lower posterior mean of herd-test-week variance for HD than for DH could indicate that management to prevent mastitis (and other factors influencing susceptibility) is fairly stable across time, whereas management (and other factors) related to recoverability is more variable over time. This could suggest that there are possibilities to enhance recoverability by improvements in herd management (Welderufael et al., 2017). The posterior mean of cow related variances indicates less variation in recoverability from mastitis than the variation in susceptibility to mastitis among cows. Between traits, the high posterior mean of cow effect correlation (rc = -0.90) indicates that highly susceptible cows are in general less likely to recover fast. Relatively, the trait correlation between cow-parity interaction effects was much smaller than the cow effect correlation, indicating even highly susceptible cows in parity one could recover relatively faster in the other next parity.
The cow effect correlations across parities but within traits were positive and ranged from low to moderate. For the HD trait, the weakest posterior mean of cow effect correlation across parities was observed between parities 1 and 3, indicating that susceptibility in parity 1 is distinct from susceptibility in parity 3. For the DH trait, the weakest posterior mean of correlation across parities was observed between parities 2 and 3, suggesting recoverability in parities 2 and 3 should not be considered as the same trait. In general, we would expect parities to be similar, but this data shows larger variation in cow-parity interaction than the cow effect variation as we presented it in Table 2. The cow-parity interaction variance is larger than the cow variance, i.e., the larger part of cow variance is parity specific. Results from the trivariate analysis where parities are considered as different traits, also point to relatively low agreement between parities (Table 2). So, the data does not support the general expectation that parities may be similar, and this can explain the absence of common association signals across parities in the association results. A possible confounding effects of multiple mastitis pathogens may have contributed to the lack of consistent association signals across parities and traits. Sørensen et al. (2009) reported that genetic correlations among pathogen-specific mastitis traits are far from 1 (ranged from 0.45 to 0.77), suggesting that mastitis caused by different pathogen should be considered as different traits.
SNP Variants Associated With Susceptibility to – and Recoverability From Mastitis
Mastitis is a complex trait. Numerous functional candidate genes could be involved in both directions of the disease, susceptibility to – and recoverability from mastitis. In the current study, most of the significant association signals in parity 1 and in the other parities (parity 2 or 3) detected on the same chromosome do not overlap, suggesting that genetic regulation of mastitis could also be parity dependent. Effect of genes has been shown to vary depending on parity or age of cows (Wojdak-Maksymiec et al., 2013). Wojdak-Maksymiec et al. (2013) reported that allele T of the gene TNF-α was associated with a lower number of mastitis cases in lower parities and a higher number of mastitis cases in higher parities. Similarly, the causal variants or genes could be different for susceptibility to – and recoverability from mastitis as association signals present on the same chromosome do not overlap. Our results support the findings that QTLs even for highly correlated traits (e.g., SCC and CM) present on the same chromosome do not overlap (Sender et al., 2013). Most of the significant variants discovered in the current study for both susceptibility to – and recoverability from mastitis lies within or nearby genes that have been annotated for their role in immune systems. But we observe no overlap of signals neither across parities nor traits. Here, therefore, we discuss a few potential candidates for causal genes separately for parities and traits.
Most of the suspected candidate genes for susceptibility to mastitis are found in chromosomes that are frequently reported (e.g., on the cattle QTL data base) for their association with mastitis traits defined from SCCs. The SNP rs43732911 on BTA7, an intron variant and in significant association with susceptibility to mastitis in the full dataset, is located within a potential candidate protein coding gene MARCH3 (E3 ubiquitin-protein ligase MARCH3). The gene MARCH3 encodes an E3 ubiquitin-protein ligase enzyme that may be involved in regulation of the endosomal transport pathway and it has been implicated in diverse biological functions, such as immune regulation, protein quality control, and membrane trafficking (Fukuda et al., 2006). The SNP rs109785134 on BTA5, an intron variant in significant association with susceptibility to mastitis in parity 2, is located within a protein coding gene STAB2 (stabilin-2 precursor). Studies in pigs (Howard et al., 2015) and in mice (Schledzewski et al., 2011) showed that the gene STAB2 is involved in clearing of metabolic waste from blood circulation. The protein encoded by this gene functions in angiogenesis, lymphocyte homing, cell adhesion, or receptor scavenging (GeneCards; D’Souza et al., 2013). The receptor has been shown to bind and endocytose ligands such as hyaluronan, low density lipoprotein, Gram-positive and Gram-negative bacteria, and advanced glycosylation end products (GeneCards). The SNP rs109785134 on BTA13, a missense variant and in significant association with susceptibility to mastitis in parity 3, is located within the protein coding gene SLX4IP (SLX4 interacting protein). SLX4-deficient mice have been shown to develop epithelial cancers and to have a contracted hematopoietic stem cell pool (Hodskinson et al., 2014).
Most of the candidate genes for recoverability from mastitis identified by the intron variants have been reported for their association with biological processes of adaptation and immune systems. The SNP rs110414316 on BTA15, an intron variant and in significant association with recoverability from mastitis in the full dataset is located in a protein coding gene ATG16L2 (autophagy related 16 like 2). The gene ATG16L2 has been reported to play a role in autophagy (Yin et al., 2014). The SNP rs42550814 on BTA15:4668361, an intron variant and in significant association with recoverability from mastitis in parity 1, is located within a protein coding gene PDGFD (platelet derived growth factor D). The protein encoded by the gene PDGFD plays an important role in wound healing and macrophage recruitment (Uutela et al., 2004). In a transgenic mice experiment (Uutela et al., 2004), the gene has been shown to induce macrophage recruitment, to elevate interstitial fluid pressure in dermis and resulted to wound healing. Even though, the type of wound is not from bacterial infection like as is in mastitis, the gene plays a significant role in wound recovery and we think we have found a possible candidate for causal gene in recoverability. The SNP rs109126926 on BTA1, an intron variant and in significant association with recoverability from mastitis in parity 2, is located within a protein coding gene PTX3 (pentraxin 3). The gene PTX3 may involve in the innate immune response against intra-amniotic infection and inflammation (Cruciani et al., 2010). The expression of this protein is induced by inflammatory cytokines in response to inflammatory stimuli in several mesenchymal and epithelial cell types, particularly endothelial cells and mononuclear phagocytes (GeneCards). Cytokines, an important group of inflammatory mediators, play an important role in the host innate immune response to infection (Fu et al., 2013). The protein encoded by the gene plays a role in angiogenesis and tissue remodeling which is very important part of recovery, and the protein serves as a biomarker for several inflammatory conditions (GeneCards). Lutzow et al. (2008) found PTX3 to be upregulated after Staphylococcus aureus infection, which is one of the main pathogens for mastitis. This gene has been reported in several studies for its role in innate immune response and regulation of inflammation (Lutzow et al., 2008; Brenaut et al., 2014). A review (Garlanda et al., 2016) on gene targeted mice and genetic associations in humans suggest that PTX3 plays an important role in resistance against different pathogens including Escherichia coli. Therefore, we suggest that this gene is a possible candidate for the causal gene in recoverability from mastitis.
We have also identified some potential candidate genes from the nearby gene cluster, in addition to the genes identified by within gene SNP variants. The SNP rs41255569 on BTA7, an upstream gene variant and in significant association with susceptibility to mastitis in parity 3, is mapped very near to a functionally known candidate genes MAST3 (microtubule associated serine/threonine kinase 3) and IFI30 (lysosomal thiol reductase). Both genes have been reported for their role in antigen processing. In a gene expression assays (Labbe et al., 2008), the gene MAST3 showed abundant expression in antigen-presenting cells and in lymphocytes. It has been also shown that knockdown of MAST3 decreased the presence of Toll-like receptor-4-dependent NF-kappaB that initiate the innate immune response during pathogen invasion (Labbe et al., 2008). The enzyme (a lysosomal thiol reductase) encoded by the gene IFI30 has also an important role in MHC class II-restricted antigen processing (GeneCards). The enzyme also facilitates MHC class I-restricted recognition of exogenous antigens containing disulfide bonds by CD8+ T-cells or cross-presentation (GeneCards).
In Figure 4 we present a 1Mb gene cluster surrounding the significantly associated variant in the region. This illustrates that complex traits could be regulated by several clusters of genes surrounding association signals and confirms the fact that mastitis is a complex trait and is determined by many genes with small effect limiting the ability of GWAS to identify causal genes (Hayes et al., 2010).
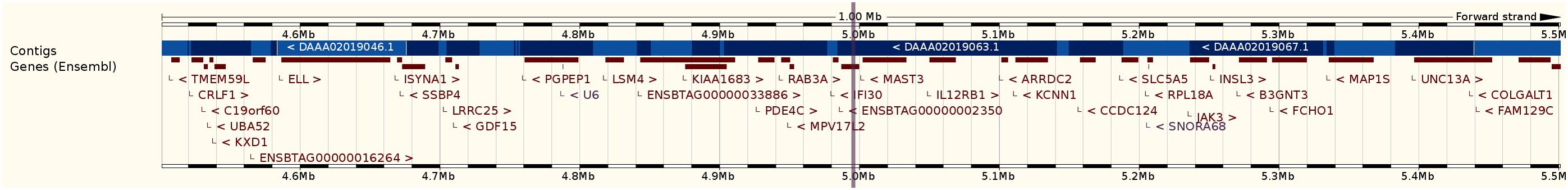
FIGURE 4. 1 Mb gene clusters on BTA7 surrounding significantly associated rs41255569 variant (located at about 5.00 Mb) with susceptibility to mastitis in parity 3.
Similarly, the SNP rs29012637 on BTA7, an intergenic variant, was significantly associated with recoverability from mastitis, and is mapped very near to the gene EPS15L1 (epidermal growth factor receptor pathway substrate 15 like 1). The gene EPS15L1 has been reported as an essential gene for T lymphocyte development in Zebrafish (Seiler et al., 2015). As the gene is highly conserved between zebrafish, mouse and human protein, it is likely to have similar functions in other mammals (Seiler et al., 2015), including cattle. Therefore, the gene EPS15L1 could be considered as a potential candidate gene for recoverability from mastitis because it plays an important role in promoting production of lymphocytes, which are main components in the immune system.
Biscarini et al. (2016) used an alternative approach to detect regions of the genome associated with susceptibility to infectious and metabolic diseases in dairy cows. Their approach was based on runs of homozygosity (ROH), where stretches of homozygous DNA are compared in cases and controls and those associated with susceptibility to disease are supposed to be more frequent in cases than in controls. The SNP rs41618899 in the current study and in significant association with susceptibility to mastitis in parity 3 is located within a previously reported ROH [stretching from 25878820 to 30099199 bps on BTA12 (Biscarini et al., 2016)] associated with infectious diseases including mastitis. Similarly, the SNP rs41565600 in significant association with recoverability from mastitis in parity 3 is located within a previously reported ROH [stretching from 2991449 to 4655753 bps on BTA7 (Biscarini et al., 2016)] associated with different infectious diseases including mastitis.
From the annotations related to immune responses, it is plausible to suspect MARCH3 (involved in regulation of the endosomal transport pathway), MAST3 (highly expressed in antigen-presenting cells and in lymphocytes) and STAB2 (involved in lymphocyte homing, cell adhesion, and receptor scavenging) as candidate for the causal genes for susceptibility to mastitis. For recoverability from mastitis we suggest EPS15L1 (essential for lymphocyte development), PDGFD (involves in macrophage recruitment and wound healing), and PTX3 (involved in regulating inflammation) as candidates for the causal genes. Regardless of the gene annotations related to immune responses, we were not fully convinced to suggest these genes as strong candidates as we were not able to confirm previously identified SNP variants or regions of the genome. However, this is the first GWAS study for recoverability from mastitis and our results need to be validated. Moreover, most of the functional annotations were derived from gene expression studies in mice and humans and gene functions may be different in cattle. There have also been discrepancies between gene-expression platforms and analyses methods reported to be obstacles in mapping loci that might underlie functional trait variation (de Koning and Haley, 2005). The findings in the current study could be considered as a starting point for further investigations in identifying causal genetic variants or chromosomal regions for both susceptibility to – and recoverability from mastitis and especially for the latter as this is the first study to report SNP variants and chromosomal regions.
Conclusion
This study is the first in reporting SNP variants for their association with recoverability from mastitis in Danish Holstein cows. We have identified 29 significant SNP variants (14 for susceptibility to – and 15 for recoverability from mastitis) determined at a significance level of P-value < 10-4. Among these detected SNP variants, most of them are in genes known for their role in immunity and wound healing. On the bovine genome, locations of association signals were different for different parities, suggesting that effect of genes are parity dependent. Similarly, association signals for mastitis susceptibility were mapped in different locations from associations for recoverability from mastitis, suggesting that susceptibility to – and recoverability from mastitis are regulated by different genes.
Author Contributions
BW performed the genome-wide association analysis and wrote the draft manuscript. PL and LJ contributed to the research hypothesis and discussion of the results. D-JK contributed to the research hypothesis, analysis, discussion of the results, and writing the manuscript. WF contributed to the research hypothesis, analysis of phenotype data, and discussion of the results. All authors approved the final version of the manuscript.
Funding
BW benefited from a joint grant from the European Commission and Swedish University of Agricultural Sciences (SLU), within the framework of the Erasmus-Mundus joint doctorate EGS-ABG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00141/full#supplementary-material
Footnotes
- ^http://www.animalgenome.org/cgi-bin/QTLdb/BT/index, accessed September 2017
- ^https://www.ensembl.org
- ^http://www.genecards.org
References
Abdel-Shafy, H., Bortfeldt, R. H., Reissmann, M., and Brockmann, G. A. (2014). Short communication: validation of somatic cell score-associated loci identified in a genome-wide association study in German Holstein cattle. J. Dairy Sci. 97, 2481–2486. doi: 10.3168/jds.2013-7149
Aken, B. L., Ayling, S., Barrell, D., Clarke, L., Curwen, V., Fairley, S., et al. (2016). The Ensembl gene annotation system. Database 2016:baw093. doi: 10.1093/database/baw093
Aulchenko, Y. S., De Koning, D. J., and Haley, C. (2007). Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics 177, 577–585. doi: 10.1534/genetics.107.075614
Biscarini, F., Biffani, S., Morandi, N., Nicolazzi, E. L., and Stella, A. (2016). Using runs of homozygosity to detect genomic regions associated with susceptibility to infectious and metabolic diseases in dairy cows under intensive farming conditions. arXiv:1601.07062. Available at: https://arxiv.org/pdf/1601.07062.pdf
Brenaut, P., Lefevre, L., Rau, A., Laloe, D., Pisoni, G., Moroni, P., et al. (2014). Contribution of mammary epithelial cells to the immune response during early stages of a bacterial infection to Staphylococcus aureus. Vet. Res. 45:16. doi: 10.1186/1297-9716-45-16
Carlén, E., Schneider, M. D., and Strandberg, E. (2005). Comparison between linear models and survival analysis for genetic evaluation of clinical mastitis in dairy cattle. J. Dairy Sci. 88, 797–803. doi: 10.3168/jds.S0022-0302(05)72744-2
Carlén, E., Strandberg, E., and Roth, A. (2004). Genetic parameters for clinical mastitis, somatic cell score, and production in the first three lactations of Swedish Holstein cows. J. Dairy Sci. 87, 3062–3070. doi: 10.3168/jds.S0022-0302(04)73439-6
R Core Team (2016). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna: R Core Team.
Cruciani, L., Romero, R., Vaisbuch, E., Kusanovic, J. P., Chaiworapongsa, T., Mazaki-Tovi, S., et al. (2010). Pentraxin 3 in amniotic fluid: a novel association with intra-amniotic infection and inflammation. J. Perinat. Med. 38, 161–171. doi: 10.1515/Jpm.2009.141
de Koning, D. J., and Haley, C. S. (2005). Genetical genomics in humans and model organisms. Trends Genet. 21, 377–381. doi: 10.1016/j.tig.2005.05.004
D’Souza, S., Park, S. Y., and Kim, I. S. (2013). Stabilin-2 acts as an engulfment receptor for the phosphatidylserine-dependent clearance of primary necrotic cells. Biochem. Biophys. Res. Commun. 432, 412–417. doi: 10.1016/j.bbrc.2013.01.133
Franzén, J., Thorburn, D., Urioste, J. I., and Strandberg, E. (2012). Genetic evaluation of mastitis liability and recovery through longitudinal analysis of transition probabilities. Genet. Select. Evol. 44:10. doi: 10.1186/1297-9686-44-10
Fu, Y. H., Zhou, E. S., Liu, Z. C., Li, F. Y., Liang, D. J., Liu, B., et al. (2013). Staphylococcus aureus and Escherichia coli elicit different innate immune responses from bovine mammary epithelial cells. Vet. Immunol. Immunopathol. 155, 245–252. doi: 10.1016/j.vetimm.2013.08.003
Fukuda, H., Nakamura, N., and Hirose, S. (2006). MARCH-III is a novel component of endosomes with properties similar to those of MARCH-II. J. Biochem. 139, 137–145. doi: 10.1093/jb/mvj012
Garlanda, C., Jaillon, S., Doni, A., Bottazzi, B., and Mantovani, A. (2016). PTX3, a humoral pattern recognition molecule at the interface between microbe and matrix recognition. Curr. Opin. Immunol. 38, 39–44. doi: 10.1016/j.coi.2015.11.002
Gianola, D., and Foulley, J. (1983). Sire evaluation for ordered categorical data with a threshold model. Genet. Select. Evol. 15, 201–224. doi: 10.1186/1297-9686-15-2-201
Goddard, M. E., and Hayes, B. J. (2009). Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10, 381–391. doi: 10.1038/nrg2575
Hadfield, J. D. (2010). MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R Package. J. Stat. Softw. 33, 1–22. doi: 10.18637/jss.v033.i02
Halasa, T., Huijps, K., Osteras, O., and Hogeveen, H. (2007). Economic effects of bovine mastitis and mastitis management: a review. Vet. Q. 29, 18–31. doi: 10.1080/01652176.2007.9695224
Hayes, B. J., Pryce, J., Chamberlain, A. J., Bowman, P. J., and Goddard, M. E. (2010). Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in holstein cattle as contrasting model traits. PLoS Genet. 6:e1001139. doi: 10.1371/journal.pgen.1001139
Heringstad, B., Klemetsdal, G., and Ruane, J. (2000). Selection for mastitis resistance in dairy cattle: a review with focus on the situation in the Nordic countries. Livest. Prod. Sci. 64, 95–106. doi: 10.1016/S0301-6226(99)00128-1
Hirschhorn, J. N., and Daly, M. J. (2005). Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108. doi: 10.1038/nrg1521
Hodskinson, M. R. G., Silhan, J., Crossan, G. P., Garaycoechea, J. I., Mukherjee, S., Johnson, C. M., et al. (2014). Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell 54, 472–484. doi: 10.1016/j.molcel.2014.03.014
Hogeveen, H., Huijps, K., and Lam, T. J. (2011). Economic aspects of mastitis: new developments. N. Z. Vet. J. 59, 16–23. doi: 10.1080/00480169.2011.547165
Howard, J. T., Jiao, S. H., Tiezzi, F., Huang, Y. J., Gray, K. A., and Maltecca, C. (2015). Genome-wide association study on legendre random regression coefficients for the growth and feed intake trajectory on Duroc Boars. BMC Genet. 16:59. doi: 10.1186/s12863-015-0218-8
Labbe, C., Goyette, P., Lefebvre, C., Stevens, C., Green, T., Tello-Ruiz, M. K., et al. (2008). MAST3: a novel IBD risk factor that modulates TLR4 signaling. Genes Immun. 9, 602–612. doi: 10.1038/gene.2008.57
Lutzow, Y. C. S., Donaldson, L., Gray, C. P., Vuocolo, T., Pearson, R. D., Reverter, A., et al. (2008). Identification of immune genes and proteins involved in the response of bovine mammary tissue to Staphylococcus aureus infection. BMC Vet. Res. 4:18. doi: 10.1186/1746-6148-4-18
Madsen, P., and Jensen, J. (2013). A User’s Guide to DMU. A Package for Analysing Multivariate Mixed Models. Version 6, Release 5.2. Tjele: University of Aarhus.
Meredith, B. K., Kearney, F. J., Finlay, E. K., Bradley, D. G., Fahey, A. G., Berry, D. P., et al. (2012). Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 13:21. doi: 10.1186/1471-2156-13-21
Nilsen, H., Olsen, H. G., Hayes, B., Nome, T., Sehested, E., Svendsen, M., et al. (2009). Characterization of a QTL region affecting clinical mastitis and protein yield on BTA6. Anim. Genet. 40, 701–712. doi: 10.1111/j.1365-2052.2009.01908.x
Pirola, Y., Della Vedova, G., Bonizzoni, P., Stella, A., and Biscarini, F. (2013). “Haplotype-based prediction of gene alleles using pedigrees and SNP genotypes,” in Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics ACM, New York, NY, 33.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Rupp, R., and Boichard, D. (2003). Genetics of resistance to mastitis in dairy cattle. Vet. Res. 34, 671–688. doi: 10.1051/vetres:2003020
Sahana, G., Guldbrandtsen, B., Thomsen, B., and Lund, M. S. (2013). Confirmation and fine-mapping of clinical mastitis and somatic cell score QTL in Nordic Holstein cattle. Anim. Genet. 44, 620–626. doi: 10.1111/age.12053
Schledzewski, K., Geraud, C., Arnold, B., Wang, S. J., Grone, H. J., Kempf, T., et al. (2011). Deficiency of liver sinusoidal scavenger receptors stabilin-1 and-2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J. Clin. Invest. 121, 703–714. doi: 10.1172/JCI44740
Seiler, C., Gebhart, N., Zhang, Y., Shinton, S. A., Li, Y. S., Ross, N. L., et al. (2015). Mutagenesis screen identifies agtpbp1 and eps15L1 as essential for T lymphocyte development in Zebrafish. PLoS One 10:e0131908. doi: 10.1371/journal.pone.0131908
Sender, G., Korwin-Kossakowska, A., Pawlik, A., Hameed, K. G. A., and Oprzadek, J. (2013). Genetic basis of mastitis resistance in dairy cattle - a review. Ann. Anim. Sci. 13, 663–673. doi: 10.2478/aoas-2013-0043
Sodeland, M., Kent, M. P., Olsen, H. G., Opsal, M. A., Svendsen, M., Sehested, E., et al. (2011). Quantitative trait loci for clinical mastitis on chromosomes 2,6, 14 and 20 in Norwegian Red cattle. Anim. Genet. 42, 457–465. doi: 10.1111/j.1365-2052.2010.02165.x
Sørensen, L. P., Madsen, P., Mark, T., and Lund, M. S. (2009). Genetic parameters for pathogen-specific mastitis resistance in Danish Holstein cattle. Animal 3, 647–656. doi: 10.1017/S1751731109003899
Strillacci, M. G., Frigo, E., Schiavini, F., Samore, A. B., Canavesi, F., Vevey, M., et al. (2014). Genome-wide association study for somatic cell score in Valdostana Red Pied cattle breed using pooled DNA. BMC Genet. 15:106. doi: 10.1186/s12863-014-0106-7
Tiezzi, F., Parker-Gaddis, K. L., Cole, J. B., Clay, J. S., and Maltecca, C. (2015). A genome-wide association study for clinical mastitis in first parity us holstein cows using single-step approach and genomic matrix re-weighting procedure. PLoS One 10:e0114919. doi: 10.1371/journal.pone.0114919
Uutela, M., Wirzenius, M., Paavonen, K., Rajantie, I., He, Y. L., Karpanen, T., et al. (2004). PDGF-D induces macrophage recruitment, increased interstitial pressure, and blood vessel maturation during angiogenesis. Blood 104, 3198–3204. doi: 10.1182/blood-2004-04-1485
Vazquez, A. I., Gianola, D., Bates, D., Weigel, K. A., and Heringstad, B. (2009). Assessment of Poisson, logit, and linear models for genetic analysis of clinical mastitis in Norwegian Red cows. J. Dairy Sci. 92, 739–748. doi: 10.3168/jds.2008-1325
Welderufael, B. G., Janss, L. L. G., de Koning, D. J., Sorensen, L. P., Lovendahl, P., and Fikse, W. F. (2017). Bivariate threshold models for genetic evaluation of susceptibility to and ability to recover from mastitis in Danish Holstein cows. J. Dairy Sci. 100, 4706–4720. doi: 10.3168/jds.2016-11894
Wilmink, J. (1987). Adjustment of test-day milk, fat and protein yields for age, season and stage of lactation. Livest. Prod. Sci. 16, 335–348. doi: 10.1016/0301-6226(87)90003-0
Wojdak-Maksymiec, K., Szyda, J., and Strabel, T. (2013). Parity-dependent association between TNF-alpha and LTF gene polymorphisms and clinical mastitis in dairy cattle. BMC Vet. Res. 9:114. doi: 10.1186/1746-6148-9-114
Yin, L. L., Liu, J. H., Dong, H. Q., Xu, E. H., Qiao, Y. C., Wang, L., et al. (2014). Autophagy-related gene16L2, a potential serum biomarker of multiple sclerosis evaluated by bead-based proteomic technology. Neurosci. Lett. 562, 34–38. doi: 10.1016/j.neulet.2013.12.070
Keywords: dairy cow, genome-wide association study, mastitis, recoverability, susceptibility
Citation: Welderufael BG, Løvendahl P, de Koning D-J, Janss LLG and Fikse WF (2018) Genome-Wide Association Study for Susceptibility to and Recoverability From Mastitis in Danish Holstein Cows. Front. Genet. 9:141. doi: 10.3389/fgene.2018.00141
Received: 29 September 2017; Accepted: 04 April 2018;
Published: 24 April 2018.
Edited by:
Peter Dovc, University of Ljubljana, SloveniaReviewed by:
Eveline M. Ibeagha-Awemu, Agriculture and Agri-Food Canada (AAFC), CanadaFilippo Biscarini, Consiglio Nazionale delle Ricerche (CNR), Italy
Copyright © 2018 Welderufael, Løvendahl, de Koning, Janss and Fikse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. G. Welderufael, YmVyaWh1LndlbGRlcnVmYWVsQHNsdS5zZQ==