 Yan Zhao
Yan Zhao Xing Chen
Xing Chen Jun Yin
Jun Yin- School of Information and Control Engineering, China University of Mining and Technology, Xuzhou, China
Increasing evidence has indicated that microRNAs (miRNAs) are associated with numerous human diseases. Studying the associations between miRNAs and diseases contributes to the exploration of effective diagnostic and treatment approaches for diseases. Unfortunately, the use of biological experiments to reveal the potential associations between miRNAs and diseases is time consuming and costly. Therefore, it is very necessary to use simple and efficient calculation models to predict potential disease-related miRNAs. Considering the limitations of other previous methods, we proposed a novel computational model of Symmetric Nonnegative Matrix Factorization for MiRNA-Disease Association prediction (SNMFMDA) to reveal the relation of miRNA-disease pairs. SNMFMDA could be applied to predict miRNAs associated with new diseases. Compared to the direct use of the integrated similarity in previous computational models, the integrated similarity need to be interpolated by symmetric non-negative matrix factorization (SymNMF) before application in SNMFMDA, and the relevant probability of disease-miRNA was obtained mainly through Kronecker regularized least square (KronRLS) method in our model. What's more, the AUC of global leave-one-out cross validation (LOOCV) reached 0.9007, and the AUC based on local LOOCV was 0.8426. Besides, the mean and the standard deviation of AUCs achieved 0.8830 and 0.0017 respectively in 5-fold cross validation. All of the above results demonstrated the superior prediction performance of SNMFMDA. We also conducted three different case studies on Esophageal Neoplasms, Breast Neoplasms and Lung Neoplasms, and 49, 49, and 48 of the top 50 of their predicted miRNAs respectively were confirmed by databases or related literatures. It could be expected that SNMFMDA would be a model with the ability to predict disease-related miRNAs efficiently and accurately.
Introduction
MicroRNAs (miRNAs) are a class of endogenous non-coding RNAs with regulatory functions found in eukaryotes, which are approximately 20–25 nucleotides in length (Ambros, 2001). There are evidences manifesting that miRNAs are one of the most abundant gene regulatory molecules in multicellular organisms, which might affect the expression of many protein-coding genes and play an important regulatory role in animals and plants (Bartel, 2004). With more and more researchers being interested in miRNAs, the researches on miRNAs have been further deepened, and the number of discovered miRNAs is gradually increasing in recent years. The latest database records 24,521 microRNA loci in 206 species and 30,424 mature miRNAs after processing (Kozomara and Griffiths-Jones, 2014). Recently, it has been verified that miRNAs are crucial constituent in cells and may make an important impact in many important biological processes, including proliferation (Cheng et al., 2005), development (Karp and Ambros, 2005), differentiation (Miska, 2005), viral infection (Miska, 2005) and so on. So it is taken for granted that there are associations between miRNAs and the generation as well as development of a number of human diseases (Alvarez-Garcia and Miska, 2005). For instance, by targeting BCL6 corepressor like BCORL1, the migration and invasion of hepatocellular carcinoma (HCC) cells are restrained by mir-876-5p, which provides a new idea for the treatment of HCC (Xu et al., 2018). And it has been confirmed that miR-485-5p inhibits the development and improves the chemosensitivity of breast cancer by regulating survivin, which provides a potential method for addressing the chemoresistance of breast cancer (Wang et al., 2018). Therefore, it is of great significance to research the relations between diseases and miRNAs, which contributes to study the pathogenesis of the disease at the molecular level and makes a big difference in the early diagnosis of human diseases (Jiang et al., 2010). Using biological experiments to identify potential disease-related miRNAs is time-consuming and costly (Jiang et al., 2013), so it is imperative to use low-cost, high-efficiency methods to predict miRNAs associated with diseases. In recent years, since a large amount of biological data has been collected and organized into different databases, it is feasible and necessary to develop computational model to reveal potential disease-miRNA associations based on these databases (Chen, 2015).
In the last couple of years, more and more computational models for miRNA-disease associations prediction have been developed (Chen et al., 2017b). In the view of that miRNAs with similar functions tend to be involved with phenotypically similar diseases and vice versa, numerous computational methods have been proposed recently (Bandyopadhyay et al., 2010). Chen et al. (2016b) presented the model of Within and Between Score for MiRNA-Disease Association prediction (WBSMDA) to predict potential miRNAs associated with diseases. In this method, Within-Score and Between-Score about miRNAs and diseases were calculated by integrating miRNA functional similarity, disease semantic similarity, known miRNA-disease associations and Gaussian interaction profile kernel similarity, and then these two scores were combined to acquire the relation probability of miRNA-disease pair. Unfortunately, how to more reasonably integrate Within-scores and Between-score to calculate relevant probabilities remained unresolved. What's more, Chen et al. (Chen and Yan, 2014) also proposed Regularized Least Squares for MiRNA-Disease Association (RLSMDA) to reveal the unknown relations between miRNAs and diseases. RLSMDA was a semi-supervised method, so it didn't need negative samples. However, the optimal values of parameters in RLSMDA had not yet been obtained, which might affect the prediction performance. By implementing random walk on the miRNA–miRNA functional similarity network, Chen et al. (2012) developed another model named Random Walk with Restart for MiRNA–Disease Association (RWRMDA) to identify miRNAs related with diseases. The outstanding prediction performance of RWRMDA had been confirmed by a number of experiments, but there was still a main limitation in this model. RWRMDA couldn't be applied to predict the relations between miRNAs and new diseases without any known associated miRNAs. Later, Chen et al. (2017a) introduced a novel model called Ranking-based KNN for miRNA-Disease Association prediction (RKNNMDA) to uncover the potential associations between miRNAs and diseases by applying K-Nearest Neighbors (KNN) algorithm to obtain k-nearest-neighbors both for miRNAs and diseases. After resorting the k-nearest-neighbors based on the Support Vector Machine (SVM) ranking model, they got the ranking of association probability of disease-miRNA pairs by implementing weighted voting. RKNNMDA was capable of being implemented on new diseases, which overcame the biggest limitation of RWRMDA. It was a pity that there were also some limitations in this method. RKNNMDA could not be used to score all miRNAs based on the same criteria, especially for miRNAs with more known related diseases. Mørk et al. (2014) proposed a reliable calculation model of miRNA-Protein-Disease (miRPD) which didn't directly predict the miRNAs related with diseases but through proteins. The relations between miRNAs and diseases were uncovered by integrating the predicted and known miRNA–protein associations with the associations of protein–disease text mined from the literature. And the prediction performance of miRPD would be further improved if more involved datasets were taken into account. Xuan et al. (2015) developed a novel method of MIRNAs associated with Diseases Prediction (MIDP) to reveal the associations between miRNAs and diseases by implementing random walk on a miRNA functional similarity network where the similarity scores of miRNAs pairs were obtained through their related diseases. What's more, Xuan et al. also presented its extension method MIDPE to predict the miRNAs related with new diseases. In addition, by constructing a high-dimensional vector space to store the distribution information on miRNAs and diseases, Pasquier et al. (Pasquier and Gardes, 2016) proposed MiRAI to identify the miRNAs associated with diseases based on the similarity of the high-dimensional vectors composed of the distribution information on miRNAs and diseases. Xuan et al. (2013) presented an effective model named Human Disease-related MiRNA Prediction (HDMP) where the calculation method of miRNA functional similarity was improved by taking more related information into account. miRNAs in the same family or cluster were assigned higher weights since they were more likely to be associated with diseases with phenotype similarity. In this method, the sub-scores of the miRNA's k neighbors were equal to the product of the neighbor's weight and the miRNA functional similarity, and then by adding the sub-scores of k neighbors, the relevance score of a miRNA-disease pair was obtained. In addition, Chen et al. (2018a) proposed Network Distance Analysis for MiRNA-Disease Association prediction (NDAMDA) to detect the miRNAs associated with diseases. Compared to other methods, the improvement of NDAMDA lied in that in addition to the direct network distance between two studied diseases (miRNAs), the respective mean distances for each of them and all the rest of diseases (miRNAs) were taken into consideration. By implementing the matrix completion algorithm to update the adjacency matrix which recorded the known associations of disease-miRNA pairs in HMDD and then uncovering the unknown relations, Li et al. (2017) developed a method of Matrix Completion for MiRNA-Disease Association prediction model (MCMDA) without the need of negative samples. Compared with other computational models, the biggest advantage of MCMDA was that it only required known miRNA-disease associations, which also led to that MCMDA couldn't be introduced to predict potential associated miRNAs for new diseases or potential associated diseases for new miRNAs. Furthermore, the optimal parameters of this method were still unknown. In addition, by integrating miRNA functional similarity, disease semantic similarity, Gaussian interaction profile kernel similarity, and miRNA-disease associations confirmed by experiments into a heterogeneous graph, Chen et al. (2016c) presented a model of Heterogeneous Graph Inference for MiRNA-Disease Association prediction (HGIMDA) to reveal the unknown relations of miRNA-disease pairs by incorporating related data into a heterogeneous graph and summarizing all paths with the length equal to three to calculate the association probability of disease-miRNA pair. Unfortunately, limitations also existed in this method that for those miRNAs with more known related diseases, scores made by HGIMDA were generally higher than those miRNAs with less. Later, Chen et al. (2018b) proposed another method called Graph Regression for MiRNA-Disease Association prediction (GRMDA). In this method, by using two matrix decomposition methods to extract important correlation properties and filter noise, graph regression was performed synchronously in three potential spaces including the associated space, miRNA similarity space, and disease similarity space to reveal the potential disease-miRNAs associations. However, there were still some problems of GRMDA to be settled. For example, according to the size of the matrix, how to choose the optimal parameters in SVD and PLS remained unsolved. By optimizing the existing method for maximizing the flow of information, which was mainly used to prioritize disease-associated protein-coding genes, Yu et al. (2017) developed a combinatorial prioritization algorithm to predict the miRNA-disease associations. This method didn't require negative samples, which solved the problem that negative microRNA-disease associations were difficult to obtain.
In this paper, we proposed Symmetric Nonnegative Matrix Factorization for MiRNA-Disease Association prediction (SNMFMDA) to predict potential miRNA-disease associations. The process was mainly divided into two steps. Firstly, we used symmetric non-negative matrix factorization (SymNMF) to interpolate the integrated similarity matrix. Secondly, based on interpolated integrated similarity matrix, we utilized Kronecker regularized least square (KronRLS) method to obtained disease-miRNA association score matrix. We implemented global and local Leave-One-Out Cross Validation (LOOCV) and 5-fold cross validation to assess the prediction performance of SNMFMDA. As shown in the results, the AUC values of global LOOCV, local LOOCV, and 5-fold cross validation of SNMFMDA reached 0.9007, 0.8426, and 0.8830 ± 0.0017 respectively, which verified the excellent prediction performance of SNMFMDA.
Materials and Methods
Human miRNA-Disease Association
In this paper, we obtained known human disease-miRNA associations from HMDD v2.0, which recorded 5430 experimentally verified associations between 383 diseases and 495 miRNAs. To better represent whether there were known associations between diseases and miRNAs, we defined a nd × nm adjacency matrix A, where nd and nm corresponded to the number of diseases and miRNAs respectively. If the relation between disease d(i) and miRNA m(j) had been verified, the value of the element A(d(i), d(j)) of the matrix was 1, otherwise 0.
MiRNA Functional Similarity
On the basis of the assumption that functionally similar miRNAs tend to be associated with similar diseases and vice versa, miRNA functional similarities were calculated in this paper (Wang et al., 2010), and we could download them from http://www.cuilab.cn/fles/images/cuilab/misim.zip. For the sake of better describing the functional similarity between miRNAs, we defined the miRNA functional similarity matrix FS, where the element FS(m(i), m(j)) represented the functional similarity score between miRNA m(i) and m(j) (For the specific calculation process of miRNAs functional similarity, please see Supplementary Material).
Disease Semantic Similarity Model 1
To describe the association between diseases, the Directed Acyclic Graphs (DAGs) were built. DAG(D) = (D, T(D), E(D)) was applied to indicate disease D, where T(D) was a set of nodes composed of node D itself and its ancestor nodes and E(D) was a set consisting of edges directly from parent nodes to the child nodes (Wang et al., 2010). The contribution of disease d to the semantic value of disease D in DAG(D) and the semantic value of disease D were defined as follows:
where ▵ is the semantic contribution factor. The contribution of disease D to its own semantic value was 1, and the contribution of other diseases to the semantic value of disease D was negatively related to the distance between the disease and disease D, so the diseases in the same layer might have the same contribution to the semantic value of disease D.
Here, based on the model in the paper (Xuan et al., 2013), we constructed disease semantic similarity matrix SS1, whose element SS1(d(i), d(j)) indicated the semantic similarity score between disease d(i) and d(j). Based on the assumption that the more DAGs the two diseases overlapped, the greater their semantic similarity would be. The disease semantic similarity between disease d(i) and d(j) was calculated as follows:
Disease Semantic Similarity Model 2
For these diseases that appeared in the same layer of the DAG(A), according to the above definition of the disease semantic similarity model 1, they had the same contribution to the semantic value of the disease A. However, they might appear in different number of disease DAGs. For example, for two diseases d(i) and d(j) that appeared in the same layer of DAG(A), disease d(i) occurred in more disease DAGs, while d(j) appeared in less. It was clear that the contributions of the two diseases to the semantic value of disease A were different and the disease d(i) should have a less contribution to the semantic value of the disease A than d(j). Therefore, it was unreasonable to simply calculate the contribution to the semantic value of disease according to the definition of the disease semantic similarity model 1. Here, according to the model in the paper (Xuan et al., 2013), we defined the disease semantic similarity model 2 to supplement model 1. In the second model, diseases appeared in the same layer of DAG(A) might not necessarily ensure that they had the same contribution to the semantic value of disease A. The contribution of disease D to the semantic value of disease A was calculated as follows:
The semantic value of disease A was calculated as follows:
Similar to the disease semantic similarity matrix SS1, the element SS2(d(i), d(j)) of the disease semantic similarity matrix SS2 was calculated as follows:
Here, SS2(d(i), d(j)) was the disease semantic similarity between disease d(i) and d(j). Combining the two models of disease semantic similarity, we could calculate the final disease semantic similarity matrix SS as follow:
Gaussian Interaction Profile Kernel Similarity
On the basis of the assumption that functionally similar miRNAs were more likely to be associated with similar diseases and vice versa, by taking the topological information of known miRNA-disease association network into account, we defined Gaussian interaction profile kernel similarity for diseases to describe the similarities between diseases based on the model in the paper (van Laarhoven et al., 2011). Here, we applied binary vector IP(d(i)) to represent the ith row of the adjacency matrix A, which recorded the association information of disease d(i) with all miRNAs. The Gaussian interaction profile kernel similarity matrix for diseases was defined as KD. The element KD(d(i), d(j)) indicated the Gaussian interaction profile kernel similarity between disease d(i) and d(j) and could be calculated as follows:
Here, the role of parameter γd is to control the kernel bandwidth and it could be obtained by normalizing another new bandwidth parameter by the average number of associated miRNAs for all the diseases.
Here, we set the value of to 1. Similarly, the Gaussian interaction profile kernel similarity matrix KM was defined as follows:
The binary vector IP(m(i)) represents the ith column of the adjacency matrix A. Similar to γd, parameters γm was calculated as follows:
Here, the value of was set to 1.
Integrated Similarity for miRNAs and Diseases
As we know, many diseases can be described by a DAG. Based on the assumption that two diseases with large overlapping parts of their DAGs are considered to have large semantic similarity, we could calculate the semantic similarity between diseases, but we could not get DAG for all diseases, so for those diseases without DAG, we could not calculate the semantic similarity between them and other diseases. Therefore, we constructed integrated similarity matrix SD for diseases by integrating disease semantic similarity matrix and Gaussian interaction profile kernel similarity matrix in the following way according to the model in this paper (Chen et al., 2016b):
Similar to disease, the integrated similarity matrix SM was calculated as follow:
SNMFMDA
Motivated by the paper (Chen and Li, 2017), in this paper, we proposed SNMFMDA to predict potential miRNA-disease associations and the flow chart of the algorithm is shown in Figure 1. First step, we used SymNMF to interpolate the integrated similarity matrix SM and SD. Second step, based on interpolated integrated similarity matrix SM, SD, we utilized KronRLS method to obtained score matrix S with the same dimension as the adjacency matrix A, and each element of S was the associated probabilities of the corresponding disease-miRNA pairs. The two-step process was as follows:
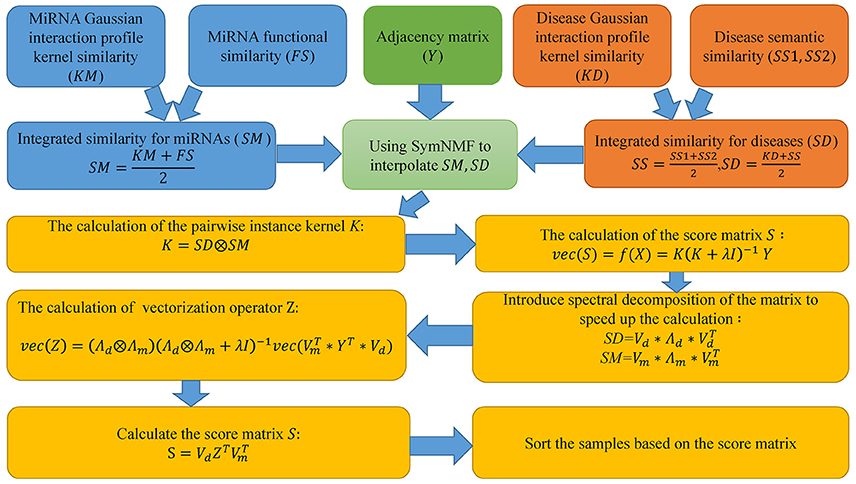
Figure 1. The flowchart of SNMFMDA included three steps: the integration of data; the calculation of the score matrix; the sorting of samples.
SymNMF
As an unsupervised learning method, non-negative matrix factorization (NMF) was extremely versatile and it had gradually become one of the most popular multidimensional data processing tools in signal processing, semantic analysis of documents and image engineering (He et al., 2011). In our model, we improved the integrated similarity by introducing SymNMF, which was a special kind of nonnegative matrix factorization. For the matrix SD, our purpose was to find a matrix P with the same size as the integrated similarity matrix SD, which also satisfied the following requirement:
The specific process was as follows: The first step was initialization and we constructed a random matrix P0 whose elements were all positive as the initialization of the matrix P. Pi indicated the matrix corresponding to P in the beginning of the ith update, and the norm Ei in ith update could be computed as follow:
The second step was update. Here, we temporarily marked P as Pnew after each update. The specific process was as follows:
where A.*B and A./B were the entrywise product (i.e., Hadamard product) and entrywise division respectively. The value of α should be less than 1 but close to 1, here, we set α = 0.999. Then compared Enew to Ei, if EN was smaller, update process ended, otherwise, updated P as follows:
After that, the procedure went back to formula (15) and started the next update. Using SymNMF to perform interpolation on the similarity matrix SM was similar to SD.
KronRLS
First of all, we needed to build a list D = [d(1), d(2), …d(nd)] for diseases. Similarly, list M = [m(1), m(2), …m(nm)] was constructed to denoted nm miRNAs. All the columns of adjacency matrix A were stitched together to form a n-dimensional column vector Y where n = nd × nm indicated the total number of disease-miRNA pairs. And then we built another n-dimensional column vectors X whose element Xi represented the disease-miRNA pair corresponding to the ith element in Y. The purpose of the RLS algorithm was to find the mapping function f from vector X to score matrix S by minimizing the following function.
where ||f||K is a norm of function f on the Hilbert space related with the kernel K. λ is a regularization parameter that determine the trade-off between prediction error and model complexity, here, we set it to 1. The representative theorem ensured that the Equation (23) had a closed form solution:
Here, a is also an n-dimensional vector and it could be get by solving the following equation:
where I is the identity matrix and K is named as the pairwise instance kernel to represent the similarity of two data points in the Hilbert space. To be specific, for two disease-miRNA pairs (di, mj) and (dw, mz), K((di, mj), (dw, mz)) indicates the similarity between the two disease-miRNA pairs. And the kernel could be calculated as follow:
where SD ⊗ SM is the Kronecker product of SD and SM, and the relation probabilities of all disease-miRNA pairs could be obtained according to the kernel as follow:
Here, vec(·) is a vectorization operator that combine all the columns of a matrix into a column vector.
In order to solve the problem more effectively, we introduced spectral decomposition of the matrix to speed up the calculation. The decompositions of integrated similarity SD, SM and K were defined as follow:
Here, the dimension of the matrix ∨d(∨m, ∨) is the same as SD(SM, K), and each of its columns is the eigenvector of the matrix SD(SM, K). Matrix ∧d(∧m, ∧) is a diagonal matrix whose diagonal element ∧dii(∧mii, ∧ii) is the eigenvalue of SD(SM, K) corresponding to the ith column [i.e., the ith eigenvector of SD(SM, K)]. Then, the Kronecker product of SD and SM could be calculated as follows:
where:
Here, we introduced a property of the Kronecker product:
By integrating the above formulas, the score matrix S could be calculated as follows:
where:
Results
Performance Evaluation
In this study, based on the 5,430 confirmed associations between 383 diseases and 495 miRNAs recorded in HMDD v2.0, local and global LOOCV were applied to test the prediction performance of SNMFMDA. In LOOCV, each of the 5,430 known associations (positive samples) was left out in turn as the test sample and the remaining 5,429 known associations were considered as training samples, while the miRNA-disease pairs without verified association were viewed as candidate samples (unknown samples), and then we applied SNMFMDA to calculated the association probability of candidate samples and test sample. In global LOOCV, the score of test sample was ranked with all of the candidate samples, while we sort it with all the unknown samples that contained the studied diseases in local LOOCV. When the ranking of test sample was higher than the given threshold, we affirmed that SNMFMDA had correctly predicted the sample. We set the true positive rate (TPR, sensitivity) as the vertical axis and the false positive rate (FPR, 1-specificity) as the horizontal axis. When the thresholds took different values, they correspond to different points in the coordinate system. The bight composed of all these points was the Receiver operating characteristics (ROC) curve. Here, Sensitivity was the ratio of the number of correctly predicted test samples to the total number of positive samples, and specificity was the percentage of candidate samples whose ranking were lower than the given threshold to all of the unknown samples. The area under the ROC curve (AUC) was calculated to evaluate the reliability of SNMFMDA. The AUC value of 0.5 meant that the computational model was equivalent to random prediction, and AUC = 1 indicated that the prediction performance of the calculation model was excellent. In other words, when the value of AUC was greater than 0.5 and less than 1, the larger the value, the better the prediction performance.
The comparison of the prediction performance between a couple of computational methods based on the AUC value of global and local LOOCV respectively was shown in Figure 2. As a consequence, the AUC of SNMFMDA was 0.9007, while the AUC values of HGIMDA (Chen et al., 2016c), MCMDA (Li et al., 2017), MaxFlow (Yu et al., 2017), RLSMDA (Chen and Yan, 2014), HDMP (Xuan et al., 2013), WBSMDA (Chen et al., 2016b) were respectively 0.8781, 0.8749, 0.8624, 0.8426, 0.8366, 0.8030 in global LOOCV. In local LOOCV, SNMFMDA obtained AUC of 0.8426, which were clearly better than HGIMDA (0.8077), MCMDA (0.7718), MaxFlow (0.7774), RLSMDA (0.6953), HDMP (0.7702), WBSMDA (0.8031), MiRAI (0.6299), MIDP (0.8196), and RWRMDA (0.7891). Both RWRMDA and MIDP weren't capable to predict potential related miRNAs for all diseases at the same time, so we could only evaluate their prediction performance with local LOOCV instead of global LOOCV. Besides, the association probabilities of candidate samples calculated by MiRAI had a high-positive correlation with the number of known associations of corresponding diseases. The more known miRNAs associated with a disease, the greater the disease-related candidate samples' association probabilities would be. Thus, it wasn't reasonable to compare the association probabilities of candidate samples corresponding to different diseases. Therefore, we couldn't apply global LOOCV to evaluate the prediction performance of RWRMDA, MIDP, and MiRAI. What's more, as could be seen from Figure 2, the value of the AUC for local LOOCV of MiRAI was relatively small. This was because that the core of MiRAI was collaborative filtering which caused its prediction accuracy to heavily depend on the number of known miRNA-disease associations. The database used in our method had 383 diseases but there were few known miRNAs associated with each disease. Therefore, the predictive performance of MiRAI based on this database was far worse than that in the original literature where the training database contained more verified associations for each disease.
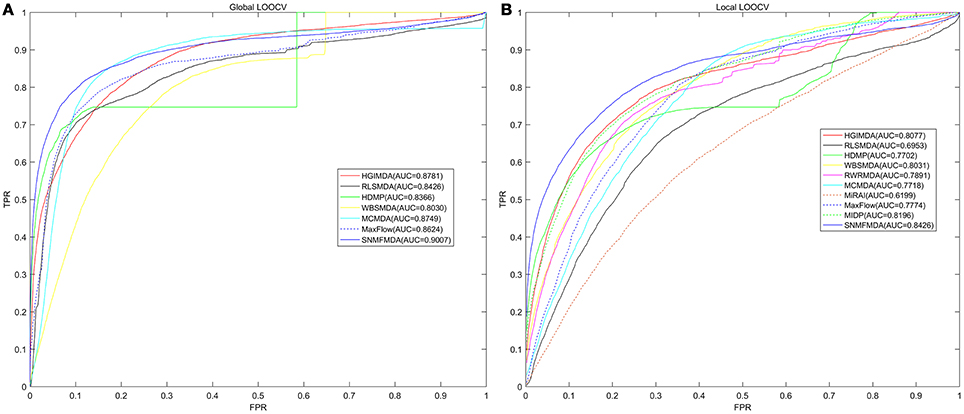
Figure 2. Comparison of prediction performance between SNMFMDA and other computation models (HGIMDA, RLSMDA, HDMP, WBSMDA, MCMDA, MaxFlow, MiRAI, MIDP) based on global (A) and local (B) cross validation results. As shown in the figure, the AUC value of global and local LOOCV of SNMFMDA reached 0.9007 and 0.8426, which fully proved the superior prediction performance of SNMFMDA.
In addition, we also applied 5-fold cross-validation to evaluate the predictive performance of SNMFMDA. All known miRNA-disease associations were randomly divided into five equal parts, and each part was in turn treated as the test sample while the other four parts were treated as the training samples. Besides, in order to reduce the influence of the division of known associations on prediction accuracy, we performed 100 random divisions. It could be seen from the results that the mean and standard deviation of the AUC in 5-fold cross validation respectively reached 0.8830 and 0.0017, which was obviously better than MCMDA (0.8767 ± 0.0011), MaxFlow (0.8579 ± 0.001), RLSMDA (0.8569 ± 0.0020), HDMP (0.8342 ± 0.0010), and WBSMDA (0.8185 ± 0.0009). In summary, all cross-validation results further proved the superior prediction performance of SNMFMDA.
Case Studies
In addition to cross-validation, we also used case studies to evaluate the prediction accuracy of our computational model. Here, we applied three different case studies on Esophageal Neoplasms (EN), Breast Neoplasms (BN), and Lung Neoplasms (LN) to test the prediction performance from different aspects. In the first kind, we could obtain 5,430 known miRNA-disease associations (positive samples) from HMDD v2.0 (Li et al., 2014) for 383 diseases and 495 miRNAs, and the remaining 184,155 samples were unknown samples. In order to predict the potential associations of miRNA-disease pairs from unknown samples, we scored them by SNMFMDA and ranked them. Finally, we verified the top 50 potential associated miRNAs for the investigated disease by two other databases dbDEMC (Yang et al., 2010) and miR2Disease (Jiang et al., 2009) that recorded a number of verified miRNA-disease associations. In order to assess SNMFMDA's prediction power for new diseases without any known associated miRNAs, the second kind of case study was implemented. Here, when we studied a disease, we first removed all its known associations based on the HMDD v2.0 that was, all the 1 s in the row corresponding to this disease in the adjacency matrix were turned to 0. After that we used SNMFMDA to score all miRNAs for the investigated disease and then sorted these miRNAs. Finally, we verified whether the associations between the disease and the top 50 miRNAs were verified by the three databases dbDEMC, miR2Disease and HMDD v2.0. Because there are a number of databases in which known miRNA-diseases associations were recorded, when the prediction accuracy of computation method based on a certain database was good, it couldn't explain that the prediction performance of the method was superior. To prove the applicability of our model to different databases, we carried out the third type of case studies. The third case study was similar to the first, and the difference between them was that the dataset on which SNMFMDA was based was not the same. There were 1895 verified associations between 137 diseases and 271 miRNAs recorded in the database HMDD v1.0 used in the third case study. Finally, we applied three databases (HMDD v2.0, miR2Disease and dbDEMC) to verify the top 50 predicted miRNAs for the investigated disease after utilizing our calculation model.
As one of the most common tumors in the world, Esophageal Neoplasms (EN) has the top 10 morbidity and mortality in all cancers (He et al., 2012). According to the latest estimates of related departments in US, in 2018, there would be 12,850 patients dying from EN, accounting for 4% of all patients dying of cancer (Siegel et al., 2018). Although the treatment method have been improved, the damage of EN to human have not been significantly reduced (He et al., 2012). The survival rate of patients with EN was less than 25% in the last five years (Kim et al., 2011). Research showed that if EN could be diagnosed early, its mortality rate was expected to drop to 10% (Daly et al., 2000), so finding better and more efficient diagnosis and treatment was imperative (Xie et al., 2013). More and more researches indicated that there was a close relationship between miRNAs and the development of human diseases (Alvarez-Garcia and Miska, 2005). A number of associations between miRNAs and EN had been verified. For instance, by sponging miR-200a, which was functionally similar to a competitive endogenous RNA, lncRNA MALAT1 adjusted the expression of ZEB1 and ZEB2 to facilitate the invasion and migration of EN cells by mean of inducing epithelial-mesenchymal transition (Zhang et al., 2017). We used SNMFMDA to perform the first case study on EN, and the results showed that 47 of the top 50 predicted EN-related miRNAs were verified by other two databases dbDEMC and miR2Disease. For the remaining three miRNAs that had not been verified by the above two databases, there were studies showing that serum expression levels of mir-218 (15th in the prediction list) in EN patients were significantly lower than those in healthy people, and the levels were related to tumor differentiation, staging, and lymph node metastasis. For this reason, mir-218 was highly likely to be the target of the early diagnosis of EN, which provided a new idea for the detection of this cancer (Jiang et al., 2015). In EN cells, mir-122 (43th in the prediction list) targeted pyruvate kinase M2 (PKM2), and Tanshinone IIA could limit the expression of PKM2 by promoting the expression of mir-122, which in turn restricted the growth of EN cells (Zhang et al., 2016). As could be seen from the verification results, only one of the top 50 predicted miRNAs had not been validated by the database or literature (see Table 1).
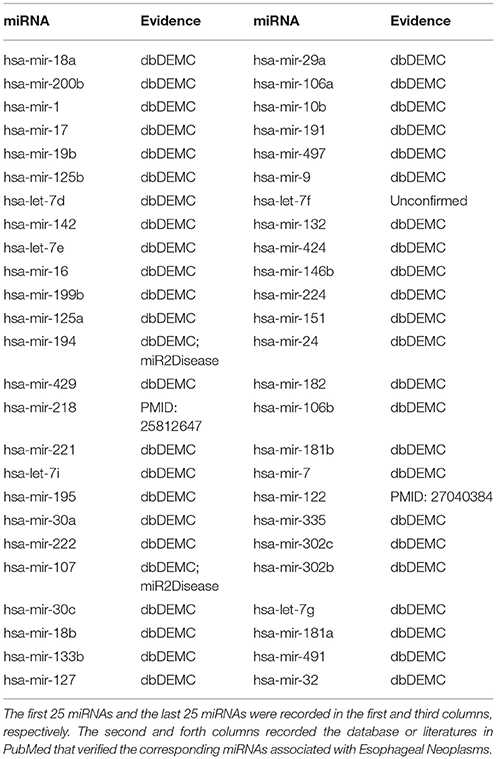
Table 1. Prediction of the top 50 predicted miRNAs associated with Esophageal Neoplasms.
In order to facilitate further validation and research, we have provided the complete prediction list of potential miRNAs associated with all the 383 human diseases in HMDD v2.0 (see Supplementary Table 1).
Breast Neoplasms (BN) is a type of cancer with high morbidity and mortality among women in the United States (Kelsey and Horn-Ross, 1993). According to the prediction of relevant departments, 40,920 women would die from BN in the United States in 2018, accounting for 14% of the total cancer deaths (Siegel et al., 2018). According to the current medical level, the only way to improve the cure rate and reduce the mortality rate of BN lies in the early detection and timely treatment (Tao et al., 2015). In order to improve the diagnostic efficiency, the researchers have put forward many methods including the prediction of potential relevant miRNAs of BN. It has been proved that the expression levels of mir-21 and mir-146a in plasma samples of BN patients were obviously higher than that of healthy volunteers and we could identify whether a patient had BN based on the expression level of mir-21 and mir-146a in plasma (Kumar et al., 2013). We implemented the second case study on BN and 45 of the top 50 miRNAs potentially associated with BN were verified by databases (HMDD, dbDEMC and miR2Disease). Among the remaining 5 miRNAs that were not validated by the databases, the expression of mir-142 (12th in the prediction list) was dysregulated in BN cells and mir-142 could restricted the invasion of BN cells by simultaneously targeting WASL, Integrin Alpha V, and Additional Cytoskeletal Elements (Schwickert et al., 2015). The expression of mir-378a-3p (20th in the prediction list) was lower in BN tissues, and it acted on the endocrine resistance mechanism of BN by regulating the expression of GOLT1A (Ikeda et al., 2015). It was also verified that there was excessive expression of mir-302f (48th in the prediction list) in HER2-postive BN (Kang et al., 2014). What's more, researches also confirmed that there was overexpression of mir-744 (49th in the prediction list) in BN cell and mir-744 played a part in the drug resistance of BN (Chen et al., 2016a). The above results (see Table 2) showed that 49 of the top 50 potential BN-associated miRNAs predicted by SNMFMDA were validated, which indicated that the prediction performance of SNMFMDA was still reliable when it was applied to new diseases.

Table 2. Prediction of the top 50 predicted miRNAs associated with Breast Neoplasms.
Lung Neoplasms (LN) is one of the most deadly cancers (Liu and Wei, 2018), and its poor prognosis have also caused great harm to human health (Zhao et al., 2018). According to the estimation of the American Cancer Society, in 2018, new cases of bronchus and LN would reach 121,680, accounting for 14% of all newly diagnosed cancer patients, and it was estimated that there would be 83550 patients dying from lung cancer, more than a quarter of the total deaths (Siegel et al., 2018). The early diagnosis and treatment of LN is very difficult, fortunately, more and more studies have shown that miRNAs are closely related to the development, progression, and progression of LN (Zhao et al., 2018). For example, researches showed that there was significantly higher expression of mir-221 in LN patients than that in healthy people, and biological analysis indicated that the target of mir-221 was most likely related to the formation and development of LN (Zhu et al., 2017). Besides, mir-221 was very likely to become a non-aggressive biomarker for the diagnosis of LN (Zhu et al., 2017). As an embryo-expressing lung miRNA, mir-127 had been shown to be closely linked to the poor prognosis of LN (Shi et al., 2017). Therefore, the prediction of miRNAs associated with LN could enable us to understand the pathogenesis of cancer and might provide novel diagnostic methods and treatment approaches. We applied SNMFMDA to perform the third case study on LN to test the prediction power of the model when it is applied to another database HMDD v1.0. The prediction result showed that 44 of the top 50 potential LN-associated miRNAs were verified by other databases (dbDEMC, miR2Diseaes, HMDD v2.0). For the remaining 6 miRNAs that weren't verified by the three databases, studies showed that the mir-92 (6th in the prediction list) family was less expressed in cisplatin-resistant cells, which indicated that the mir-92 family played a part in the regulation of cisplatin resistance in non-small cell lung cancer (Zhao et al., 2015). Some researches confirmed that the overexpression of mir-194 (20th in the prediction list) produced an effect on the expression of Mpl/ERK pathway proteins and restrained the mitosis and proliferation of non-small cell lung cancer cells by targeting Human nuclear distribution C (hNUDC), which provided a novel strategy for the treatment of LN (Zhou et al., 2016). Studies confirmed that mir-372-3p (38th in the prediction list) was obviously overexpressed in lung squamous cell carcinoma cells and limited the expression of FGF9 by binding to it, which contributed to the proliferation of lung squamous cell carcinoma cells (LSCC). In contrast, the low expression of mir-372-3p or high expression of FGF9 were conducive to inhibit the growth and invasion of LSCC cells (Wang et al., 2017). The expression level of mir-320 (46th in the prediction list) in non-small cell lung cancer (NSCLC) cells was lower than the level in normal cells, and mir-320 limited cell growth in NSCLC cells through targeting fatty acid synthase (Lei et al., 2016). Based on the above results (see Table 3), 48 of the top 50 potential LN-associated miRNAs predicted by SNMFMDA were validated, which indicated that the prediction performance of the model based on other datasets was also very reliable.
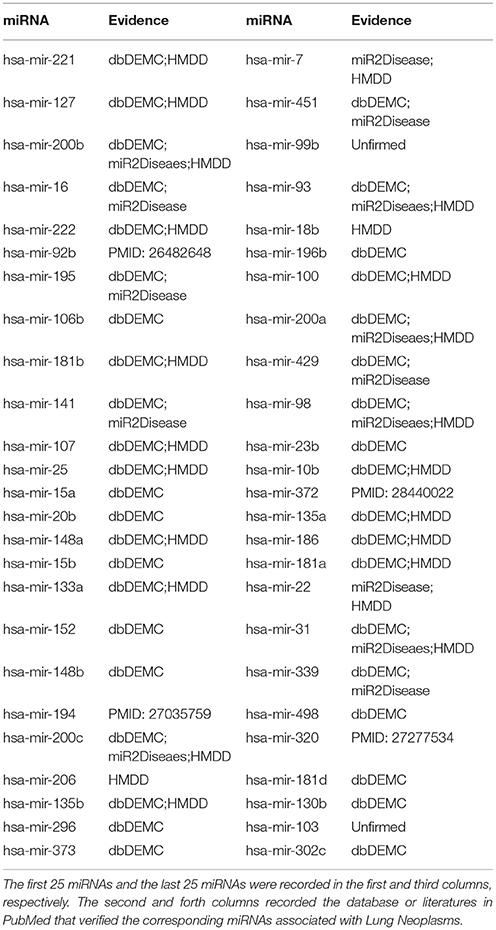
Table 3. Prediction of the top 50 predicted miRNAs associated with Lung Neoplasms based on known associations in HMDD v1.0.
Discussion
As accumulating studies have demonstrated that miRNAs play an extremely important role in human physiological processes, researches on the association between miRNAs and diseases have attracted more and more attention. Since it is time-consuming and costly to use biological experiments to reveal potential miRNA-disease associations, many computational models have been proposed to predict disease-related miRNAs in recent years. In the paper, we developed a novel model of SNMFMDA to reveal the relation of miRNA-disease pairs by integrating the known miRNA-disease associations recorded in HMDD v2.0, miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity for diseases and miRNAs. SNMFMDA overcame the limitation of many previous models that they were incapable of predicting miRNAs associated with new diseases. As shown in the prediction results, the AUC values of global LOOCV, local LOOCV, and 5-fold cross validation reached 0.9007, 0.8426, and 0.8830 ± 0.0017 respectively. As we all know, models with global AUC value above 0.9 were rare, so SNMFMDA is a model with higher credibility. In the future, our model would be an effective tool to reveal potential disease-related miRNAs, which is conducive to the diagnosis and treatment of diseases.
The superior prediction performance of the model was mainly due to the following aspects. Firstly, the database on which SNMFMDA was based was reliable and the model could be used to predict miRNAs potentially associated with new diseases by introducing the information of disease similarity. Secondly, we used SymNMF to interpolate the integrated similarity, while many of the previous methods used the integrated similarity directly. Finally, we introduced spectral decomposition to speed up the calculation of the Kronecker product in our model. Certainly, there are still some limitations to be resolved in the future. For example, SNMFMDA might not score miRNAs using the same criteria, especially for those with more known related diseases. Although the prediction accuracy of SNMFMDA is obviously higher than many previous calculation methods, if the biological database on which our model is based can be further improved, SNMFMDA's prediction performance would be better. The calculation of disease similarity and miRNA similarity used in our model may not be the most perfect method, and we expect to add more biological data sets in future calculations to improve the accuracy of similarity calculations. Besides, SNMFMDA involved calculating the Kronecker product of two matrices. The solution of the Kronecker product of two matrices was equivalent to that each element of the previous matrix multiplied by the next matrix, so the sizes of the Kronecker product was much larger than the first two matrices. Therefore, calculating the Kronecker product often led to memory problems in computer. In addition, our model SNMFMDA did not consider miRNA-protein association and miRNA-cellular pathway association, which significantly affected the prediction performance of the model.
Author Contributions
YZ implemented the experiments, analyzed the result, and wrote the paper. XC conceived the project, developed the prediction method, designed the experiments, and revised the paper. JY analyzed the result and revised the paper. All authors read and approved the final manuscript.
Funding
XC was supported by National Natural Science Foundation of China under Grant No. 61772531.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00324/full#supplementary-material
References
Alvarez-Garcia, I., and Miska, E. A. (2005). MicroRNA functions in animal development and human disease. Development 32, 4653–662. doi: 10.1242/dev.02073
Ambros, V. (2001). microRNAs: tiny regulators with great potential. Cell 107, 823–826. doi: 10.1016/S0092-8674(01)00616-X
Bandyopadhyay, S., Mitra, R., Maulik, U., and Zhang, M. Q. (2010). Development of the human cancer microRNA network. Silence 1:6. doi: 10.1186/1758-907X-1-6
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Chen, H., and Li, J. (2017). “A flexible and robust multi-source learning algorithm for drug repositioning,” in Proceedings of the 8th ACM International Conference on Bioinformatics, (Boston, MA: Computational Biology, and Health Informatics; ACM), 510–515.
Chen, X. (2015). Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 5:13186. doi: 10.1038/srep13186
Chen, X., Liu, M. X., and Yan, G. Y. (2012). RWRMDA: predicting novel human microRNA-disease associations. Mol. Biosyst. 8, 2792–2798. doi: 10.1039/c2mb25180a
Chen, X., Lu, P., Wang, D. D., Yang, S. J., Wu, Y., Shen, H. Y., et al. (2016a). The role of miRNAs in drug resistance and prognosis of breast cancer formalin-fixed paraffin-embedded tissues. Gene 595, 221–226. doi: 10.1016/j.gene.2016.10.015
Chen, X., Wang, L. Y., and Huang, L. (2018a). NDAMDA: network distance analysis for MiRNA-disease association prediction. J. Cell. Mol. Med. 22, 2884–2895. doi: 10.1111/jcmm.13583
Chen, X., Wu, Q. F., and Yan, G. Y. (2017a). RKNNMDA: ranking-based KNN for MiRNA-Disease Association prediction. RNA Biol. 14, 952–962. doi: 10.1080/15476286.2017.1312226
Chen, X., Xie, D., Zhao, Q., and You, Z. H. (2017b). MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinformatics. doi: 10.1093/bib/bbx130. [Epub ahead of print].
Chen, X., Yan, C. C., Zhang, X., You, Z. H., Deng, L., Liu, Y., et al. (2016b). WBSMDA: within and between score for MiRNA-Disease Association prediction. Sci. Rep. 6:21106. doi: 10.1038/srep21106
Chen, X., Yan, C. C., Zhang, X., You, Z. H., Huang, Y. A., and Yan, G. Y. (2016c). HGIMDA: heterogeneous graph inference for miRNA-Disease Association prediction. Oncotarget 7, 65257–65269. doi: 10.18632/oncotarget.11251
Chen, X., and Yan, G. Y. (2014). Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 4:5501. doi: 10.1038/srep05501
Chen, X., Yang, J. R., Guan, N. N., and Li, J. Q. (2018b). GRMDA: graph regression for MiRNA-Disease Association Prediction. Front. Physiol. 9:92. doi: 10.3389/fphys.2018.00092
Cheng, A. M., Byrom, M. W., Shelton, J., and Ford, L. P. (2005). Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 33, 1290–1297. doi: 10.1093/nar/gki200
Daly, J. M., Fry, W. A., Little, A. G., Winchester, D. P., Mckee, R. F., Stewart, A. K., et al. (2000). Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J. Am. Coll. Surg. 190, 562–572. doi: 10.1016/S1072-7515(00)00238-6
He, B., Yin, B., Wang, B., Xia, Z., Chen, C., and Tang, J. (2012). MicroRNAs in esophageal cancer (review). Mol. Med. Rep. 6, 459–465. doi: 10.3892/mmr.2012.975
He, Z., Xie, S., Zdunek, R., Zhou, G., and Cichocki, A. (2011). Symmetric nonnegative matrix factorization: algorithms and applications to probabilistic clustering. IEEE Trans. Neural Netw. 22, 2117–2131. doi: 10.1109/TNN.2011.2172457
Ikeda, K., Horie-Inoue, K., Ueno, T., Suzuki, T., Sato, W., Shigekawa, T., et al. (2015). miR-378a-3p modulates tamoxifen sensitivity in breast cancer MCF-7 cells through targeting GOLT1A. Sci. Rep. 5:13170. doi: 10.1038/srep13170
Jiang, Q., Hao, Y., Wang, G., Juan, L., Zhang, T., Teng, M., et al. (2010). Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 4(Suppl. 1):S2. doi: 10.1186/1752-0509-4-S1-S2
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting human microRNA-disease associations based on support vector machine. Int. J. Data Min. Bioinform. 8, 282–293. doi: 10.1504/IJDMB.2013.056078
Jiang, Q., Wang, Y., Hao, Y., Juan, L., Teng, M., Zhang, X., et al. (2009). miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 37, D98–D104. doi: 10.1093/nar/gkn714
Jiang, Z., Song, Q., Yang, S., Zeng, R., Li, X., Jiang, C., et al. (2015). Serum microRNA-218 is a potential biomarker for esophageal cancer. Cancer Biomark. 15, 381–389. doi: 10.3233/CBM-150480
Kang, H. S., Kim, J., Jang, S. G., Kwon, S. Y., Park, Y. S., Green, J. E., et al. (2014). MicroRNA signature for HER2-positive breast and gastric cancer. Anticancer Res. 34, 3807–3810.
Karp, X., and Ambros, V. (2005). Developmental biology. Encountering microRNAs in cell fate signaling. Science 310, 1288–1289. doi: 10.1126/science.1121566
Kelsey, J. L., and Horn-Ross, P. L. (1993). Breast cancer: magnitude of the problem and descriptive epidemiology. Epidemiol. Rev. 15, 7–16. doi: 10.1093/oxfordjournals.epirev.a036118
Kim, T., Grobmyer, S. R., Smith, R., Ben-David, K., Ang, D., Vogel, S. B., et al. (2011). Esophageal cancer–the five year survivors. J. Surg. Oncol. 103, 179–183. doi: 10.1002/jso.21784
Kozomara, A., and Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Kumar, S., Keerthana, R., Pazhanimuthu, A., and Perumal, P. (2013). Overexpression of circulating miRNA-21 and miRNA-146a in plasma samples of breast cancer patients. Indian J. Bioche. Biophys. 50, 210–214.
Lei, T., Zhu, Y., Jiang, C., Wang, Y., Fu, J., Fan, Z., et al. (2016). MicroRNA-320 was downregulated in non-small cell lung cancer and inhibited cell proliferation, migration and invasion by targeting fatty acid synthase. Mol. Med. Rep. 14, 1255–1262. doi: 10.3892/mmr.2016.5370
Li, J. Q., Rong, Z. H., Chen, X., Yan, G. Y., and You, Z. H. (2017). MCMDA: matrix completion for MiRNA-disease association prediction. Oncotarget 8, 21187–21199. doi: 10.18632/oncotarget.15061
Li, Y., Qiu, C., Tu, J., Geng, B., Yang, J., Jiang, T., et al. (2014). HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 42, D1070–D1074. doi: 10.1093/nar/gkt1023
Liu, L., and Wei, S. (2018). [Research progress of KRAS mutation in non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi 21, 419–424. doi: 10.3779/j.issn.1009-3419.2018.05.11
Miska, E. A. (2005). How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 15, 563–568. doi: 10.1016/j.gde.2005.08.005
Mørk, S., Pletscher-Frankild, S., Palleja Caro, A., Gorodkin, J., and Jensen, L. J. (2014). Protein-driven inference of miRNA-disease associations. Bioinformatics 30, 392–397. doi: 10.1093/bioinformatics/btt677
Pasquier, C., and Gardès, J. (2016). Prediction of miRNA-disease associations with a vector space model. Sci. Rep. 627036. doi: 10.1038/srep27036
Schwickert, A., Weghake, E., Brüggemann, K., Engbers, A., Brinkmann, B. F., Kemper, B., et al. (2015). microRNA miR-142-3p inhibits breast cancer Cell invasiveness by synchronous targeting of WASL, integrin Alpha, V., and additional Cytoskeletal elements. PLoS ONE 10:e0143993. doi: 10.1371/journal.pone.0143993
Shi, L., Wang, Y., Lu, Z., Zhang, H., Zhuang, N., Wang, B., et al. (2017). miR-127 promotes EMT and stem-like traits in lung cancer through a feed-forward regulatory loop. Oncogene 36, 1631–1643. doi: 10.1038/onc.2016.332
Siegel, R. L., Miller, K. D., and Jemal, A. (2018). Cancer statistics. CA Cancer J. Clin. 68, 7–30. doi: 10.3322/caac.21442
Tao, Z., Shi, A., Lu, C., Song, T., Zhang, Z., and Zhao, J. (2015). Breast cancer: epidemiology and etiology. Cell Biochem. Biophys. 72, 333–338. doi: 10.1007/s12013-014-0459-6
van Laarhoven, T., Nabuurs, S. B., and Marchiori, E. (2011). Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 27, 3036–3043. doi: 10.1093/bioinformatics/btr500
Wang, D., Wang, J., Lu, M., Song, F., and Cui, Q. (2010). Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26, 1644–1650. doi: 10.1093/bioinformatics/btq241
Wang, M., Cai, W. R., Meng, R., Chi, J. R., Li, Y. R., Chen, A. X., et al. (2018). miR-485-5p suppresses breast cancer progression and chemosensitivity by targeting survivin. Biochem. Biophys. Res. Commun. 50, 48–54. doi: 10.1016/j.bbrc.2018.04.129
Wang, Q., Liu, S., Zhao, X., Wang, Y., Tian, D., and Jiang, W. (2017). MiR-372-3p promotes cell growth and metastasis by targeting FGF9 in lung squamous cell carcinoma. Cancer Med. 6, 1323–1330. doi: 10.1002/cam4.1026
Xie, Z., Chen, G., Zhang, X., Li, D., Huang, J., Yang, C., et al. (2013). Salivary microRNAs as promising biomarkers for detection of esophageal cancer. PLoS ONE 8:e57502. doi: 10.1371/journal.pone.0057502
Xu, Q., Zhu, Q., Zhou, Z., Wang, Y., Liu, X., Yin, G., et al. (2018). MicroRNA-876-5p inhibits epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma by targeting BCL6 corepressor like 1. Biomed. Pharmacother. 103, 645–652. doi: 10.1016/j.biopha.2018.04.037
Xuan, P., Han, K., Guo, M., Guo, Y., Li, J., Ding, J., et al. (2013). Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 8:e70204. doi: 10.1371/annotation/a076115e-dd8c-4da7-989d-c1174a8cd31e
Xuan, P., Han, K., Guo, Y., Li, J., Li, X., Zhong, Y., et al. (2015). Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 31, 1805–1815. doi: 10.1093/bioinformatics/btv039
Yang, Z., Ren, F., Liu, C., He, S., Sun, G., Gao, Q., et al. (2010). dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genomics 11(Suppl. 4):S5. doi: 10.1186/1471-2164-11-S4-S5
Yu, H., Chen, X., and Lu, L. (2017). Large-scale prediction of microRNA-disease associations by combinatorial prioritization algorithm. Sci. Rep. 7:43792. doi: 10.1038/srep43792
Zhang, H. S., Zhang, F. J., Li, H., Liu, Y., Du, G. Y., and Huang, Y. H. (2016). Tanshinone A inhibits human esophageal cancer cell growth through miR-122-mediated PKM2 down-regulation. Arch. Biochem. Biophys. 598, 50–56. doi: 10.1016/j.abb.2016.03.031
Zhang, Q. Q., Cui, Y. H., Wang, Y., Kou, W. Z., Cao, F., Cao, X. J., et al. (2017). [Mechanism of long non-coding RNA-metastasis associated lung adenocarcinoma transcript 1 induced invasion and metastasis of esophageal cancer cell EC-109]. Zhonghua Zhong Liu Za Zhi 39, 405–411. doi: 10.3760/cma.j.issn.0253-3766.2017.06.002
Zhao, H., Kuang, L., Wang, L., Ping, P., Xuan, Z., Pei, T., et al. (2018). Prediction of microRNA-disease associations based on distance correlation set. BMC Bioinformatics 19:141. doi: 10.1186/s12859-018-2146-x
Zhao, J., Fu, W., Liao, H., Dai, L., Jiang, Z., Pan, Y., et al. (2015). The regulatory and predictive functions of miR-17 and miR-92 families on cisplatin resistance of non-small cell lung cancer. BMC Cancer 15:731. doi: 10.1186/s12885-015-1713-z
Zhou, L., Di, Q., Sun, B., Wang, X., Li, M., and Shi, J. (2016). MicroRNA-194 restrains the cell progression of non-small cell lung cancer by targeting human nuclear distribution protein C. Oncol. Rep. 35, 3435–3444. doi: 10.3892/or.2016.4708
Keywords: microRNA, disease, association prediction, matrix factorization, Kronecker regularized least square
Citation: Zhao Y, Chen X and Yin J (2018) A Novel Computational Method for the Identification of Potential miRNA-Disease Association Based on Symmetric Non-negative Matrix Factorization and Kronecker Regularized Least Square. Front. Genet. 9:324. doi: 10.3389/fgene.2018.00324
Received: 03 July 2018; Accepted: 30 July 2018;
Published: 21 August 2018.
Edited by:
Quan Zou, Tianjin University, ChinaReviewed by:
Jiawei Luo, Hunan University, ChinaJialiang Yang, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2018 Zhao, Chen and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xing Chen, eGluZ2NoZW5AYW1zcy5hYy5jbg==