 Giovana T. Torrezan1,2†
Giovana T. Torrezan1,2† Fernanda G. dos Santos R. de Almeida1†
Fernanda G. dos Santos R. de Almeida1† Márcia C. P. Figueiredo1
Márcia C. P. Figueiredo1 Bruna D. de Figueiredo Barros1Cláudia A. A. de Paula1Renan Valieris3
Bruna D. de Figueiredo Barros1Cláudia A. A. de Paula1Renan Valieris3 Jorge E. S. de Souza4,5,6Rodrigo F. Ramalho1
Jorge E. S. de Souza4,5,6Rodrigo F. Ramalho1 Felipe C. C. da Silva1Elisa N. Ferreira1,7Amanda F. de Nóbrega8
Felipe C. C. da Silva1Elisa N. Ferreira1,7Amanda F. de Nóbrega8 Paula S. Felicio9
Paula S. Felicio9 Maria I. Achatz8,10Sandro J. de Souza2,6,11Edenir I. Palmero9,12
Maria I. Achatz8,10Sandro J. de Souza2,6,11Edenir I. Palmero9,12 Dirce M. Carraro1,2*
Dirce M. Carraro1,2*- 1Laboratory of Genomics and Molecular Biology, International Research Center, CIPE/A.C. Camargo Cancer Center, São Paulo, Brazil
- 2National Institute for Science and Technology in Oncogenomics and Therapeutic Innovation, São Paulo, Brazil
- 3Laboratory of Bioinformatics and Computational Biology, International Research Center, CIPE/A.C. Camargo Cancer Center, São Paulo, Brazil
- 4Instituto de Bioinformática e Biotecnologia−2bio, Natal, Brazil
- 5Instituto Metrópole Digital, Federal University of Rio Grande do Norte, Natal, Brazil
- 6Bioinformatics Multidisciplinary Environment, Federal University of Rio Grande do Norte, Natal, Brazil
- 7Research and Development, Fleury Group, São Paulo, Brazil
- 8Oncogenetics Department, A.C. Camargo Cancer Center, São Paulo, Brazil
- 9Molecular Oncology Research Center, Barretos Cancer Hospital, São Paulo, Brazil
- 10Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Department of Health and Human Services, Bethesda, MD, United States
- 11Brain Institute, Federal University of Rio Grande do Norte, Natal, Brazil
- 12Barretos School of Health Sciences, Dr. Paulo Prata – FACISB, Barretos, Brazil
Pathogenic variants in known breast cancer (BC) predisposing genes explain only about 30% of Hereditary Breast Cancer (HBC) cases, whereas the underlying genetic factors for most families remain unknown. Here, we used whole-exome sequencing (WES) to identify genetic variants associated to HBC in 17 patients of Brazil with familial BC and negative for causal variants in major BC risk genes (BRCA1/2, TP53, and CHEK2 c.1100delC). First, we searched for rare variants in 27 known HBC genes and identified two patients harboring truncating pathogenic variants in ATM and BARD1. For the remaining 15 negative patients, we found a substantial vast number of rare genetic variants. Thus, for selecting the most promising variants we used functional-based variant prioritization, followed by NGS validation, analysis in a control group, cosegregation analysis in one family and comparison with previous WES studies, shrinking our list to 23 novel BC candidate genes, which were evaluated in an independent cohort of 42 high-risk BC patients. Rare and possibly damaging variants were identified in 12 candidate genes in this cohort, including variants in DNA repair genes (ERCC1 and SXL4) and other cancer-related genes (NOTCH2, ERBB2, MST1R, and RAF1). Overall, this is the first WES study applied for identifying novel genes associated to HBC in Brazilian patients, in which we provide a set of putative BC predisposing genes. We also underpin the value of using WES for assessing the complex landscape of HBC susceptibility, especially in less characterized populations.
Introduction
Hereditary breast cancer (HBC) corresponds to ~5–10% of all breast cancer cases (Honrado et al., 2005). The most common breast cancer predisposing syndrome is hereditary breast and ovarian cancer syndrome (HBOC) that is related to pathogenic germline variants in BRCA1 (OMIM 113705) and BRCA2 (OMIM 600185) genes (Anglian Breast Cancer Study, 2000). These genes correspond to ~20–25% of all HBC (Anglian Breast Cancer Study, 2000; Kean, 2014; Silva et al., 2014). Besides BRCA1/2 genes, pathogenic variants in other high- and moderate-risk genes, such as TP53, CHEK2, ATM, STK11, PALB2, among others, also lead to an increased breast cancer (BC) risk, revealing a high complexity in breast cancer predisposition (Elledge and Allred, 1998; Meijers-Heijboer et al., 2002; Walsh and King, 2007).
To date, over 35 genes have been suggested to carry high and/or moderate BC risk variants (OMIM, 20151; Shiovitz and Korde, 2015). However, only a minority of these genes have an established significant association demonstrated by both stringent burden testing and statistical analyses (Easton et al., 2015). Moreover, despite extensive sequencing efforts, variants in known BC susceptibility genes are present in <30% of BC cases with positive family history or an early age of onset (Shiovitz and Korde, 2015; Chandler et al., 2016), meaning that the underlying genetic factors for most HBC remain unknown.
In the past few years, advances in next-generation sequencing (NGS), specially whole-exome sequencing (WES), have led to the identification of causative variants in several rare familial syndromes, including hereditary cancer (Comino-Méndez et al., 2011; Seguí et al., 2015). Up to the present time, more than 16 different WES studies (both family-based and case studies) have been carried out for HBC, and a few novel BC susceptibility genes were identified: XRCC2, RINT1, RECQL, and FANCM (Chandler et al., 2016). Nevertheless, the small number of novel major BC autosomal dominant predisposing genes disclosed in these studies has pointed to the possible existence of very rare, or even particular, high and moderate penetrant variants. Conversely, other forms of inheritance, such as recessive and oligogenic transmission of cancer predisposition, cannot be discarded (Sokolenko et al., 2015). In this sense, further WES investigation in different families or populations is crucial for expanding the catalog of breast tumor predisposing genes.
In two previous studies of our group, we screened young BC women (Carraro et al., 2013) and women with clinical criteria of HBOC (Silva et al., 2014) for pathogenic variants in the complete coding sequence of BRCA1, BRCA2, and TP53 genes, and for CHEK2 c.1100delC point mutation, detecting 22–26% of pathogenic variant carriers. Both studies disclosed a large number of women negative for pathogenic variants in the most important genes associated with BC risk, claiming for the necessity of identifying rare and/or novel BC predisposing genes. Thus, the aim of the current study was to investigate, by WES, breast cancer patients with clinical criteria for HBOC and without pathogenic variants in major breast cancer predisposing genes, using rigorous functional criteria for selection of detected variants, in order to identify the most promising new HBC-causing genes.
Materials and Methods
Patients and Controls
WES was performed in 17 patients from A.C. Camargo Cancer Center (15 unrelated patients and two siblings) diagnosed with BC and fulfilling one or more of the following criteria of HBOC syndrome: early onset BC (<36 years); bilateral BC; breast plus another primary related tumor (ovary, fallopian tube or primary peritoneal tumors). These patients were selected from previous studies (Carraro et al., 2013; Silva et al., 2014) from our group and were negative for pathogenic variants in BRCA1/2, TP53, and CHEK2 c.1100delC. Two patients (including the two sisters) were carriers of variants of uncertain clinical significance (VUS) in BRCA1 gene. The detailed inclusion criteria from both studies were described previously (Carraro et al., 2013; Silva et al., 2014). One affected woman of one family participated in the cosegregation study for specific candidate variants.
Five germline BRCA1-mutation carriers that were submitted to WES in the same platform were included for variant filtering. For validation of selected variants, target NGS validation was applied in 25 healthy women without family history of cancer, considered here as a control group. Additionally, a selected number of candidate genes were screened in an independent group of 42 patients at risk for HBC from a distinct project, obtained from Barretos Cancer Hospital (Barretos, São Paulo, Brazil). Figure 1 depicts the study design and workflow, describing the projects steps and the analysis performed in each patients and controls groups.
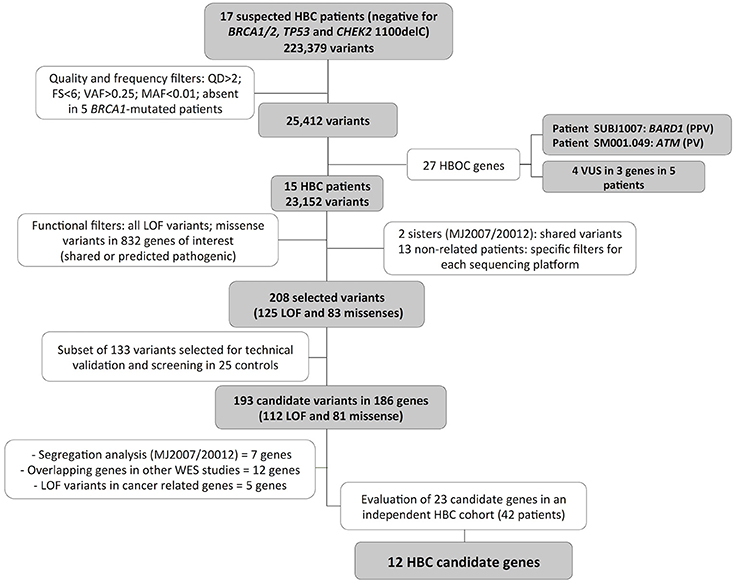
Figure 1. Variants selection workflow. WES data from 17 breast cancer patients were analyzed using quality, frequency, and functional based filters, resulting in 186 breast cancer predisposing candidate genes. A final 23 candidate genes were entirely investigated for LOF or possibly pathogenic variants in 42 additional BC patients suspected of HBC and negative for mutations in the major predisposing genes, resulting in 12 final HBC candidate genes. FS, FisherStrand; QD, QualByDepth; PPV, probably pathogenic variant; PV, pathogenic variant; HBOC, Hereditary Breast and Ovarian Cancer; LOF, loss of function; SA, segregation analysis; WES, whole exome sequencing.
All participants signed an informed consent. This study was performed in accordance with the Helsinki Declaration and was approved by the A.C. Camargo Cancer Center (1754/13) and Barretos Hospital (916/2015) ethics committees.
DNA Isolation
Genomic DNA was obtained from A.C. Camargo Cancer Center Biobank. In brief, DNA was extracted from peripheral leukocytes by Puregene®-DNA purification Kit (Qiagen, Hilden, Germany), according to manufacturer's instructions. DNA concentration, purity and integrity were assessed by spectrophotometry (Nanodrop 2000—Thermo Fisher Scientific, Waltham, MA) and fluorometry (Qubit—Life Technologies, Foster City, CA, USA).
Whole Exome Sequencing
For the 17 patients of the discovery set, WES was performed using the SOLiD and/or Ion Proton platforms. For SOLiD exomes, libraries were prepared using SOLiD™ Fragment Library Barcoding Kit (Life Technologies) and SureSelect Human All Exon V4 Kit 50 Mb (Agilent Technologies), according to the manufacturer's instructions. Sequencing of paired-end libraries (50 X 75 bp) was performed in a Solid 5500XL System (Life Technologies). For Ion Proton exomes, libraries were prepared using Ion Xpress™ Plus Fragment Library Kit and Ion TargetSeq™ Exome Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. Each Ion Proton exome library was sequenced on Ion Proton instrument using Ion PI Sequencing 200 Kit v3 and Ion PI Chip v3 (Thermo Fisher Scientific). The resulting sequences were mapped to the reference genome (GRCh37/hg19). Base Calling and alignment were performed by SOLiD™ BioScope 1.2™ Software (Life Technologies) (SOLID data) and by Torrent Suite v4.2 server (Ion Proton data). Variant calling and annotation were done by GATK (Genome Analysis Toolkit) pipeline made available by the Broad Institute. The data obtained in this study is available at Sequence Read Archive (SRP120031).
Variants Selection and Prioritization
For variant filtering, identified variants were annotated with VarSeq (Golden Helix) against reference databases (RefSeq, 1000Genomes, ESP6500, ExAC, dbSNP, and ClinVar). First, for quality filtering, we selected variants with QD > 2 (QD = variant call confidence normalized by depth of sample reads supporting a variant), FS < 6 (FS = strand bias estimated by GATK using Fisher's Exact Test), base coverage ≥ 10x, variant allele frequency (VAF) > 0.25. For four patients with data from both Solid and Ion Proton, only variants detected in both platforms were selected. For one patient with data exclusively from Ion Proton, variants occurring in regions of homopolymer > 4 bases were excluded. Qualified variants were excluded if present in five BRCA1-mutation carriers patients analyzed by WES in Solid 5500, and variants present in population databases with frequency > 1% (minor allele frequency [MAF] > 0.01), as well as variants present in more than three unrelated patients. Finally, a recently public available Brazilian database of WES from 609 healthy individuals (Abraom—Brazilian genomic variants; http://abraom.ib.usp.br/) was also used for manually excluding population-specific variants (MAF > 0.01).
Next, for a function-based prioritization, we selected variants leading to loss of function in any gene (frameshift indels, stop codon, and canonical splice site variants) and missense or in-frame indels variants in 832 genes of interest. These genes were selected from commercial panels targeting somatic and germline cancer mutated genes, consensus cancer genes previously described (Futreal et al., 2004) and genes from DNA repair pathways (from KEGG and Putnam et al., 2016) (Supplementary Table 1). For the two related patients, any shared missense or in-frame indels variants in these 832 genes were selected. For the 15 unrelated patients, we selected only variants predicted to be damaging in at least four out of six variant effect prediction software. For these analyses, the results from the following tools were obtained using VarSeq: SIFT, Polyphen v2, Functional Analysis through Hidden Markov Models (FATHAMM and FATHAMM-MKL), MutationAssessor and MutationTaster. Additionally, we analyzed the potential effect on splicing of the selected LOF and missense variants using dbscSNV annotations (cut-off > 0.6 in ADA and/or RF scores).
Sanger Validation
Two pathogenic variants (PV) or probably pathogenic variants (PPV) in BARD1 and ATM were validated by Sanger sequencing. Briefly, 50 ng of leukocyte DNA was submitted to PCR performed with GoTaq Green Master Mix (Promega), cleaned with ExoSAP-IT (USB Corporation) and sequenced in both directions with BigDye Terminator v3.1 (Life Technologies) using an ABI 3130xl DNA sequencer (Life Technologies), according to manufacturer's instructions. The sequencing results were aligned using CLCBio Genomics Workbench Software (CLCBio, Qiagen). Primer sequences are available under request.
Targeted NGS Validation
A subset of 139 variants (Supplementary Table 2) selected from exome data were validated by multiplex targeted NGS using a custom Ion AmpliSeq panel. Primers were designed using Ion AmpliSeq Designer v3.0.1 (Life Technologies). Libraries were prepared with 20 ng of DNA from each patient using Ion AmpliSeq™ Library Kit 2.0 (Life Technologies). Sequencing was performed using either Ion PGM or Ion Proton platforms, according to the manufacturer's instructions. Sequencing reads mapped to the human genome reference (hg19) using Torrent Suite Browser 4.0.1. On average 166,697 mapped reads were obtained per sample, yielding a mean targeted base coverage of 156X (ranging from 54 to 450). Variants were identified using the VariantCaller v4.0.r73742 plugin and confirmed using CLC Genomics Workbench software (Qiagen). The identified variants were considered if base coverage was ≥10x and VAF > 25%.
To filter out genetic variants common in Brazilian population, the validated variants were evaluated in control group of 25 healthy women by using the same panel. For that, pools of five equimolar genomic DNA samples were prepared by containing 4 ng of each patient (five patients per pool). Libraries preparation, sequencing and mapping were performed as described above. On average 928,194 mapped reads were obtained per pool (mean targeted base coverage 1114X; ranging from 990 to 1,314). Variant calls were obtained using the VariantCaller v4.0.r73742 plugin applying the following filter parameters: VAF > 2%; variant coverage ≥10X.
Cosegregation Analysis
For one family in which a segregation analysis was feasible, DNA from one additional affected individual was obtained. The cosegregation study of specific variants was performed using the same custom gene panel and protocol described previously or with amplicon based library construction and sequencing in Ion Proton platform.
Independent Cohort Validation
For screening the HBC predisposing candidate genes selected in this study an independent cohort comprised of 42 breast cancer patients at risk for HBC from Barretos Cancer Hospital was used. These samples were analyzed through WES in a parallel study using Nextera Rapid Capture Expanded Exome and NextSeq 500 System (Illumina, San Diego, CA). In these data, we assessed the entire coding regions of the 23 genes disclosed in this study for the presence of rare and possibly pathogenic variants, using the same criteria as in our discovery cohort.
Results
In this study we used WES to disclose variants contributing to BC increased risk in patients fulfilling stringent clinical criteria indicating a genetic predisposition to BC and that were negative for pathogenic variants in four major BC genes (BRCA1/2, TP53, and CHEK2 1100delC). The clinical features and family history of cancer for the 17 selected patients are described in Supplementary Table 3.
For the WES, an average of 46,307,427 sequence reads was obtained for each patient and 75.7% (average) of the target bases were covered by 10 or more reads (Supplementary Table 4). More than 200,000 variants were identified in these patients. To prioritize the identified variants, we applied several filters focusing on quality, frequency and function of the identified alterations. The workflow of the variant prioritization is depicted in Figure 1 and the details of used filters are described in the Materials and Methods section.
Regarding frequency filters, we excluded variants with a minor allele frequency (MAF) >1% in public databases or those present in five germline BRCA1-mutation carriers sequenced in our facility, assuming that these variants represent benign or low-penetrance variants. Following these initial data filtering, 25,412 were identified.
Variants in Moderated and High Penetrance Breast Cancer Genes
Initially, we used WES data to search for rare variants in 27 well-established and emerging HBC predisposing genes (the four previously evaluated genes (BRCA1/2, TP53, and CHEK2 c.1100delC) and 23 additional genes): ATM, BARD1, BLM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, FANCC, FANCM, MLH1, MSH2, MUTYH, NBN, NF1, PALB2, PMS2, PTEN, RAD51C, RAD51D, STK11, TP53, FAM175A, MRE11, RAD51B, RECQL, and RINT1 (Nielsen et al., 2016). In this analysis, we identified two patients harboring frameshift indel variants (one in ATM and one in BARD1) and five patients (including the two sisters) with variants of uncertain clinical significance (VUS) (Table 1). In three patients (MJ2037 and MJ2007/2012) we confirmed the BRCA1 VUS previously detected by Sanger sequencing. All variants detected in these genes were classified according to the ACMG guidelines (Richards et al., 2015).
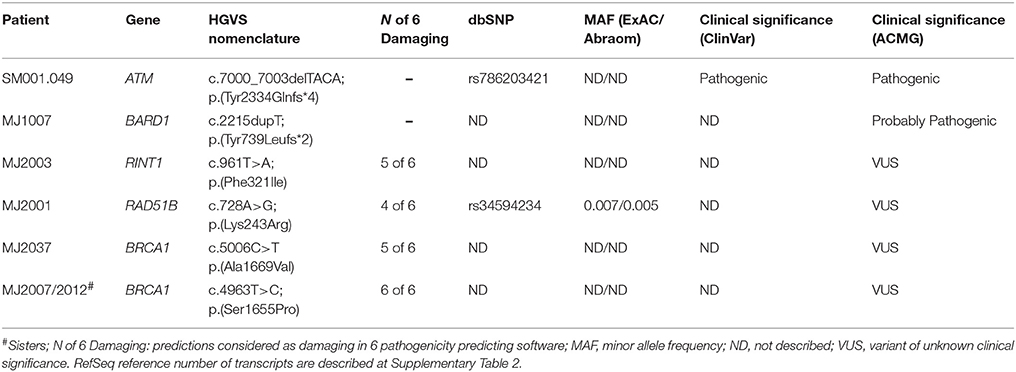
Table 1. Pathogenic and VUS detected in 27 known HBC genes.
The ATM p.(Tyr2334Glnfs*4) variant is described as pathogenic in ClinVar database. The BARD1 p.(Tyr739Leufs*2) is not described in any database and was classified as probably pathogenic, since it is a rare truncating variant leading to partial loss of the second BRCT domain and the phosphobinding region. These two variants were confirmed by Sanger sequencing in the proband and, for ATM, also in one affected relative (Supplementary Figure 1).
Four rare missense variants identified in our patients were classified as probably damaging by at least four prediction software, and three of them are not described in any population database. Three of them are located in recognized functional domains of the affected proteins: BRCA1 p.Ala1699Val and p.Ser1655Pro are located at the C-terminal BRCT domain, responsible for BRCA1 interaction with others DNA repair proteins and RINT1 p.Phe321Ile is located at the functional TIP20 domain.
Candidate Selection for Novel Breast Cancer Predisposing Genes
Next, for the 15 patients without any probable pathogenic variant (excluding ATM and BARD1 mutated patients) we applied a functional-based variant prioritization. Candidate variants were selected according to the predicted impact in the protein function and affected gene, including all loss-of-function variants (nonsense, frameshift indels, and splice site) as well as missense and in-frame indels occurring in a list of 832 cancer-related genes (DNA repair and cancer related genes—Supplementary Table 1). For the two sisters (MJ2007 and MJ2012), all variants shared between the two were selected as candidates. For the 13 unrelated patients, we selected missense variants predicted to be damaging by at least 4 out of 6 prediction software.
After filtering, we obtained a total of 208 variants, including 125 LOF and 83 missenses (Supplementary Table 2). In order to technically validate our variant selection workflow, a subset of these 208 variants (133 out of 208) was submitted for technical validation by targeted NGS in the same WES samples and, of these, 126 were validated (95%) (Supplementary Table 4). Using this same custom panel, we evaluated 25 control samples of healthy Brazilian women without cancer for filtering common polymorphisms in our population. Eight variants were detected in at least one control sample and where then excluded from our candidates list, resulting in 193 candidate variants (118 validated and 75 not evaluated).
For the family of the two affected sisters, one additional affected aunt diagnosed with ovarian cancer at age 45 was available for segregation analysis (Figure 2). We analyzed 17 variants that were shared between the two sisters and 8 variants were also present in the aunt, including the VUS variant in BRCA1 (Table 2).
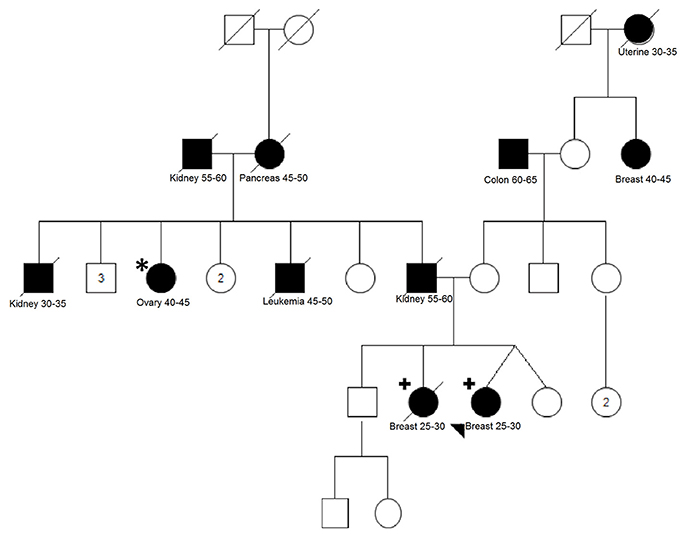
Figure 2. Pedigree of family MJ2007/MJ20012. Two breast cancer affected sisters (indicated with the plus sign) were analyzed using WES. Variants shared between the sisters were screened in one affected paternal aunt (indicated with an asterisk). Filled-in symbols indicate individuals affected by cancer. Cancer type, cancer age of onset or age appear underneath each individual. Numbers inside the symbols indicate the number of unaffected siblings not shown individually in the pedigree.
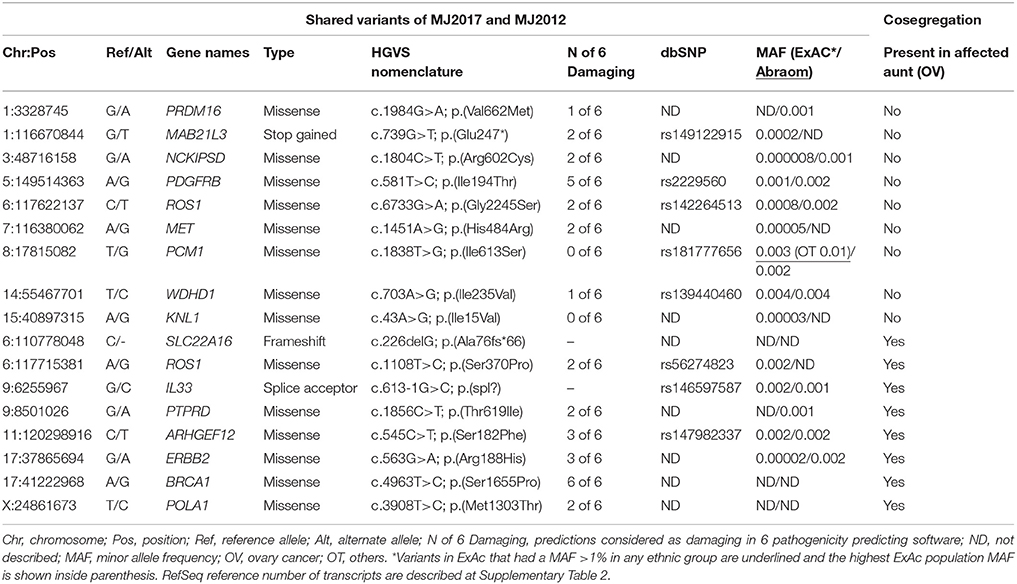
Table 2. Cosegregation analysis of variants detected in the sisters MJ2007 and MJ2012.
Then, the remaining 186 genes prioritized in our study were compared to candidate genes reported in eight previous WES studies of HBC (Snape et al., 2012; Thompson et al., 2012; Gracia-Aznarez et al., 2013; Hilbers et al., 2013; Kiiski et al., 2014; Wen et al., 2014; Noh et al., 2015; Kim et al., 2017) and 12 common genes were identified, 9 of them presenting LOFs variants in at least one study (Table 3). For two genes the same LOF variants were identified in our and at a second study (PZP p.Arg680* and KRT76 p.Glu276*).
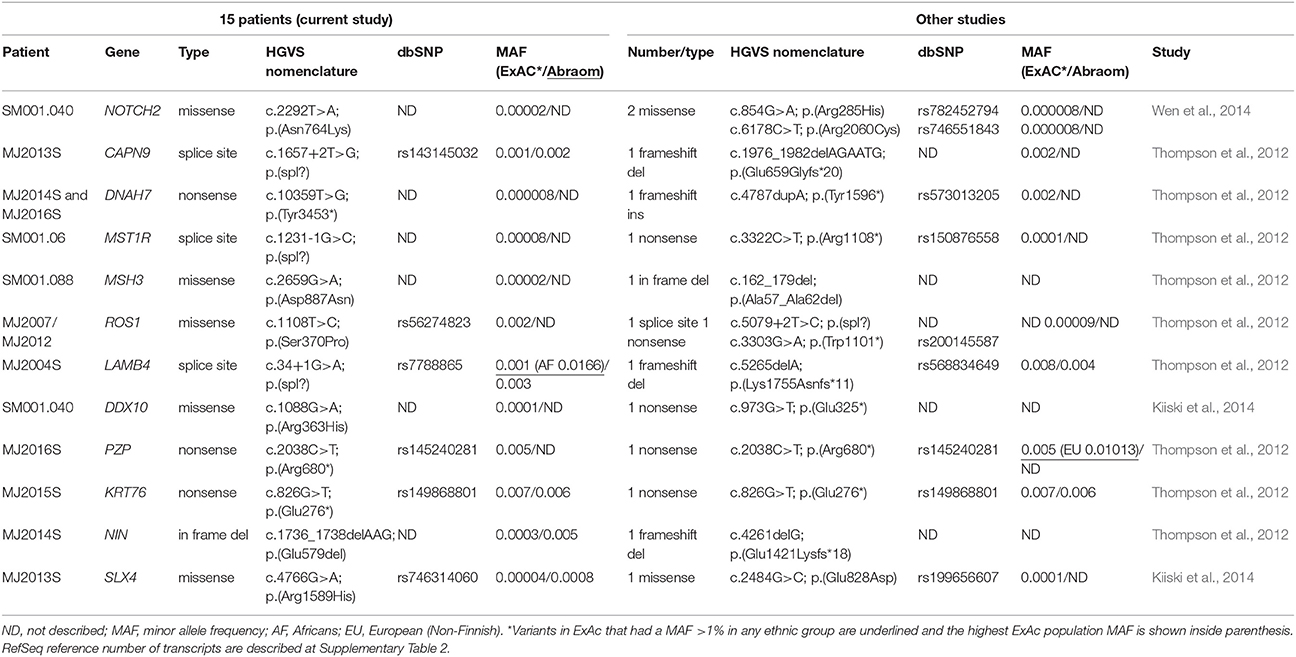
Table 3. Overlapping genes selected as candidates in other WES studies.
Thus, from the 193 final candidate variants, we selected 23 candidate genes of BC predisposition: 7 novel candidate genes segregating in the 3 members of the MJ2007/2012 family (SLC22A16, ROS1, IL33, PTPRD, ARHGEF12, ERBB2, POLA1), five cancer-related genes harboring LOF variants (GALNT3, RAF1, PICALM, KL, ERCC1) and 12 genes overlapping with candidate genes identified in other studies (CAPN9, KRT76, PZP, DNAH7, MST1R, LAMB4, NIN, MSH3, SLX4, DDX1, NOTCH2, and ROS1—ROS1 was also selected in the segregating genes list). The entire coding region of the 23 genes were evaluated in an independent Brazilian cohort.
Assessing 23 Candidate Genes in an Independent Cohort of Patients at Risk for HBC
To select the most promising candidate genes, we analyzed the 23 candidate genes disclosed in our study in an independent cohort of 42 Brazilian women at risk for HBC. These patients were all negative for pathogenic variants in BRCA1/2, TP53, and ATM genes. In these data, we assessed the entire coding regions of the selected genes for the presence of rare (MAF < 1%) and possibly pathogenic variants, selecting all LOF variants and missense variants predicted to be pathogenic in at least 3 out of 6 algorithms.
In this cohort, we detected 16 variants in 12 of the 23 candidate genes (Table 4). NOTCH2 gene was the one with more variants, harboring three missense; ERBB2 and DNAH7 harbored two missenses each. Only one LOF variant was detected, affecting ERCC1 gene, which was the same variant detected in our discovery cohort (c.875G>A; p.Trp292*). The remaining genes presented one rare missense variant each.
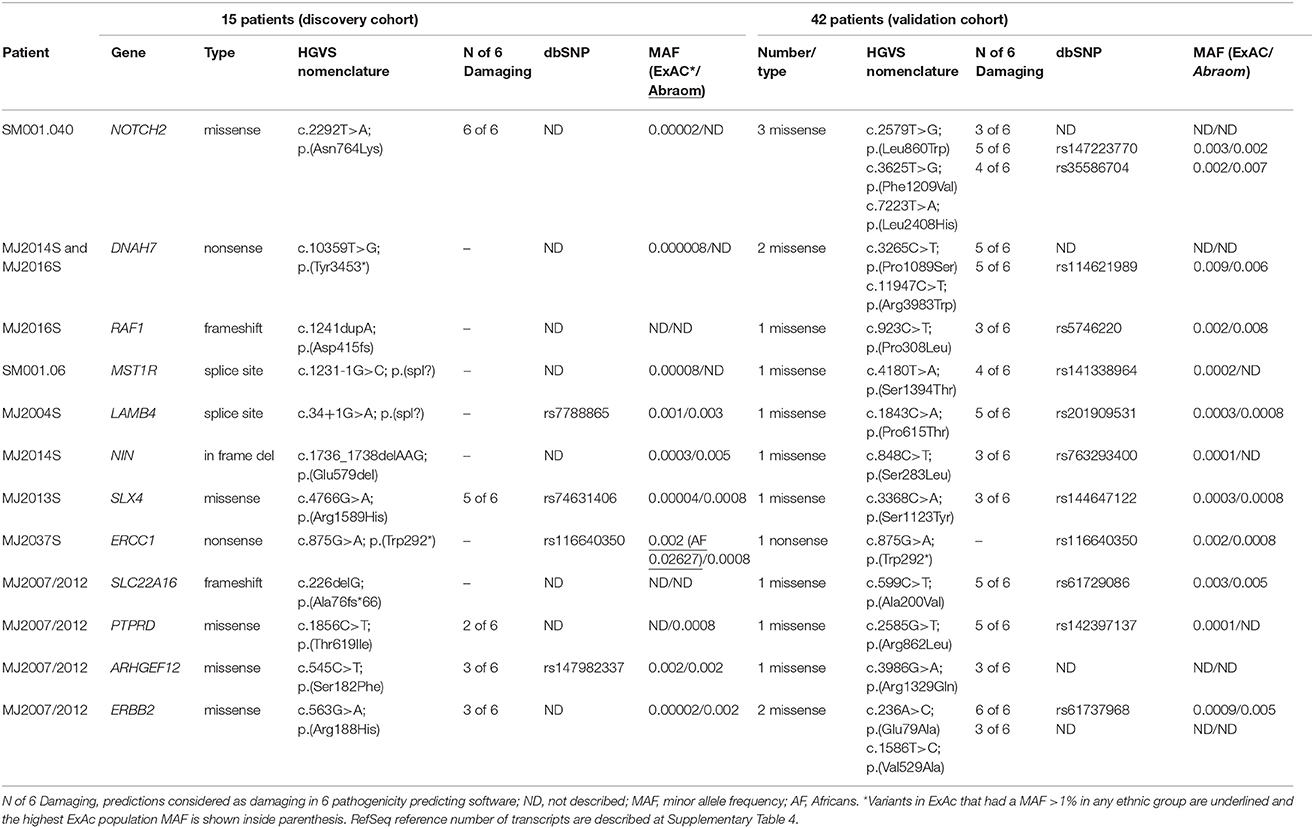
Table 4. Variants detected in 12 candidate genes at the discovery cohort and at an independent cohort.
Discussion
Recently, the use of WES in clinical genetics has been proven to be an effective alternative for establishing the genetic basis of Mendelian diseases, particularly in diseases where multiple genes can be affected (Trujillano et al., 2016). Moreover, in both clinical and research settings, WES has been applied to elucidate the genetic cause of cancer predisposition. In this sense, WES offers the opportunity to concomitantly investigate several known cancer risk genes as well as to identify novel cancer predisposing genes. Thus, in this study we used WES to disclose variants contributing to BC increased risk in patients that were negative for pathogenic variants in three major BC genes—BRCA1/2 and TP53 genes—and the most common point mutation in CHEK2 gene (c.1100delC). For this, we used stringent clinical criteria for selecting patients with strong indicative of harboring a genetic predisposition to BC, such as early onset BC (<36 years); bilateral BC; or the presence of a second primary related tumor.
First, by evaluating known BC predisposing genes, we could establish the causative variants in two probands. One of them harbored an ATM truncating pathogenic variant and the other a novel BARD1 truncating variant, considered as probably pathogenic. The BARD1 p.(Tyr739Leufs*2) variant is predicted to cause partial loss of the second functional BRCT domain and the phosphobinding region. Several studies suggest that both BRCT repeats are necessary for BARD1 normal function (Birrane et al., 2007; Irminger-Finger et al., 2016) and truncating variants in this region have been previously reported in association with HBC (De Brakeleer et al., 2010). Additionally, compatible with the probable pathogenic role of this variant, our proband presented triple negative BC and BARD1 pathogenic variants were recently described to be related to this molecular subtype (De Brakeleer et al., 2016).
Besides these LOF variants, we identified four rare missense VUS in three HBC genes (BRCA1, RINT1, and RAD51B). The identification of VUS in genetic testing represent a challenging concern for genetic counselors due to uncertainty in clinical decision making, which can lead to more intensive management than necessary in most of the times or, more rarely, in inappropriate prevention measures (Plon et al., 2011). The recently introduction of NGS gene panels in genetic testing have increased the number of patients diagnosed with VUS, emphasizing the urgent need for better pathogenicity predictions models and collaborative efforts to increase observational data that can aid a posteriori classification to variants, such as cosegregation analysis, personal and family history, co-occurrence with pathogenic variants, and histological and molecular features of tumors (Spurdle et al., 2012).
In the 15 patients without known pathogenic variants, we could identify more than 25,000 novel or rare variants (MAF < 1%), thus several filtering strategies were applied to prioritize those more likely to be related to HBC. Since the majority of hereditary cancer predisposing genes harbor an excess of loss-of function variants, we focused on this type of overtly deleterious variants, regardless of the affected gene. Furthermore, most BC risk genes are involved in DNA repair and genomic integrity pathways (Shiovitz and Korde, 2015; Nielsen et al., 2016), and prioritizing variants in these genes is a rational approach that have been used successfully in previous studies (Mantere et al., 2016). As so, we have also focused on missense variants in a defined set of cancer-related and DNA repair genes. By doing that, we were able to reduce our candidate genes list to a few hundreds.
Importantly, for one family with two sisters affected by BC at young ages (29 years), we could improve the selection by retaining only shared variants and also perform segregation analysis of the candidate variants in an aunt affected by ovarian cancer. From this analysis, eight cosegregating variants emerged, including a BRCA1 VUS. Besides BRCA1 gene, only ERBB2 has been previously implicated in BC predisposition, although with conflicting data about the increased risk conferred by some alleles (Breyer et al., 2009; Wang et al., 2013). Regarding the two LOF variants found to be cosegregating in this family (genes SLC22A16 and IL33), no relation between both genes and BC could be recognized in the literature.
One possible explanation for the results observed in this family and that could also be responsible for the cancer predisposition in other patients of our study is the polygenic model. In this model, which has been suggested and reviewed by different authors (Oldenburg et al., 2007; Shiovitz and Korde, 2015), moderate and low penetrance alleles would act in synergy and play a predominant role. Additionally, the high number of affected relatives with different tumor types in both maternal and paternal sides of this family can be a confounding factor for understanding the phenotypes and cosegregation results. Unfortunately, most affected family members of this family were deceased, limiting additional investigations and the interpretation of our findings.
To gain further insight on the relevance of our identified candidate genes, we evaluated the most promising ones in an independent cohort comprising 42 Brazilian HBC women. Several rare and possibly damaging variants were identified in this cohort, providing additional evidence of the potential role in BC predisposition of some new genes. Of those, we highlight four genes related to cancer development and progression (NOTCH2, ERBB2, MST1R, and RAF1) and two DNA repair genes (ERCC1 and SLX4). Interestingly, ERCC1 and SLX4 are partners that act in the repair of interstrand cross-links and are also required for homology-directed repair of DNA double-strand breaks. Additionally, ERCC1 is also involved in the nucleotide excision repair pathway (McNeil and Melton, 2012). Both genes have been investigated regarding BC susceptibility, with some common ERCC1 variants being identified as risk alleles in Chinese population (Yang et al., 2013) and rare truncating and possibly damaging variants in SLX4 being described in some high risk HBOC patients (Bakker et al., 2013; Shah et al., 2013). Remarkably, in the ERCC1 gene we identified the same nonsense variant in both discovery and validation cohorts (p.Trp292*), while in SLX4 one of the rare missense identified in our cohorts (p.Ser1123Tyr) was previously described in one HBC patient (Shah et al., 2013).
Some limitations of our study are inherent to WES method since predisposition variants can be located in non-coding or not captured regions of the genome, such as promoter or deep intronic pathogenic variants. Moreover, although the strategic filtering applied here is necessary to reduce the number of proposed candidates, it can result in the omission of the causative variant (for example, by excluding protein-impacting synonymous variants). Additionally, large genomic rearrangements have been implicated in HBC, and even though specific bioinformatics pipelines can be applied in WES data to extract these results, these analyses were not performed in our study. Finally, when it comes to interpreting the potential effect of our candidate variants in splicing, both coding as well as splice site variants can cause splicing alterations that lead to in-frame functional proteins instead of frameshift truncated ones, and functional assays would be necessary to validate bioinformatics predictions.
Considering the evidence presented here, we can neither conclude that these variants identified in the 15 patients negative for known pathogenic variant are the definitive cause of BC predisposition nor determine the magnitude of the risk that these genes could present. Nevertheless, our results provide a set of novel putative BC predisposing genes and reinforce WES as useful tool for assessing the complex landscape of HBC predisposition. Importantly, this represents the first WES data of a HBC cohort from South America and the analysis of an admixed population such as the Brazilian can reveal unique features compared to other Western populations. In this sense, the WES data generated in our study, as well as other previous and future studies, can be reanalyzed in the future and possibly identify genetic overlaps between families, aiding to gene discoveries (Chandler et al., 2016). Finally, the assignment of a novel gene or specific variant as a true BC predisposition factor requires solid phenotypic evidence from cosegregation analysis, in vitro and in vivo functional assays and genotyping large series of case and controls from distinct populations. The efforts for discovery and validation of novel HBC genes will continue to provide insights into disease mechanisms, eventually leading to the development of more effective therapies and improved management of affected families.
Author Contributions
GT, FdS, EF, and DC: conceived and designed the experiments; GT, FdA, MF, BB, CdP, and EF: performed and analyzed the experiments; RV, JdS, RR, and SdS: performed bioinformatics analysis; AdN, MA, PF, and EP: assessed clinical data, selected, and recruited the patients; SdS, EP, and DC: contributed reagents, materials, and analysis tools; GT, FdA, and DC: wrote and edited the paper. All authors have read and approved the final manuscript.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo [2008/57887-9, 2013/23277-8 and 2013/24633-2], Conselho Nacional de Desenvolvimento Científico e Tecnológico [408833/2006-8], FINEP-CT-INFRA (02/2010) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior [23038.004629/2014-19].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the patients and relatives for participating in the study and the A.C. Camargo biobank for sample processing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00161/full#supplementary-material
Footnotes
1. ^Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), 2018. Available online at: https://omim.org/
References
Anglian Breast Cancer Study (2000). Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br. J. Cancer 83, 1301–1308. doi: 10.1054/bjoc.2000.1407
Bakker, J. L., van Mil, S. E., Crossan, G., Sabbaghian, N., De Leeneer, K., Poppe, B., et al. (2013). Analysis of the novel fanconi anemia gene SLX4/FANCP in familial breast cancer cases. Hum. Mutat. 34, 70–73. doi: 10.1002/humu.22206
Birrane, G., Varma, A. K., Soni, A., and Ladias, J. A. (2007). Crystal structure of the BARD1 BRCT domains. Biochemistry 46, 7706–7712. doi: 10.1021/bi700323t
Breyer, J. P., Sanders, M. E., Airey, D. C., Cai, Q., Yaspan, B. L., Schuyler, P. A., et al. (2009). Heritable variation of ERBB2 and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 18, 1252–1258. doi: 10.1158/1055-9965.EPI-08-1202
Carraro, D. M., Koike Folgueira, M. A., Garcia Lisboa, B. C., Ribeiro Olivieri, E. H., Vitorino Krepischi, A. C., de Carvalho, A. F., et al. (2013). Comprehensive analysis of BRCA1, BRCA2 and TP53 germline mutation and tumor characterization: a portrait of early-onset breast cancer in Brazil. PLoS ONE 8:e57581. doi: 10.1371/journal.pone.0057581
Chandler, M. R., Bilgili, E. P., and Merner, N. D. (2016). A Review of whole-exome sequencing efforts toward hereditary breast cancer susceptibility gene discovery. Hum. Mutat. 37, 835–846. doi: 10.1002/humu.23017
Comino-Méndez, I., Gracia-Aznárez, F. J., Schiavi, F., Landa, I., Leandro-García, L. J., Letón, R., et al. (2011). Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 43, 663–667. doi: 10.1038/ng.861
De Brakeleer, S., De Grève, J., Desmedt, C., Joris, S., Sotiriou, C., Piccart, M., et al. (2016). Frequent incidence of BARD1-truncating mutations in germline DNA from triple-negative breast cancer patients. Clin. Genet. 89, 336–340. doi: 10.1111/cge.12620
De Brakeleer, S., De Grève, J., Loris, R., Janin, N., Lissens, W., Sermijn, E., et al. (2010). Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat. 31, 1175–1185. doi: 10.1002/humu.21200
Easton, D. F., Pharoah, P. D., Antoniou, A. C., Tischkowitz, M., Tavtigian, S. V., Nathanson, K. L., et al. (2015). Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 372, 2243–2257. doi: 10.1056/NEJMsr1501341
Elledge, R. M., and Allred, D. C. (1998). Prognostic and predictive value of p53 and p21 in breast cancer. Breast Cancer Res. Treat. 52, 79–98. doi: 10.1023/A:1006163101948
Futreal, P. A., Coin, L., Marshall, M., Down, T., Hubbard, T., Wooster, R., et al. (2004). A census of human cancer genes. Nat. Rev. Cancer 4, 177–183. doi: 10.1038/nrc1299
Gracia-Aznarez, F. J., Fernandez, V., Pita, G., Peterlongo, P., Dominguez, O., de la Hoya, M., et al. (2013). Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS ONE 8:e55681. doi: 10.1371/journal.pone.0055681
Hilbers, F. S., Meijers, C. M., Laros, J. F., van Galen, M., Hoogerbrugge, N., Vasen, H. F. A., et al. (2013). Exome sequencing of germline DNA from non-BRCA1/2 familial breast cancer cases selected on the basis of aCGH tumor profiling. PLoS ONE 8:e55734. doi: 10.1371/journal.pone.0055734
Honrado, E., Benítez, J., and Palacios, J. (2005). The molecular pathology of hereditary breast cancer: genetic testing and therapeutic implications. Mod. Pathol. 18, 1305–1320. doi: 10.1038/modpathol.3800453
Irminger-Finger, I., Ratajska, M., and Pilyugin, M. (2016). New concepts on BARD1: regulator of BRCA pathways and beyond. Int. J. Biochem. Cell Biol. 72, 1–17. doi: 10.1016/j.biocel.2015.12.008
Kean, S. (2014). The “ other ” breast cancer genes. Science 343, 1457–1459. doi: 10.1126/science.343.6178.1457
Kiiski, J. I., Pelttari, L. M., Khan, S., Freysteinsdottir, E. S., Reynisdottir, I., Hart, S. N., et al. (2014). Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. U.S.A. 111, 15172–15177. doi: 10.1073/pnas.1407909111
Kim, Y. C., Soliman, A. S., Cui, J., Ramadan, M., Hablas, A., Abouelhoda, M., et al. (2017). Unique features of germline variation in five Egyptian familial breast cancer families revealed by exome sequencing. PLoS ONE 12:e0167581. doi: 10.1371/journal.pone.0167581
Mantere, T., Winqvist, R., Kauppila, S., Grip, M., Jukkola-Vuorinen, A., Tervasmäki, A., et al. (2016). Targeted next-generation sequencing identifies a recurrent mutation in MCPH1 associating with hereditary breast cancer susceptibility. PLoS Genet. 12:e1005816. doi: 10.1371/journal.pgen.1005816
McNeil, E. M., and Melton, D. W. (2012). DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 40, 9990–10004. doi: 10.1093/nar/gks818
Meijers-Heijboer, H., van den Ouweland, A., Klijn, J., Wasielewski, M., de Snoo, A., Oldenburg, R., et al. (2002). Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 31, 55–59. doi: 10.1038/ng879
Nielsen, F. C., van Overeem Hansen, T., and Sørensen, C. S. (2016). Hereditary breast and ovarian cancer: new genes in confined pathways. Nat. Rev. Cancer 16, 599–612. doi: 10.1038/nrc.2016.72
Noh, J. M., Kim, J., Cho, D. Y., Choi, D. H., Park, W., and Huh, S. J. (2015). Exome sequencing in a breast cancer family without BRCA mutation. Radiat. Oncol. J. 33, 149–154. doi: 10.3857/roj.2015.33.2.149
Oldenburg, R. A., Meijers-Heijboer, H., Cornelisse, C. J., and Devilee, P. (2007). Genetic susceptibility for breast cancer: how many more genes to be found? Crit. Rev. Oncol. Hematol. 63, 125–149. doi: 10.1016/j.critrevonc.2006.12.004
Plon, S. E., Cooper, H. P., Parks, B., Dhar, S. U., Kelly, P. A., Weinberg, A. D., et al. (2011). Genetic testing and cancer risk management recommendations by physicians for at-risk relatives. Genet. Med. 13, 148–154. doi: 10.1097/GIM.0b013e318207f564
Putnam, C. D., Srivatsan, A., Nene, R. V., Martinez, S. L., Clotfelter, S. P., Bell, S. N., et al. (2016). A genetic network that suppresses genome rearrangements in Saccharomyces cerevisiae and contains defects in cancers. Nat. Commun. 11256, 1–14. doi: 10.1038/ncomms11256
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi: 10.1038/gim.2015.30
Seguí, N., Mina, L. B., Lázaro, C., Sanz-Pamplona, R., Pons, T., Navarro, M., et al. (2015). Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 149, 563–566. doi: 10.1053/j.gastro.2015.05.056
Shah, S., Kim, Y., Ostrovnaya, I., Murali, R., Schrader, K. A., Lach, F. P., et al. (2013). Assessment of SLX4 mutations in hereditary breast cancers. PLoS ONE 8:e66961. doi: 10.1371/journal.pone.0066961
Shiovitz, S., and Korde, L. A. (2015). Genetics of breast cancer: a topic in evolution. Ann. Oncol. 26, 1291–1299. doi: 10.1093/annonc/mdv022
Silva, F. C., Lisboa, B. C. G., Figueiredo, M. C. P., Torrezan, G. T., Santos, É. M. M., Krepischi, A. C., et al. (2014). Hereditary breast and ovarian cancer: assessment of point mutations and copy number variations in Brazilian patients. BMC Med. Genet. 15:55. doi: 10.1186/1471-2350-15-55
Snape, K., Ruark, E., Tarpey, P., Renwick, A., Turnbull, C., Seal, S., et al. (2012). Predisposition gene identification in common cancers by exome sequencing: insights from familial breast cancer. Breast Cancer Res. Treat. 134, 429–433. doi: 10.1007/s10549-012-2057-x
Sokolenko, A. P., Suspitsin, E. N., Kuligina, E. S., Bizin, I. V., Frishman, D., and Imyanitov, E. N. (2015). Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 369, 274–288. doi: 10.1016/j.canlet.2015.09.014
Spurdle, A. B., Healey, S., Devereau, A., Hogervorst, F. B. L., Monteiro, A. N., Nathanson, K. L., et al. (2012). ENIGMA–evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 33, 2–7. doi: 10.1002/humu.21628
Thompson, E. R., Doyle, M. A., Ryland, G. L., Rowley, S. M., Choong, D. Y. H., Tothill, R. W., et al. (2012). Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 8:e1002894. doi: 10.1371/journal.pgen.1002894
Trujillano, D., Bertoli-Avella, A. M., Kumar Kandaswamy, K., Weiss, M. E., Köster, J., Marais, A., et al. (2016). Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182. doi: 10.1038/ejhg.2016.146
Walsh, T., and King, M.-C. (2007). Ten genes for inherited breast cancer. Cancer Cell 11, 103–105. doi: 10.1016/j.ccr.2007.01.010
Wang, H., Liu, L., Lang, Z., Guo, S., Gong, H., Guan, H., et al. (2013). Polymorphisms of ERBB2 and breast cancer risk: a meta-analysis of 26 studies involving 35,088 subjects. J. Surg. Oncol. 108, 337–341. doi: 10.1002/jso.23386
Wen, H., Kim, Y. C., Snyder, C., Xiao, F., Fleissner, E. A., Becirovic, D., et al. (2014). Family-specific, novel, deleterious germline variants provide a rich resource to identify genetic predispositions for BRCAx familial breast cancer. BMC Cancer 14:470. doi: 10.1186/1471-2407-14-470
Keywords: cancer predisposition genes, hereditary breast cancer, whole-exome sequencing, germline pathogenic variants, cancer susceptibility, DNA repair genes
Citation: Torrezan GT, de Almeida FGdSR, Figueiredo MCP, Barros BDdF, de Paula CAA, Valieris R, de Souza JES, Ramalho RF, da Silva FCC, Ferreira EN, de Nóbrega AF, Felicio PS, Achatz MI, de Souza SJ, Palmero EI and Carraro DM (2018) Complex Landscape of Germline Variants in Brazilian Patients With Hereditary and Early Onset Breast Cancer. Front. Genet. 9:161. doi: 10.3389/fgene.2018.00161
Received: 16 November 2017; Accepted: 17 April 2018;
Published: 07 May 2018.
Edited by:
Luis G. Carvajal-Carmona, University of California, Davis, United StatesReviewed by:
Tracy A. O'Mara, QIMR Berghofer Medical Research Institute, AustraliaJohn Frederick Pearson, University of Otago, New Zealand
Copyright © 2018 Torrezan, de Almeida, Figueiredo, Barros, de Paula, Valieris, de Souza, Ramalho, da Silva, Ferreira, de Nóbrega, Felicio, Achatz, de Souza, Palmero and Carraro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirce M. Carraro, ZGlyY2UuY2FycmFyb0BhY2NhbWFyZ28ub3JnLmJy
†These authors have contributed equally to this work.