 Anna Zlotina
Anna Zlotina Artem Kiselev1
Artem Kiselev1 Alexey Sergushichev
Alexey Sergushichev Anna Kostareva
Anna Kostareva- 1Almazov National Medical Research Centre, Saint Petersburg, Russia
- 2ITMO University, Saint Petersburg, Russia
- 3Department of Women’s and Children’s Health, Center for Molecular Medicine, Karolinska Institute, Solna, Sweden
“Heart–hand” type syndromes represent a group of rare congenital conditions that combine cardiac pathology (structural defect or arrhythmic disorder) and limb abnormality. Significant clinical variability and genetic heterogeneity typical for such syndromes complicate correct diagnosis, prognosis, and appropriate genetic counseling of the affected families. By now, only single genes have been unambiguously determined as a genetic cause of heart–hand syndromes and phenotypically similar conditions. In the present study, we report on a 25-year-old Russian female patient with a clinical picture resembling ulnar-mammary syndrome (UMS). Principal clinical manifestations included heart septal fibrosis and non-sustained left ventricular tachycardia combined with fifth finger camptodactyly, hypoplastic breast, abnormal teeth, and mental retardation. Target Sanger sequencing and array-based comparative genome hybridization confirmed the lack of pathogenic mutations and large-scale deletions in TBX3 (12q24.21), the only gene known to be associated with UMS cases to date. Based on the results of whole-exome sequencing, 14 potential candidate variants were identified. Among them, a novel missense variant in SYNM gene (exon 1, c.173C > T, p.A58V), encoding intermediate filament protein synemin was characterized. Until the present, no association between SYNM mutations and congenital clinical syndromes has been reported. At the same time, taking into account synemin tissue-specific expression profiles and available data on abnormal knock-out mice phenotypes, we propose SYNM as a candidate gene contributing to the UMS-like phenotype. Further comprehensive functional studies are required to evaluate possible involvement of SYNM in genesis of complex heart-limb pathology.
Introduction
Congenital heart disorders can represent isolated anomalies or be a part of complex syndromic phenotypes. “Heart–hand” syndromes (HHSs) are a group of rare congenital clinical conditions, where patients in addition to cardiac pathology (congenital heart defect and/or arrhythmic disorder) present with various abnormalities of limb skeleton, as well as additional dysmorphia (Holt and Oram, 1960; Ruiz de la Fuente and Prieto, 1980; Hollister and Hollister, 1981; Silengo et al., 1990; Šinkovec et al., 2005). Genetic basis of “heart–hand” type syndromes and phenotypically similar pathologies remains poorly understood with only a few causative genes or chromosomal loci identified. In particular, TBX5 gene defects were shown to be responsible for the most common prototypical heart–hand syndrome type I, or Holt-Oram syndrome (HOS, MIM1 142900) characterized by cardiac septal defects, conduction system disease and radial ray anomaly of forelimb skeleton (Basson et al., 1997; Li et al., 1997). TBX5 belongs to an evolutionary conserved gene family encoding transcription factors with a DNA-binding domain, T-box, and plays various roles in developmental processes, including cardiogenesis and specification of forelimb identity (Rodriguez-Esteban et al., 1999; Bruneau et al., 2001). Truncation, missense, and splice site mutations in TBX5 are well-described in HOS cases with a mutation type and position defining the severity of cardiac and skeletal phenotype (reviewed in Packham and Brook, 2003). Besides, the phenotype expression of Holt-Oram syndrome can be caused by interstitial chromosomal deletions in chromosomes 6 and 14 (Adamopoulos et al., 2004; Le Meur et al., 2005).
Mutations in another transcription factor gene – TFAP2B – expressed in neural crest cells were shown to be responsible for a phenotypically distinct autosomal dominant disorder, Char syndrome (MIM 169100) characterized by patent ductus arteriosus, fifth digit middle phalangeal hypoplasia, and additional facial dysmorphism (Char, 1978; Satoda et al., 2000). By contrast, Slovenian type of heart–hand syndrome (HHS IV, MIM 610140) combining conduction system disease, atrial and ventricular tachyarrhythmias, dilated cardiomyopathy, and brachydactyly proved to be a laminopathy caused by particular mutations in LMNA gene encoding a structural intermediate filament (IF) protein of a nuclear lamina (Šinkovec et al., 2005; Renou et al., 2008; Zaragoza et al., 2017).
Ulnar-mammary syndrome (UMS, also referred to as Schinzel syndrome or Pallister UMS, MIM 181450) represents a similar autosomal dominant condition also involving hands and the heart. Key UMS clinical features usually include ulnar ray defects (ranging from fifth finger deformities to complete absence of the ulna), hypoplasia of mammary and apocrine glands, abnormal teeth, genital hypoplasia, and puberty delay. At the same time, additional manifestations comprise myocardial pathology, namely cardiac conduction abnormality and/or congenital heart defect (Meneghini et al., 2006; Linden et al., 2009). The syndrome is associated with mutations in TBX3 gene (locus 12q23-24.1), another member of T-box gene family (Bamshad et al., 1997, 1999). Indeed, a particular vital role of TBX3 was shown for limb and mammary gland development, differentiation of cardiac conduction system and heart looping and growth (reviewed in Washkowitz et al., 2012). In addition to single nucleotide mutations, there are several reported cases of contiguous microdeletions at 12q24.21 locus encompassing both TBX3 and TBX5 genes and giving rise to phenotypes that combine UMS and Holt-Oram syndrome features (Borozdin et al., 2006; Alby et al., 2013; Bogarapu et al., 2014; Iwanicka-Pronicka et al., 2016).
Numerous phenotypically overlapping clinical cases were reported with undetermined genetic basis (Ruiz de la Fuente and Prieto, 1980; Hollister and Hollister, 1981; Silengo et al., 1990; Morava et al., 2003; Demura et al., 2010; Nanda et al., 2010). Identification of new deleterious genetic variants, candidate genes and modifiers, which became possible due to high-throughput sequencing approaches and array-based comparative genome hybridization (array-CGH), proves to be helpful for meeting the diagnostic challenge and enables new insights into molecular and cellular mechanisms underlying combined limb-heart malformations (Liu et al., 2017; Zaragoza et al., 2017).
In the present study, we report on a 25-year-old woman with a clinical picture resembling UMS. Main clinical manifestations included pathology of 5th digits, hypoplasia of the mammary glands, mental retardation, and heart septal fibrosis combined with non-sustained ventricular tachycardia. Sanger sequencing and array-CGH allowed to exclude causative role of TBX3. Based on the results of whole-exome sequencing (WES), we describe a novel missense variant in SYNM gene encoding IF protein synemin and discuss its potential involvement in the patient’s phenotype.
Materials and Methods
Standard karyotyping was carried out on GTG-banded metaphase chromosomes obtained from phytohemagglutinin-stimulated peripheral blood lymphocytes. Oligonucleotide array-based CGH was performed using Agilent 8x60K array platform with median probe spacing 41 kb (SurePrint G3 Human CGH Microarray, Agilent Technologies, Santa Clara, CA, United States). The data obtained was processed and analyzed using CytoGenomics Software (v3.0.1.1, Agilent Technologies). Copy number variations (CNVs) were called using an aberration detection statistical algorithm ADM-2, with a sensitivity threshold of 6.0.
Bidirectional Sanger sequencing was applied to search for single-nucleotide genetic variants in TBX3 gene (Gene ID: 6926, NG_008315.12) using BigDye Terminator Sequencing Kit (Applied Biosystems) and Genetic Analyzer AB3100 (Applied Biosystems/Hitachi, Japan). WES DNA-library was prepared using the SureSelectXT Human All Exon v6 r2 (60 Mbp) target enrichment kit (Agilent Technologies, Santa Clara, CA, United States). Sequencing run was carried out with SBSv4 chemistry and the Illumina HiSeq instrument (Illumina, San Diego, CA, United States). Alignment was performed using Burrows-Wheeler Aligner (BWA-MEM-0.7.1, Li and Durbin, 2009) with GRCh37/hg19 human genome assembly as a reference after that the data processing (Picard 2.8.3) and variant calling (GATK 3.7.) was performed according to Broad institute GATK Best Practice. Variant annotation was done using Annovar (Wang et al., 2010). Variant population frequencies were evaluated based on ExAc3 and gnomAD4 resources, functional prediction was made based on dbNSFP (v3.3a). Data on tissue-specific gene expression profiles were taken from UniProt5, GNF gene expression atlas, Human Protein Atlas6. Additionally, variants were evaluated based on expression rank in heart tissue according to GTEx dataset. Average target region coverage was ∼ x150 with 95% of the target region being covered to a depth of 20 or more.
Written informed consent was obtained from the patient for the genetic study and publication of images. The study was performed according to Helsinki Declaration and study approval was obtained from Institutional Ethical Review Board at the Almazov National Medical Research Centre in St. Petersburg.
Results
Clinical Case
A 25-year-old woman was hospitalized due to frequent premature ventricular beats of high grade (17,000 per day) and repeated episodes of bidirectional non-sustained ventricular tachycardia without syncope. Echocardiography revealed enlarged left ventricular dimension and local ventricular wall thinning. Upon routine clinical examination bilateral symmetrical hand abnormality was noted, namely the fifth finger camptodactyly (Figure 1A). Additionally, hypoplasia of the breast with inverted nipples was observed (Figure 1C). Facial features included wide-set eyes, a broad nasal tip and thin upper lip vermilion and strabismus (Figure 1B). Dental abnormalities were represented by tooth malalignment and hypoplasia involving canines and back teeth (Figures 1C,D). No defects were documented in her lower limbs. Apart from physical defects, intellectual deficit was noted and included mild mental retardation and learning disabilities. Family history reported that proband’s mother died due to congenital heart defect and congestive heart failure at the age of 30. Grandmother from mother side was not affected. No other relatives were available for examination. Due to the lack of family data, it is hard to conclude the mode of inheritance unambiguously. However, keeping in mind the mother’s phenotype, the dominant inheritance could be suggested (Supplementary Figure S1).
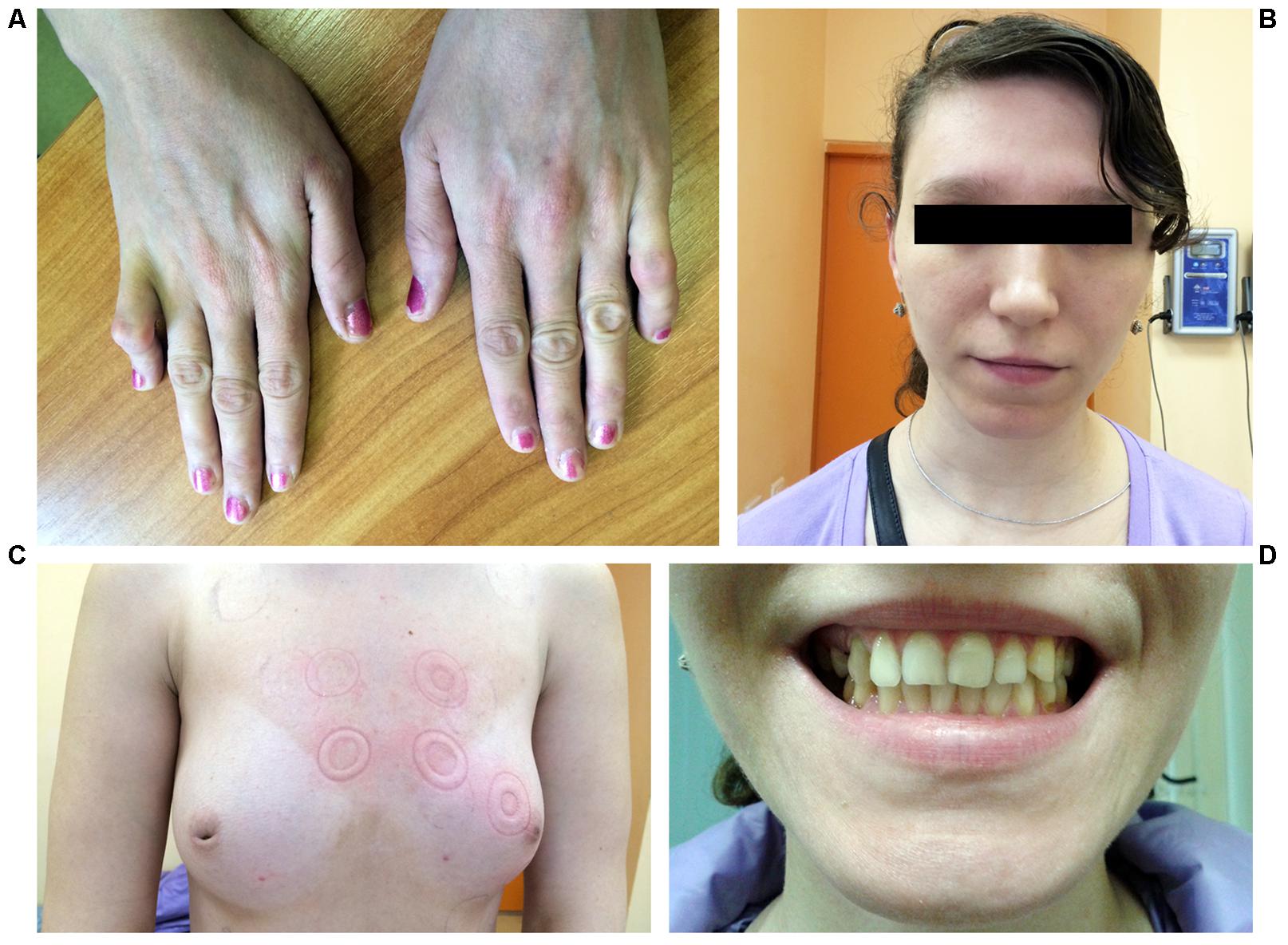
FIGURE 1. Photographs representing phenotype of the patient. (A) Hands demonstrating fifth finger camptodactyly. (B) Facial features include wide-set eyes, strabismus, a broad nasal tip, and thin upper lip vermilion. (C) Hypoplastic breast with inverted nipples. (D) Dental abnormalities involving canines and back teeth.
Genetic Studies
Standard cytogenetic analysis showed normal female karyotype. Taking into consideration the patient’s clinical phenotype similar to UMS, the next step was to screen the TBX3 locus for genome variations. However, bidirectional Sanger sequencing of TBX3 protein-coding regions including 3′- and 5′- flanking intronic sequences did not reveal any known pathogenic mutations or variants of uncertain significance. High-resolution microarray-CGH analysis allowed to exclude a whole-gene TBX3 deletion as well as other causative microimbalances over ∼100–150 kb in size. These findings imply that the patient’s syndromic phenotype is unlikely to be caused by TBX3 deficiency and is rather due to another genetic defect.
To search for candidate genes, WES was performed. The detailed workflow of filtering strategy with total numbers of variants left after each step is depicted as a flowchart (Figure 2). The called variants were filtered according to their exonic function and population frequencies so that deep intronic variants, exonic synonymous substitutions and all variants with allele frequency 0.1% and higher were excluded from the further analysis. As a result, rare protein-changing variants (missense, frameshifts, nonsense, and predicted splice sites) were further evaluated based on gene functions, clinical annotations, mode of inheritance and prediction of variant functional effect (Supplementary Table S1). We did not identify any genes responsible for “hear-hand” syndromes or syndromic conditions with overlapping cardiopathology, limb skeletal manifestation or malformed breast, including TBX5 (Holt-Oram syndrome), LMNA (Slovenian type of heart–hand syndrome), TFAP2B (Char syndrome), TP63 [Limb-mammary syndrome (MIM 603543), ADULT syndrome (MIM 103285)].

FIGURE 2. Flowchart representing a strategy for filtering of genetic variants identified by whole-exome sequencing (WES).
As a next step we focused on the genes highly expressed in tissues/organs affected and known to be involved in their morphogenesis, pathogenesis and functioning. After the filtering process, 14 candidate variants were selected (Supplementary Table S2). Among them, a heterozygous missense variant in SYNM gene (exon 1, c.173C > T, p.A58V), encoding IF protein synemin was selected as a favorable candidate and validated by Sanger sequencing (Figures 3A,B). In contrast to other IF proteins that tend to be tissue-specific, synemin was detected in a broad spectrum of tissues and organs including heart, breast, adipose tissue, bone cells, and brain. Though no clinical mutations in SYNM have been reported by now, the data on severe cardiac and osteopenic phenotypes in SYNM knockouts have been accumulated (for details, see section “Discussion”).
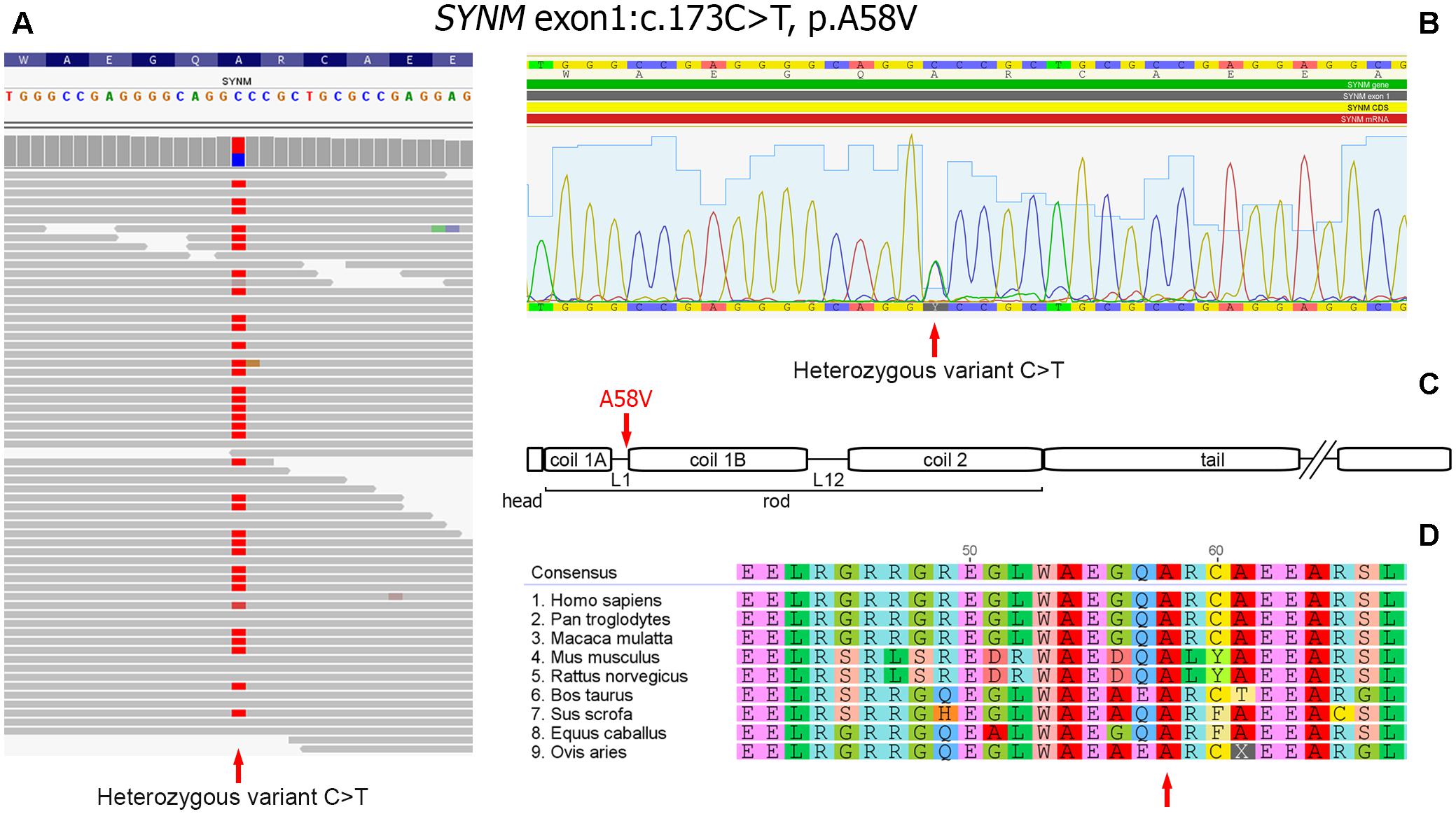
FIGURE 3. Heterozygous missence variant in SYNM c.173C > T (p.A58V). (A) Identification of the variant by WES. The variant position is pointed by an arrow. (B) Validation of WES results by Sanger sequencing: a fragment of sequencing chromatogram. Heterozygous C/T variant is pointed by an arrow. (C) Position of the amino acid change in synemin protein (shown by an arrow). Protein domain structure is depicted according to UniProt (http://www.uniprot.org). (D) Evolutionary conservation analysis of the amino-acid position across mammalian species (shown by an arrow).
Synemin presents all functional domains typical for IFs (Bellin et al., 1999; Mizuno et al., 2001; Titeux et al., 2001) and the identified genetic variant is mapped to the rod domain, on the border of a short polypeptide linker L1 and α-helical segment 1B (Figure 3C). The variant is currently absent in publicly available databases of normal or clinical SNPs such as Clinvar, dbSNP, ExAC, 1000 Genomes. Alignment of synemin protein sequences demonstrated evolutionary conservation at this amino acid position across mammalian species (Figure 3D). According to SIFT, FATHMM, MetaLR and M-CAP functional prediction tools, the variant is evaluated as deleterious. In dbSNP database, we found a rare unclassified missence variant at the same nucleotide position of SYNM (rs1367107502, MAF: 0.00002, TOPMED project), where, however, another allele (C > A) and residue (p.A58D) change took place. Based on ACMG guideline for the interpretation of sequence variants (Richards et al., 2015), the variant described here should be classified as a variant with unknown significance (VOUS) and needs to be further functionally tested using appropriate animal or cell culture models.
Discussion
Ulnar-mammary syndrome represents a rare congenital multi-systemic disorder characterized by incomplete penetrance and significant intra- and inter-familial clinical variability (Bamshad et al., 1999; Wollnik et al., 2002; Linden et al., 2009), which complicates accurate diagnosis and appropriate genetic counseling. The presence of limb abnormalities, especially involving the digits of the hand’s ulnar ray, and mammary gland hypoplasia implies the consideration of UMS. In the present study we describe a female patient with UMS-like phenotype manifestations including abnormalities of hand fifth digits, breast, teeth, and cardiac phenotype. The latter in form of septal fibrosis and non-sustained ventricular tachyarrhythmia is consistent with UMS phenotype, though heart pathology is rather rare component of the syndrome. In particular, two confirmed cases of a ventricular septal defect and one case of conduction abnormality in the form of Wolff–Parkinson–White syndrome were previously reported in UMS pediatric patients (Meneghini et al., 2006; Linden et al., 2009). Some clinical manifestations of our patient such as mental retardation and strabismus are not typically described as a part of UMS syndrome. To the best of our knowledge, the only UMS case combined with mental retardation was not caused by a TBX3 point mutation, but by a contiguous 1.28 Mb deletion at 12q24.2 chromosomal region (Klopocki et al., 2006). With regard to eye anomalies, congenital unilateral anophthalmia of unknown etiology was also noted once in a UMS patient (Linden et al., 2009).
To date, TBX3 is the only gene associated with UMS disorder. More than 20 TBX3 pathogenic mutations or large-scale gene aberrations have been reported in association with UMS cases with haploinsufficiency being regarded as a main disease-causing mechanism (Bamshad et al., 1999; Wollnik et al., 2002; Klopocki et al., 2006; Linden et al., 2009; Alby et al., 2013; Tanteles et al., 2017). At the same time, in our patient the lack of TBX3 mutation was confirmed by target Sanger sequencing, WES and CGH-microarray results. Notably, single cases of unclassified clinical conditions phenotypically similar to UMS have been earlier reported, where a TBX3 defect was not identified as a genetic cause (Morava et al., 2003). Therefore, such conditions might be regarded as an UMS-like disorder with apparently another genetic basis, possibly linked to other transcription factors or structural genes mutations.
In present study no morbid or unclassified variants were revealed in the genes underlying common heart–hand syndromes. Given the several criteria for variant evaluation such as absence of population frequency and prediction of functional effects, as well as protein tissue-specific expression profiles and literature data, we propose SYNM, encoding IF synemin, as a potentially novel candidate gene contributing to UMS-like condition. High level of synemin expression is shown in various types of tissues including those that are involved in the patient’s syndromic phenotype. In particular, initially synemin was described as an IF protein abundant in all muscle cells where in case of striated muscles it predominantly localized in the region of Z-disk, costamers and intercellular junctions, including intercalated disks of cardiomyocytes (Granger and Lazarides, 1980; Bilak et al., 1998; Bellin et al., 2001; Hirako et al., 2003). Since then, a list of synemin-positive cell types has been significantly extended and included mammary glands, adipose tissue, osteoblasts, some neural cells (The Human Protein Atlas; Hirako et al., 2003; Moorer et al., 2016; reviewed in Paul and Skalli, 2016).
Despite the absence of so far reported cases of SYNM mutations and their clinical phenotype, there are comprehensive data on abnormalities in synemin knock-out mice (Li et al., 2014; García-Pelagio et al., 2015, 2018; Moorer et al., 2016). Taking into consideration the prominent cardiac phenotype of our patient, it is of importance to note that the mice lacking synemin (synm-/-) demonstrate structural and functional abnormalities in the heart and myopathic changes (García-Pelagio et al., 2015, 2018). Absence of synemin in mice causes left ventricular remodeling, contractile and systolic dysfunction at 3 and 12–16 months of age with subsequent left ventricular hypertrophy and dilatation (García-Pelagio et al., 2018). In vitro examination of cardiomyocytes isolated from such knock-outs demonstrated the decreased calcium transients and contractility. Finally, synemin-null heart was characterized by alterations in a level of some signaling molecules (PKA-RII, ERK, and p70S6K) vital for cardiomyocyte function, which conforms to the previous knowledge on A-kinase anchoring properties of synemin in the heart (Russell et al., 2006). Depending on tissue origin, being unable to self-assemble into filaments, synemin co-polymerizes with other IF representatives, namely with desmin and vimentin (Bellin et al., 1999; Titeux et al., 2001). Thus, some other skeletal and cardiac muscle pathologies such as desmin-related myopathies are accompanied by altered synemin expression and cell distribution (Carlsson et al., 2002; Olivé et al., 2003).
In light of skeleton pathology, recent comprehensive study of synemin-null mice model demonstrated a prominent role of synemin in bone physiology (Moorer et al., 2016). That is, the animals were shown to suffer from osteopenia due to a substantial reduction in trabecular bone mass accompanied by the reduced osteoblast activity and number in vivo (Moorer et al., 2016). Ex vivo experiments on the isolated primary osteoblasts confirmed impaired proliferation of synemin -/- cells as compared to the wild-type but, surprisingly, revealed their elevated osteogenic differentiation capacity, thus, pointing to synemin involvement in osteoblast differentiation (Moorer et al., 2016). Similar results implying a role of synemin in a cell self-maintenance/differentiation balance were obtained for muscle satellite cells (Li et al., 2014) and glioblastoma cells (Pitre et al., 2012).
Accumulated data on synemin expression profiles, interacting partners and knock-out phenotypes demonstrate both structural and signaling roles of synemin during development or in adult tissues, including the role in structure and function of cardiac muscle and bone formation. Until the present, co-occurrence of synemin mutations with congenital clinical syndromes such as combined heart-limb pathology has not been yet reported. At the same time, the involvement of IF proteins in heart–hand syndrome phenotypes has already been shown. In particular, two specific mutations in LMNA gene encoding a nuclear lamina protein were identified as a genetic cause of the HHS type IV characterized by tachyarrhythmia, cardiomyopathy, and brachydactyly (Renou et al., 2008; Zaragoza et al., 2017). The findings obtained in the present study support a possible association between an IF gene and combined heart–hand malformation. The potential molecular mechanism underlying this association could involve cell proliferation/differentiation process in progenitor cells during development similar to that reported for LMNA mutations responsible for different laminopathy conditions (Malashicheva et al., 2015).
Concluding Remarks
Here, we report on a non-typical case of UMS with prominent cardiac manifestation and mild mental retardation. In contrast to common UMS cases, the described phenotype is not associated with TBX3 mutation or large-scale deletion. The results of WES data analysis and a review of literature data pointed to an IF gene, SYNM, as a potential new candidate gene contributing to the UMS-like condition. To prove a possible association between SYNM mutation and UMS-like condition further functional cell studies are planned.
Data Availability
The dataset of analyzed whole-exome sequencing results is included in the manuscript and the supplementary files. The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
EP and AaK patient workup and genetic counseling. AZ and AmK genetic analysis. AmK, AS, AZ, and AaK NGS data processing and analysis. AZ and AaK design of the study. AaK coordination of the study. AZ drafted the manuscript. AZ, AmK, AS, EP, and AaK final revision and approval of the manuscript. All authors agreed to be accountable for all aspects of the work.
Funding
This work was financially supported by Russian Science Foundation (RSF) (Grant No. 17-75-10125).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Dr. Olga Pavlova, specialist of Research Resource Center “Molecular and Cell Technologies” (St. Petersburg State University) for assistance in exome-library preparation and Dr. Dmitrii Polev, leading specialist of Research Resource Center “Biobank” (St. Petersburg State University) for great help in exome sequencing run and data handing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00209/full#supplementary-material
FIGURE S1 | Pedigree chart of the reported family. Gray color indicates mother’s phenotype partially overlapping with the proband’s clinical picture. Question mark indicates unavailable data on the phenotype.
TABLE S1 | Whole-exome sequencing data: a list of genetic variants filtered according to their exonic function and population allele frequencies. As a result, 402 rare protein-changing variants (missense, frameshifts, nonsense and predicted splice sites) with AF < 0.001 (ExAc and gnomAD databases) were selected.
TABLE S2 | Whole-exome sequencing data: a final list of 14 candidate genetic variants filtered based on CADD prediction scores, expression rank in the heart and tissue-specific expression in mammary glands, bone and brain (GTEx dataset, Uniprot, GNF gene expression atlas, Human Protein Atals).
Footnotes
- ^Online Mendelian Inheritance (OMIM) available at: https://omim.org/
- ^https://www.ncbi.nlm.nih.gov/gene/6926
- ^Exome Aggregation Consortium available at: http://exac.broadinstitute.org/
- ^Genome Aggregation Database http://gnomad.broadinstitute.org/
- ^The Universal Protein Resource available at: http://www.uniprot.org
- ^The Human Protein Atlas available at: https://www.proteinatlas.org/
References
Adamopoulos, S., Kokkinou, S., Parissis, J. T., and Kremastinos, D. T. (2004). New insight into “heart–hand” syndromes: a newly discovered chromosomal abnormality in a family with “heart–hand” syndrome. Int. J. Cardiol. 97, 129–132. doi: 10.1016/j.ijcard.2004.05.001
Alby, C., Bessieres, B., Bieth, E., Attie-Bitach, T., Fermont, L., Citony, I., et al. (2013). Contiguous gene deletion of TBX5 and TBX3 leads to a varible phenotype with combined features of holt-oram and ulnar-mammary syndromes. Am. J. Med. Genet. A 161, 1797–1802. doi: 10.1002/ajmg.a.36054
Bamshad, M., Le, T., Watkins, W. S., Dixon, M. E., Kramer, B. E., Roeder, A. D., et al. (1999). The spectrum of mutations in TBX3: genotype/phenotype relationship in ulnar-mammary syndrome. Am. J. Hum. Genet. 64, 1550–1562. doi: 10.1086/302417
Bamshad, M., Lin, R. C., Law, D. J., Watkins, W. C., Krakowiak, P. A., Moore, M. E., et al. (1997). Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat. Genet. 16, 311–315. doi: 10.1038/ng0797-311
Basson, C. T., Bachinsky, D. R., Lin, R. C., Levi, T., Elkins, J. A., Soults, J., et al. (1997). Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 15, 30–35. doi: 10.1038/ng0197-30
Bellin, R. M., Huiatt, T. W., Critchley, D. R., and Robson, R. M. J. (2001). Synemin may function to directly link muscle cell intermediate filaments to both myofibrillar Z-lines and costameres. J. Biol. Chem. 276, 32330–32337. doi: 10.1074/jbc.M104005200
Bellin, R. M., Sernett, S. W., Becker, B., Ip, W., Huiatt, T. W., and Robson, R. M. (1999). Molecular characteristics and interactions of intermediate filament protein synemin. J. Biol. Chem. 274, 29493–29499. doi: 10.1074/jbc.274.41.29493
Bilak, S. R., Sernett, S. W., Bilak, M. M., Bellin, R. M., Stromer, M. H., Huiatt, T. W., et al. (1998). Properties of the novel intermediate filament protein synemin and its identification in mammalian muscle. Arch. Biochem. Biophys. 355, 63–76. doi: 10.1006/abbi.1998.0702
Bogarapu, S., Bleyl, S. B., Calhoun, A., Viskochil, D., Saarel, E. V., Everitt, M. D., et al. (2014). Phenotype of a patient with contiguous deletion of TBX5 and TBX3: expanding the disease spectrum. Am. J. Med. Genet. A 164, 1304–1309. doi: 10.1002/ajmg.a.36447
Borozdin, W., Bravo-Ferrer Acosta, A. M., Seemanova, E., Leipoldt, M., Bamshad, M. J., Unger, S., et al. (2006). Contiguous hemizygous deletion of TBX5, TBX3, and RBM19 resulting in a combined phenotype of Holt-Oram and ulnar-mammary syndromes. Am. J. Med. Genet. A 140A, 1880–1886. doi: 10.1002/ajmg.a.31340
Bruneau, B. G., Nemer, G., Schmitt, J. P., Charron, F., Robitaille, L., Caron, S., et al. (2001). A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106, 709–721. doi: 10.1016/S0092-8674(01)00493-7
Carlsson, L., Fischer, C., Sjöberg, G., Robson, R. M., Sejersen, T., and Thornell, L.-E. (2002). Cytoskeletal derangements in hereditary myopathy with a desmin L345P mutation. Acta Neuropathol. 104, 493–504. doi: 10.1007/s00401-002-0583-z
Char, F. (1978). Peculiar facies with short philtrum, duck-bill lips, ptosis and low-set ears–a new syndrome? Birth Defects Orig. Artic. Ser. 14, 303–305.
Demura, M., Yoneda, T., Karashima, S., Higashikata, T., Mabuchi, H., Kawano, M., et al. (2010). A possible new syndrome with double endocrine tumors in association with an unprecedented type of familial heart-hand syndrome: a case report. J. Med. Case Rep. 4:347. doi: 10.1186/1752-1947-4-347
García-Pelagio, K. P., Chen, L., Joca, H. C., Ward, C., Jonathan Lederer, W., and Bloch, R. J. (2018). Absence of synemin in mice causes structural and functional abnormalities in heart. J. Mol. Cell. Cardiol. 114, 354–363. doi: 10.1016/j.yjmcc.2017.12.005
García-Pelagio, K. P., Muriel, J., O’Neill, A., Desmond, P. F., Lovering, R. M., Lund, L., et al. (2015). Myopathic changes in murine skeletal muscle lacking synemin. Am. J. Physiol. Cell Physiol. 308, C448–C462. doi: 10.1152/ajpcell.00331.2014
Granger, B. L., and Lazarides, E. (1980). Synemin: a new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell 22, 727–738. doi: 10.1016/0092-8674(80)90549-8
Hirako, Y., Yamakawa, H., Tsujimura, Y., Nishizawa, Y., Okumura, M., Usukura, J., et al. (2003). Characterization of mammalian synemin, an intermediate filament protein present in all four classes of muscle cells and some neuroglial cells: co-localization and interaction with type III intermediate filament proteins and keratins. Cell Tissue Res. 313, 195–207. doi: 10.1007/s00441-003-0732-2
Hollister, D. W., and Hollister, W. G. (1981). The “long-thumb” brachydactyly syndrome. Am. J. Med. Genet. 8, 5–16. doi: 10.1002/ajmg.1320080103
Holt, M., and Oram, S. (1960). Familial heart disease with skeletal malformations. Br. Heart J. 22, 236–242. doi: 10.1136/hrt.22.2.236
Iwanicka-Pronicka, K., Socha, M., Ję1drzejowska, M., Krajewska-Walasek, M., and Jamsheer, A. (2016). Life-threatening cardiac episode in a Polish patient carrying contiguous gene microdeletion of the TBX5 and the TBX3 genes. Springerplus 5:1638. doi: 10.1186/s40064-016-3275-1
Klopocki, E., Neumann, L. M., Tönnies, H., Ropers, H. H., Mundlos, S., and Ullmann, R. (2006). Ulnar-mammary syndrome with dysmorphic facies and mental retardation caused by a novel 1.28Mb deletion encompassing the TBX3 gene. Eur. J. Hum. Genet. 14, 1274–1279. doi: 10.1038/sj.ejhg.5201696
Le Meur, N., Goldenberg, A., Michel-Adde, C., Drouin-Garraud, V., Blaysat, G., Marret, S., et al. (2005). Molecular characterization of a 14q deletion in a boy with features of Holt-Oram syndrome. Am. J. Med. Genet. 134, 439–442. doi: 10.1002/ajmg.a.30660
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Q. Y., Newbury-Ecob, R. A., Terrett, J. A., Wilson, D. I., Curtis, A. R., Yi, C. H., et al. (1997). Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29. doi: 10.1038/ng0197-21
Li, Z., Parlakian, A., Coletti, D., Alonso-Martin, S., Hourde, C., Joanne, P., et al. (2014). Synemin acts as a regulator of signalling molecules during skeletal muscle hypertrophy. J. Cell Sci. 127, 4589–4601. doi: 10.1242/jcs.143164
Linden, H., Williams, R., King, J., Blair, E., and Kini, U. (2009). Ulnar mammary syndrome and TBX3: expanding the phenotype. Am. J. Med. Genet. A 149, 2809–2812. doi: 10.1002/ajmg.a.33096
Liu, Z., Yin, N., Gong, L., Tan, Z., Yin, B., Yang, Y., et al. (2017). Microduplication of 7q36.3 encompassing the SHH long-range regulator (ZRS) in a patient with triphalangeal thumb-polysyndactyly syndrome and congenital heart disease. Mol. Med. Rep. 15, 793–797. doi: 10.3892/mmr.2016.6092
Malashicheva, A., Bogdanova, M., Zabirnyk, A., Smolina, N., Ignatieva, E., Freilikhman, O., et al. (2015). Various lamin A/C mutations alter expression profile of mesenchymal stem cells in mutation specific manner. Mol. Genet. Metab. 115, 118–127. doi: 10.1016/j.ymgme.2015.04.006
Meneghini, V., Odent, S., Platonova, N., Egeo, A., and Merlo, G. R. (2006). Novel TBX3 mutation data in families with Ulnar-Mammary syndrome indicate a genotype-phenotype relationship: mutations that do not disrupt the T-domain are associated with less severe limb defects. Eur. J. Med. Genet. 49, 151–158. doi: 10.1016/j.ejmg.2005.04.021
Mizuno, Y., Thompson, T. G., Guyon, J. R., Lidov, H. G. W., Brosius, M., Imamura, M., et al. (2001). Desmuslin, an intermediate filament protein that interacts with -dystrobrevin and desmin. Proc. Natl. Acad. Sci. U.S.A. 98, 6156–6161. doi: 10.1073/pnas.111153298
Moorer, M. C., Buo, A. M., Garcia-Pelagio, K. P., Stains, J. P., and Bloch, R. J. (2016). Deficiency of the intermediate filament synemin reduces bone mass in vivo. Am. J. Physiol. Cell Physiol. 311, C839–C845. doi: 10.1152/ajpcell.00218.2016
Morava,É, Czakó, M., Kárteszi, J., Cser, B., Weissbecker, K., and Méhes, K. (2003). Ulnar/fibular ray defect and brachydactyly in a family: a possible new autosomal dominant syndrome. Clin. Dysmorphol. 12, 161–165. doi: 10.1097/01.mcd.0000072164.33788.0a
Nanda, S., Longo, S., and Arastu, M. I. (2010). Unicuspid aortic valve, hand anomalies: a heart-hand syndrome. Am. J. Med. Sci. 339, 296–299. doi: 10.1097/MAJ.0b013e3181c5ba64
Olivé, M., Goldfarb, L., Dagvadorj, A., Sambuughin, N., Paulin, D., Li, Z., et al. (2003). Expression of the intermediate filament protein synemin in myofibrillar myopathies and other muscle diseases. Acta Neuropathol. 106, 1–7. doi: 10.1007/s00401-003-0695-0
Packham, E. A., and Brook, J. D. (2003). T-box genes in human disorders. Hum. Mol. Genet. 12, 37R–44R. doi: 10.1093/hmg/ddg077
Paul, M., and Skalli, O. (2016). Synemin Molecular Features and the Use of Proximity Ligation Assay to Study Its Interactions, 1st Edn. New York, NY: Elsevier
Pitre, A., Davis, N., Paul, M., Orr, A. W., and Skalli, O. (2012). Synemin promotes AKT-dependent glioblastoma cell proliferation by antagonizing PP2A. Mol. Biol. Cell 23, 1243–1253. doi: 10.1091/mbc.E11-08-0685
Renou, L., Stora, S., Yaou, R. B., Volk, M., Sinkovec, M., Demay, L., et al. (2008). Heart-hand syndrome of Slovenian type: a new kind of laminopathy. J. Med. Genet. 45, 666–671. doi: 10.1136/jmg.2008.060020
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rodriguez-Esteban, C., Tsukui, T., Yonei, S., Magallon, J., Tamura, K., and Izpisua Belmonte, J. C. (1999). The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature 398, 814–818. doi: 10.1038/19769
Ruiz de la Fuente, S., and Prieto, F. (1980). Heart-hand syndrome. III. A new syndrome in three generations. Hum. Genet. 55, 43–47.
Russell, M. A., Lund, L. M., Haber, R., McKeegan, K., Cianciola, N., and Bond, M. (2006). The intermediate filament protein, synemin, is an AKAP in the heart. Arch. Biochem. Biophys. 456, 204–215. doi: 10.1016/j.abb.2006.06.010
Satoda, M., Zhao, F., Diaz, G. A., Burn, J., Goodship, J., Davidson, H. R., et al. (2000). Mutations in TFAP2B cause Char syndrome, a familial form of patent ductus arteriosus. Nat. Genet. 25, 42–46. doi: 10.1038/75578
Silengo, M. C., Biagioli, M., Guala, A., Lopez-Bell, G., and Lala, R. (1990). Heart-hand syndrome II. A report of Tabatznik syndrome with new findings. Clin. Genet. 38, 105–113. doi: 10.1111/j.1399-0004.1990.tb03556.x
Šinkovec, M., Petroviè, D., Volk, M., and Peterlin, B. (2005). Familial progressive sinoatrial and atrioventricular conduction disease of adult onset with sudden death, dilated cardiomyopathy, and brachydactyly. A new type of heart-hand sydrome?. Clin. Genet. 68, 155–160. doi: 10.1111/j.1399-0004.2005.00476.x
Tanteles, G. A., Nicolaou, N., Syrimis, A., Metaxa, R., Nicolaou, M., Christophidou-Anastasiadou, V., et al. (2017). Novel TBX3 mutation in a family of Cypriot ancestry with ulnar-mammary syndrome. Clin. Dysmorphol. 26, 61–65. doi: 10.1097/MCD.0000000000000170
Titeux, M., Brocheriou, V., Xue, Z., Gao, J., Pellissier, J. F., Guicheney, P., et al. (2001). Human synemin gene generates splice variants encoding two distinct intermediate filament proteins. Eur. J. Biochem. 268, 6435–6448. doi: 10.1046/j.0014-2956.2001.02594.x
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Washkowitz, A. J., Gavrilov, S., Begum, S., and Papaioannou, V. E. (2012). Diverse functional networks of Tbx3 in development and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 273–283. doi: 10.1002/wsbm.1162
Wollnik, B., Kayserili, H., Uyguner, O., Tukel, T., and Yuksel-Apak, M. (2002). Haploinsufficiency of TBX3 causes ulnar-mammary syndrome in a large Turkish family. Ann. Genet. 45, 213–217. doi: 10.1016/S0003-3995(02)01144-9
Keywords: heart–hand syndromes, ulnar-mammary syndrome, TBX3, SYNM, intermediate filaments, ventricular tachycardia
Citation: Zlotina A, Kiselev A, Sergushichev A, Parmon E and Kostareva A (2018) Rare Case of Ulnar-Mammary-Like Syndrome With Left Ventricular Tachycardia and Lack of TBX3 Mutation. Front. Genet. 9:209. doi: 10.3389/fgene.2018.00209
Received: 16 April 2018; Accepted: 25 May 2018;
Published: 15 June 2018.
Edited by:
Jumana Y. Al-Aama, King Abdulaziz University, Saudi ArabiaReviewed by:
Natalia Dmitrieva, National Heart, Lung, and Blood Institute (NHLBI), United StatesChristopher Huang, University of Cambridge, United Kingdom
Copyright © 2018 Zlotina, Kiselev, Sergushichev, Parmon and Kostareva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Zlotina, YW5uYS16bG90aW5hQHlhbmRleC5ydQ== Anna Kostareva, YWtvc3RhcmV2YUBob3RtYWlsLmNvbQ==