Abstract
Mutations in the NPHS1, NPHS2, LAMB2, and the WT1 genes are responsible for causing nephrotic syndrome (NS) in two third of the early onset cases. This study was carried out to assess the frequencies of mutations in these genes in a cohort of pediatric NS patients. A total of 64 pediatric familial or sporadic SRNS cases were recruited. Among these, 74% had a disease onset of up to 3 years of age. We found one homozygous frameshift mutation in the NPHS1 gene in one CNS case and two homozygous mutations in the NPHS2 gene. Six mutations in four cases in the LAMB2 gene were also identified. No mutation was detected in the WT1 gene in isolated SRNS cases. LAMB2 gene missense mutations were segregating in NS cases with no extra-renal abnormalities. Analysis of the population genomic data (1000 genome and gnomAD databases) for the prevalence estimation revealed that NS is more prevalent than previously determined from clinical cohorts especially in Asian population compared with overall world populations (prevalence worldwide was 1in 189036 and in South-Asian was 1in 56689). Our results reiterated a low prevalence of mutations in the NPHS1, NPHS2, LAMB2, and WT1 genes in the studied population from Pakistan as compared to some European population that showed a high prevalence of mutations in these genes. This is a comprehensive screening of the genes causing early onset NS in sporadic and familial NS cases suggesting a more systematic and robust approach for mutation identification in all the 45 disease-causing genes in NS in our population is required.
Introduction
Nephrotic syndrome (NS) is a common kidney disorder in children characterized by proteinuria, hematuria, hyperalbuminemia, and hypercholesterolemia. Pediatric NS is a heterogeneous disease that manifests early in life and termed as congenital (CNS), infantile, and childhood-onset NS according to the age of onset. A majority shows favorable response to the steroid treatment while about 20–30% of the children establish steroid resistant NS (SRNS) and rapidly progress to renal failure. Patients with inherited NS show poor renal survival but have a low disease-recurrence rate after kidney transplantation (Conlon et al., 1999).
Immunosuppression therapy has shown to be effective in 8–10% of the genetic cases (Bierzynska et al., 2015). Mutations in around 45 genes have been associated with familial and sporadic NS (Bierzynska et al., 2015). Several approaches are established for preferential screening of the genes based on the age of onset, inheritance patterns and histopathology. In a large study with isolated sporadic NS occurring within the first year of life, two third of the cases were due to the mutations in the NPHS1, NPHS2, WT1, and LAMB2 genes (Hinkes et al., 2007). Mutations in the NPHS1 and NPHS2 genes together share a large proportion of mutations that cause NS in children. The other two genes, WT1 and LAMB2 have also been associated with the syndromic or complex forms (Löwik et al., 2009; Zenker et al., 2009).
NS is a major glomerular disease in Pakistani children accounted for 50–74% of all the cases presented with glomerular diseases in major kidney centers in the country (Ali et al., 2011; Imtiaz et al., 2016). Among these 30% of the cases develop steroid resistance (Mubarak et al., 2009) while 10–20% of the NS cases show steroid resistance according to published studies (Ruf et al., 2004; Weber et al., 2004). In a previous study, we observed low frequencies of mutations in the NPHS1 and NPHS2 genes in pediatric NS (Abid et al., 2012). Therefore, it was essential to perform a systematic screening of other genes causing NS in children. In view of the observations, presented by Hinkes et al. (2007), the current study was designed to ascertain the prevalence of mutations in the LAMB2, WT1, NPHS1, and the NPHS2 genes to further explore the spectrum of disease-genes in Pakistani children with early onset NS.
Furthermore, we used publicly available whole genome/exome data to calculate population based estimates of the prevalence of NS worldwide and in our population. This study provides insight into the NS phenotypes and the prevalence and significance of particular alleles especially that are prevalent in our population.
Materials and methods
Patient recruitment and data collection
A total of 64 NS patients (ranging from congenital and childhood-onset) were selected from a cohort recruited from the pediatric nephrology department of the Sindh Institute of Urology and Transplantation Karachi. Among them 14 patients were of early onset NS (congenital to 3 years of age). The research protocol was approved by the Institutional Review Board and conformed with the tenets of the Declaration of Helsinki. Written informed consent was obtained from the parents of all the subjects. Patients with CNS, infantile and childhood onset NS including familial and sporadic cases that are younger than 16 years of age, resistant to standard steroid therapy were selected in this study.
NS was diagnosed by the presence of edema, urinary protein excretion equal to or greater than 40 mg/m2/h and serum albumin below 2.5 g/l. Renal failure was designated when estimated glomerular filtration rate (eGFR) was <90 ml/min by the Schwartz formula (Schwartz and Work, 2009). All the patients received standard steroid therapy on initial presentation. The clinical response to steroid therapy was classified as described as: (1) steroid sensitive; i.e., complete remission of proteinuria during the steroid therapy persisting for at least 12 weeks after therapy; (2) steroid dependent; i.e., remission of proteinuria during therapy but recurrence when the dosage was reduced below a critical level or relapse of proteinuria within the first 3 months after the end of therapy and (3) resistant; i.e., no remission of proteinuria during 4 consecutive weeks of daily steroid therapy.
Mutation analysis
Blood samples were collected in ACD vacutainer tubes. Genomic DNA was extracted using the standard phenol-chloroform extraction procedure. Mutation analysis was performed by direct DNA sequencing of 29 exons of the NPHS1 gene, 8 exons of the NPHS2 gene, 11 exons of the WT1 gene and 33 exons of the LAMB2 gene. Genomic sequences of the genes were obtained from the Ensembl genome browser (Ensembl ID's: ENSG00000161270 for the NPHS1, ENSG00000116218 for the NPHS2, ENSG00000184937 for the WT1 and ENSG00000172037 for the LAMB2 genes respectively) and exon-specific intronic primers were designed in the forward and reverse directions and were obtained commercially (Eurofins MWG Operon, Germany). Each exon was individually amplified by PCR in a 25 μl reaction volume using 1 μg of genomic DNA under standard PCR conditions. Amplified PCR products were purified using the PCR clean-up kit (Promega Wizard®, Promega Corporation, Madison, WI, USA). The sequencing reaction was performed using the BigDye terminator cycle sequencing kit, V3.1 (Applied Biosystems®, California, USA). Sequencing products were purified using the Centri-Sep spin columns (Princeton Separation®) and were analyzed on an automated DNA analyzer (ABI, 3100). Each mutation was confirmed by repeat sequencing in both the forward and reverse orientations.
Bioinformatics analysis
Each identified variant was assessed for the clinical interpretation of pathogenicity by using InterVar (http://wintervar.wglab.org; Li and Wang, 2017) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar; Landrum et al., 2016). To assess the damaging effects of missense mutations not previously reported in the Human Gene Mutation Database (HGMD public; http://www.hgmd.cf.ac.uk accessed 30-09-2016), in silico bioinformatic tools like PolyPhen-2 (v2; http://genetics.bwh.harvard.edu/pph2/index.shtml), SIFT (http://sift.jcvi.org/), and Condel (http://bg.upf.edu/fannsdb/; González-Pérez and López-Bigas, 2011) were employed. Splice-site mutations were predicted by NetGene 2 (http://www.cbs.dtu.dk/services/NetGene2/) server. A multiple sequence alignment (MSA) across different species was constructed using the CLUSTAL OMEGA tool (http://www.ebi.ac.uk/Tools/msa/clustalo/) which utilized BLOSUM algorithm for this purpose (Goujon et al., 2010). MSA was then used to calculate the evolutionary conservation scores of amino acid positions where mutations were found (Landau et al., 2005; Ashkenazy and Kliger, 2010) and also to construct the phylogenetic tree with MEGA 5 program (Tamura et al., 2011).
In an attempt to investigate the potential impact of mutations, an in-silico prediction on wild type and mutant protein was performed using the online protein modeling I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER) and were further analyzed for comparison for respective mutations using PyMOL Molecular Graphics System (Version 1.7.4.4 2010 Schrodinger, LLC; http://www.pymol.org). In the wild type protein model, the identified missense mutations were made by using the Discovery Studio Version 3.1. The amino acid change is visualized in the rotamer form with side chain orientations incorporated from Dunbrack backbone dependent rotamer library with maximum probabilities. For c.2673dupCA mutation in NPHS1 gene, translation analysis was done by using Expasy translate tool.
The data for the estimation of prevalence and carrier frequencies (CF) were downloaded from the 1000 genome browser (http://1000genomes.org/ accessed 05-10-2017; The 1000 Genome Project Consortium et al., 2015) by using the Allele Frequency Calculator tool (http://grch37.ensembl.org/Multi/Tools/AlleleFrequency accessed 05-10-2017) and genome Aggregation database (gnomAD http://gnomad.broadinstitute.org/ accessed 07-10-2017; Lek et al., 2016) in VCF format. Known mutations within the NPHS1, NPHS2, and the LAMB2 genes that were listed in the HGMD/ClinVar were annotated. All other non-synonymous/indel variants in the coding regions with MAF <0.01 were selected and were scored for potential pathogenicity using Condel tool. Using the reference allele count and alternate allele count, prevalence (1/q2) and carrier frequency (1/2pq) were calculated according to the Hardy-Weinberg Equilibrium for each gene variants by using the sum of all alternate NPHS1, NPHS2, and LAMB2 alleles (also for known plus unknown scored as pathogenic according to Condel). Calculations for combined three genes frequencies were done by adding the p2 values of each gene's frequencies.
Results
A total of 64 patients including early-onset (from congenital to 3 years of age) and childhood-onset NS (disease onset after 3 years of age) were screened for mutations in the NPHS1, NPHS2, LAMB2, and WT1genes. In the first part of the study, 14 early-onset cases were screened for mutation in the NPHS1 and the NPHS2 genes only. Three patients aged 0–3 years in this group were identified to have homozygous mutations in the NPHS1 and the NPHS2 genes. In the second part of the study, a total of 61 patients were included. Among these, 11 patients were from first part and 50 patients were selected from a previous cohort that was negative for the NPHS1 and the NPHS2 gene mutations (Abid et al., 2012). All these (n = 61) were then screened for mutations in the LAMB2 and the WT1 genes.
Clinical characteristics of the patients are given in Table 1. Clinical data were obtained for all the cases. Early-onset cases include congenital and infantile onset cases as well as children with disease onset of 2–3 years of age. Family history was positive in 20% of the cases, whereas, 80% cases were sporadic NS. Histopathological findings were available in majority of the cases that predominantly showed FSGS and MCD. Renal failure was established in 8 patients, of these three expired.
Table 1
| Total number of children | 64 |
| Age of onset | Since birth−16 years |
| Females (%) | 25 (39.0%) |
| Males (%) | 39 (60.9%) |
| Male to Female ratio | 1.52:1 |
| CLASSIFICATION OF NS | |
| Early-onset NS (CNS/infantile/2–3years) (%) | 47 (73.5%) |
| Childhood NS (%) | 17 (26.5%) |
| RENAL BIOPSY FINDINGS | |
| FSGSa | 16 |
| MCDb | 16 |
| IgMNc | 4 |
| MesPGNd | 4 |
| MGNe | 2 |
| FAMILY HISTORY | |
| Positive (%) | 13 (20.3%) |
| Negative (%) | 51 (79.7%) |
| OUTCOME | |
| ESRDf/CRFg | 5 (9.43%) |
| Lost to follow up | 21 (18.8%) |
| Expired | 3 (5.66%) |
Clinical characteristics of children with Nephrotic Syndrome.
Focal segmental glomerular sclerosis
minimal change disease
IgM nephropathy
mesengial proliferative glomerulonephritis
membranous glomerulonephritis
end stage renal disease
chronic renal failure.
Mutation screening
We identified one homozygous mutation in the NPHS1 gene in one CNS case, two homozygous mutations in the NPHS2 gene in two early onset cases. Homozygous and compound heterozygous mutations were detected in four childhood onset cases in the LAMB2 gene (Table 2, Figure 1). No mutation was discovered the WT1 (entire coding sequence) gene screening.
Table 2
| Patient | Sex | Family history | Age Onset (yrs) | Biopsy | cDNA | Protein | Response to therapy | Renal outcome/Time follow up | dbSNP/HGMD ID/ClinVar In silico prediction |
|---|---|---|---|---|---|---|---|---|---|
| LAMB2 | |||||||||
| NS001 | M | No | 10 | IgMNa | c.1678T/A c.3071C/T | p.F560I p.P1024L | SRNSb | Expired with ESRDc | Not reported/not reported/pathogenic rs368506627/not reported/uncertain significance |
| NS032 | F | yes | 12 | d FSGS | c.2974A/G c.3443G/A | p.I992V p.R1148H | SRNS | partial remission with CyAe | rs529614319/not reported/pathogenic rs138774635/not reported/benign |
| NS050 | M | No | 13.5 | FSGS | c.3144G/T | p.Q1048H | SRNS | No response | Not reported/not reported/likely pathogenic |
| NS113 | M | No | 1 | FSGS | c.4667C/T | p.A1556V | SRNS | Lost to follow up | rs774045808/CM066905/pathogenic |
| NPHS1 | |||||||||
| NS301 | M | yes | CNS | – | c.2673dupCA | SRNS | No response | Not reported/not reported/pathogenic | |
| NPHS2 | |||||||||
| NS313 | M | No | 2 | FSGS | c.795-2A/G Splice-site | – | SRNS | Lost to follow up | Not reported/not reported/pathogenic splice-site aborted |
| NS304 | M | No | 3 | MesPGN | c.708_713delAGAGAG | – | SRNS | ESRD | Not reported/not reported/pathogenic |
| NPHS2 RISK ALLELE | |||||||||
| NS228 | M | No | 2 | MesPGNf | c.755G/A | p.R229Q | SRNS | No response | rs61747728/CM023107/likely pathogenic and risk factor |
| NS245 | F | No | CNS | FSGS | c.755G/A | p.R229Q | SRNS | Partial remission | rs61747728/CM023107/likely pathogenic and risk factor |
List of homozygous/compound heterozygous mutations identified.
IgM nephropathy
Steroid resistant nephrotic syndrome
end stage renal disease
focal segmental glomerular sclerosis
Cyclosporine A
mesengial proliferative glomerulonephritis.
Figure 1
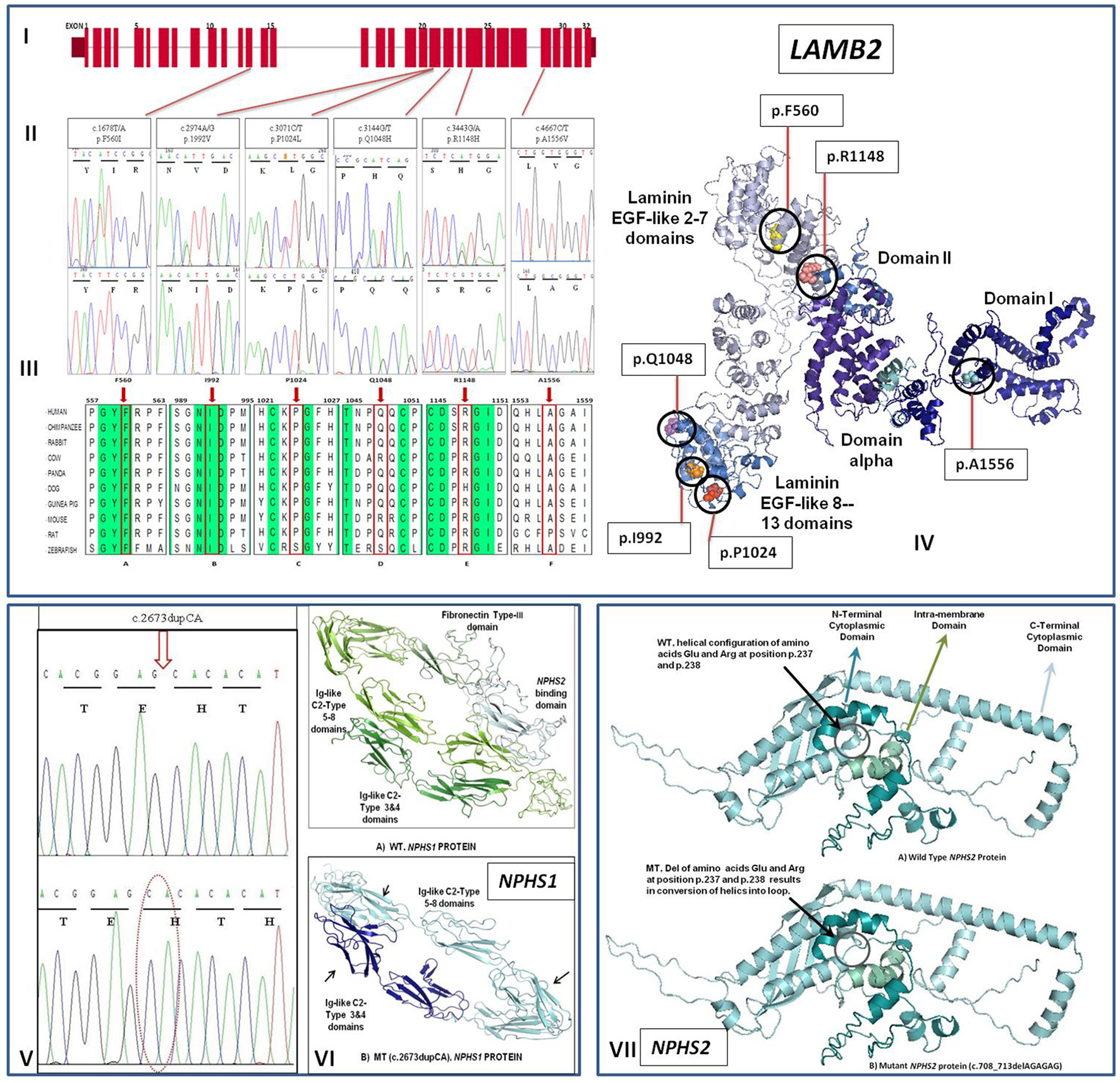
(I) Exon structure of human LAMB2 gene contains 32 exons. (II) Sequence electropherogram showing mutated (upper panel) and normal (lower panel) sequences. (III) Phylogenetic tree and MSA for LAMB2 gene showing sequence alignment of particular amino acid and its conservation in other orthologs. Conserved sequences are highlighted. (a) and (b) shows the highly conserved amino acids indicated by red frame and arrow at position 560 and 992, respectively while (c–f) shows that amino acids at position 1,024, 1,048, 1,148, and 1,556 are mostly conserved in other species. (IV) An illustration of Molecular distribution of LAMB2 missense mutations in relation to the functional domains of the protein. (V) Representative electropherogram (lower panel) of mutation (c.2673dupCA) in the NPHS1 gene compared with wild type sequence (upper panel). (VI) Conformational changes in the overall configuration of the mutant nephrin protein marked with arrows. (VII) Conformational changes in the overall configration of the mutant podocin protein (mutant positions are encircled). WT wild type, MT mutant protein.
Analysis of the NPHS1 gene revealed only one mutation (c.2673dupCA) in one congenital case that inherited one mutant copy of the gene from each parent (Table 2, Figure 1V). Translation analysis for homozygous c.2673dupCA mutation demonstrated that CA duplication at position 2673 in exon 20 results in shifting of the reading frame from position 893 with the introduction of premature stop codon at position 904 leads to protein truncation. Comparision of crystal diagram with wild type protein model (Figure 1VI) suggested the loss of both fibronectin type III and NPHS2 binding domains in the mutated protein which include some extracellular region, transmembrane, and cytosolic region. These domains are responsible for the interaction with surrounding molecules to maintain the integrity of Slit diaphgram. Possibly loss of two these interacting domains may result in over all disruption of filtration barrier leading to proteinuria and steriod resistant nephrotic syndrome. A novel splice-site mutation (c.795-2A/G) which abolishes the splice-site in the NPHS2 gene was identified in one child aged 2 years of age. The patient did not respond to standard steroid therapy and lost to follow up after 2 years of treatment. A homozygous deletion (c.708_713delAGAGAG) in the NPHS2 gene was present in another early-onset NS patient. Deletion of two amino acids (R237, E238) as a result of this mutation changed the helical configuration into loop structure in the NPHS2 protein structure (Figure 1VII). Two early onset cases also had p.R229Q mutation which is attributed as a risk factor for NS.
We did not find any mutation in the WT1 gene in isolated early onset NS cases. The LAMB2 gene screening revealed six homozygous or compound heterozygous mutations in four infantile and late-onset sporadic NS cases (Table 2, Figures 1I–IV). All LAMB2 gene mutations were missense mutations. Among these three mutations were novel mutations (Table 2). All mutations were conserved in nature and were predicted to be pathogenic by In-Silico analysis (Figures 1I–IV). Only one mutation was identified in an infantile case. None of the CNS case showed mutation in the LAMB2 gene. Pathogenicity, clinical correlation of mutations and responses to therapy of respective patients are given in Table 2. Ten infantile and early onset NS cases also exhibited single variants in the NPHS1 and the LAMB2 genes (Table 3). None of the above mutations were found in 50 healthy controls. Minor allele frequencies (MAF) of these variants when reported was > 0.01 with no homozygous detected in both 1000-genome and Genome Aggregation Database (gnomAD).
Table 3
| Patient | Sex | Family history | Age onset (yrs) | Biopsy | Nucleotide change | Amino acid change | Response to therapy | Renal Outcome/Time to follow up | dbSNP/HGMD ID/ClinVar In-silico prediction |
|---|---|---|---|---|---|---|---|---|---|
| LAMB2 GENE | |||||||||
| NS003 | F | No | 2 | MCDb | c.3935G/A | p.R1312Q | SRNS | Maintained on ACEI 5 years follow up | rs759882519/not reported/likely pathogenic |
| NS008 | M | No | 5 | – | c.3152C/T | p.P1051L | SRNS | In remission | rs543606035/not reported/benign |
| NS094 | F | Yes | 2 | MCD | c.3787G/A | p.E1263L | SRNS | Switched to Tac due to CyA toxicity, partial remission | Not reported/not reported/likely pathogenic |
| NS118 | M | No | 3 | FSGSa | c.2974A/G | p.I992V | SRNS | In remission after CyA | rs529614319/not reported/pathogenic |
| NS125 | F | No | 3 | FSGS | c.2516T/A | p.L839H | SRNS | ESRDd | Not reported/not reported/pathogenic |
| NS134 | F | No | 5.5 | FSGS | c.1193C/T | p.T398I | SRNS | rs77500937/not reported/likely benign | |
| NS166 | F | No | 5 | FSGS | c.5027G/A | p.G1676E | SRNS | In remission after CyA | Not reported/not reported/pathogenic |
| NS144 | F | No | 1.4 | – | c.5108G/A | p.R1703H | SRNS | rs771531508/not reported/uncertain significance | |
| NS155 | No | 3 | – | c.4163G/A | p.R1388Q | SRNS | rs146522641/not reported/benign | ||
| NPHS1 GENE | |||||||||
| NS1401 | M | yes | 14 | FSGS | c.563A/T | p.N188I | SRNS | ESRD | rs145125791/CM020470/likely benign |
| NPHS2 GENE | |||||||||
| NS304 | M | no | 3 | MesPGNc | c.708_713delAGAGAG | p.L236LfsX147 | SRNS | ESRD | Not reported/not reported/pathogenic |
List of heterozygous mutations identified.
Focal segmental glomerular sclerosis
minimal change disease
mesengial proliferative glomerulonephritis
end stage renal disease.
Estimation of prevalence and carrier frequencies
We extracted NPHS1, NPHS2, and LAMB2 genes reference and alternate allele counts from two databases; 1000-Genome and Genome Aggregation Database (gnomAD). 1000-Genome browser was used to download the phase 3 data (all population as well as Punjabi from Pakistan; PJL). Data contain 2504 individuals (5008 chromosomes) from 26 populations, whereas, Punjabi population data contain 96 individuals (192 chromosomes) from Lahore, Pakistan. Similarly, all population and South Asian (SAS) allele frequency data was also extracted from the gnomAD database. gnomAD database contains genome/exome sequencing data of 123,136 individuals (all populations) and 15391 exomes data from South-Asian region (SAS, including Pakistanis). We extracted allele frequencies of those variants reported to be disease causing in various studies and are annotated in HGMD and ClinVar databases as known disease causing. In silico analyses was performed and we identified 56 known mutations as likely pathogenic. Among these, 18 variants were found in the NPHS1 gene, 12 in the NPHS2 gene and 26 variants were noted in the LAMB2 gene (Table 4). Additionally, we extracted all rare coding region variants that were non-synonymous, non-sense, frameshifting, and were not previously identified as disease causing and scored by using in-silico tools. A total of 23 such rare variants (9 in NPHS1, 10 in NPHS2, and 4 in the LAMB2 genes) were found in the variant data (Table 5) that were predicted to be deleterious by In-silico analysis.
Table 4
| Gene | GRCH37 position | NCBI dbSNP ID | Ref/Alt | Consequence | gnomADe | MAF (%) | gnomAD-SAS allele count | MAF (%) | 1K G allele count | MAF (%) | 1KG-PJL allele count | MAF (%) | SIFT | PPH2 | MA | FATHMM | CONDEL | CONDEL LABEL | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NPHS2 | 1:179520493 | rs571452152 | A/G | p.Tyr255His | 1/245830 | 0.0004 | 0/30780 | 0 | 1/5008 | 0.019 | – | – | 0.01 | 0.574 | 2.95 | −6.26 | 0.6718112 | D |
| 2 | NPHS2 | 1:179520354 | rs768932711 | G/A | p.Pro369Leu | 2/245844 | 0.0008 | 2/30778 | 0.006 | – | – | – | 0.15 | 0.001 | 0 | −6 | 0.5486173 | D | |
| 3 | NPHS2 | 1:179520418 | rs754243843 | T/G | p.Asn348His | 1/245938 | 0.0004 | 1/30782 | 0.003 | – | – | – | 0.04 | 0.45 | 1.46 | −6.3 | 0.621615 | D | |
| 4 | NPHS2 | 1:179520429 | rs751105124 | A/G | p.Phe344Ser | 2/245958 | 0.0008 | 2/30780 | 0.006 | – | – | – | 0 | 0.007 | 1.15 | −6.15 | 0.6076879 | D | |
| 5 | NPHS2 | 1:179521804 | rs770495227 | C/A | p.Arg269Ser | 1/245354 | 0.0004 | 1/30744 | 0.003 | – | – | – | 0.21 | 0.029 | 0.56 | −6.24 | 0.5838985 | D | |
| 6 | NPHS2 | 1:179526298 | rs771256385 | G/T | p.Ser201Tyr | 1/246000 | 0.0004 | 1/30782 | 0.003 | – | – | – | 0.02 | 0.044 | 0.8 | −6.4 | 0.6003373 | D | |
| 7 | NPHS2 | 1:179526308 | rs765185151 | C/G | p.Glu198Gln | 1/245982 | 0.0004 | 1/30778 | 0.003 | – | – | – | 0.35 | 0.47 | 0.75 | −3.54 | 0.5275746 | D | |
| 8 | NPHS2 | 1:179533863 | rs779163992 | T/G | p.Ile114Leu | 1/246156 | 0.0004 | 1/30782 | 0.003 | – | – | – | 0.26 | 0.025 | 1.21 | −6.25 | 0.6123593 | D | |
| 9 | NPHS2 | 1:179533901 | rs777738678 | C/G | p.Cys101Ser | 1/246108 | 0.0004 | 1/30782 | 0.003 | – | – | – | 0 | 0.131 | 2.52 | −5.92 | 0.6424912 | D | |
| 10 | NPHS2 | 1:179526338 | rs757665750 | C/A | p.Glu188Ter | 2/245860 | 0.0008 | 2/30780 | 0.006 | – | – | – | – | – | – | – | – | ||
| 11 | NPHS2 | 1:179530438 | rs778201387 | CT/C | p.Arg146GlufsTer35 | 1/246138 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | – | – | – | – | ||
| 12 | NPHS2 | 1:179533920 | rs759842258 | A/AT | p.Ser95IlefsTer8 | 1/246070 | 0.0004 | 1/30770 | 0.003 | – | – | – | – | – | – | – | – | ||
| 13 | LAMB2 | 3:49158675 | rs769627057 | T/C | p.Tyr1794Cys | 1/246080 | 0.0004 | 1/30780 | 0.003 | – | – | 0 | 0.998 | 2.69 | 0.53 | 0.57388975 | D | ||
| 14 | LAMB2 | 3:49159680 | rs779155013 | A/G | p.Leu1566Pro | 1/245622 | 0.0004 | 1/30772 | 0.003 | – | – | 0 | 0.963 | 2.59 | 0.92 | 0.53606262 | D | ||
| 15 | LAMB2 | 3:49161398 | rs760892618 | C/T | p.Cys1187Tyr | 1/244248 | 0.0004 | 1/30774 | 0.003 | – | – | 0 | 0.999 | 4.61 | 1.39 | 0.71407755 | D | ||
| 16 | LAMB2 | 3:49161433 | rs750381148 | C/A | p.Gln1175His | 3/244722 | 0.001 | 3/30773 | 0.009 | – | – | 0.04 | 0.999 | 2.08 | −0.03 | 0.52880462 | D | ||
| 17 | LAMB2 | 3:49161479 | rs559556131 | C/T | p.Gly1160Asp | 3/244552 | 0.001 | 3/30780 | 0.009 | – | – | 0 | 1 | 4.67 | −1.34 | 0.85034712 | D | ||
| 18 | LAMB2 | 3:49162278 | rs766539657 | T/G | p.Ser989Arg | 1/246238 | 0.0004 | 1/30782 | 0.003 | – | – | 0 | 0.241 | 2.88 | 0.06 | 0.61882555 | D | ||
| 19 | LAMB2 | 3:49162280 | rs754732425 | C/T | p.Cys988Tyr | 3/246234 | 0.001 | 2/30782 | 0.006 | – | – | 0 | 0.999 | 4.6 | −3.67 | 0.77695588 | D | ||
| 20 | LAMB2 | 3:49162299 | rs530751136 | G/A | p.Arg982Trp | 2/246208 | 0.0008 | 0/30782 | 0 | 1/5008 | 0.019 | 1/192 | 0.52 | 0.02 | 0.861 | 2.31 | 0.06 | 0.55138327 | D |
| 21 | LAMB2 | 3:49162302 | rs778680962 | C/T | p.Gly981Ser | 1/246222 | 0.0004 | 1/30782 | 0.003 | – | – | 0.06 | 1 | 2.14 | 0.16 | 0.5256399 | D | ||
| 22 | LAMB2 | 3:49162334 | rs763062098 | G/A | p.Pro970Leu | 1/246106 | 0.0004 | 1/30782 | 0.003 | – | – | 0.14 | 0.999 | 2.51 | −0.05 | 0.58191315 | D | ||
| 23 | LAMB2 | 3:49162504 | rs749808119 | G/A | p.Ala940Val | 2/243584 | 0.0008 | 1/30766 | 0.003 | – | – | 0.03 | 0.999 | 3.59 | −0.14 | 0.70961854 | D | ||
| 24 | LAMB2 | 3:49162716 | rs375392013 | C/T | p.Arg897His | 2/240634 | 0.0008 | 1/30782 | 0.003 | – | – | 0.21 | 0.999 | 2.16 | −0.02 | 0.53696553 | D | ||
| 25 | LAMB2 | 3:49162762 | rs751697643 | G/A | p.His882Tyr | 1/246028 | 0.0004 | 1/30780 | 0.003 | – | – | 0.02 | 0.664 | 3.89 | −0.15 | 0.74173607 | D | ||
| 26 | LAMB2 | 3:49162811 | rs771584138 | C/G | p.Gln865His | 1/245372 | 0.0004 | 1/30780 | 0.003 | – | – | 0.12 | 0.894 | 2.16 | 0 | 0.5357451 | D | ||
| 27 | LAMB2 | 3:49163579 | rs764955129 | G/A | p.Pro722Leu | 1/244568 | 0.0004 | 1/30770 | 0.003 | – | – | 0.02 | 0.248 | 2.69 | 0.7 | 0.56367037 | D | ||
| 28 | LAMB2 | 3:49166738 | rs751844883 | C/T | p.Ser513Asn | 2/245150 | 0.0008 | 2/30758 | 0.006 | – | – | 0.05 | 0.911 | 2.26 | 0.07 | 0.54547393 | D | ||
| 29 | LAMB2 | 3:49168184 | rs747053168 | T/C | p.His342Arg | 1/246178 | 0.0004 | 1/30782 | 0.003 | – | – | 0.04 | 0.63 | 2.19 | 0.03 | 0.53820621 | D | ||
| 30 | LAMB2 | 3:49168388 | rs775204900 | C/T | p.Gly304Ser | 5/246088 | 0.002 | 1/30782 | 0.003 | – | – | 0.04 | 1 | 2.92 | −0.2 | 0.63898086 | D | ||
| 31 | LAMB2 | 3:49168417 | rs536441871 | G/T | p.Pro294His | 1/245872 | 0.0004 | 1/30776 | 0.003 | 1/5008 | 0.09 | 0/192 | 0 | 0.05 | 0.982 | 3.5 | 0.06 | 0.68772865 | D |
| 32 | LAMB2 | 3:49168439 | rs780152505 | C/T | p.Gly287Arg | 3/276512 | 0.001 | 1/30772 | 0.003 | – | – | 0 | 1 | 3.78 | −0.3 | 0.738678 | D | ||
| 33 | LAMB2 | 3:49168498 | rs772368832 | C/T | p.Arg267Gln | 7/273782 | 0.0025 | 1/30718 | 0.003 | – | – | 0.52 | 0.591 | 0.98 | −0.96 | 0.52262762 | D | ||
| 34 | LAMB2 | 3:49169033 | rs147986864 | G/A | p.Arg195Trp | 9/275688 | 0.003 | 1/30768 | 0.003 | – | – | 0 | 0.979 | 2.55 | −1.13 | 0.63595261 | D | ||
| 35 | LAMB2 | 3:49169603 | rs758877377 | G/C | p.Ile135Met | 2/246268 | 0.0008 | 2/30782 | 0.006 | – | – | 0.01 | 0.961 | 3.19 | −1.3 | 0.70191895 | D | ||
| 36 | LAMB2 | 3:49169732 | rs752886136 | C/T | p.Arg119Gln | 1/246262 | 0.0004 | 1/30782 | 0.003 | – | – | 0.59 | 0.767 | 3.01 | −0.96 | 0.69118365 | D | ||
| 37 | LAMB2 | 3:49169807 | rs767544919 | C/T | p.Arg94Gln | 3/246272 | 0.001 | 2/30782 | 0.006 | – | – | 0.18 | 0.999 | 2.06 | −0.96 | 0.58205729 | D | ||
| 38 | LAMB2 | 3:49169958 | rs776819202 | C/T | p.Gly72Asp | 1/245660 | 0.0004 | 1/30782 | 0.003 | – | – | 0.11 | 0.306 | 3.06 | −0.96 | 0.69687401 | D | ||
| 39 | NPHS1 | 19:36321826 | rs777436326 | G/C | p.Pro1172Ala | 1/246246 | 0.0004 | 0/30782 | 0 | – | – | – | – | 0.32 | 0.563 | 0.9 | −2.97 | 0.52476152 | D |
| 40 | NPHS1 | 19:36330157 | rs757169332 | G/A | p.Leu1031Phe | 1/246246 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | 0.01 | 0.34 | 1.59 | −0.85 | 0.55029744 | D |
| 41 | NPHS1 | 19:36330288 | rs746380730 | T/C | p.Tyr987Cys | 1/246264 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | 0.04 | 0.982 | 2.76 | 0.43 | 0.58656495 | D |
| 42 | NPHS1 | 19:36333170 | rs748705495 | A/C | p.Val840Gly | 1/244238 | 0.0004 | 1/30756 | 0.003 | – | – | – | – | 0 | 0.975 | 1.55 | −0.91 | 0.54873772 | D |
| 43 | NPHS1 | 19:36334444 | rs750854389 | C/A | p.Gly755Val | 1/246270 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | 0.07 | 0.286 | 1.94 | −1.27 | 0.54028481 | D |
| 44 | NPHS1 | 19:36336347 | rs762869410 | C/T | p.Gly618Asp | 1/245808 | 0.0004 | 1/30778 | 0.003 | – | – | – | – | 0.04 | 0.599 | 2.22 | −1.08 | 0.59841388 | D |
| 45 | NPHS1 | 19:36336356 | rs751046394 | C/A | p.Arg615Leu | 2/245746 | 0.008 | 2/30782 | 0.006 | – | – | – | – | 0.19 | 0.192 | 1.25 | −0.99 | 0.53620728 | D |
| 46 | NPHS1 | 19:36336408 | rs758946523 | G/A | p.Pro598Ser | 1/234914 | 0.0004 | 1/30781 | 0.003 | – | – | – | – | 0.64 | 0.551 | 1.12 | −1.02 | 0.53008798 | D |
| 47 | NPHS1 | 19:36336592 | rs764351102 | G/A | p.Ser579Tyr | 1/245252 | 0.0004 | 1/30707 | 0.003 | – | – | – | – | 0.09 | 0.798 | 2.34 | −1.2 | 0.60973898 | D |
| 48 | NPHS1 | 19:36339005 | rs749319334 | G/A | p.Arg460Trp | 6/276520 | 0.002 | 1/30766 | 0.003 | – | – | – | – | 0.19 | 0.011 | 1.74 | −1.04 | 0.54867583 | D |
| 49 | NPHS1 | 19:36339010 | rs768870360 | C/G | p.Gly458Ala | 1/245546 | 0.0004 | 1/30764 | 0.003 | – | – | – | – | 0 | 0.999 | 2.11 | −2.79 | 0.56791611 | D |
| 50 | NPHS1 | 19:36339250 | rs199735886 | C/T | p.Arg407Gln | 16/246214 | 0.006 | 2/30782 | 0.006 | – | – | – | – | 0.23 | 0.271 | 1.36 | −0.96 | 0.54186854 | D |
| 51 | NPHS1 | 19:36339691 | rs746481345 | G/A | p.Pro340Ser | 1/246208 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | 0.1 | 1 | 1.94 | −4.43 | 0.57397387 | D |
| 52 | NPHS1 | 19:36339983 | rs761786407 | C/T | p.Val303Met | 1/240840 | 0.0004 | 1/30778 | 0.003 | – | – | – | – | 0.08 | 0.571 | 1 | −0.91 | 0.52460566 | D |
| 53 | NPHS1 | 19:36340149 | rs752311438 | G/T | p.Gln277Lys | 1/245308 | 0.0004 | 1/30780 | 0.003 | – | – | – | – | 1 | 0.88 | 1.05 | −1.04 | 0.52596441 | D |
| 54 | NPHS1 | 19:36340525 | rs779764581 | C/A | p.Gln213His | 2/246176 | 0.0008 | 2/30782 | 0.006 | – | – | – | – | 0.05 | 0.571 | 1.1 | −2.08 | 0.52665949 | D |
| 55 | NPHS1 | 19:36341334 | rs779291027 | T/C | p.Ile180Met | 3/246262 | 0.001 | 3/30782 | 0.009 | – | – | – | – | 0.15 | 0.609 | 1.1 | −0.9 | 0.52979925 | D |
| 56 | NPHS1 | 19:36342391 | rs761152159 | G/C | p.Pro81Arg | 1/239854 | 0.0004 | 1/30736 | 0.003 | – | – | – | – | 0.01 | 0.996 | 2.47 | 0.88 | 0.52561198 | D |
List of known mutant alleles in the NPHS1, NPHS2, and the LAMB2 genes found in the gnomADe and 1000Genome data.
D, deleterious
Table 5
| Gene | Position | dbSNP ID | Ref/Alt allele | Protein consequence | gnomADe | MAF (%) | gnomAD-SAS allele count | MAF (%) | 1K Allele count | MAF (%) | 1KG-PJL allele count | MAF (%) | SIFT | PPH2 | MA | FATHMM | CONDEL | CONDEL LABEL | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | LAMB2 | 3:49158944 | rs760355583 | G/A | p.Gln1728Ter | 1/246228 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | – | – | – | – | – | |
| 2 | LAMB2 | 3:49166461 | rs759042337 | G/A | p.Arg575Ter | 7/241974 | 0.0028 | 4/30762 | 0.013 | – | – | – | – | – | – | – | – | – | |
| 3 | LAMB2 | 3:49168473 | rs769460144 | A/A | p.Tyr275Ter | 1/244402 | 0.0004 | 1/30750 | 0.003 | – | – | – | – | – | – | – | – | – | |
| 4 | LAMB2 | 3:49167271 | rs780041521 | C/T | c.1405+1G>A | 4/244056 | 0.0016 | 1/30746 | 0.003 | – | – | – | – | – | – | – | – | – | |
| 5 | NPHS2 | 1:179520496 | rs763818901 | G/A | p.Arg322Ter | 1/245804 | 0.0004 | 1/30782 | 0.003 | – | – | – | – | – | – | – | – | – | – |
| 6 | NPHS2 | 1:179526186 | rs748812981 | C/A | p.Arg238Ser | 4/276486 | 0.0016 | 1/30736 | 0.003 | – | – | – | – | 0 | 0.998 | 3.545 | −4.37 | 0.68850199 | D |
| 7 | NPHS2 | 1:179526191 | rs146906190 | C/G | p.Glu237Gln | 205/276570 | 0.074 | 4/30748 | 0.013 | 1/5008 | 0.019 | 0/192 | 0 | 0 | 0.998 | 3.545 | −4.37 | 0.68850199 | D |
| 8 | NPHS2 | 1:179526362 | rs74315347 | C/T | p.Val180Met | 3/245560 | 0.0012 | 1/30778 | 0.003 | – | – | – | – | 0.02 | 0.577 | 1.005 | −6.26 | 0.605115534 | D |
| 9 | NPHS2 | 1:179530462 | rs74315342 | C/T | p.Arg138Gln | 159/277072 | 0.057 | 2/30782 | 0.006 | – | – | – | – | 0.02 | 0.999 | 2.28 | −6.29 | 0.641951573 | D |
| 10 | NPHS2 | 1:179533825 | rs771320565 | CT/C | p.Lys126ArgfsTer9 | 1/246163 | 0.0004 | 1/30781 | 0.003 | – | – | – | – | – | – | – | – | – | – |
| 11 | NPHS2 | 1:179520587 | rs776016942 | C/T | c.874-1G>A | 1/245424 | 0.0004 | 1/30778 | 0.003 | – | – | – | – | – | – | – | – | – | – |
| 12 | NPHS2 | 1:179520493 | rs571452152 | A/G | p.Tyr323His | 1/245830 | 0.0004 | 0/30780 | 0 | 1/5008 | 0.019 | 1/192 | 0.5 | 0.01 | 0.574 | 2.945 | −6.26 | 0.671811197 | D |
| 13 | NPHS2 | 1:179526301 | rs542500942 | G/A | p.Ala200Val | 5/246006 | 0.002 | 4/30780 | 0.013 | 1/5008 | 0.019 | 1/192 | 0.5 | 0.03 | 0.449 | 1.47 | −3.69 | 0.554213306 | D |
| 14 | NPHS2 | 1:179544873 | rs545872093 | G/C | p.Pro43Ala | – | – | – | 17/5008 | 0.39 | 1/192 | 0.5 | 0.86 | 0 | −0.205 | −5.84 | 0.529596199 | D | |
| 15 | NPHS1 | 19:36330221 | rs762184939 | G/C | p.Tyr1009Ter | 2/246268 | 0.0008 | 2/30780 | 0.006 | – | – | – | – | – | – | – | – | – | – |
| 16 | NPHS1 | 19:36339690 | rs386833861 | G/T | p.Pro340His | 3/246204 | 0.0012 | 3/30782 | 0.009 | – | – | – | 0.04 | 1 | 1.935 | −4.45 | 0.57449583 | D | |
| 17 | NPHS1 | 19:36339995 | rs753476209 | G/A | p.Arg299Cys | 2/240156 | 0.0008 | 1/30768 | 0.003 | – | – | – | 0.02 | 0.912 | 1.77 | −1 | 0.549329816 | D | |
| 18 | NPHS1 | 19:36340176 | rs749341977 | G/A | p.Arg268Ter | 6/275332 | 0.002 | 0/30772 | 0 | – | – | – | – | – | – | – | – | – | |
| 19 | NPHS1 | 19:36341889 | rs386833945 | G/A | p.Pro167Leu | 1/246044 | 0.0004 | 1/30780 | 0.003 | – | – | – | 1 | 1 | 2.005 | −3.06 | 0.561752557 | D | |
| 20 | NPHS1 | 19:36339610 | rs386833865 | G/A | p.Arg367Cys | 10/246176 | 0.004 | 4/30782 | 0.013 | – | – | – | – | 0.01 | 0.964 | 1.845 | −1.07 | 0.547062581 | D |
| 21 | NPHS1 | 19:36342241 | rs386833934 | G/A | p.Ala107Val | 3/244336 | 0.0012 | 2/30718 | 0.006 | – | – | – | – | 0.01 | 0.98 | 3.22 | 0.59 | 0.631325147 | D |
| 22 | NPHS1 | 19:36330189 | rs749003854 | C/A | p.Gly1020Val | 6/246268 | 0.002 | 6/30782 | 0.019 | – | – | – | – | 0 | 1 | 3.755 | −0.89 | 0.773421163 | D |
| 23 | NPHS1 | 19:36321958 | rs267606919 | G/A | p.Arg1160Ter | 25/246216 | 0.01 | 12/30782 | 0.038 | – | – | – | – | – | – | – | – | – | – |
List of variants alleles in the NPHS1, NPHS2, and LAMB2 genes (scored as likely pathogenic by using in silico tools) found in in gnomADe and 1000Genome data.
An overall NS carrier frequency (CF) of 1:272 (all populations) and 1:278 (SAS population) with a prevalence of 1:295369 (all populations) and 1:308642 (SAS population) was calculated from the known mutant alleles (Table 6) by using the Hardy-Weinberg equation. Inclusion of rare predicted deleterious (but not reported in clinical cohorts; Table 5) alleles to the known mutant alleles list increased the CF to 1:218 (all populations), 1:120 (SAS population) and prevalence to 1:189036 (all populations), and 1:56689 (SAS population; Table 7).
Table 6
| gnomAD | gnomAD-SAS | 1KG | 1KG PJL | gnomAD + 1KG | gnomAD-SAS + 1KG PJL | |
|---|---|---|---|---|---|---|
| NPHS2 | ||||||
| Allele Count | 380/256102 | 15/30772 | 20in5008 | 3/192 | 400/261110 | 18/30964 |
| Mutant Allele Feq (%) | 0.148 | 0.048 | 0.399 | 1.560 | 0.153 | 0.058 |
| Prevalence (1 in) | 456538 | 4340278 | 62814 | 4109 | 427186 | 2972652 |
| Carrier Frq (1 in) | 338 | 1042 | 126 | 33 | 327 | 863 |
| LAMB2 | ||||||
| Allele Count | 13/244165 | 7/30760 | 0/5008 | 192 | 13/249173 | 7in30952 |
| Mutant Allele Feq (%) | 0.005 | 0.022 | 0.000 | 0.000 | 0.005 | 0.0220 |
| Prevalence (1 in) | 400000000 | 20661157 | – | – | 400000000 | 20661157 |
| Carrier Frq (1 in) | 10001 | 2273 | – | – | 10001 | 2273 |
| NPHS1 | ||||||
| Allele Count | 58/251247 | 31/30772 | 0in5008 | 192 | 58/256255 | 31in30964 |
| Mutant Allele Feq (%) | 0.023 | 0.100 | 0.000 | 0.000 | 0.022 | 0.100 |
| Prevalence (1 in) | 18903592 | 1000000 | – | – | 20661157 | 1000000 |
| Carrier Frq (1 in) | 2174 | 501 | – | – | 2273 | 501 |
| NPHS2+LAMB2+NPHS1 | ||||||
| Allele Count | 451/250505 | 53/30768 | 20/5008 | 3/192 | 471/255513 | 56in30960 |
| Mutant Allele Feq (%) | 0.18 | 0.172 | 0.399 | 1.56 | 0.184 | 0.180 |
| Prevalence (1 in) | 308642 | 338021 | 62814 | 4109 | 295369 | 308642 |
| Carrier Frq (1 in) | 278 | 291 | 126 | 33 | 272 | 278 |
Carrier frequencies and prevalence based on NPHS1, NPHS2, and LAMB2 known mutations found in the gnomADe and 1000Genome data.
Table 7
| gnomAD | gnomAD-SAS | 1KG | 1KG-PJL | gnomAD + 1KG | gnomAD-SAS + 1KG PJL | |
|---|---|---|---|---|---|---|
| NPHS2 | ||||||
| Allele Count | 395/251020 | 29/30775 | 21/5008 | 3/192 | 416/256028 | 32/30967 |
| Mutant Allele Feq. (%) | 0.157 | 0.094 | 0.419 | 1.560 | 0.162 | 0.103 |
| Prevalence (1 in) | 405696 | 1131734 | 56960 | 4109 | 381039 | 942596 |
| Carrier Frq. (1 in) | 319 | 532 | 120 | 33 | 309 | 486 |
| LAMB2 | ||||||
| Allele Count | 72/246450 | 40/30768 | 2/5008 | 1/192 | 74/251458 | 41/30960 |
| Mutant Allele Feq. (%) | 0.029 | 0.130 | 0.039 | 0.52 | 0.029 | 0.1320 |
| Prevalence (1 in) | 11890606 | 591716 | 6574622 | 36982 | 11890606 | 573921 |
| Carrier Frq. (1 in) | 1725 | 385 | 1283 | 97 | 1725 | 379 |
| NPHS1 | ||||||
| Allele Count | 100/248788 | 53/30772 | 0/5008 | 0/192 | 100/253796 | 53/30964 |
| Mutant Allele Feq. (%) | 0.040 | 0.172 | 0.000 | 0.000 | 0.039 | 0.171 |
| Prevalence (1 in) | 6250000 | 338021 | – | – | 6574622 | 341986 |
| Carrier Frq. (1 in) | 1251 | 291 | – | – | 1283 | 293 |
| NPHS2+LAMB2+NPHS1 | ||||||
| Allele Count | 567/248753 | 127/30771 | 23/5008 | 4/192 | 590/253761 | 0.0042 |
| Mutant Allele Feq. (%) | 0.227 | 0.412 | 0.459 | 2.08 | 0.23 | 0.420 |
| Prevalence (1 in) | 194065 | 58912 | 47465 | 2311 | 189036 | 56689 |
| Carrier Frq. (1 in) | 221 | 122 | 109 | 25 | 218 | 120 |
Carrier frequency and prevalence based on NPHS1, NPHS2, and LAMB2 known plus rare predicted to be pathogenic variants found in the gnomAD and the 1000Genome data.
Discussion
SRNS cases are challenging in the manner of highly variable clinical outcomes where 50% of the children develop ESRD within 15 years of life (Mekahli et al., 2009; Zagury et al., 2013). It constitutes the second most frequent cause of ESRD in the first two decades of life (North-American Pediatric Renal Trials and Collaborative Studies, NAPRTCS, 2008). In a previous analysis we have shown that mutations in the NPHS1 and the NPHS2 were not very common among CNS, SRNS, and familial cases in our population (Abid et al., 2012). Our results showed a low prevalence of disease causing mutations in the NPHS1 (22% early onset, 5.5% overall) and NPHS2 (3.3% early onset and 3.4% overall) genes in the studied NS population from Pakistan as compared to the other populations (Sadowski et al., 2015). To further extend the spectrum of the disease causing mutations in other NS causing genes, we selected a cohort of patients in whom the NPHS1 and NPHS2 genes have been excluded. We identified 50 such cases from our previous cohort including 14 new cases recruited for the NPHS1 and NPHS2 gene screening. Hinkes et al. (2007) have shown that 85% of the SRNS cases with congenital onset and 66% with infantile onset can be explained by mutations in the NPHS1, NPHS2, WT1, and LAMB2 genes. Matejas et al. (2010) indicated that the analysis of LAMB2 gene which is mutated in Pierson syndrome could be included in the diagnostics of early onset NS in the absence of extra renal abnormalities. Based on the screening algorithm presented by Hinkes et al. (2007) and Matejas et al. (2010) we selected LAMB2 and WT1 genes along with the NPHS1 and the NPHS2 genes for screening in our cohort.
Mutations identified in the LAMB2 gene (Table 2) in our cohort indicated that the LAMB2 gene should be included in the analysis algorithm as previously opposed by Santín et al. (2011) who excluded this gene from the algorithm introduced for molecular-genetic diagnostics of non-syndromic childhood onset primary SRNS. They also proposed the analysis of exons 8 and 9 of the WT1 gene in this algorithm; however upon screening of the whole WT1 gene, we did not find a single mutation in our isolated NS cases. Lipska et al. (2014) reported WT1 missense mutations associated with diffuse mesangial sclerosis (74%), early-onset SRNS and rapid progression to ESRD. In a cohort of Saudi Arabian families with childhood NS, disease causing mutations in the NPHS1 gene were identified in 12% (6/49) cases and in NPHS2 mutations were found in 22%(11/49) cases (Al-Hamed et al., 2013). Similarly, screening of 36 SRNS patients from the UK renal registry, molecular analysis revealed causative NPHS1 and NPHS2 gene mutations in 14% (5/36) and 8% (3/36) of the patients (McCarthy et al., 2013). One interesting observation of these two studies was the absence of LAMB2 and WT1 gene mutation in the two later studies.
Several recent studies have also reported gene panel screening with all the known genes in large cohorts using the next generation sequencing (NGS) technology (Bullich et al., 2015; Sadowski et al., 2015). These reports present a single gene cause of the disease in 29–36% of the patients. In comparison to these International cohort results, Wang et al. (2017) have identified a single gene cause of NS in 28.3% cases in Chinese cohort. The commonly mutated genes were ADCK4 (6.67%), NPHS1 (5.83%), WT1 (5.83%), and NPHS2 (3.33%) among Chinese populations. The differences between the Chinese study (Wang et al., 2017) and the Sadowski et al. (2015) study are significant, where the most common disease causing gene in Chinese cohort was ADCK4. No NPHS2 gene mutation was detected in the CNS or infantile NS cases in the Chinese cohort consistent with the results of Japanese, Korean, African-American SRNS populations and Pakistanis (Chernin et al., 2008; Cho et al., 2008; Wang et al., 2017). This was in contrary to some other reports, where mutations in the NPHS2 gene was found in 12% in Spanish population (Santín et al., 2011), 5.7–12.7% in an International cohort (Bullich et al., 2015; Sadowski et al., 2015), 37.5% in a European cohort of CNS and infantile onset NS (Hinkes et al., 2007).
The data indicate marked ethnic and geographical differences in the etiology of disease. The exact cause of this wide variation in the prevalence of genetic abnormalities in different studies is not clear, but may reflect ethnic differences, the use of different inclusion criteria, the differences in the methodologies used for the detection of mutations in these genes, and the number of genes screened for mutations. In the current study, we included only those cases of pediatric NS known to have high prevalence of genetic abnormalities. The observed low prevalence of nephrin, podocin, laminin B2 and Wilm's tumor-1gene mutations in CNS, infantile, and childhood SRNS is in marked contrast to the findings from the studies from Europe, and US, where the prevalence of these gene mutations are high (Hinkes et al., 2007; Matejas et al., 2010). However, our findings are concordant with the findings from a few regional studies, such as studies from China, Korea, Japan, and African American children (Sako et al., 2005; Chernin et al., 2008; Cho et al., 2008; Wang et al., 2017). The later study did not find any mutations in NPHS2 and exons 8 and 9 of the WT1 genes in African American cohort.
We did not find any significant correlation of the mutations in either gene with the age of the patients, clinical subtypes, gender, histology of the lesions, prognosis, or family history. Similar findings have been reported by most other investigators as well. This most probably is due to the small number of patients with each distinct type of mutations. Large studies with longer follow up may be helpful in this context.
We calculated the NS prevalence based on frequencies of genes in large population databases; the 1000 Genome and gnomAD. We have found an overall carrier frequency of 1 in 218 worldwide and 1 in 120 in South-Asian populations. Worldwide prevalence was found to be 1 in 189,036 and 1 in 56,689 in South Asian populations (Table 7). According to the literature, idiopathic nephrotic syndrome has a reported incidence of two to seven cases per 100,000 children and a prevalence of 16 cases per 100,000 (Eddy and Symons, 2003). There is also an epidemiological evidence of a higher incidence of nephrotic syndrome in children from South Asia (McKinney et al., 2001). The current estimates of prevalence of NS in three genes population data showed a much higher prevalence in SAS populations than the clinical studies estimations; however the worldwide data showed a much lesser prevalence according to current population data. There is no proper registry maintained for the prevalence of NS in Pakistani children. However, some individual single center studies presented local prevalence (Ali et al., 2011; Mubarak et al., 2012; Shah et al., 2015; Imtiaz et al., 2016). According to these estimates, NS is the most prevalent clinical presentation represented 50–74% of all the children presented with renal diseases (Ali et al., 2011; Imtiaz et al., 2016). The most common histo-pathological presentation was minimal change disease (MCD; 24.09–30.3%) followed by FSGS (18.3–38%) in these subjects (Ali et al., 2011; Mubarak et al., 2012; Imtiaz et al., 2016).
There are a few limitations of this study. A small sized patient cohort screening may reduce the chances of identification of disease mutations. However our cohort is a heterogeneous cohort with patients collected from all major ethnicities in Pakistan. SIUT is a tertiary care center, where patients are referred from all over Pakistan representing all the major ethnicities in the country. We only screened four genes reported to cause NS in children as we hypothesized this study according to the observations of some previously conducted studies (Hinkes et al., 2007; Zenker et al., 2009; Matejas et al., 2010; Santín et al., 2011). There are now more than 40 genes reported to cause different types of NS (Lipska et al., 2014; Bullich et al., 2015; Sadowski et al., 2015). It is therefore, necessary to design a comprehensive study to screen all the known disease causing gene for a better understanding of the disease etiology in this cohort.
In conclusion, we have observed a low prevalence of disease causing mutations in three major disease causing genes in our NS population and no mutations in the WT1 gene in early onset NS cases. The data indicated that genetic screening strategies put forward for several international populations may not be appropriate for this cohort. Therefore, a comprehensive screening for all the known genes is required which will help in improved management of the disease and will provide a reference population for gene screening within evolutionary related populations of this region.
Statements
Ethics statement
This study was carried out in accordance with the recommendations of Centre for Biomedical Ethics and Culture. The protocol was approved by the Centre for Biomedical Ethics and Research. Written informed consent was obtained from the parents of the subjects in accordance with the Declaration of Helsinki.
Author contributions
AA and SK conceived and designed the study. AA, SS, and MS performed the study. AA analyzed and prepared the manuscript. AL and SH provided sample and clinical data.
Acknowledgments
We are grateful to the patients who participated in the study. This work was supported by a core grant to SIUT.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Abid A. Khaliq S. Shahid S. Lanewala A. Mubarak M. Hashmi S. et al . (2012). A spectrum of novel NPHS1 and NPHS2 gene mutations in pediatric nephrotic syndrome patients from Pakistan. Gene502, 133–137. 10.1016/j.gene.2012.04.063
2
Al-Hamed M. H. Al-Sabban E. Al-Mojalli H. Al-Harbi N. Faqeih E. Al Shaya H. et al . (2013). A molecular genetic analysis of childhood nephrotic syndrome in a co.hort of Saudi Arabian families. J. Hum. Genet. 58, 480–4809. 10.1038/jhg.2013.27
3
Ali A. Ali M. U. Akhtar S. Z. (2011). Histological pattern of paediatric renal diseases in northern Pakistan. J. Pak. Med. Assoc. 61, 653–658.
4
Ashkenazy H. Kliger Y. (2010). Reducing phylogenetic bias in correlated mutation analysis. Protein Eng. Des. Sel. 23, 321–326. 10.1093/protein/gzp078
5
Bierzynska A. Soderquest K. Koziell A. (2015). Genes and podocytes - new insights into mechanisms of podocytopathy. Front. Endocrinol. 23:226. 10.3389/fendo.2014.00226
6
Bullich G. Trujillano D. Santín S. Ossowski S. Mendizábal S. Fraga G. et al . (2015). Targeted next-generation sequencing in steroid-resistant nephrotic syndrome: mutations in multiple glomerular genes may influence disease severity. Eur. J. Hum. Genet. 23, 1192–1199. 10.1038/ejhg.2014.252
7
Chernin G. Heeringa S. F. Gbadegesin R. Liu J. Hinkes B. G. Vlangos C. N. et al . (2008). Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 23, 1455–1460. 10.1007/s00467-008-0861-7
8
Cho H. Y. Lee J. H. Choi H. J. Lee B. H. Ha I. S. Choi Y. et al . (2008). WT1 and NPHS2 mutations in Korean children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 23, 63–70. 10.1007/s00467-007-0620-1
9
Conlon P. J. Lynn K. Winn M. P. Quarles L. D. Bembe M. L. Pericak-Vance M. et al . (1999). Spectrum of disease in familial focal and segmental glomerulosclerosis. Kidney Int. 56, 1863–1871. 10.1046/j.1523-1755.1999.00727.x
10
Eddy A. A. Symons J. M. (2003). Nephrotic syndrome in childhood. Lancet. 3, 629–639. 10.1016/S0140-6736(03)14184-0
11
González-Pérez A. López-Bigas N. (2011). Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 88, 440–449. 10.1016/j.ajhg.2011.03.004
12
Goujon M. McWilliam H. Li W. Valentin F. Squizzato S. Paern J. et al . (2010). A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699. 10.1093/nar/gkq313
13
Hinkes B. G. Mucha B. Vlangos C. N. Liu J. Hasselbacher K. Hangan D. et al . (2007). Nephrotic syndrome in the first year of life: Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Paediatrics. 119, e907–e919. 10.1542/peds.2006-2164
14
Imtiaz S. Nasir K. Drohlia M. F. Salman B. Ahmad A. (2016). Frequency of kidney diseases and clinical indications of pediatric renal biopsy: A single center experience. Indian J. Nephrol. 26, 199–205. 10.4103/0971-4065.159304
15
Landau G. M. Parida L. Weimann O. (2005). Gene proximity analysis across whole genomes via PQ trees. J. Comput. Biol. 12, 1289–1306. 10.1089/cmb.2005.12.1289
16
Landrum M. J. Lee J. M. Benson M. Brown G. Chao C. Chitipiralla S. et al . (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868. 10.1093/nar/gkv1222
17
Lek M. Karczewski K. J. Minikel E. V. Samocha K. E. Banks E. Fennell T. et al . (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291. 10.1038/nature19057
18
Li Q. Wang K. (2017). InterVar: clinical interpretation of genetic variants by the 2015 acmg-amp guidelines. Am. J. Hum. Genet. 100, 267–280. 10.1016/j.ajhg.2017.01.004
19
Lipska B. S. Ranchin B. Iatropoulos P. Gellermann J. Melk A. Ozaltin F. et al . (2014). Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. 85, 1169–1178. 10.1038/ki.2013.519
20
Löwik M. M. Groenen P. J. Levtchenko E. N. (2009). Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis. Eur. J. Pediatr. 168, 1291–1304. 10.1007/s00431-009-1017-x
21
Matejas V. Hinkes B. Alkandari F. Al-Gazali L. Annexstad E. Aytac M. B. et al . (2010). Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 31, 992–1002. 10.1002/humu.21304
22
McCarthy H. J. Bierzynska A. Wherlock M. Ognjanovic M. Kerecuk L. Hegde S. et al . (2013). Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 8, 637–648. 10.2215/CJN.07200712
23
McKinney P. A. Feltbower R. G. Brocklebank J. T. Fitzpatrick M. M. (2001). Time trends and ethnic patterns of childhood nephrotic syndrome in Yorkshire, U. K. Pediatr. Nephrol. 1, 1040–1044. 10.1007/s004670100021
24
Mekahli D. Liutkus A. Ranchin B. Yu A. Bessenay L. Girardin E. et al . (2009). Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: a multicenter study. Pediatr. Nephrol. 24, 1525–1532. 10.1007/s00467-009-1138-5
25
Mubarak M. Lanewala A. Kazi J. I. Akhter F. Sher A. Fayyaz A. et al . (2009). Histological spectrum of childhood nephrotic syndrome in Pakistan. Clin. Exp. Nephrol. 13, 589–593. 10.1007/s10157-009-0216-0
26
Mubarak M. Kazi J. I. Shakeel S. Lanewala A. Hashmi S. (2012). The spectrum of histopathological lesions in children presenting with steroid-resistant nephrotic syndrome at a single center in Pakistan. Sci. World J.2012:681802. 10.1100/2012/681802
27
Ruf R. G. Lichtenberger A. Karle S. M. Haas J. P. Anacleto F. E. Schultheiss M. et al . (2004). Patients with mutations in NPHS2 (p006Fdocin) do not respond to standard steroid treatment of nephrotic syndrome. J. Am. Soc. Nephrol.15, 722–732. 10.1097/01.ASN.0000113552.59155.72
28
Sadowski C. E. Lovric S. Ashraf S. Pabst W. L. Gee H. Y. Kohl S. et al . (2015). A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol.26, 1279–1289. 10.1681/ASN.2014050489
29
Sako M. Nakanishi K. Obana M. Yata N. Hoshii S. Takahashi S. et al . (2005). Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients with congenital nephrotic syndrome. Kidney Int. 67, 1248–1255. 10.1111/j.1523-1755.2005.00202.x
30
Santín S. Bullich G. Tazón-Vega B. García-Maset R. Giménez I. Silva I. et al . (2011). Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol.6, 1139–1148. 10.2215/CJN.05260610
31
Schwartz G. J. Work D. F. (2009). Measurement and estimation of GFR in children and adolescents. Clin. J. Am. Soc. Nephrol. 4, 1832–1843. 10.2215/CJN.01640309
32
Shah S. S. Akhtar N. Sunbleen F. Ur Rehman M. F. Ahmed T. (2015). Histopathological patterns in paediatric idiopathic steroid resistant nephrotic syndrome. J. Ayub Med. Coll. Abbottabad. 27, 633–636. 10.5812/nephropathol.8997
33
Tamura K. Peterson D. Peterson N. Stecher G. Nei M. Kumar S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. 10.1093/molbev/msr121
34
The 1000 Genome Project Consortium Auton A. Brooks L. D. Durbin R. M. Garrison E. P. Kang H. M. et al . (2015). A global reference for human genetic variation. Nature526, 68–74. 10.1038/nature15393
35
Wang F. Zhang Y. Mao J. Yu Z. Yi Z. Yu L. et al . (2017). Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 32, 1181–1192. 10.1007/s00467-017-3590-y
36
Weber S. Grobouval O. Esquivel E. L. Morinière V. Tête M. J. Legendre C. et al . (2004). NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. 66, 571–579. 10.1111/j.1523-1755.2004.00776.x
37
Zagury A. Oliveira A. L. Montalvão J. A. Novaes R. H. Sá V. M. Moraes C. A. et al . (2013). Steroid-resistant idiopathic nephrotic syndrome in children: Long-term follow-up and risk factors for end-stage renal disease. J. Bras. Neurol. 35, 191–199. 10.5935/0101-2800.20130031
38
Zenker M. Machuca E. Antignac C. (2009). Genetics of nephrotic syndrome: new insights into molecules acting at the glomerular filtration barrier. J. Mol. Med. 87:849–857. 10.1007/s00109-009-0505-9
Summary
Keywords
nephrotic syndrome, NPHS1 , NPHS2 , LAMB2 , WT1
Citation
Abid A, Shahid S, Shakoor M, Lanewala AA, Hashmi S and Khaliq S (2018) Screening of the LAMB2, WT1, NPHS1, and NPHS2 Genes in Pediatric Nephrotic Syndrome. Front. Genet. 9:214. doi: 10.3389/fgene.2018.00214
Received
14 April 2018
Accepted
25 May 2018
Published
22 June 2018
Volume
9 - 2018
Edited by
Jordi Pérez-Tur, Instituto de Biomedicina de Valencia (IBV), Spain
Reviewed by
Michal Malina, Newcastle Hospitals, United Kingdom; Muhammad Jawad Hassan, National University of Sciences and Technology, Pakistan
Updates
Copyright
© 2018 Abid, Shahid, Shakoor, Lanewala, Hashmi and Khaliq.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiysha Abid aiyshaabid@gmail.com; aiysha.abid@siut.org
This article was submitted to Genetic Disorders, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.