 Min Xu1
Min Xu1 Hui Xiong1Yanfang Han1Chijun Li1Shaozhen Mai1,2Zhongzhou Huang1Xuechen Ai1Zhixuan Guo1Fanqin Zeng1
Hui Xiong1Yanfang Han1Chijun Li1Shaozhen Mai1,2Zhongzhou Huang1Xuechen Ai1Zhixuan Guo1Fanqin Zeng1 Qing Guo1*
Qing Guo1*- 1Department of Dermatology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
- 2Department of Dermatology, The Eighth Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
Neurofibromatosis type I is a rare neurocutaneous syndrome resulting from loss-of-function mutations of NF1. The present study sought to determine a correlation between mutation regions on NF1 and the risk of developing optic pathway glioma (OPG) in patients with neurofibromatosis type I. A total of 215 patients with neurofibromatosis type I, from our clinic or previously reported literature, were included in the study after applying strict inclusion and exclusion criteria. Of these, 100 patients with OPG were classified into the OPG group and 115 patients without OPG (aged ≥ 10 years) were assigned to the Non-OPG group. Correlation between different mutation regions and risk of OPG was analyzed. The mutation clustering in the 5′ tertile of NF1 was not significantly different between OPG and Non-OPG groups (P = 0.131). Interestingly, patients with mutations in the cysteine/serine-rich domain of NF1 had a higher risk of developing OPG than patients with mutations in other regions [P = 0.019, adjusted odds ratio (OR) = 2.587, 95% confidence interval (CI) = 1.167–5.736], whereas those in the HEAT-like repeat region had a lower risk (P = 0.036, adjusted OR = 0.396, 95% CI = 0.166–0.942). This study confirms a new correlation between NF1 genotype and OPG phenotype in patients with neurofibromatosis type I, and provides novel insights into molecular functions of neurofibromin.
Introduction
Neurofibromatosis type I (MIM entry: 162200) is a rare disease with autosomal dominant inheritance that belongs to a neurocutaneous syndrome characterized by café-au-lait spots, skin freckling, neurofibroma, Lisch nodules, and optic pathway glioma (OPG) (Jett and Friedman, 2010). Its incidence and prevalence are 1/2500 and 1/4000, respectively (Pasmant et al., 2012). Neurofibromatosis type I results from loss-of-function mutations of the tumor suppressor NF1 gene encoding neurofibromin (Trovó-Marqui and Tajara, 2006). Neurofibromin has several domains and special structures; its main role is to act as a negative regulator in the Ras pathway (Ratner and Miller, 2015). Although the NF1 sequence has been known for nearly 30 years, the correlation between genotype and phenotype in neurofibromatosis type I remains poorly understood. This is largely due to low incidence, the considerable length of NF1, diffused distribution of mutations, and the age-dependent variability of manifestations (Wallace et al., 1990). So far, the few published genotype–phenotype correlations have indicated a severe phenotype in patients with whole-gene deletion and the absence of cutaneous neurofibroma in patients with a 3-bp inframe deletion (c.2970–2972 delAAT) or missense mutation (p.Arg1809Cys) (Cnossen et al., 1997; Upadhyaya et al., 2007; Mautner et al., 2010; Pinna et al., 2014).
Recently, two studies suggested that patients with mutations in the 5′ tertile (exon 1–21) of NF1 had a higher risk of developing OPG (Sharif et al., 2011; Bolcekova et al., 2013). OPG is a type of pilocytic astrocytoma located in the cerebral optic pathway, which can impair patients’ vision and visual field (Listernick et al., 2007). However, approximately 50–75% of patients present no symptoms at the time OPG is diagnosed (Sylvester et al., 2006), making it important to evaluate the risk of OPG in asymptomatic patients. Notably, a subsequent study based on stricter inclusion criteria, revealed that mutation clustering in the 5′ tertile was common to both the OPG and control groups (Hutter et al., 2016). These conflicting results show that further studies are required to establish a firm correlation between NF1 genotype and OPG.
Previous studies were limited by either small samples or poor inclusion criteria, both of which could lead to biased results. In the present effort, we used strict inclusion criteria, which we applied also to patients from previous studies, to obtain a sufficiently large cohort. Furthermore, the region comprising the 5′ tertile is fairly vast, which might have affected the accuracy of previous results. Instead, here we applied an established practice common to the study of hereditary diseases (Hateboer et al., 2000; Tartaglia et al., 2002; Aartsma-Rus et al., 2006; Mori-Yoshimura et al., 2012), and focused specifically on correlations between mutations in regions encoding protein domains or other regions of the NF1 gene and the OPG phenotype in patients with neurofibromatosis type I.
Materials and Methods
Literature Search
A thorough search of English-language literature for relevant papers which focused on the genotype–phenotype correlation or described clinical manifestations and gene mutations in patients with neurofibromatosis type I was performed. Databases such as PubMed and Google Scholar, as well as university library resources were searched. Search terms included “(OPG OR optic pathway glioma OR optic glioma) AND (nf1 OR neurofibromatosis) AND (genotype OR mutation),” “(glioma OR optic nerve tumor OR optic nerve glioma) AND (nf1 OR neurofibromatosis) AND (genotype OR mutation),” “(nf1 OR neurofibromatosis) AND (OPG OR optic pathway glioma OR optic glioma),” and “(nf1 OR neurofibromatosis) AND (glioma OR optic nerve tumor OR optic nerve glioma).” All related papers were read through to evaluate whether the reported patients were compatible with the present study’s inclusion criteria.
Study Subjects
Strict inclusion criteria included: (1) conformity to NIH diagnostic criteria for neurofibromatosis type I (Conference, 1988); (2) brain radiology (computed tomography or magnetic resonance imaging) to diagnose or exclude OPG; (3) NF1 gene test to determine pathogenic germline mutations; (4) age of patients without OPG of 10 years or more (rare patients with neurofibromatosis type I could develop OPG after that point) (Nicolin et al., 2009; Hutter et al., 2016).
Exclusion criteria included: (1) inconclusive radiological diagnosis of OPG; (2) wrong assessment of gene mutations, due to cDNA changes not conforming to predicted amino acid alterations or the original cDNA base in the reported position not conforming to the corresponding base in the reference sequence; (3) existence of two variants of NF1 in a patient for which the pathogenic one could not be determined according to present evidence; (4) patients in a study which only focused on a certain point mutation, because these patients were selected artificially.
After selection following the above inclusion and exclusion criteria, five patients from our clinic and 210 patients described in the literature were included in this study. Of these, 100 patients were classified in the OPG group and 115 in the Non-OPG group. The five patients from our clinic were scanned to obtain brain magnetic resonance imaging and 2 mL of blood was collected for NF1 gene testing using next-generation sequencing (NGS) in an Illumina HiSeq 2000 system (Illumina Corporation, San Diego, CA, United States), and small mutations were verified using Sanger sequencing among probands’ family members. Multiplex ligation-dependent probe amplification (MLPA) was used to detect the large rearrangement.
In summary, small mutations and large rearrangements of NF1 were detected in 208 and 7 patients, respectively. Among the 208 patients with small mutations, the most common methods for ascertaining mutations were Sanger sequencing (114, 54.81%) and NGS (55, 26.44%), whereas the sequencing methods of the remaining 39 patients (18.75%) were not reported in the literatures. Among the seven patients with large rearrangements, the most common methods for detecting mutations were MLPA (6, 85.71%), followed by single nucleotide polymorphism array (1, 14.29%).
Mutation Analysis
Each NF1 gene mutation was verified and analyzed in Mutalyzer1 and all mutation information (c.DNA position and base changes) was transformed to novel nomenclature using transcript number NG_009018.1 (NF1_v002; Anastasaki et al., 2017). Furthermore, mutations were also analyzed using MutationTaster2, which predicted amino acid alterations. Exons were divided into 5′ tertile (exon 1–21), middle tertile (exon 22–38), and 3′ tertile (exon 39–57) for mutation clustering analysis (Sharif et al., 2011). The splice site mutation was classified to the consensus exon for statistical analysis. Additionally, to detect a correlation between NF1 genotype and OPG phenotype, several gene regions were analyzed. These included the cysteine/serine-rich domain (CSRD, residues 543–909), tubulin-binding domain (TBD, residues 1095–1197), GTPase-activating protein-related domain (GRD, residues 1198–1530), Sec14-like domain (Sec14, residues 1560–1705), pleckstrin homology-like domain (PH, residues 1716–1816), HEAT-like repeat regions (HLR, residues 1825–2428), C-terminal domain (CTD, residues 2260–2817), nuclear localization signal region (NLS, residues 2534–2550), and syndecan-binding region (SBR, residues 2619–2719) (Vandenbroucke et al., 2004; Bonneau et al., 2009; Luo et al., 2014). The bipartite module with phospholipid binding activity consisted of Sec14 and PH; therefore, the area of Sec14-PH combined the regions of Sec14 and PH (D’Angelo et al., 2006). Patients with whole-gene deletions were not included in the analysis of mutation clustering in different tertiles or all gene regions.
Statistical Analysis
This study was designed as a case control study. Data were analyzed using SPSS version 24.0 (IBM Corporation, Chicago, IL, United States) and GraphPad PRISM version 7.01 (GraphPad Software, San Diego, CA, United States). Constituent ratio and sector diagram were used to describe the distribution of mutation types. The number of patients in each group is presented in bar graphs. Contingency tables and Chi-square test were used for categorical variables. When any expected value on contingency tables was below five, results were corrected for continuity. When any expected value on contingency tables was below one or the total sample number was less than 40, Fisher’s exact test was used. Logistic regression was used to evaluate the risk of developing OPG due to mutations in each region, and crude odds ratios (ORs) were reported with 95% confidence intervals (CIs). Mutations in regions with significantly high or low risk of developing OPG were analyzed using multiple logistic regression, and adjusted ORs were also reported with 95% CI. P-values < 0.05 were considered statistically significant.
Results
Distribution of Mutation Types and Mutation-Containing Regions in Patients
The number of patients with frameshift mutation, nonsense mutation, splice site mutation, missense mutation, inframe mutation, large deletion, whole-gene deletion, and atypical splicing mutation was 33 (33%), 31 (31%), 17 (17%), 13 (13%), 2 (2%), 3 (3%), 1 (1%), and 0, respectively, in the OPG group and 38 (33.04%), 34 (29.57%), 23 (20%), 14 (12.17%), 2 (1.74%), 0, 3 (2.61%), and 1 (0.87%), respectively, in the Non-OPG group. The distribution of mutation types did not differ significantly between the two groups (P = 0.696, Figure 1A). The distribution of mutation-containing regions for the two groups is shown in Figure 1B, and details are presented in Supplementary Table S1.
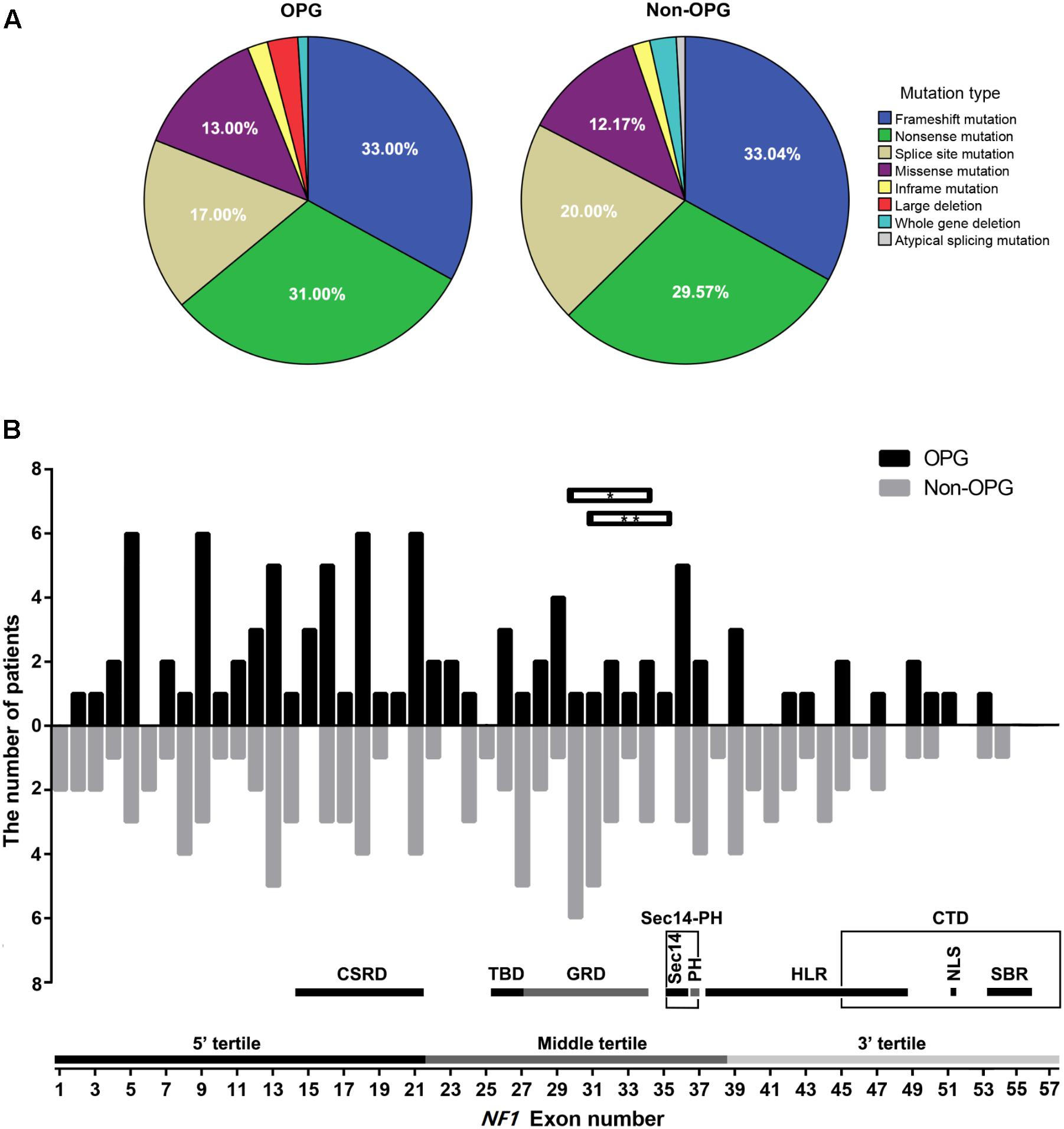
FIGURE 1. Distribution of mutation types and mutation-containing regions on the NF1 gene in patients. (A) Percentage of different mutation types in the OPG and Non-OPG groups. (B) Bar graph showing the number of patients whose mutations were located within different exons in the OPG and Non-OPG groups. Splice site mutation was included in the consensus exon. Whole-gene deletion is not shown in this figure. Horizontal bar with∗ indicates one OPG patient with a large deletion of exons 30–34. Horizontal bar with∗∗ indicates one OPG patient with a large deletion of exons 31–35. A schematic diagram showing the number of exons contained in different regions or tertiles is presented. CSRD, cysteine/serine-rich domain; TBD, tubulin-binding domain; GRD, GTPase-activating protein-related domain; Sec14, Sec14-like domain; PH, pleckstrin homology-like domain; Sec14-PH, module combining Sec14 and PH; HLR, HEAT-like repeat regions; CTD, C-terminal domain; NLS, nuclear localization signal region; SBR, syndecan-binding region.
Mutation Clustering in the 5′ Tertile Occurs in Both OPG and Non-OPG Groups
Having assembled a large sample using strict inclusion criteria, we verified whether mutation clustering conformed to that in previous studies by comparing the number of patients from both groups, whose mutations were located in different tertiles (Sharif et al., 2011; Bolcekova et al., 2013; Hutter et al., 2016). Mutation clustering in the 5′ tertile was observed in both the OPG group and Non-OPG group, but the difference between them was not statistically significant (P = 0.131, Figure 2A). The total number of patients from both groups, presenting mutations located in the 5′ tertile, middle tertile, or 3′ tertile was 101 (47.87%, 95% CI = 41.07–54.66%), 73 (34.60%, 95% CI = 28.13–41.67%), and 37 (17.53%, 95% CI = 12.36–22.71%), respectively (Figure 2B). Thus, mutation clustering in the 5′ tertile was characteristic of all patients and not only of those in the OPG group.
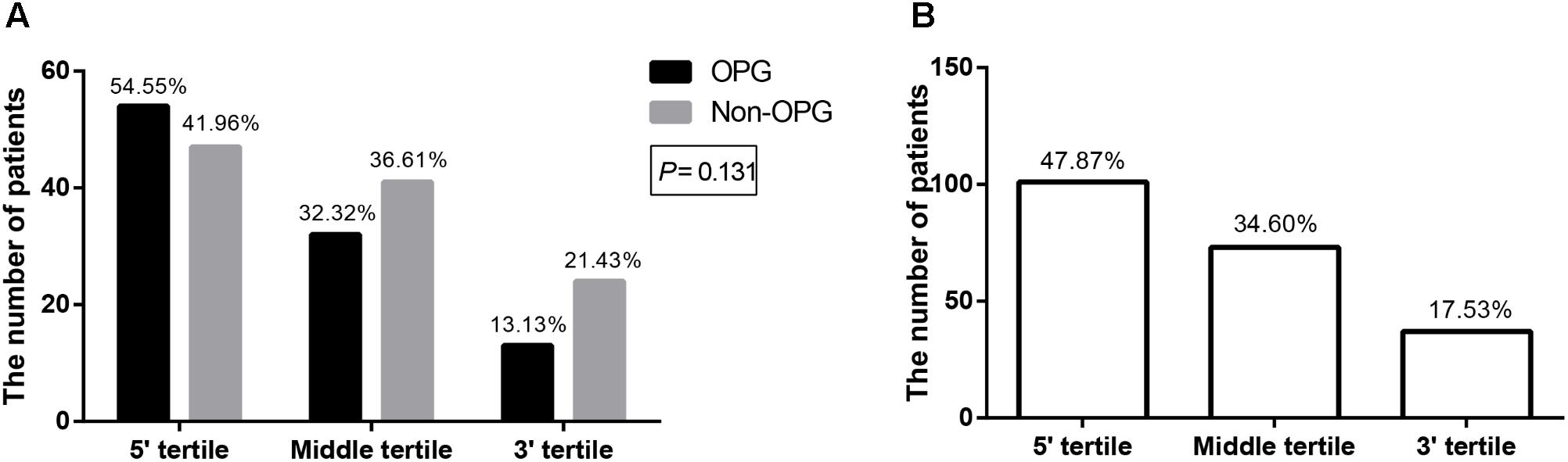
FIGURE 2. Bar graph depicting the number of patients with mutations located in different tertiles of NF1. The number of patients with mutations located in different tertiles of NF1 in the OPG group versus Non-OPG group (A) and total patients (B). The number above each bar represents the percentage in the relevant group.
Patients With Mutations in the CSRD Are at Higher Risk of Developing OPG, Those in the HLR Are at Lower Risk
A protein domain is defined as the functional region of a protein; accordingly, mutations affecting protein domain regions can directly disrupt the function of a protein (Welti et al., 2011). We analyzed all common domain regions of NF1 to evaluate the OPG risk of mutations in these regions (Table 1). Patients with mutations in the CSRD correlated with a higher risk of developing OPG than patients with mutations in other regions (P = 0.019, adjusted OR = 2.587, 95% CI = 1.167–5.736). Patients with mutations in Sec14-PH displayed a tendency toward a higher risk of developing OPG than patients with mutations in other regions; however, this trend was not statistically significant (P = 0.057, adjusted OR = 3.712, 95% CI = 0.961–14.332). Instead, patients with mutations in the HLR had a lower risk of developing OPG than patients with mutations in other regions (P = 0.036, adjusted OR = 0.396, 95% CI = 0.166–0.942). The remaining gene regions did not reveal any significantly different mutation distributions between the two groups. Thus, the risk of developing OPG in patients with neurofibromatosis type I correlated with mutations in the CSRD and HLR.
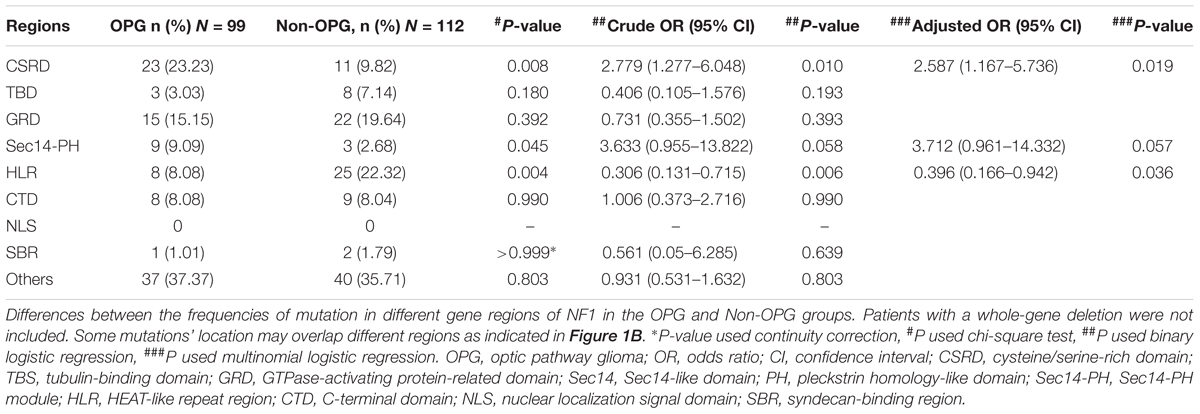
TABLE 1. Mutations in different NF1 gene regions and their risk of developing OPG.
Distribution of Mutation Types in the CSRD and HLR
As different mutation types exert a different effect on protein structure and function (Richards et al., 2015), we analyzed the distribution of mutation types in gene regions where disruptions would lead to high or low risk of developing OPG (Table 2). In spite of the higher frequency of nonsense mutations within the CSRD in the OPG group and higher frequency of splicing site mutations within the HLR in the Non-OPG group, no significant differences were detected (P > 0.05). Thus, there was not enough evidence to suggest that a certain mutation type contributed in a predominant way to the high or low risks of developing OPG in these two gene regions.
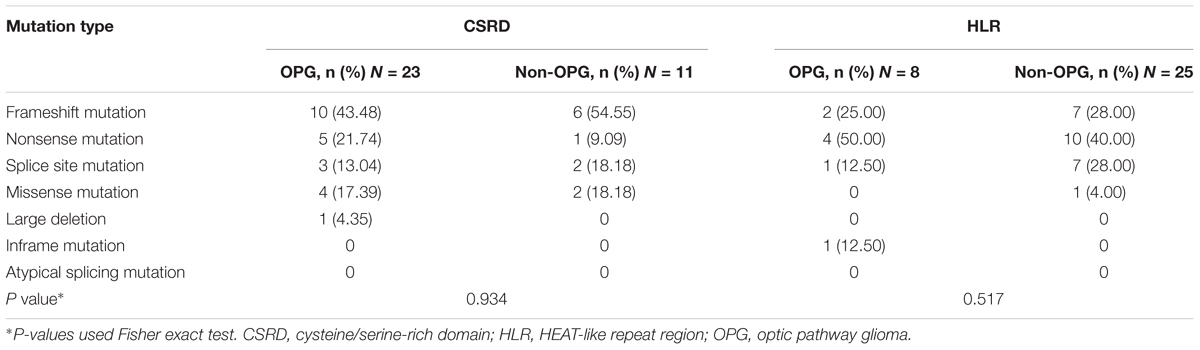
TABLE 2. Distribution of mutation types in the CSRD and HLR between the OPG and Non-OPG groups.
Discussion
The impact of a specific gene mutation on the corresponding protein has been shown to vary with the location of the mutation (Hateboer et al., 2000; Tartaglia et al., 2002; Aartsma-Rus et al., 2006; Mori-Yoshimura et al., 2012). In this study, a large cohort of patients assembled using strict inclusion and exclusion criteria was analyzed to evaluate the correlation between mutation-containing regions and the risk of developing OPG. Results indicated mutation clustering in the 5′ tertile of NF1 existed in both the OPG group and Non-OPG group; however, our study newly reported that patients with mutations in the CSRD and HLR of NF1 exhibited a higher and lower risk, respectively, of developing OPG than patients with mutations in other regions, irrespective of mutation type.
Optic pathway glioma is the most common central nervous system tumor in patients with neurofibromatosis type I. Several studies have attempted to reveal a correlation between genotype and phenotype in neurofibromatosis patients with OPG (Sharif et al., 2011; Bolcekova et al., 2013; Hutter et al., 2016); however, results have been mostly inconclusive, probably due to the small number of participants and varying inclusion criteria. Furthermore, inclusion criteria may have not been sufficiently rigorous in these studies, and included lack of a radiology-based diagnosis of OPG or the presence of Non-OPG patients younger than 10 years. These, may have resulted in wrong classification of patients in the OPG or Non-OPG groups, thus causing bias in the results (Sharif et al., 2011; Bolcekova et al., 2013). Here, we included a larger sample size using strict inclusion and exclusion criteria. Our results demonstrated no correlation between mutations clustering in the 5′ tertile of NF1 and the risk of OPG, thus confirming the study by Hutter et al. (2016).
The region comprising the 5′ tertile is fairly vast and may affect the accuracy of the results. To obtain more precise results, we analyzed correlations between mutations either in regions encoding protein domains or other regions of NF1 and the OPG phenotype. CSRD is a unique domain located at the N-terminus of neurofibromin and its function is not entirely clear. CSRD, with its three cysteine pairs (residues 622/632, 673/680, and 714/721), could be phosphorylated by cAMP-dependent protein kinase A (PKA) and protein kinase C (PKC) (Marchuk et al., 1991; Fahsold et al., 2000; Mangoura et al., 2006). PKC-dependent phosphorylation of the CSRD could enhance the biological effect of neurofibromin through interactions with the actin cytoskeleton and the allosteric effect on its Ras-GAP domain (GRD), which could reduce Ras activity and suppress tumorigenesis (Mangoura et al., 2006). Mutations affecting the CSRD might limit the biological effect of neurofibromin, possibly explaining why mutations in the CSRD were associated with a higher risk of developing OPG. Phosphorylation of the CSRD regulates GRD activity, but mutations in the latter did not increase the risk of developing OPG to the same extent as did mutations in the former. This observation is hard to reconcile at present, but suggests that heterozygous mutations in the CSRD might be more harmful than heterozygous mutations in the GRD in the tumorigenesis of OPG, which should be investigated further.
Sec14-PH was reported as a bipartite module of neurofibromin responsible for binding to cellular phospholipids and possibly the membrane localization of neurofibromin (Welti et al., 2007, 2008). Our results showed a tendency toward a higher risk of developing OPG in patients with mutations in the Sec14-PH; however, this trend was not statistically significant and might require verification in a larger cohort. Besides regions whose disruption conferred a higher risk of OPG, mutations in the HLR appeared to associate with a lower risk of developing OPG compared with mutations in other regions. HLR is a newly discovered structure, responsible for interactions between proteins and different molecules in a variety of proteins (Kajava et al., 2004; Jeong et al., 2013; Hadpech et al., 2017). The structure of the HLR is highly similar to that of HEAT repeats with repetitive arrays of short amphiphilic α-helices, and it exists in a wide variety of eukaryotic proteins, allowing them to adapt to both hydrophilic and hydrophobic environments (Yoshimura and Hirano, 2016). Until now, little has been known about the molecular function of the HLR in neurofibromin. Our results indicate that, contrary to the CSRD, mutations in this region were associated with a lower risk of OPG than mutations in other regions. This finding suggests that the HLR might perform a negative feedback function on neurofibromin in astrocytes. We believe that this hypothesis warrants further consideration in future experiments. It should be noted that the risk associated with mutations in the CSRD and HLR of NF1 can also be observed in participants examined in previous studies focusing on correlation between mutations in the 5′ tertile and OPG phenotype (Supplementary Table S2) (Bolcekova et al., 2013; Hutter et al., 2016).
Although different mutation types exert different effects on protein structure and function (Richards et al., 2015), we failed to observe any significant correlation between mutation type and OPG phenotype in mutations located in the CSRD or HLR. However, any tendency toward a correlation observed for some mutation types, requires verification with a larger sample, because subdivision per mutation type created groups of too small a size.
In the present study, we successfully decreased the bias resulting from diagnosis by tightening the inclusion and exclusion criteria. Nevertheless, some level of bias resulted from the different procedures and techniques used to quantify the pathogenic mutation. Even though more than half the patients were examined using Sanger sequencing, often regarded as gold standard for molecular diagnostics (Rehm, 2013), possible bias resulting from the different methods and procedures should be kept in mind. The other limitations of this study include: (1) no consideration of racial difference as a possible variable for broadening sample size; (2) lack of a clear correlation between the risk of developing OPG and other mutations not located in the CSRD or HLR, but which also affected the function of these regions.
Conclusion
The present study newly revealed that neurofibromatosis type I patients with mutations located in the CSRD and HLR of the NF1 gene had a higher and lower risk than patients with mutations in other regions, respectively, of developing OPG. Thus in clinic, we should pay more attention to patients with mutations in CSRD to evaluate the development of OPG. This study suggests that a precise strategy is important in the analysis of genotype–phenotype correlations of NF1 and provides novel insights into the molecular functions of neurofibromin.
Ethics Statement
This study was carried out in accordance with recommendations of ‘the Ethics Committee of Sun Yat-sen Memorial Hospital’ with written informed consent from all our subjects. All our patients gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the ‘the Ethics Committee of Sun Yat-sen Memorial Hospital’.
Author Contributions
MX designed the study and drafted the manuscript. MX, HX, and YH searched for previously reported cases and analyzed the data. CL, SM, ZH, XA, and ZG verified the genetic data. FZ and QG supervised the study and assisted in revising the manuscript.
Funding
This study was sponsored by grants from the National Natural Science Foundation of China (grant numbers 81573044, 81602762); and the Natural Science Foundation of Guangdong Province (grant number 2016A030310185).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to all the participants in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00270/full#supplementary-material
Footnotes
References
Aartsma-Rus, A., Van Deutekom, J. C., Fokkema, I. F., Van Ommen, G. J., and Den Dunnen, J. T. (2006). Entries in the leiden duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34, 135–144. doi: 10.1002/mus.20586
Anastasaki, C., Le, L. Q., Kesterson, R. A., and Gutmann, D. H. (2017). Updated nomenclature for human and mouse neurofibromatosis type 1 genes. Neurol. Genet. 3:e169. doi: 10.1212/NXG.0000000000000169
Bolcekova, A., Nemethova, M., Zatkova, A., Hlinkova, K., Pozgayova, S., Hlavata, A., et al. (2013). Clustering of mutations in the 5′ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma 60, 655–665. doi: 10.4149/neo-2013-084
Bonneau, F., Lenherr, E. D., Pena, V., Hart, D. J., and Scheffzek, K. (2009). Solubility survey of fragments of the neurofibromatosis type 1 protein neurofibromin. Protein Expr. Purif. 65, 30–37. doi: 10.1016/j.pep.2008.12.001
Conference, N. C. D. (1988). Neurofibromatosis: conference statement. Arch. Neurol. 45, 575–578. doi: 10.1001/archneur.1988.00520290115023
Cnossen, M. H., van der Est, M. N., Breuning, M. H., van Asperen, C. J., Breslau-Siderius, E. J., van der Ploeg, A. T., et al. (1997). Deletions spanning the neurofibromatosis type 1 gene: implications for genotype-phenotype correlations in neurofibromatosis type 1? Hum. Mutat. 9, 458–464. doi: 10.1002/(SICI)1098-1004(1997)9:5<458::AID-HUMU13>3.0.CO;2-1
D’Angelo, I., Welti, S., Bonneau, F., and Scheffzek, K. (2006). A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 7, 174–179. doi: 10.1038/sj.embor.7400602
Fahsold, R., Hoffmeyer, S., Mischung, C., Gille, C., Ehlers, C., Kücükceylan, N., et al. (2000). Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am. J. Hum. Genet. 66, 790–818. doi: 10.1086/302809
Hadpech, S., Nangola, S., Chupradit, K., Fanhchaksai, K., Furnon, W., Urvoas, A., et al. (2017). Alpha-helicoidal HEAT-like repeat proteins (alphaRep) selected as interactors of HIV-1 nucleocapsid negatively interfere with viral genome packaging and virus maturation. Sci. Rep. 7:16335. doi: 10.1038/s41598-017-16451-w
Hateboer, N., Veldhuisen, B., Peters, D., Breuning, M. H., San-Millán, J. L., Bogdanova, N., et al. (2000). Location of mutations within the PKD2 gene influences clinical outcome. Kidney Int. 57, 1444–1451. doi: 10.1046/j.1523-1755.2000.00989.x
Hutter, S., Piro, R. M., Waszak, S. M., Kehrer-Sawatzki, H., Friedrich, R. E., Lassaletta, A., et al. (2016). No correlation between NF1 mutation position and risk of optic pathway glioma in 77 unrelated NF1 patients. Hum. Genet. 135, 469–475. doi: 10.1007/s00439-016-1646-x
Jett, K., and Friedman, J. M. (2010). Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 12, 1–11. doi: 10.1097/GIM.0b013e3181bf15e3
Jeong, J. H., Kim, Y. S., Rojviriya, C., Cha, H. J., Ha, S. C., and Kim, Y. G. (2013). The structure of TON1937 from archaeon Thermococcus onnurineus NA1 reveals a eukaryotic HEAT-like architecture. Int. J. Biol. Macromol. 61, 433–438. doi: 10.1016/j.ijbiomac.2013.07.010
Kajava, A. V., Gorbea, C., Ortega, J., Rechsteiner, M., and Steven, A. C. (2004). New HEAT-like repeat motifs in proteins regulating proteasome structure and function. J. Struct. Biol. 146, 425–430. doi: 10.1016/j.jsb.2004.01.013
Listernick, R., Ferner, R. E., Liu, G. T., and Gutmann, D. H. (2007). Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann. Neurol. 61, 189–198. doi: 10.1002/ana.21107
Luo, G., Kim, J., and Song, K. (2014). The C-terminal domains of human neurofibromin and its budding yeast homologs Ira1 and Ira2 regulate the metaphase to anaphase transition. Cell Cycle 13, 2780–2789. doi: 10.4161/15384101.2015.945870
Mangoura, D., Sun, Y., Li, C., Singh, D., Gutmann, D. H., Flores, A., et al. (2006). Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 25, 735–745. doi: 10.1038/sj.onc.1209113
Mautner, V. F., Kluwe, L., Friedrich, R. E., Roehl, A. C., Bammert, S., Högel, J., et al. (2010). Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J. Med. Genet. 47, 623–630. doi: 10.1136/jmg.2009.075937
Marchuk, D. A., Saulino, A. M., Tavakkol, R., Swaroop, M., Wallace, M. R., Andersen, L. B., et al. (1991). CDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1 gene product. Genomics 11, 931–940. doi: 10.1016/0888-7543(91)90017-9
Mori-Yoshimura, M., Monma, K., Suzuki, N., Aoki, M., Kumamoto, T., Tanaka, K., et al. (2012). Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. J. Neurol. Sci. 318, 100–105. doi: 10.1016/j.jns.2012.03.016
Nicolin, G., Parkin, P., Mabbott, D., Hargrave, D., Bartels, U., Tabori, U., et al. (2009). Natural history and outcome of optic pathway gliomas in children. Pediatr. Blood Cancer 53, 1231–1237. doi: 10.1002/pbc.22198
Pinna, V., Lanari, V., Daniele, P., Consoli, F., Agolini, E., Margiotti, K., et al. (2014). P.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 23, 1068–1071. doi: 10.1038/ejhg.2014.243
Pasmant, E., Vidaud, M., Vidaud, D., and Wolkenstein, P. (2012). Neurofibromatosis type 1: from genotype to phenotype. J. Med. Genet. 49, 483–489. doi: 10.1136/jmedgenet-2012-100978
Ratner, N., and Miller, S. J. (2015). A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 15, 290–301. doi: 10.1038/nrc3911
Rehm, H. L. (2013). Disease-targeted sequencing: a cornerstone in the clinic. Nat. Rev. Genet. 14, 295–300. doi: 10.1038/nrg3463
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sharif, S., Upadhyaya, M., Ferner, R., Majounie, E., Shenton, A., Baser, M., et al. (2011). A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet. 48, 256–260. doi: 10.1136/jmg.2010.081760
Sylvester, C. L., Drohan, L. A., and Sergott, R. C. (2006). Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type 1. Curr. Opin. Ophthalmol. 17, 7–11. doi: 10.1097/01.icu.0000193070.32369.37
Tartaglia, M., Kalidas, K., Shaw, A., Song, X., Musat, D. L., van der Burgt, I., et al. (2002). PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 70, 1555–1563. doi: 10.1086/340847
Trovó-Marqui, A. B., and Tajara, E. H. (2006). Neurofibromin: a general outlook. Clin. Genet. 70, 1–13. doi: 10.1111/j.1399-0004.2006.00639.x
Upadhyaya, M., Huson, S. M., Davies, M., Thomas, N., Chuzhanova, N., Giovannini, S., et al. (2007). An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 80, 140–151. doi: 10.1086/510781
Vandenbroucke, I., Van Oostveldt, P., Coene, E., De Paepe, A., and Messiaen, L. (2004). Neurofibromin is actively transported to the nucleus. FEBS Lett. 560, 98–102. doi: 10.1016/S0014-5793(04)00078-X
Wallace, M. R., Marchuk, D. A., Andersen, L. B., Letcher, R., Odeh, H. M., Saulino, A. M., et al. (1990). Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249, 181–186. doi: 10.1126/science.2134734
Welti, S., D’Angelo, I., and Scheffzek, K. (2008). Structure and function of neurofibromin. Monogr. Hum. Genet. 16, 113–128. doi: 10.1159/000126561
Welti, S., Fraterman, S., D’Angelo, I., Wilm, M., and Scheffzek, K. (2007). The sec14 homology module of neurofibromin binds cellular glycerophospholipids: mass spectrometry and structure of a lipid complex. J. Mol. Biol. 366, 551–562. doi: 10.1016/j.jmb.2006.11.055
Welti, S., Kuhn, S., D’Angelo, I., Brügger, B., Kaufmann, D., and Scheffzek, K. (2011). Structural and biochemical consequences of NF1 associated nontruncating mutations in the Sec14-PH module of neurofibromin. Hum. Mutat. 32, 191–197. doi: 10.1002/humu.21405
Keywords: neurofibromatosis type I, NF1, genotype, mutation, optic pathway glioma, phenotype
Citation: Xu M, Xiong H, Han Y, Li C, Mai S, Huang Z, Ai X, Guo Z, Zeng F and Guo Q (2018) Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 9:270. doi: 10.3389/fgene.2018.00270
Received: 03 May 2018; Accepted: 03 July 2018;
Published: 24 July 2018.
Edited by:
Musharraf Jelani, King Abdulaziz University, Saudi ArabiaReviewed by:
Mohamed Saad Zaghloul, Cairo University, EgyptSergiusz Jozwiak, Medical University of Warsaw, Poland
Copyright © 2018 Xu, Xiong, Han, Li, Mai, Huang, Ai, Guo, Zeng and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Guo, Z3VvcWluZ3N5c3VAc2luYS5jb20=