 Cláudia P. Godinho
Cláudia P. Godinho Paulo J. Dias
Paulo J. Dias Elise Ponçot
Elise Ponçot Isabel Sá-Correia
Isabel Sá-Correia- iBB-Institute for Bioengineering and Biosciences, Department of Bioengineering, Instituto Superior Técnico, Universidade de Lisboa, Lisbon, Portugal
Pleiotropic drug resistance (PDR) family of ATP-binding cassette (ABC) transporters play a key role in the simultaneous acquisition of resistance to a wide range of structurally and functionally unrelated cytotoxic compounds in yeasts. Saccharomyces cerevisiae Pdr18 was proposed to transport ergosterol at the plasma membrane, contributing to the maintenance of adequate ergosterol content and decreased levels of stress-induced membrane disorganization and permeabilization under multistress challenge leading to resistance to ethanol, acetic acid and the herbicide 2,4-D, among other compounds. PDR18 is a paralog of SNQ2, first described as a determinant of resistance to the chemical mutagen 4-NQO. The phylogenetic and neighborhood analysis performed in this work to reconstruct the evolutionary history of ScPDR18 gene in Saccharomycetaceae yeasts was focused on the 214 Pdr18/Snq2 homologs from the genomes of 117 strains belonging to 29 yeast species across that family. Results support the idea that a single duplication event occurring in the common ancestor of the Saccharomyces genus yeasts was at the origin of PDR18 and SNQ2, and that by chromosome translocation PDR18 gained a subtelomeric region location in chromosome XIV. The multidrug/multixenobiotic phenotypic profiles of S. cerevisiae pdr18Δ and snq2Δ deletion mutants were compared, as well as the susceptibility profile for Candida glabrata snq2Δ deletion mutant, given that this yeast species has diverged previously to the duplication event on the origin of PDR18 and SNQ2 genes and encode only one Pdr18/Snq2 homolog. Results show a significant overlap between ScSnq2 and CgSnq2 roles in multidrug/multixenobiotic resistance (MDR/MXR) as well as some overlap in azole resistance between ScPdr18 and CgSnq2. The fact that ScSnq2 and ScPdr18 confer resistance to different sets of chemical compounds with little overlapping is consistent with the subfunctionalization and neofunctionalization of these gene copies. The elucidation of the real biological role of ScSNQ2 will enlighten this issue. Remarkably, PDR18 is only found in Saccharomyces genus genomes and is present in almost all the recently available 1,000 deep coverage genomes of natural S. cerevisiae isolates, consistent with the relevant encoded physiological function.
Introduction
Several ATP-binding cassette (ABC) transporters that catalyze the ATP-dependent active solute transport across cell membranes in yeasts are associated with multidrug/multixenobiotic resistance (MDR/MXR) (Jungwirth and Kuchler, 2006; Monk and Goffeau, 2008; Piecuch and Obłak, 2014). Although these transporters are usually considered drug/xenobiotic pumps, evidence is arising supporting the idea that their involvement in MDR/MXR may result from their specific and, in general, not yet determined biological role in the active transport of physiological substrates (Prasad and Panwar, 2004; Cabrito et al., 2011; Prasad et al., 2016; Godinho et al., 2018). Moreover, the presence of a large number of ABC transporters involved in MDR/MXR in the genomes of yeasts and other organisms, from bacteria to man, also strongly suggests that these transporters may play important physiological roles even in the absence of the cytotoxic compounds to which they confer resistance. For example, the yeast ABC-MDR/MXR family of transporters that includes the Pleiotropic Drug Resistance (PDR) transporters, perform endogenous activities beyond their proposed role as drug exporters, in particular as lipid transporters (Balzi and Goffeau, 1995; Borst et al., 2000; Pomorski et al., 2004; Panwar et al., 2008; Van Meer et al., 2008; Prasad et al., 2016). Saccharomyces cerevisiae genome encodes 10 PDR proteins: Adp1, Aus1, Pdr5, Pdr10, Pdr11, Pdr12, Pdr15, Pdr18, Snq2, and YOL075c (Decottignies and Goffeau, 1997; Paulsen et al., 1998; Paumi et al., 2009). A combined phylogeny and neighborhood analysis of the evolution of these ABC transporters in nine yeast species belonging to the subphylum Saccharomycotina has shown that Pdr18 is a paralog of Snq2 and that SNQ2 and PDR18 genes reside in unshared chromosomal environments (Seret et al., 2009). However, the small number of yeast species genomes available when this study was performed did not allow a firm conclusion concerning the hypothesized gene duplication event at the origin of these two PDR gene sub-lineages. In fact, it was doubtful whether the duplication event remounted to the whole genome duplication (WGD) event or if it was an independent event that occurred post-WGD (Seret et al., 2009).
The S. cerevisiae plasma membrane transporter Pdr18 was described as a MDR/MXR determinant required for ergosterol transport at the plasma membrane level (Cabrito et al., 2011; Teixeira et al., 2012; Godinho et al., 2018). Pdr18 expression was found to lead to increased yeast tolerance to the herbicides 2,4-dichlorophenoxyacetic acid (2,4-D), 2-methyl-4-chlorophenoxyacetic acid (MCPA), and barban, the agricultural fungicide mancozeb, the metal cations Zn2+, Mn2+, Cu2+, and Cd2+ (Cabrito et al., 2011) and to ethanol (Teixeira et al., 2012) and acetic acid (Godinho et al., 2018). The involvement of Pdr18 in the maintenance of yeast plasma membrane ergosterol content under 2,4-D or acetic acid stresses was related with its role as a determinant of resistance to multiple stresses in yeast (Cabrito et al., 2011; Teixeira et al., 2012; Godinho et al., 2018). A coordinated response involving the transcriptional activation of PDR18 and several ergosterol biosynthetic genes was found to occur in response to acetic acid stress, strongly suggesting the involvement of Pdr18 in ergosterol homeostasis in stressed yeast cells (Godinho et al., 2018). Moreover, the proposed role for Pdr18 in ergosterol homeostasis was demonstrated to be important to counteract acetic acid-induced decrease of plasma membrane lipid order, increase of plasma membrane non-specific permeability and decrease of transmembrane electrochemical potential (Godinho et al., 2018).
The PDR18 paralog gene SNQ2 was first described based on its involvement in yeast resistance to the chemical mutagens 4-nitroquinoline 1-oxide (4-NQO) and triaziquone (Servos et al., 1993). Later, several other publications extended the range of compounds to which SNQ2 expression confers increased tolerance in yeast (Servos et al., 1993; Hirata et al., 1994; Mahé et al., 1996a,b; Miyahara et al., 1996; Kolaczkowski et al., 1998; Ververidis et al., 2001; Piper et al., 2003; Wehrschütz-Sigl et al., 2004; van Leeuwen et al., 2012; Ling et al., 2013; Nishida et al., 2013; Snider et al., 2013; Tsujimoto et al., 2015). Although no role in lipid homeostasis was demonstrated for Snq2 transporter, it was shown that Snq2 is involved in the alleviation of estradiol toxicity in S. cerevisiae (Mahé et al., 1996a). For this reason, it was hypothesized that Snq2 could also have affinity for lipid transport, especially for the estradiol structurally related molecule ergosterol (Mahé et al., 1996a; Kuchler et al., 1997).
Gene duplication is considered to be one of the most important forces driving the evolution of genetic functional innovation and genes encoding membrane transporters are one of the functional gene categories that exhibit high number of duplication events (Ohno, 1970; Zhang, 2003; Taylor and Raes, 2004). In yeast, for example, this is the case of genes encoding proteins of the Major Facilitator Superfamily (MFS) of transporters (Dias et al., 2010; Dias and Sá-Correia, 2013, 2014). Also, transporters from the PDR family were involved in multiple gene duplications and gene losses occurring during their evolutionary history (Seret et al., 2009; Kovalchuk and Driessen, 2010). After gene duplication, it is possible the occurrence of the inactivation of one of the copies (pseudogenization), the maintenance of the two copies (dosage effect), the adoption of part of the function or of the expression pattern of their parental gene (subfunctionalization), or the acquisition of a related or new function (neofunctionalization) (Zhang, 2003; Conant and Wolfe, 2008). The duplicate genes are maintained in the genome depending upon their function, mode of duplication, expression rate and the organism taxonomic lineage (Taylor and Raes, 2004).
To better understand the duplication event that gave rise to the PDR18 and SNQ2 paralog genes, the evolutionary history of PDR18 was reconstructed in this work by combining phylogenetic tree building methodology with gene neighborhood analysis in 117 strains genomes belonging to 29 species across the Saccharomycetaceae family. A systematic multidrug/multixenobiotic phenotypic profiling of S. cerevisiae deletion mutants for PDR18 or SNQ2 genes was also performed. Given that the genomes of the post-WGD Candida glabrata species encode only one Pdr18/Snq2 homolog (CgSNQ2) the susceptibility profiling for the Cgsnq2Δ deletion mutant was also examined to get additional insights into the functional divergence of S. cerevisiae Pdr18 and Snq2 and the common ancestral gene on the origin of the post-WGD single duplication event.
Materials and Methods
Identification of the Homologs of S. cerevisiae Pdr18 and Snq2 Proteins in Hemiascomycete Yeast Genomes
A total of 1,110,525 Open Reading Frames (ORFs) encoded in the genomes of 171 strains belonging to 68 different yeast species of the subphylum Saccharomycotina were retrieved and compiled in a local Genome DB (Dias and Sá-Correia, 2013, 2014). A second in-house database built in this work, BLASTp DB, comprises the output values of the blastp algorithm (Altschul et al., 1990) for each pairwise entry of all possible combinations between the translated ORFs compiled in the Genome DB, including length of the alignment, e-value, percentage of identity and similarity, and alignment score. The blastp algorithm used a gapped alignment with the following parameters: open gap (-1), extend gap (-1), threshold for extending hits (11), and word size (3). This approach generated a total of 328 million pairwise alignments. The ORFs encoding PDR proteins were identified through the adoption of a network traversal strategy considering the whole set of blastp pairwise relationships as a network that was subsequently traversed at a range of different e-value thresholds (Dias and Sá-Correia, 2013, 2014; Palma et al., 2017). The S. cerevisiae Pdr18 was selected to represent the PDR sensu stricto (Seret et al., 2009) transporters and was used as a starting node for network traversal. The S. cerevisiae Adp1 and Yol075c were selected to represent the PDR sensu lato transporters (Seret et al., 2009) and also used as starting nodes in independent network traversals. The disjoint sets of translated ORFs obtained from these three traversals were merged. The amino acid sequences of these ORFs were analyzed for potential false positive members of the PDR protein family, protein fragments and/or frameshifts. The remaining amino acid sequences were aligned using MUSCLE software (Edgar, 2004). Subsequently, the protdist and neighbor algorithms made available by the PHYLIP software suite (Felsenstein, 1989) were used to construct a preliminary phylogenetic tree. Using as reference the cluster of residence of the Snq2 and Pdr18 proteins, the branch comprising the homologs of these two S. cerevisiae proteins in this tree was identified. For species abbreviation a four letters code is used, composed by the first two letter of the genus and species. The number displayed after the first four letters is used to abbreviate the strain name when the genome of more than one strain from a given species was examined. To standardize the annotation used, translated ORFs are represented by small letters.
Phylogenetic Analysis and Tree Construction
The MUSCLE software suite (Edgar, 2004) was used to build a multiple alignment of the amino acid sequences of the Snq2 and Pdr18 proteins encoded in Saccharomycetaceae yeasts that was analyzed using the Jalview 2.9 software suite (Clamp et al., 2004; Waterhouse et al., 2009). The processing of these sequences did not involve any step of masking or trimming. The “read.fasta” and “write.nexus.data” functions made available by the seqinr 3.4-5 (Charif et al., 2007) and by the ape 4.1 R packages (Paradis et al., 2004; Popescu et al., 2012), respectively, were used to convert the multiple alignment in fasta format into a nexus file that was subsequently fed into MrBayes 3.2.6 (Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003), a Bayesian Markov chain Monte Carlo (MCMC) package for phylogenetic analysis. The Message Passing Interface (MPI) version of MrBayes (Altekar et al., 2004) was used to speed up phylogeny computation using Metropolis coupling of MCMC sampling. The MCMC simulations used 100,000 generations, coupling one “cold” chain together with nine heated chains. Two independent runs of MCMC sampling (each started from two distinct random trees) confirmed parameter convergence of the posterior probability distribution. The option of estimating the fixed-rate amino acid prior model made available by MrBayes was used, allowing the MCMC sampler to explore all of the nine available models by regularly proposing new ones (upon parameter convergence, each model contributes to the results in proportion to its posterior probability) and rate variation over sites was assumed to follow a gamma distribution. The remaining MrBayes MPI parameters were set to default values. The PhyML 3.0 software suite (Guindon et al., 2010) was used to construct a Maximum Likelihood (ML) derived phylogenetic tree to confirm the gathered results. The PhyML parameters and models used as input were the default ones, with exception of the method for searching the optimal phylogenetic tree, where the Subtree Pruning and Regrafting (SPR) were used as algorithm instead of Nearest Neighbor Interchange (NNI). The phylogenetic trees obtained were analyzed using the visualization software Dendroscope 3.5.7 (Huson et al., 2007; Huson and Scornavacca, 2012). The MrBayes clade credibility score and PhyML bootstrapping values calculated for each internal node of the Bayesian and ML trees, respectively, were inspected using either the FigTree 1.4.3 software suite1 or PhyloTree 0.1 package2 installed in Cytoscape 2.8.3 (Shannon et al., 2003). The bootstrap score measures the confidence level of each clade in the consensus phylogenetic tree by repeated sub-sampling data from the original data set and determining the empirical frequency that each of these clades have in the whole set of phylogenetic trees originated from these sub-samples. The clade credibility score measures the posterior probability of each clade in the consensus phylogenetic tree by determining the frequency that each of these clades has in the whole set of phylogenetic tree sampled using the estimated parameter values.
Gene Neighborhood Analysis
A chromosome block of 30 neighboring genes, 15 on each side of the pair of homologous genes under analysis, was selected to assess the conservation of the chromosome region where the members of the Snq2/Pdr18 subfamily reside (Seret et al., 2009; Dias et al., 2010). Scripting in the R language was used to retrieve 15 neighbor genes on each side of the query genes as well as the corresponding sequence clustering classification from Genome DB. The classification of each of the 30 genes neighboring each query gene was obtained using a conservative blastp e-value of E-50 to limit the number of false positive sequences gathered together with true cluster members. When dubious synteny connections between genes needed corroboration, the amino acid sequence clustering was performed at a less restrictive e-value threshold of E-40. The existence of synteny between query genes was verified through the analysis of network topology (number of shared neighbor pairs) and the biological information associated with the corresponding edges. Three sources of biological information were used as independent evidence confirming the strength of the synteny between members of the Snq2 and Pdr18 protein subfamily (Dias et al., 2010): (1) distance of the neighbors in relation to the query genes, (2) similarity of the amino acid sequences of the shared neighbors, and (3) total number of members comprised in the cluster of amino acid sequence to which the homologous neighbors belong to; sequence clusters comprising a small number of members are more reliable as synteny evidence since the probability that two homologous neighbors being in the vicinity of two query genes by chance is small.
Susceptibility Phenotypes of S. cerevisiae pdr18Δ and snq2Δ and C. glabrata snq2Δ Deletion Mutants
Strains, Media, and Growth Conditions
S. cerevisiae BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) and the derived deletion mutants pdr18Δ and snq2Δ were obtained from EUROSCARF collection. C. glabrata BPY55 (clinical isolate) and the derived deletion mutant snq2Δ built using the SAT1 flipper system were kindly provided by Professor Dominique Sanglard, Institut de Microbiologie, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland.
Cultivation of S. cerevisiae strains was performed in MM4 medium, containing 1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate (Difco, Detroit, MI, United States), 20 g/L glucose (Merck, Darmstadt, Germany), 2.65 g/L (NH4)2SO4 (Panreac AppliChem, CT, United States), 20 mg/L L-methionine, 20 mg/L L-histidine (both from Merck, Darmstadt, Germany), 60 mg/L L-leucine and 20 mg/L L-uracil (both from Sigma, St. Louis, MO, United States). C. glabrata strains were cultivated in MM medium, with the composition of MM4 medium, without supplementation with amino acids and uracil. YPD medium contained 20 g/L glucose, 20 g/L BactoTM Peptone and yeast extract (both from BD Biosciences, Franklin Lakes, NJ, United States). Solid media were prepared by the addition of 20 g/L agar (Iberagar, Barreiro, Portugal) to the different liquid media. Media pH were adjusted to 4.5 with HCl. Growth in liquid media was performed at 30°C with orbital agitation (250 rpm).
Susceptibility Tests
Growth susceptibility tests of S. cerevisiae parental strain BY4741, the corresponding pdr18Δ and snq2Δ deletion mutants and of C. glabrata BPY55 and derived deletion mutant snq2Δ to a wide range of growth inhibitory compounds was evaluated by spot assays. Yeast cell suspensions used for the spot assays were prepared from mid-exponential cell cultures grown in liquid media MM4 (S. cerevisiae), MM (C. glabrata), or YPD (both strains) by harvesting (5,000 rpm, 5 min) and resuspending the cells in sterile ddH2O to an OD600nm of 0.25 followed by four serial dilutions of 1:5 each. These cell suspensions were plated as 4 μL spots onto the surface of MM4 (S. cerevisiae) or MM (C. glabrata) or YPD (both strains) at pH 4.5 solid media, supplemented or not with the toxic compound to be tested. The plates were incubated at 30°C for 72 h and pictures were taken every 24 h. The toxic compounds tested and the selected concentrations for the screening of S. cerevisiae and C. glabrata strains in MM4 and MM are listed in Table 1, and those selected for experiments in YPD are indicated close to the respective pictures. The susceptibility of the deletion mutants was assessed by comparison of their growth performance with the growth performance of the corresponding parental strain in the presence of the different cytotoxic compounds tested. Moderately reduced growth of the deletion mutants compared with the parental strain was classified as minus (-), marked reduced growth as a double minus (--), improved growth as a plus (+) and identical growth as zero (0). Results are representative from three independent experiments.
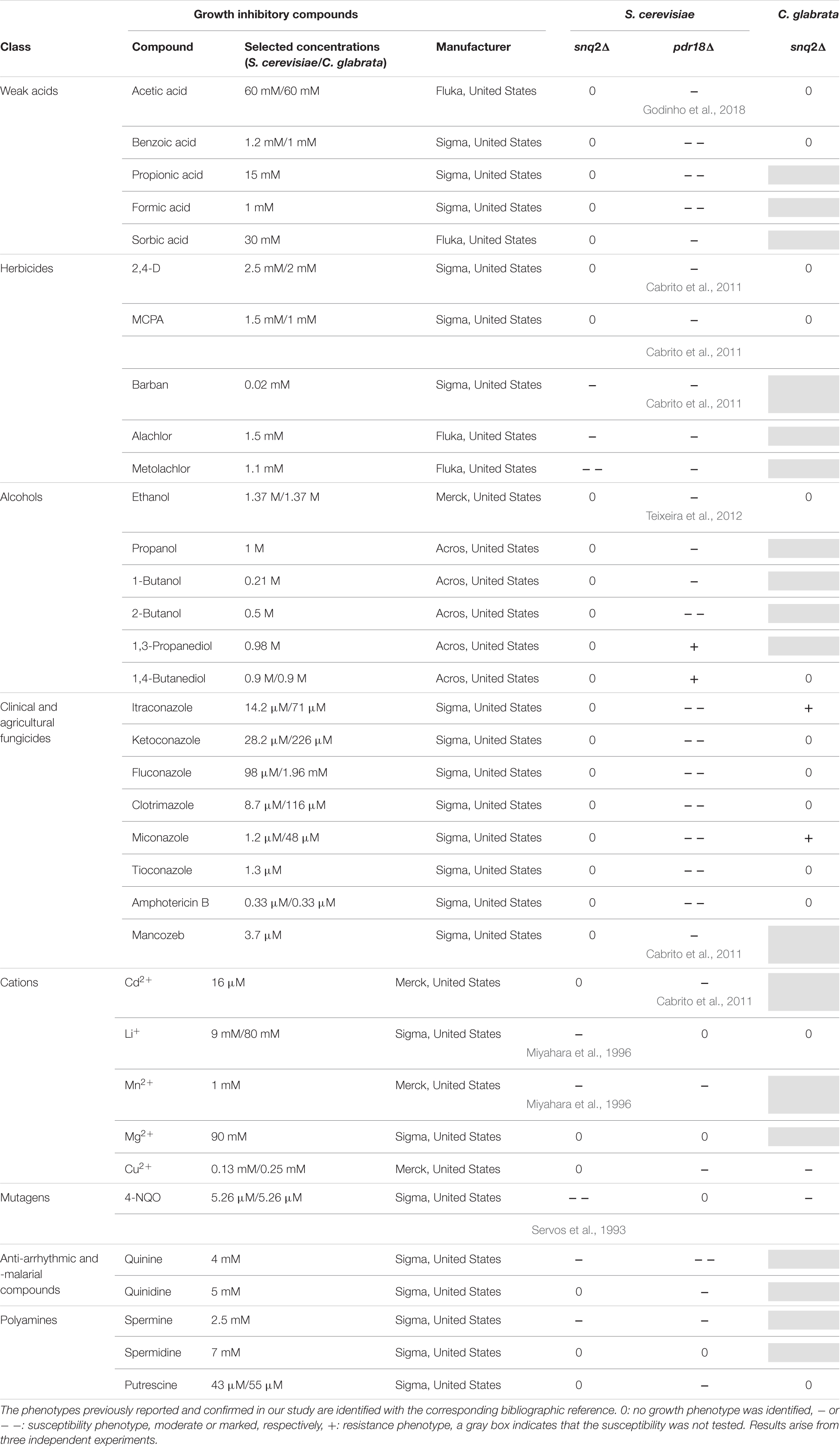
TABLE 1. Susceptibility of the deletion mutants pdr18Δ and snq2Δ of S. cerevisiae BY4741 and snq2Δ of C. glabrata BPY55 to a wide range of chemical compounds supplemented in minimal media.
Results
Identification of Snq2 and Pdr18 Proteins Encoded in the Saccharomycetaceae Yeast Genomes
To identify the PDR proteins encoded in the examined yeast genomes, the network representing the amino acid sequence similarity of the translated ORFs comprised in the Genome DB was traversed at different blastp e-value thresholds using as starting nodes Saccharomyces cerevisiae proteins Pdr18 [representing the PDR sensu stricto proteins (Seret et al., 2009)] and Yol075c and Adp1 [representing the PDR sensu lato proteins (Seret et al., 2009)]. When Pdr18 was used as starting node the analysis of the protein sets obtained at different blastp e-values indicated that a threshold of E-142 was adequate, allowing the gathering of 1,263 translated ORFs (Figure 1). Using a similar approach, a threshold of E-104 and E-84 was found to be adequate for network traversal using Yol075c and Adp1 as starting nodes, allowing the gathering of 189 and 196 translated ORFs, respectively (Figure 1). The three disjoint protein sets were merged, giving a total of 1,648 translated ORFs homologous to the yeast PDR proteins in yeast strains belonging to the subphylum Saccharomycotina. This set included members of another family of transporters (hexose transporters) that were manually removed. The corresponding amino acid sequences were aligned using MUSCLE software and the protdist and neighbor algorithms made available by the PHYLIP software suite were used to construct a preliminary phylogenetic tree. Using as reference the cluster of residence of the Snq2 and Pdr18 proteins, the phylogenetic branch comprising the homologs of these two S. cerevisiae proteins was identified. The comparison of the PDR protein set gathered in this study with those reported before encoded in the genomes of 10 yeast species (Seret et al., 2009) confirmed the co-clustering of the homologs of S. cerevisiae Snq2/Pdr18 proteins in a single branch of the phylogenetic tree. However, since it is well known that translated ORFs residing in long phylogenetic branches may correspond to protein fragments or sequence frameshifts, the blastp algorithm was used to test ORFs with dubious sequence similarity. After determining the homology of each of these proteins selected for blastp testing, a total of 214 translated ORFs, encoded in the Saccharomycetaceae yeast genomes examined, showing strong amino acid sequence similarity to the S. cerevisiae Snq2/Pdr18 transporters, were retained for further analysis (Table 2 and Supplementary Table S1).
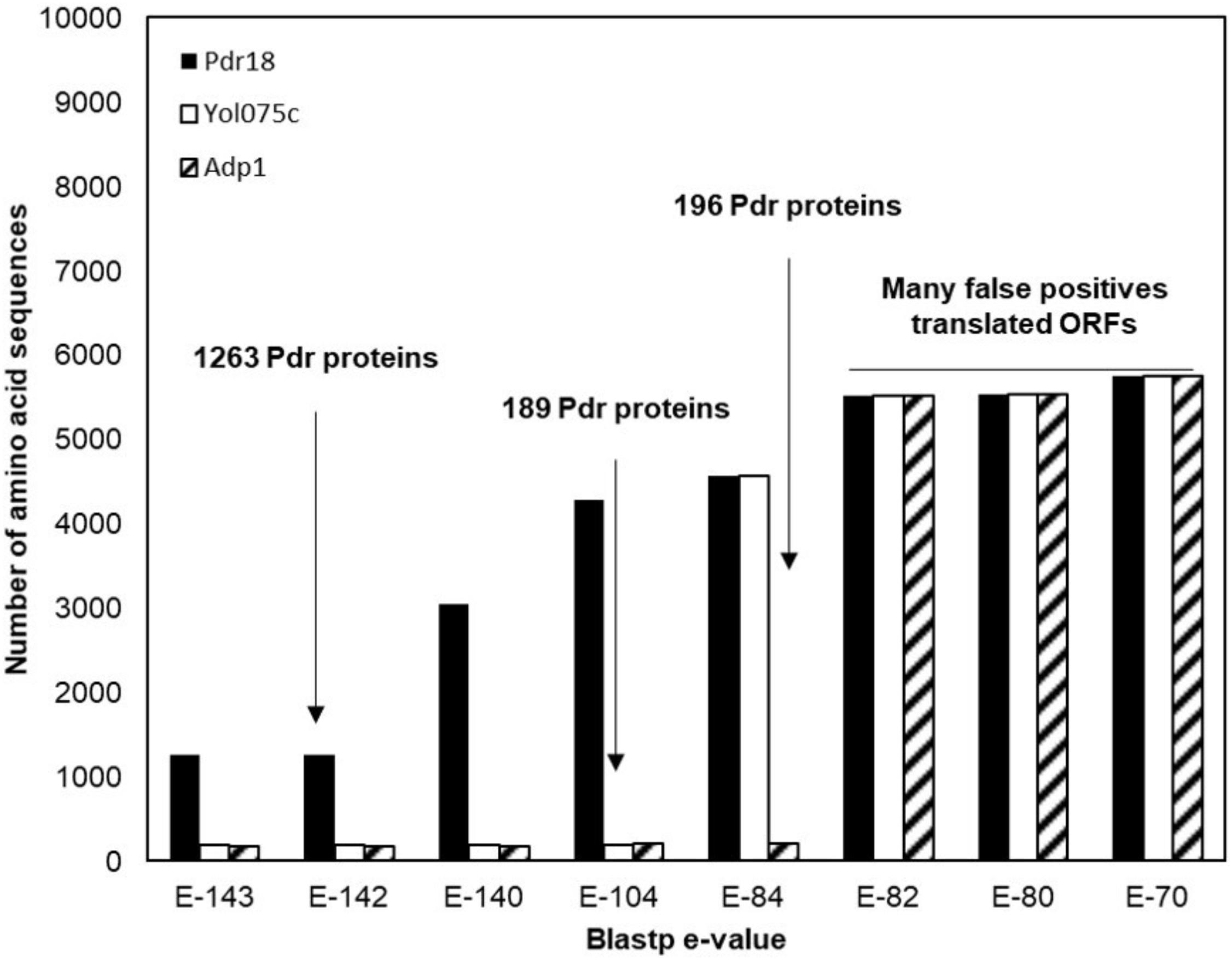
FIGURE 1. Identification of the PDR proteins encoded in the genomes of 117 strains, belonging to 29 different yeast species belonging to the Saccharomycetaceae family. Plot representing the number of amino acid sequences retrieved after constraining and traversing the pairwise similarity network at different e-values taking Pdr18, Yol075c, and Adp1 proteins as starting nodes. The blastp e-value used as threshold for network constraining is marked by an arrow.
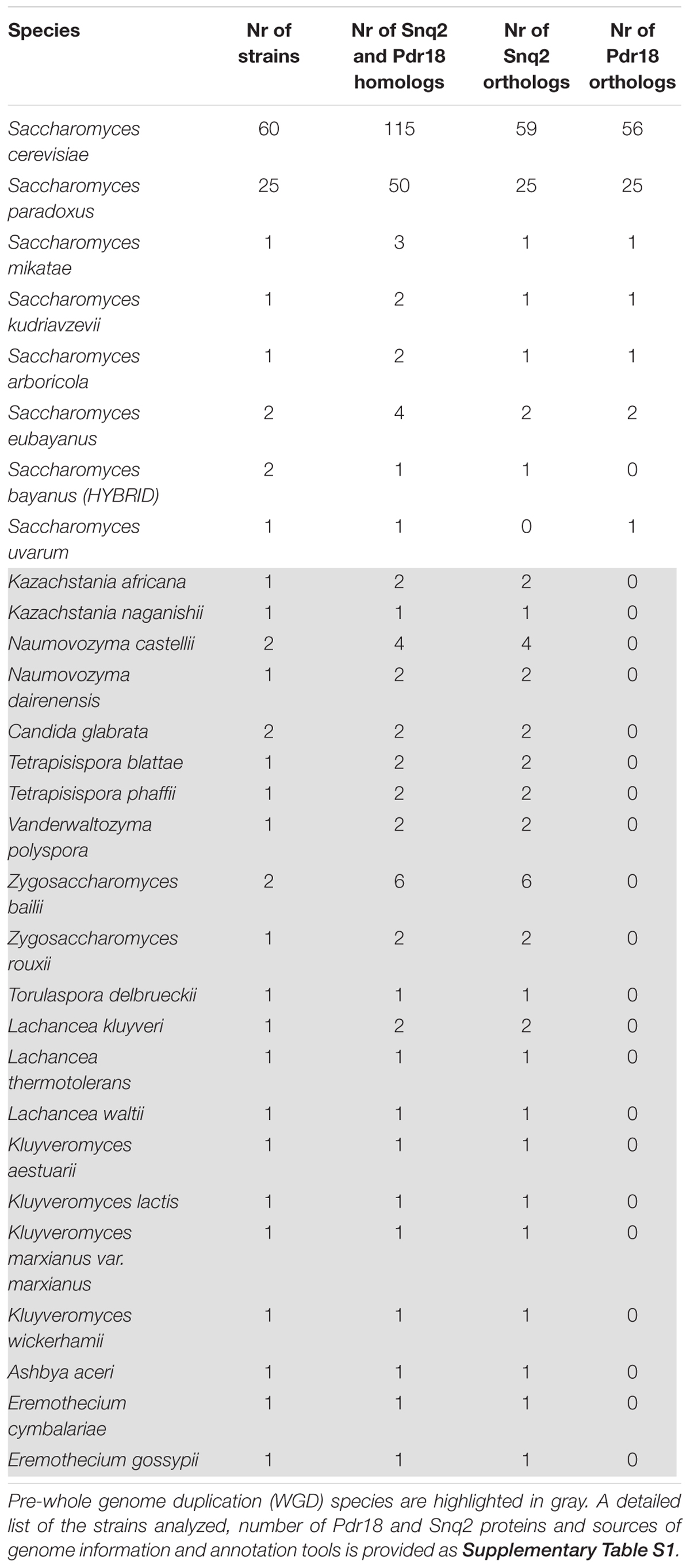
TABLE 2. Saccharomycetaceae yeast strains examined in this work, and the number of Snq2/Pdr18 homologs, Snq2 orthologs, and Pdr18 orthologs identified.
Phylogenetic Analysis of Snq2 and Pdr18 Proteins
For the phylogenetic analysis of Snq2 and Pdr18 proteins, more than one strain from a number of yeast species were used (60 strains for S. cerevisiae, 25 for Saccharomyces paradoxus, 2 for Saccharomyces eubayanus, 2 for Saccharomyces bayanus, 2 for Naumovozyma castellii, 2 for Candida glabrata, and 2 for Zygosaccharomyces bailii). All the repeated sequences were removed from the protein dataset, leaving just a representative member of each species. This resulted in 146 unique translated ORFs encoded in the Saccharomycetaceae yeast genomes that were used to construct a phylogenetic tree for Pdr18 and Snq2 proteins. For this, the 146 unique translated ORFs were aligned and the corresponding phylogenetic tree constructed using the MrBayes software suite (Figure 2). After assuring that all model parameters had converged, the corresponding consensus Bayesian phylogenetic tree was retained for further analysis. Because MrBayes software suite allowed model jumping between nine different fixed-rate amino acid models, after parameter convergence the analysis of results showed that the model Wag (Whelan and Goldman, 2001) has a contribution of 100% to the posterior probability, meaning that this model became dominant over the remaining ones during the MCMC simulation. The translated ORF lakl_1_h21010g was selected as root of the phylogenetic tree because it comprised the most divergent amino acid sequence present in the protein dataset. Complementing the Bayesian approach adopted for the construction of the phylogenetic tree, the PhyML software suite was also used to obtain a ML derived tree (not shown). The analysis of the clade credibility score and bootstrap values obtained for each internal node of the Bayesian and ML trees, respectively, indicates that the two distinct statistical approaches generated similar trees. Due to their similarity, the analysis presented in the manuscript is solely based on the Bayesian tree (Figure 2).
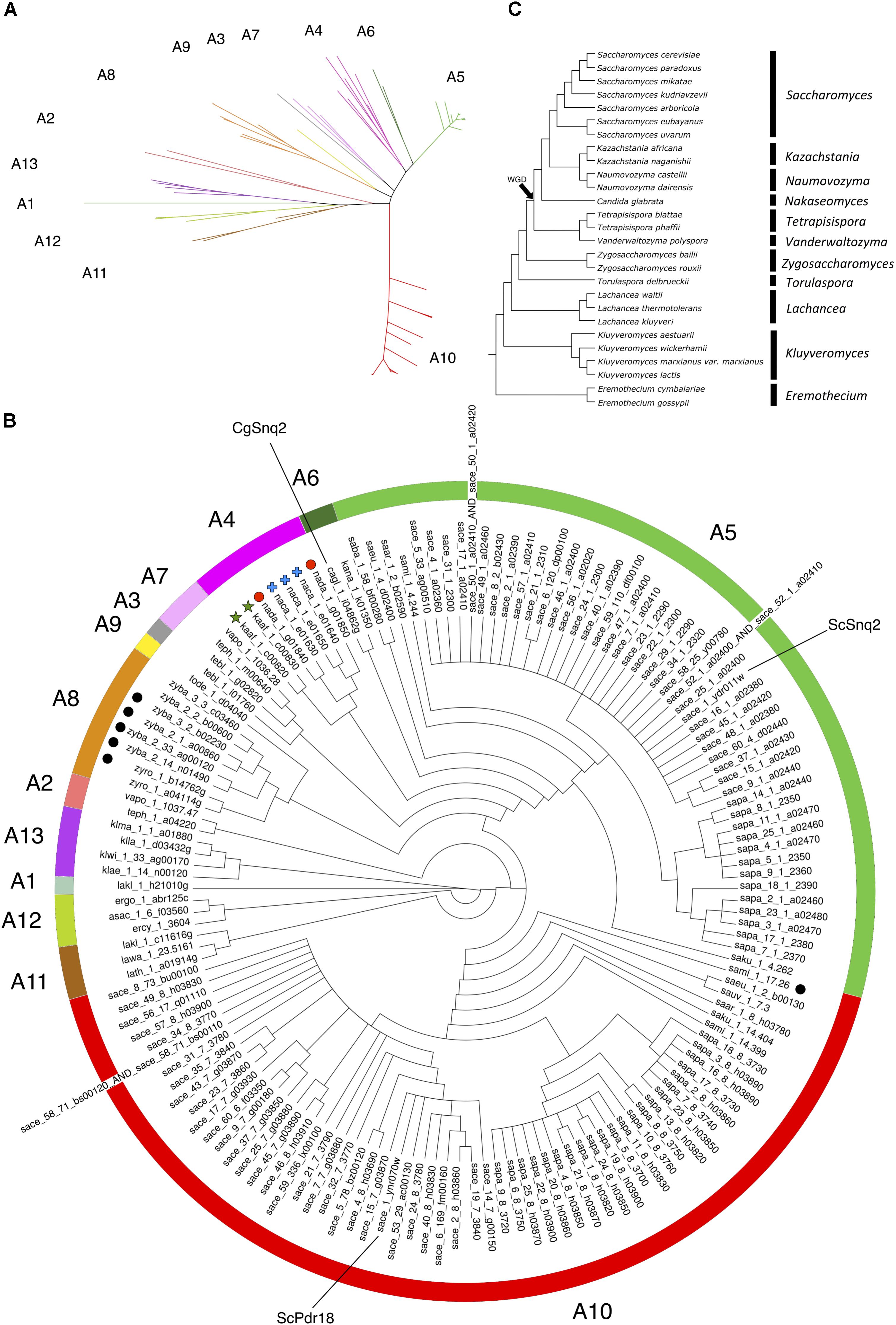
FIGURE 2. Phylogenetic tree of the Pdr18 and Snq2 homologs encoded in the genome sequences of 117 strains of 29 yeast species belonging to the Saccharomycetaceae family. (A) Radial phylogram showing the amino acid sequence similarity distances between the protein homologs of Pdr18 and Snq2. (B) Circular cladogram showing the tree topology. Singleton genes were identified with graphical labeling of the translated ORFs using black circles, and tandemly duplicated genes were identified with graphical labeling of the translated ORFs using green stars, red circles and blue crosses. The translated ORF lakl_1_h21010g was chosen as outgroup due to the strong dissimilarity of its amino acid sequence. (C) Schematic phylogenetic tree of the species (adapted from Kurtzman and Robnett, 2003).
The analysis of (i) the tree topology, (ii) the clade credibility score of the branches, and (iii) the phylogenetic distances separating the 146 unique translated ORFs led to the proposal of dividing the tree into thirteen clusters, labeled from A1 to A13 (Figures 2A,B). The Snq2 and Pdr18 homologs encoded in Lachancea, Kluyveromyces, and Eremothecium species occupy a basal position in this phylogenetic tree (Figure 2B – clusters A1, A11, A12, and A13). On the other hand, the early divergence of the Snq2 and Pdr18 homologs encoded in the genomes of the pre-WGD Zygosaccharomyces and Torulaspora species is not observed (Figure 2B – clusters A8 and A9). All translated ORFs showing strong amino acid sequence similarity to the S. cerevisiae Snq2 or Pdr18 proteins are only encoded in the genomes of yeast species classified in the Saccharomyces genus. The translated ORFs showing strong amino acid sequence similarity to either S. cerevisiae Snq2 or Pdr18 proteins are divided in two distinct branches of this phylogenetic tree, residing in the clusters A5 and A10, respectively (Figure 2B). The analysis of the phylogenetic tree also indicates that the Snq2 homologs cagl_1_i04862g and kana_1_k01350 also share strong sequence similarity (cluster 6, 74.5% and 85.9% of identity and similarity, respectively). The percentage of identity and similarity shown between these Snq2/Pdr18 homologs is unexpectedly high suggesting that a lateral gene transfer event to an ancestral strain of K. naganishii species might have mediated the acquisition of the Snq2/Pdr18 homolog encoded in this species. The homologs of the Snq2 and Pdr18 proteins encoded in yeast species of the remaining post-WGD taxonomic genera (Kazachstania, Naumovozyma, Tetrapisispora, and Vanderwaltozyma) reside in the phylogenetic clusters A2, A3, A4, and A7 (Figure 2B).
Gene Neighborhood Analysis of Pdr18 and Snq2 Orthologs in Saccharomycetaceae Yeasts
Gene neighborhood analysis of S. cerevisiae SNQ2 and PDR18 homolog genes encoded in the examined Saccharomycetaceae yeast strains genomes was performed. This study, together with the phylogenetic analysis, is useful to contribute to the elucidation of their ortholog/paralog status.
Results show that for the pre-WGD yeasts species of Torulaspora, Lachancea, Kluyveromyces, and Eremothecium genera there is a single lineage, with genes sharing strong synteny (Figures 3, 4). The main exception to this rule is the Lachancea kluyveri CBS 3082 strain because, in addition to the gene lakl_1_c11616g residing in the above mentioned conserved chromosome environment, this yeast strain also encodes one singleton gene, lakl_1_h21010g, sharing very weak synteny with Kluyveromyces lactis and Kluyveromyces marxianus var. marxianus genes (Figure 3). The pre-WGD yeast strains of the Zygosaccharomyces genus, Z. rouxii CBS 732 and Z. bailii CLIB213, both encode two SNQ2/PDR18 homolog genes (Figure 3). However, Z. bailii IST302 encodes two additional SNQ2/PDR18 homolog genes, zyba_2_14_n01490 and zyba_2_33_ag00120, lacking common neighbors with the remaining SNQ2/PDR18 homolog genes in the Saccharomycetaceae strains genomes examined (Figure 3 and Supplementary Figure S1). For this reason, these two genes were considered singletons. The high amino acid sequence identity shared by these two genes suggests that they are a paralog pair originated in a duplication event that occurred recently in the evolution of Zygosaccharomyces species.
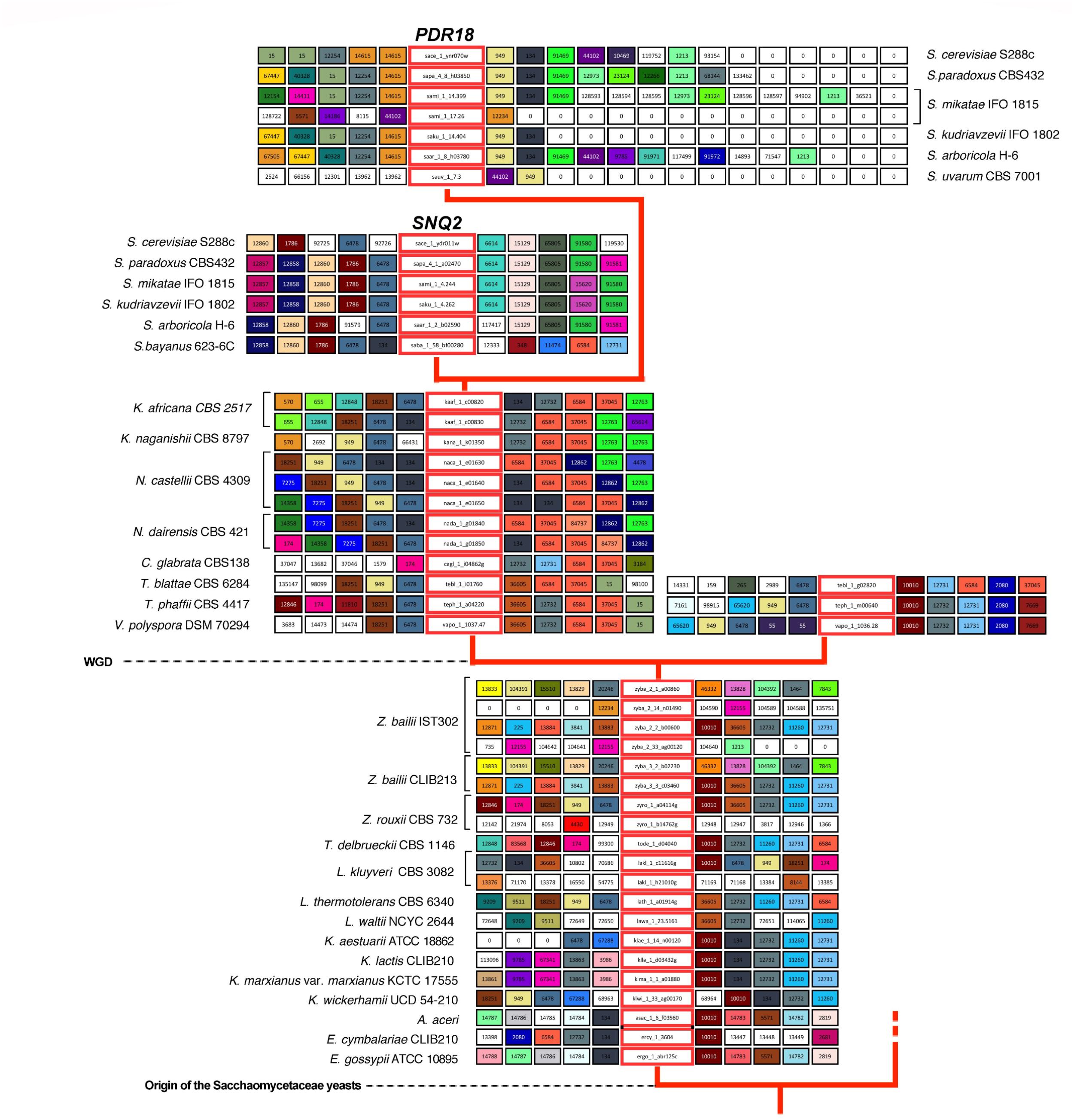
FIGURE 3. Gene neighborhood of SNQ2 and PDR18 genes and corresponding homologs encoded in the genomes examined. Genes in the central boxes represent SNQ2, PDR18 and their orthologs. Adjacent boxes represent gene neighbors and homologous neighbors are highlighted in the same color. A white box represents genes with no homologous neighbors in the represented chromosome region and white boxes with a zero represent the end of the contig/chromosome. The synteny was assessed with 15 neighbors on each side but this representation was truncated to five neighbors. In the case of PDR18 orthologs, the gene neighborhood data was not truncated in one of the sides to highlight the sub-telomeric position occupied by these genes.
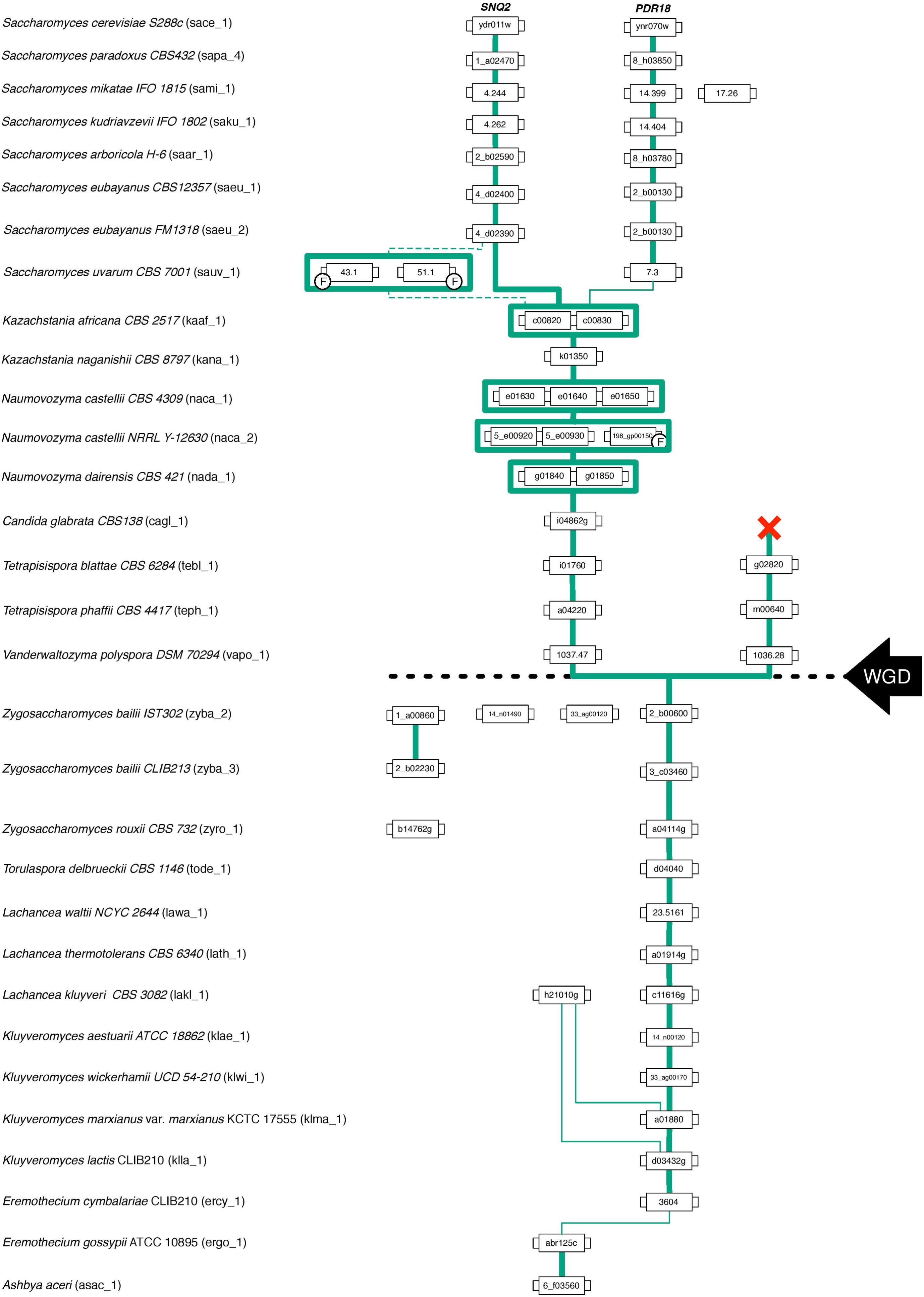
FIGURE 4. Gene lineage comprising the homologs of S. cerevisiae PDR18 and SNQ2 genes encoded in the Saccharomycetaceae species examined. Each box represents a gene and the lines connect genes sharing common neighbors. F indicates that the corresponding gene was classified as a fragment. Line thickness represents the strength of synteny between genes. The black dashed line marks the point in time where the Whole Genome Duplication (WGD) event occurred.
After the WGD event, the above described single gene lineage gave rise to two gene sub-lineages and the chromosome regions where these genes reside in pre- and early-divergent post-WGD yeast species are conserved (Figure 3). For instance, the analysis of the neighborhood of the pre-WGD Zygosaccharomyces rouxii gene zyro_1_a04114g and of the post-WGD species Vanderwaltozyma polyspora gene vapo_1_1037.47 shows the existence of 9 common neighboring genes, some of them absent from Figure 3 but that can be seen in Supplementary Figure S1. Z. rouxii gene zyro_1_a04114g also shares 9 common neighboring genes with the second gene encoded in V. polyspora (vapo_1_1036.28), belonging to the second sub-lineage of ohnolog genes originating from WGD (Figure 3 and Supplementary Figure S1).
The Candida glabrata strains’ genomes analyzed only encode one Pdr18/Snq2 homolog gene sharing strong synteny with all the homologs encoded in post-WGD species, from Naumovozyma dairenensis to Kazachstania africana (Figure 3 and Supplementary Figure S1). This sub-lineage also shows strong synteny with one of the gene sub-lineages encoded in Saccharomyces genus species, corresponding to the ScSNQ2 orthologs in Saccharomyces yeasts, supporting the ScSNQ2-ortholog status for the SNQ2/PDR18 homologs encoded in the post-WGD species, from C. glabrata to S. cerevisiae (Figures 3, 4). We also hypothesize the loss of the ohnolog gene from the second sub-lineage in the common ancestor of Saccharomyces, Nakaseomyces, Kazachstania, and Naumovozyma genera, given that all the encoded PDR18/SNQ2 homologs from C. glabrata to K. africana appear to be ScSNQ2 orthologs.
The gene neighborhood analysis of the S. cerevisiae SNQ2/PDR18 homologs encoded in the genomes of the Saccharomyces genus yeast species S. paradoxus, S. mikatae, S. kudriavzevii, and S. arboricola, shows the existence of two gene sub-lineages, one comprising the above described SNQ2 orthologs and the other comprising PDR18 orthologs. PDR18 orthologs occur exclusively in the Saccharomyces genus yeast species and the sub-lineage constituted by these orthologs shows a very strong synteny (Figure 3). However, the analysis of the gene neighborhood of the SNQ2 and PDR18 orthologs in Saccharomyces species residing in a basal phylogenetic position could not be performed due to lack of data: the translated ORF encoding the S. uvarum SNQ2 ortholog, and the PDR18 ortholog encoded in the genome of S. bayanus 623-6C were fragmented into two different contigs of small dimension. Therefore the closely related species S. eubayanus (Baker et al., 2015), whose genome had not been at first included in Genome DB, was used instead. The chromosome environment where the SNQ2 and PDR18 orthologs reside in the S. eubayanus genome was inspected by performing manual blastp pairwise comparisons of the amino acid sequence of the neighboring genes against the full protein set of S. cerevisiae. This analysis also showed that S. eubayanus CBS12357 and FM1318 strains encode a single SNQ2 ortholog and a single PDR18 ortholog sharing strong synteny with the remaining genes within each sub-lineage. These results support the notion that the chromosome environment where SNQ2 and PDR18 paralogs reside in the genome of Saccharomyces species has been conserved since their appearance in the last common ancestor of the species comprised in this taxonomic genus.
The S. cerevisiae PDR18 gene resides in the subtelomeric region of chromosome XIV (Figure 3), a region poorly conserved throughout Saccharomycetaceae yeasts’ evolution, sharing little synteny with the homologs of the SNQ2/PDR18 genes encoded in the genomes of yeast species belonging to the other post-WGD taxonomic genera. In fact, the only neighbor in the vicinity of the PDR18 gene that did not belong to large gene families and is also shared with the K. naganishii, N. castelli and N. dairenensis species is ORF YNR071C, a non-biochemically characterized member of a gene family of aldose 1-epimerases (Li et al., 2013). This ORF and the other S. cerevisiae members of this gene family, GAL10 gene and ORF YHR210C, are comprised in the cluster of amino acid similarity 949 (Figure 3). This cluster of amino acid sequence similarity comprises a total of 366 members in the 170 hemiascomycetous strains of the Genome DB (gathered at an e-value threshold of E-50). The gene neighborhood analysis showed that the members of this cluster are present in the chromosome environment where Snq2 orthologs from pre- and post-WGD species and Pdr18 orthologs from Saccharomyces genus species reside (Figure 3).
Although the gene neighborhood results suggest that the gene duplication event giving rise to the S. cerevisiae SNQ2 and PDR18 genes occurred in a recent ancestor of the genus Saccharomyces, the phylogenetic relationships between the A5 and A10 clades (Figure 2) is apparently not consistent with the proposed evolutionary scenario. To clarify this seemingly inconsistency, the amino acid sequences of the orthologs of the SNQ2/PDR18 genes encoded in the post-WGD species (left-sublineage in Figure 4) were used to construct a phylogenetic tree using the same initial parameters used in construction of the tree shown in Figure 2 (Supplementary Figure S2). The decision of constructing a new tree was based on the plausible notion that the inclusion of non-orthologous sequences might be introducing “noise” in the multiple alignment of the sequences and, consequently, distorting tree topology and introducing error in the true phylogenetic relationships established between the genes comprised in the WGD left sub-lineage. After parameter convergence, the analysis of this phylogenetic results indicated that the model Jones (Jones et al., 1992) has a contribution of 100% to the posterior probability. In fact, the analysis of this new tree clearly shows that the node joining clades A5 and A10 defines a branch comprising only genes encoded in the Saccharomyces species and that the last common ancestor of the homologs of the SNQ2 and PDR18 genes coincides with the origin of the Saccharomyces genus. The obtained clade credibility score for the orthologs of these two S. cerevisiae genes residing together in a single phylogenetic branch of this Bayesian consensus tree was calculated as being 0.6724. These results are consistent with the evolutionary scenario proposed based on the gene neighborhood analysis and confirm the previously proposed existence of a paralogous relationship between the S. cerevisiae SNQ2 and PDR18 genes but also establishing that the gene duplication event in its origin occurred more recently than formerly proposed (Seret et al., 2009), presumably coinciding with the first common ancestor of the yeast species comprised in the Saccharomyces genus, before the radiation of these species.
Susceptibility Profiling of S. cerevisiae snq2Δ and pdr18Δ and C. glabrata snq2Δ Deletion Mutants
The post-WGD C. glabrata species genome encodes a sole ScSnq2 ortholog, the CgSnq2 (Sanglard et al., 2001). The encoded gene has diverged before the hypothesized duplication event that originated S cerevisiae PDR18. For this reason, C. glabrata was the selected species to get insights into the functional divergence of the ancestral gene and S. cerevisiae PDR18 and SNQ2 genes. Since ScPdr18, ScSnq2, and CgSnq2 were reportedly involved in MDR/MXR, the functional divergence of these proteins was examined by profiling the growth susceptibility of the corresponding deletion mutants against a wide range of cytotoxic compounds, some of them already described as potential substrates for these drug/xenobiotic pumps. The susceptibility of S. cerevisiae BY4741-derived snq2Δ and pdr18Δ deletion mutant strains to a wide range of chemical compounds was screened under identical conditions in minimal medium MM4 (S. cerevisiae), ranging from weak acids, alcohols, polyamines, metal cations to herbicides, fungicides and anti-arrhythmic and -malarial compounds (Table 1). The deletion of the SNQ2 gene in S. cerevisiae was found to lead to increased susceptibility to 4-NQO, Li+ and Mn2+, in agreement with previous studies (Servos et al., 1993; Miyahara et al., 1996). Moreover, this systematic screening contributed to extend to the herbicides barban, alachlor, and metolachlor, the anti-malarial anti-arrhythmic quinine and the polyamine spermine the list of toxic compounds to which Snq2 confers protection in S. cerevisiae (Table 1).
The deletion of the PDR18 gene was found to render S. cerevisiae cells more susceptible toward almost all the compounds tested, contrasting with SNQ2 whose MDR/MXR spectrum is apparently more limited, the phenotypes only coinciding for 6 of the 35 compounds tested (Table 1). In particular, the higher toxic effect of weak acids and of fungicides that target either ergosterol biosynthesis (azoles) or the ergosterol molecule itself (amphotericin B) toward the pdr18Δ strain is evident (Table 1), consistent with the role described for this ABC transporter in acetic acid resistance and in ergosterol transport at plasma membrane (Cabrito et al., 2011; Teixeira et al., 2012; Godinho et al., 2018).
The expression of CgSNQ2 gene in C. glabrata was found not to confer either protection or increased susceptibility to stress induced by acetic and benzoic acids, the herbicides 2,4-D and MCPA, the alcohols ethanol and 1,4-butanediol, and the polyamine putrescine in minimal medium, similarly to former observations for the expression of ScSNQ2 in S. cerevisiae. This profile is considerably different from that exhibited by S. cerevisiae cells devoid of PDR18, which were more susceptible to all these compounds, except for 1,4-butanediol, toward which this deletion mutant was more tolerant (Table 1). Moreover, susceptibility phenotypes for Cgsnq2Δ deletion mutant were detected with toxic concentrations of the mutagen 4-NQO [which is a described putative substrate for S. cerevisiae efflux pump Snq2 (Servos et al., 1993)]. However, the susceptibility phenotype for the lithium cation exhibited by Scsnq2Δ mutant, was not detected for the Cgsnq2Δ mutant (Table 1). Moreover, CgSnq2 is apparently a determinant of resistance to Cu2+, while no phenotype was found for Scsnq2Δ (Table 1). The toxic effect of the azole drugs clotrimazole, ketoconazole, and fluconazole, and of amphotericin B was not alleviated by the expression of CgSNQ2 when tested in minimal medium, consistent with what was observed for S. cerevisiae ScSNQ2 expression. Surprisingly, in the presence of itraconazole and miconazole, the deletion of CgSNQ2 was apparently advantageous (Table 1).
Although the results in Table 1 suggest that CgSnq2 has no positive effect in C. glabrata BPY55 resistance to azoles, a susceptibility phenotype for the BPY55_snq2Δ mutant in the presence of the azole drugs fluconazole and ketoconazole was previously reported (Torelli et al., 2008). However, the spot assays performed in the referred study were performed in YPD medium while all the phenotypes described in Table 1 for C. glabrata were performed in minimal medium MM. Therefore, we investigated the susceptibility to azole drugs in rich medium YPD for C. glabrata strains, as well as for S. cerevisiae strains to confirm results obtained in minimal media MM4 (Figure 5). We also included the well-characterized phenotypes for Scpdr18Δ and Scsnq2Δ in the presence of acetic acid and 4-NQO, respectively, and confirmed that there is no interference of the growth medium used in the phenotypes obtained (Figure 5). The higher susceptibility of Scpdr18Δ deletion mutant, when compared to the corresponding parental strain, to the azole drugs ketoconazole, clotrimazole, miconazole, fluconazole, and to a lower extent, to itraconazole was confirmed in YPD (Figure 5). Also, as found in minimal medium MM4 (Table 1), Scsnq2Δ shows no susceptibility phenotype in the presence of the azole drugs in YPD (Figure 5). In summary, even in YPD media, CgSnq2 is not a determinant of C. glabrata tolerance to itraconazole, clotrimazole or miconazole (Figure 5), but the phenotypes previously reported for fluconazole and ketoconazole (Torelli et al., 2008) were here confirmed.
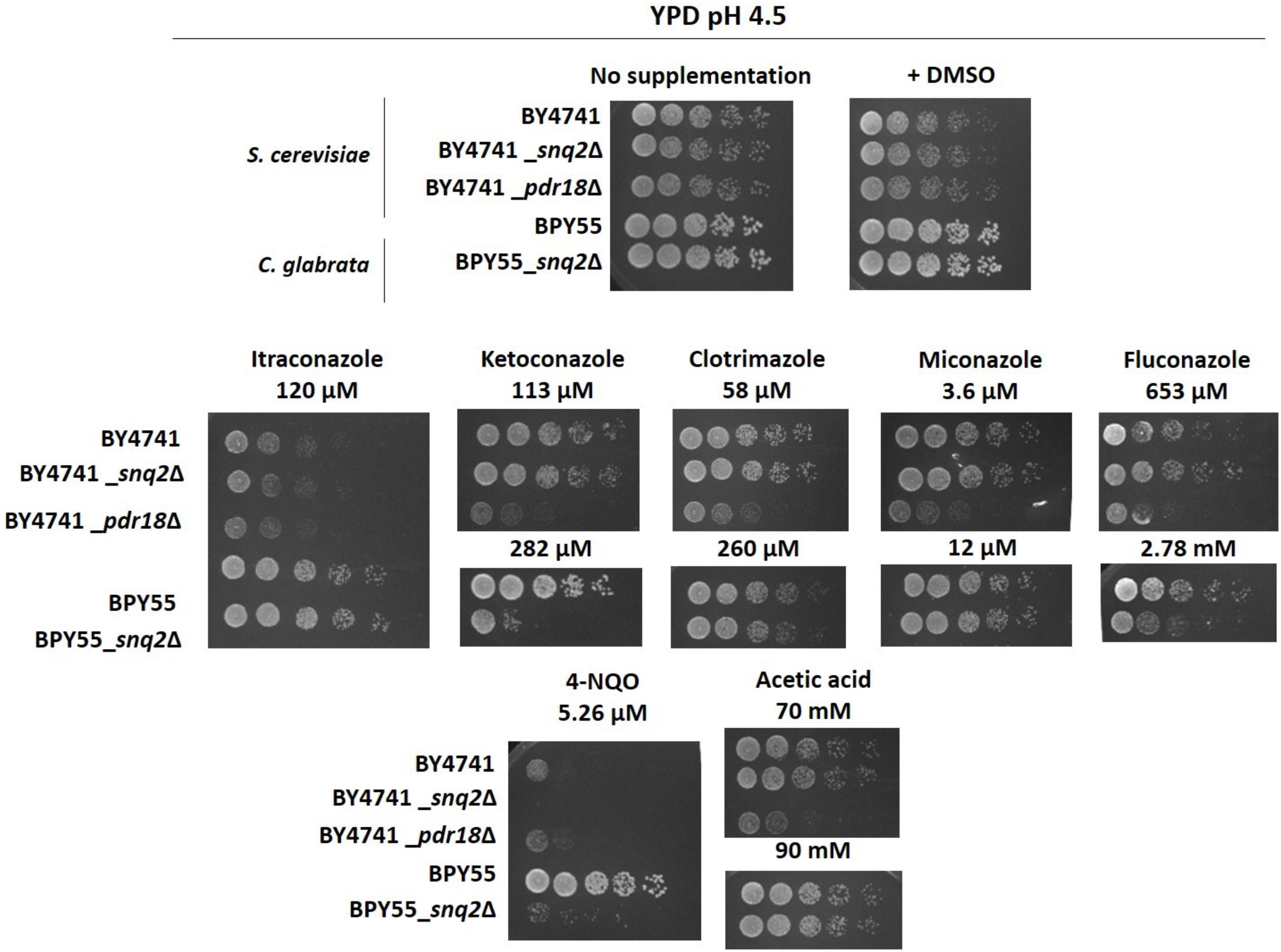
FIGURE 5. Susceptibility profiling of the S. cerevisiae pdr18Δ and snq2Δ and of C. glabrata snq2Δ deletion mutants to azole drugs, 4-NQO and acetic acid in rich media YPD. Comparison of growth by spot assays of the S. cerevisiae parental strain BY4741 and derived pdr18Δ and snq2Δ, and of C. glabrata parental strain BPY55 and derived snq2Δ mutant cell suspensions plated in solid YPD media supplemented or not with azole drugs, 4-NQO or acetic acid (at pH 4.5). All pictures were obtained after 24 h of incubation. The results are representative from three independent experiments.
Discussion
The evolutionary history of Saccharomyces cerevisiae PDR18 gene in Saccharomycetaceae yeasts was reconstructed in this study. Compared with a former analysis based on the genome sequences of only nine yeast species belonging to the Hemiascomycetes phylum (Seret et al., 2009), our study took advantage of the increasing number of yeast genomes currently available, and has examined sixteen post-WGD yeast species instead of only two, spanning six different taxonomic genera, instead of only two. This fact allowed the clarification of the evolution of S. cerevisiae PDR18 and SNQ2 genes homologs after the WGD event and led us to propose that a single gene loss event has occurred in the last common ancestor of Nakaseomyces, Naumovozyma, Kazachstania, and Saccharomyces yeasts. This event explains the “interruption” of one of the post-WGD sub-lineages given that yeast strains of the Nakaseomyces, Naumovozyma, and Kazachstania genera encode only one PDR18/SNQ2 homolog gene with high synteny between them. The probability that the duplication event has occurred during the WGD event is negligible, as it would imply the gene loss in the five yeast species belonging to the Nakaseomyces, Naumovozyma, and Kazachstania genera. Also, the gene neighborhood analysis does not provide support to the second evolutionary scenario, as only one common neighboring gene in these sub-lineages belongs to the similarity cluster 949, which is also present in the chromosome environment of many genes comprised in the other post-WGD sub-lineage proposed to give rise to the duplication event. Altogether, results do not support the ohnolog status for S. cerevisiae SNQ2 and PDR18 genes, but instead support the first proposed scenario.
Concerning the point in time where the duplication event originating the SNQ2 and PDR18 sub-lineages might have occurred in the evolution of the post-WGD yeast species, it is possible that the PDR18 gene ancestor was nurtured in one of the tandem repeats encoding PDR genes that, with exception of the K. naganishii genome, are found in the genomes of all yeast species belonging to the Kazachstania and Naumovozyma genera. Under this scenario, the amino acid sequence of the ancient gene that gave rise to PDR18 did not diverge until the last common ancestor in the origin of the Saccharomyces genus split apart from the ancestral yeast population giving rise to the Kazachstania genus. In the second scenario, the PDR18 gene ancestral was originated in an independent duplication event not related with the events on the origin of the tandem repeats observed in the genomes of the Kazachstania and Naumovozyma species. Independently of which of these evolutionary scenarios is true, subsequent genome shuffling and/or other mechanisms of genome evolution should have been responsible for the transposition of the two ancestral genes encoding Pdr18 and the neighboring gene, encoding the aldose 1-epimerase, into a new chromosome environment.
Beside the gene duplication events in the origin of the SNQ2 and PDR18 paralog genes, other gene duplication events were identified in the genomes of the protoploid Saccharomycetaceae (pre-WGD). The yeast species belonging the Zygosaccharomyces genus and the Lachancea kluyveri species encoded more than one Snq2/Pdr18 homolog in their genomes, escaping the typical pattern observed in the majority of the pre-WGD species analyzed in this study, where a sole member of the Snq2/Pdr18 protein subfamily were found encoded in the corresponding genome sequences. Interestingly, the amino acid sequences of the two singletons encoded in the genome of the Z. bailii IST302 strain showed the existence of a strong divergence in respect to the amino acid sequences of the Snq2/Pdr18 homologs encoded in the other Zygosaccharomyces strains analyzed in this study. This fact and the high amino acid sequence identity shared by these two singletons suggests that these are a paralog pair originated in a duplication event that occurred recently in the evolution of the Zygosaccharomyces yeast species. The consultation of the MIPS website comprising the aligning of the presently available genome sequences of yeast strains belonging to the Zygosaccharomyces genus showed that these two singleton genes are also absent in the interspecies hybrid Z. bailii ISA13073. This result furthers strengths the hypothesis of a recent origin of these two singletons occurring at the intraspecific level.
Since it was found that PDR18 is specific for the Saccharomyces genus, with very high conservation among the 93 Saccharomyces yeast genomes examined, the MDR/MXR profiling of this transporter in S. cerevisiae was examined for a wide number of relevant toxic compounds and the possible overlapping of the susceptibility phenotypes exhibited by the Scpdr18Δ mutant and the Scsnq2Δ mutant with its paralog gene ScSNQ2 deleted was systematically examined. ScSnq2 was found to confer resistance to a more restricted range of the toxic compounds tested, compared with ScPdr18 with a demonstrated physiological function as plasma membrane transporter in the maintenance of plasma membrane ergosterol content specially under chemical stress. This biological role was related with the decreased levels of stress-induced membrane disorganization and permeabilization and counteracting transmembrane electrochemical potential dissipation (Cabrito et al., 2011; Godinho et al., 2018) and thus with the maintenance under stress of a functional plasma membrane as a selective barrier and a suitable lipid environment for the physiological activity of the embedded proteins (del Castillo Agudo, 1992; Parks and Casey, 1995; Eisenkolb, 2002; Aguilera et al., 2006; Abe and Hiraki, 2009; Caspeta et al., 2014; Kodedová and Sychrová, 2015).
Since the anticipated functional divergence between ScPdr18, ScSnq2 and the ancestral gene on the origin of the duplication event is of relevance to understand the evolutionary process acting on the two duplicate genes, the post-WGD pathogenic species Candida glabrata was selected for a systematic analysis of the susceptibility phenotype of the corresponding deletion mutant Cgsnq2Δ. Based on the susceptibility assays performed, the sole SNQ2/PDR18 homolog in C. glabrata encoding CgSnq2 appeared to be functionally closer to ScSnq2 then to ScPdr18, playing a role in 4-NQO resistance in both species and having no impact in C. glabrata tolerance to the weak acids acetic and benzoic, the herbicides 2,4-D and MCPA, the alcohols ethanol and 1,4-butanediol, and the polyamine putrescine, to which ScPDR18 expression confers tolerance. It is noteworthy that CgSnq2 was previously found to play a role in C. glabrata BPY55 tolerance to fluconazole and ketoconazole by susceptibility assays in solid YPD medium (Torelli et al., 2008). This phenotype was confirmed in this study under similar conditions, a fact that could indicate some overlapping of the function associated to CgSnq2 and ScPdr18. However, these phenotypes were not reproduced in minimal media MM, the growth conditions used for the systematic analysis of the phenotypic profiling performed.
The apparent overlapping between ScPdr18 and CgSnq2 role observed in azole resistance in rich media can be related with species-specific adaptation of C. glabrata to these fungicides, given that this yeast species is one of the most common in nosocomial fungal infections and that azoles are one of the main families of drugs that are currently being used to treat or prevent fungal infections (Perlroth et al., 2007; Jandric and Schüller, 2011; Roetzer et al., 2011) and that the C. glabrata strain used in this work is a highly azole resistant clinical isolate (Torelli et al., 2008). The sensitivity phenotype exhibited by Cgsnq2Δ toward Cu2+ toxicity might also be related to the fact that BPY55 is a clinical isolate and that adaptation to high Cu2+ environmental concentrations is a described determinant for survival of human pathogens (Festa and Thiele, 2012; Samanovic et al., 2012; Chaturvedi and Henderson, 2014; Fu et al., 2014; García-Santamarina and Thiele, 2015).
The fact that S. cerevisiae Snq2 and Pdr18 confer resistance to a very different set of chemical compounds with little overlapping, appears to exclude the evolutionary scenario where these highly similar MDR/MXR transporters have been retained in the S. cerevisiae genome due to functional redundancy or dosage effect, suggesting as the most consistent scenarios the subfunctionalization and the neofunctionalization of the gene copies. Although subfunctionalization is commonly associated with the mere division of functions of the ancestral protein by the two duplicates, another possible model is that one of the duplicate proteins becomes more efficient at performing one of the original functions of the progenitor gene (Zhang, 2003). Considering the neofunctionalization process, in most of the cases the function adopted by one of the duplicate proteins is a related function rather than an entirely new function (Zhang, 2003; Conant and Wolfe, 2008). Although there are no detailed studies focusing on SNQ2 expression impact on yeast plasma membrane lipid content and properties, Snq2 was previously found to contribute to alleviate estradiol toxicity in S. cerevisiae, a molecule highly similar to ergosterol (Mahé et al., 1996a) and Pdr18 is described to be an ergosterol transporter at the plasma membrane (Godinho et al., 2018). The clarification of whether ScSnq2 and/or CgSnq2 may play some role on lipid homeostasis and of whether ScPdr18 is the result from subfunctionalization by function specialization or whether the biological function acquired by ScPdr18 is totally different from ScSnq2 and CgSnq2, require further work and the elucidation of SNQ2 biological function.
Pdr18 is encoded in at least 87 out of the 93 genomes from the Saccharomyces genus yeasts examined in this study: in 56 out of 60 S. cerevisiae genomes, in the 25 S. paradoxus genomes, in the genomes of S. mikatae, S. kudriavzevii, S. arboricola, and S. uvarum, and in the two S. eubayanus genomes. DNA degradation during preparation for genome sequencing is the most plausible explanation for the lack of the PDR18 gene in some of the strains. For example, PDR18 gene was not found in the genome sequence of S. cerevisiae cen.pk113-7d strain since the genome sequencing shows no coverage for the right arm of chromosome XIV, where PDR18 gene resides (Nijkamp et al., 2012). Moreover, PDR18 gene was found to be present in the genomes of 964 isolates of more than 1,000 natural S. cerevisiae isolates that were a recently examined using deep coverage genome sequencing (Peter et al., 2018). This fact is consistent with the relevant physiological function encoded by PDR18 in this yeast species (Godinho et al., 2018).
Data Availability
The dataset generated for this study can be found in TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S23282).
Author Contributions
PD and CG carried out the phylogenetic and gene neighborhood analysis. CG and EP carried out the susceptibility tests. IS-C guided and coordinated this study and together with CG and PD prepared the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by ‘Fundação para a Ciência e a Tecnologia’ (FCT) (YEASTPEC project contract ERA-IB-2/0003/2015) and Ph.D. and postdoctoral fellowships to CG (SFRH/BD/ 92252/2013) and PD (SFRH/BPD/74618/2010). Funding received by iBB from FCT (UID/BIO/04565/2013) and from Programa Operacional Regional de Lisboa 2020 (Project No. 007317) is also acknowledged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank D. Sanglard, CHUV, Lausanne, Switzerland for kindly providing the C. glabrata strains. This work was dedicated to the memory of Professor André Goffeau who coordinated the international collaborative effort that lead to the first completely sequenced yeast genome, opening the door to the field of functional genomics and genome evolution of yeasts, in particular of multidrug resistance transporter genes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00476/full#supplementary-material
Footnotes
- ^http://tree.bio.ed.ac.uk/software/figtree/
- ^http://apps.cytoscape.org/apps/phylotree
- ^https://www.helmholtz-muenchen.de/ibis/index.html
References
Abe, F., and Hiraki, T. (2009). Mechanistic role of ergosterol in membrane rigidity and cycloheximide resistance in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1788, 743–752. doi: 10.1016/j.bbamem.2008.12.002
Aguilera, F., Peinado, R. A., Millan, C., Ortega, J. M., and Mauricio, J. C. (2006). Relationship between ethanol tolerance, H+-ATPase activity and the lipid composition of the plasma membrane in different wine yeast strains. Int. J. Food Microbiol. 110, 34–42. doi: 10.1016/j.ijfoodmicro.2006.02.002
Altekar, G., Dwarkadas, S., Huelsenbeck, J. P., and Ronquist, F. (2004). Parallel metropolis coupled markov chain monte carlo for Bayesian phylogenetic inference. Bioinformatics 20, 407–415. doi: 10.1093/bioinformatics/btg427
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Baker, E., Wang, B., Bellora, N., Peris, D., Hulfachor, A. B., Koshalek, J. A., et al. (2015). The genome sequence of Saccharomyces eubayanus and the domestication of lager-brewing yeasts. Mol. Biol. Evol. 32, 2818–2831. doi: 10.1093/molbev/msv168
Balzi, E., and Goffeau, A. (1995). Yeast multidrug resistance: the PDR network. J. Bioenerg. Biomembr. 27, 71–76. doi: 10.1007/BF02110333
Borst, P., Zelcer, N., and van Helvoort, A. (2000). ABC transporters in lipid transport. Biochim. Biophys. Acta 1486, 128–144. doi: 10.1016/S1388-1981(00)00053-6
Cabrito, T. R., Teixeira, M. C., Singh, A., Prasad, R., and Sá-Correia, I. (2011). The yeast ABC transporter Pdr18 (ORF YNR070w) controls plasma membrane sterol composition, playing a role in multidrug resistance. Biochem. J. 440, 195–202. doi: 10.1042/BJ20110876
Caspeta, L., Chen, Y., Ghiaci, P., Feizi, A., Buskov, S., Hallström, B. M., et al. (2014). Altered sterol composition renders yeast thermotolerant. Science 346, 75–78. doi: 10.1126/science.1258137
Charif, D., Lobry, J., Necsulea, A., Palmeira, L., and Penel, M. S. (2007). The Seqinr Package. R Packag. Available at: https://cran.r-project.org/web/packages/seqinr/seqinr.pdf
Chaturvedi, K. S., and Henderson, J. P. (2014). Pathogenic adaptations to host-derived antibacterial copper. Front. Cell. Infect. Microbiol. 4:3. doi: 10.3389/fcimb.2014.00003
Clamp, M., Cuff, J., Searle, S. M., and Barton, G. J. (2004). The Jalview Java alignment editor. Bioinformatics 20, 426–427. doi: 10.1093/bioinformatics/btg430
Conant, G. C., and Wolfe, K. H. (2008). Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 9, 938–950. doi: 10.1038/nrg2482
Decottignies, A., and Goffeau, A. (1997). Complete inventory of the yeast ABC proteins. Nat. Genet. 15, 137–145. doi: 10.1038/ng0297-137
del Castillo Agudo, L. (1992). Lipid content of Saccharomyces cerevisiae strains with different degrees of ethanol tolerance. Appl. Microbiol. Biotechnol. 37, 647–651. doi: 10.1007/BF00240742
Dias, P. J., and Sá-Correia, I. (2013). The drug: H + antiporters of family 2 (DHA2), siderophore transporters (ARN) and glutathione: H + antiporters (GEX) have a common evolutionary origin in hemiascomycete yeasts. BMC Genomics 14:901. doi: 10.1186/1471-2164-14-901
Dias, P. J., and Sá-Correia, I. (2014). Phylogenetic and syntenic analyses of the 12-spanner drug: H + antiporter family 1 (DHA1) in pathogenic Candida species: evolution of MDR1 and FLU1 genes. Genomics 104, 45–57. doi: 10.1016/j.ygeno.2014.05.005
Dias, P. J., Seret, M.-L., Goffeau, A., Correia, I. S., and Baret, P. V. (2010). Evolution of the 12-spanner drug:H + antiporter DHA1 family in Hemiascomycetous yeasts. OMICS 14, 701–710. doi: 10.1089/omi.2010.0104
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eisenkolb, M., Zenzmaier, C., Leitner, E., and Schneiter, R. (2002). A specific structural requirement for ergosterol in long-chain fatty acid synthesis mutants important for maintaining raft domains in yeast. Mol. Biol. Cell 13, 4414–4428. doi: 10.1091/mbc.E02-02-0116
Felsenstein, J. (1989). Phylip: phylogeny inference package (version 3.2). Cladistics 5, 164–166. doi: 10.1111/j.1096-0031.1989.tb00562.x
Festa, R. A., and Thiele, D. J. (2012). Copper at the front line of the host-pathogen battle. PLoS Pathog. 8:e1002887. doi: 10.1371/journal.ppat.1002887
Fu, Y., Chang, F. M. J., and Giedroc, D. P. (2014). Copper transport and trafficking at the host-bacterial pathogen interface. Acc. Chem. Res. 47, 3605–3613. doi: 10.1021/ar500300n
García-Santamarina, S., and Thiele, D. J. (2015). Copper at the fungal pathogen-host axis. J. Biol. Chem. 290, 18945–18953. doi: 10.1074/jbc.R115.649129
Godinho, C. P., Prata, C. S., Pinto, S. N., Cardoso, C., Bandarra, N. M., Fernandes, F., et al. (2018). Pdr18 is involved in yeast response to acetic acid stress counteracting the decrease of plasma membrane ergosterol content and order. Sci. Rep. 8:7860. doi: 10.1038/s41598-018-26128-7
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hirata, D., Yano, K., Miyahara, K., and Miyakawa, T. (1994). Saccharomyces cerevisiae YDR1, which encodes a member of the ATP-binding cassette (ABC) superfamily, is required for multidrug resistance. Curr. Genet. 26, 285–294. doi: 10.1007/BF00310491
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Huson, D. H., Richter, D. C., Rausch, C., Dezulian, T., Franz, M., and Rupp, R. (2007). Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. doi: 10.1186/1471-2105-8-460
Huson, D. H., and Scornavacca, C. (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067. doi: 10.1093/sysbio/sys062
Jandric, Z., and Schüller, C. (2011). Stress response in Candida glabrata: pieces of a fragmented picture. Future Microbiol. 6, 1475–1484. doi: 10.2217/fmb.11.131
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Jungwirth, H., and Kuchler, K. (2006). Yeast ABC transporters - A tale of sex, stress, drugs and aging. FEBS Lett. 580, 1131–1138. doi: 10.1016/j.febslet.2005.12.050
Kodedová, M., and Sychrová, H. (2015). Changes in the sterol composition of the plasma membrane affect membrane potential, salt tolerance and the activity of multidrug resistance pumps in Saccharomyces cerevisiae. PLoS One 10:e0139306. doi: 10.1371/journal.pone.0139306
Kolaczkowski, M., Kolaczkowska, A., Luczynski, J., Witek, S., and Goffeau, A. (1998). In vivo characterization of the drug resistance profile of the major ABC transporters and other components of the yeast pleiotropic drug resistance network. Microb. Drug Resist. 4, 143–158. doi: 10.1089/mdr.1998.4.143
Kovalchuk, A., and Driessen, A. J. M. (2010). Phylogenetic analysis of fungal ABC transporters. BMC Genomics 11:177. doi: 10.1186/1471-2164-11-177
Kuchler, K., Egner, R., Rosenthal, F., and Mahé, Y. (1997). “The molecular basis for pleiotropic drug resistance in the yeast Saccharomyces cerevisiae: regulation of expression, intracellular trafficking and proteolytic turnover of atp binding cassette (ABC) multidrug resistance transporters,” in Molecular Mechanisms of Signalling and Membrane Transport, ed. K. Wirtz (Berlin: Springer-Verlag), 305–318.
Kurtzman, C. P., and Robnett, C. J. (2003). Phylogenetic relationships among yeasts of the “Saccharomyces complex” determined from multigene sequence analyses. FEMS Yeast Res. 3, 417–432. doi: 10.1016/S1567-1356(03)00012-6
Li, S., Ha, S. J., Kim, H. J., Galazka, J. M., Cate, J. H., Jin, Y. S., et al. (2013). Investigation of the functional role of aldose 1-epimerase in engineered cellobiose utilization. J. Biotechnol. 168, 1–6. doi: 10.1016/j.jbiotec.2013.08.003
Ling, H., Chen, B., Kang, A., Lee, J. M., and Chang, M. W. (2013). Transcriptome response to alkane biofuels in Saccharomyces cerevisiae: identification of efflux pumps involved in alkane tolerance. Biotechnol. Biofuels 6:95. doi: 10.1186/1754-6834-6-95
Mahé, Y., Lemoine, Y., and Kuchler, K. (1996a). The ATP binding cassette transporters Pdr5 and Snq2 of Saccharomyces cerevisiae can mediate transport of steroids in vivo. J. Biol. Chem. 271, 25167–25172. doi: 10.1074/jbc.271.41.25167
Mahé, Y., Parle-mcdermott, A., Nourani, A., Delahodde, A., Lamprecht, A., and Kuchler, K. (1996b). The ATP-binding cassette multidrug transporter Snq2 of Saccharomyces cerevisiae?: a novel target for the transcription factors Pdrl and Pdr3. Mol. Microbiol. 20, 109–117.
Miyahara, K., Mizunuma, M., Hirata, D., Tsuchiya, E., and Miyakawa, T. (1996). The involvement of the Saccharomyces cerevisiae multidrug resistance transporters Pdr5p and Snq2p in cation resistance. FEBS Lett. 399, 317–320. doi: 10.1016/S0014-5793(96)01353-1
Monk, B. C., and Goffeau, A. (2008). Outwitting multidrug resistance to antifungals. Science 321, 367–369. doi: 10.1126/science.1159746
Nijkamp, J. F., van den Broek, M., Datema, E., de Kok, S., Bosman, L., Luttik, M. A., et al. (2012). De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb. Cell Fact. 11:36. doi: 10.1186/1475-2859-11-36
Nishida, N., Ozato, N., Matsui, K., Kuroda, K., and Ueda, M. (2013). ABC transporters and cell wall proteins involved in organic solvent tolerance in Saccharomyces cerevisiae. J. Biotechnol. 165, 145–152. doi: 10.1016/j.jbiotec.2013.03.003
Palma, M., Dias, P. J., de Canaveira Roque, F., Luzia, L., Guerreiro, J. F., and Sá-Correia, I. (2017). The Zygosaccharomyces bailii transcription factor Haa1 is required for acetic acid and copper stress responses suggesting subfunctionalization of the ancestral bifunctional protein Haa1/Cup2. BMC Genomics 18:75. doi: 10.1186/s12864-016-3443-2
Panwar, S. L., Pasrija, R., and Prasad, R. (2008). Membrane homoeostasis and multidrug resistance in yeast. Biosci. Rep. 28, 217–228. doi: 10.1042/BSR20080071
Parks, L. W., and Casey, W. M. (1995). Physiological implications of sterol biosynthesis in yeast. Annu. Rev. Microbiol. 49, 95–116. doi: 10.1146/annurev.mi.49.100195.000523
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Paulsen, I. T., Sliwinski, M. K., Nelissen, B., Goffeau, A., and Saier, M. H. (1998). Unified inventory of established and putative transporters encoded within the complete genome of Saccharomyces cerevisiae. FEBS Lett. 430, 116–125. doi: 10.1016/S0014-5793(98)00629-2
Paumi, C. M., Chuk, M., Snider, J., Stagljar, I., and Michaelis, S. (2009). ABC transporters in saccharomyces cerevisiae and their interactors: new technology advances the biology of the ABCC (MRP) Subfamily. Microbiol. Mol. Biol. Rev. 73, 577–593. doi: 10.1128/MMBR.00020-09
Perlroth, J., Choi, B., and Spellberg, B. (2007). Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med. Mycol. 45, 321–346. doi: 10.1080/13693780701218689
Peter, J., Chiara, M., De Friedrich, A., Yue, J., Pflieger, D., Bergström, A., et al. (2018). Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556, 339–347. doi: 10.1038/s41586-018-0030-5
Piecuch, A., and Obłak, E. (2014). Yeast ABC proteins involved in multidrug resistance. Cell. Mol. Biol. Lett. 19, 1–22. doi: 10.2478/s11658-013-0111-2
Piper, P. W., Millson, S. H., Mollapour, M., Panaretou, B., Siligardi, G., Pearl, L. H., et al. (2003). Sensitivity to Hsp90-targeting drugs can arise with mutation to the Hsp90 chaperone, cochaperones and plasma membrane ATP binding cassette transporters of yeast. Eur. J. Biochem. 270, 4689–4695. doi: 10.1046/j.1432-1033.2003.03866.x
Pomorski, T., Pomorski, T., Holthuis, J. C., Holthuis, J. C., Herrmann, A., Herrmann, A., et al. (2004). Tracking down lipid flippases and their biological functions. J. Cell Sci. 117, 805–813. doi: 10.1242/jcs.01055
Popescu, A. A., Huber, K. T., and Paradis, E. (2012). Ape 3.0: new tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics 28, 1536–1537. doi: 10.1093/bioinformatics/bts184
Prasad, R., Khandelwal, N. K., and Banerjee, A. (2016). Yeast ABC transporters in lipid trafficking. Fungal Genet. Biol. 93, 25–34. doi: 10.1016/j.fgb.2016.05.008
Prasad, R., and Panwar, S. (2004). Physiological functions of multidrug transporters in yeast. Curr. Sci. 86, 62–73.
Roetzer, A., Gabaldón, T., and Schüller, C. (2011). From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol. Lett. 314, 1–9. doi: 10.1111/j.1574-6968.2010.02102.x
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Samanovic, M. I., Ding, C., Thiele, D. J., and Darwin, K. H. (2012). Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe 11, 106–115. doi: 10.1016/j.chom.2012.01.009
Sanglard, D., Ischer, F., and Bille, J. (2001). Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 45, 1174–1183. doi: 10.1128/AAC.45.4.1174-1183.2001
Seret, M. L., Diffels, J. F., Goffeau, A., and Baret, P. V. (2009). Combined phylogeny and neighborhood analysis of the evolution of the ABC transporters conferring multiple drug resistance in hemiascomycete yeasts. BMC Genomics 10:459. doi: 10.1186/1471-2164-10-459
Servos, J., Haase, E., and Brendel, M. (1993). Gene SNQ2 of Saccharomyces cerevislae, which confers resistance to 4-nitroquinoline-N-oxide and other chemicals, encodes a 169 kDa protein homologous to ATP-dependent permeases. Mol. Gen. Genet. 236, 214–218. doi: 10.1007/BF00277115
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Snider, J., Hanif, A., Lee, M. E., Jin, K., Yu, A. R., Graham, C., et al. (2013). Mapping the functional yeast ABC transporter interactome. Nat. Chem. Biol. 9, 565–574. doi: 10.1038/nchembio.1293
Taylor, J. S., and Raes, J. (2004). Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38, 615–643. doi: 10.1146/annurev.genet.38.072902.092831
Teixeira, M. C., Godinho, C. P., Cabrito, T. R., Mira, N. P., and Sá-Correia, I. (2012). Increased expression of the yeast multidrug resistance ABC transporter Pdr18 leads to increased ethanol tolerance and ethanol production in high gravity alcoholic fermentation. Microb. Cell Fact. 11:98. doi: 10.1186/1475-2859-11-98
Torelli, R., Posteraro, B., Ferrari, S., La Sorda, M., Fadda, G., Sanglard, D., et al. (2008). The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 68, 186–201. doi: 10.1111/j.1365-2958.2008.06143.x
Tsujimoto, Y., Shimizu, Y., Otake, K., Nakamura, T., Okada, R., Miyazaki, T., et al. (2015). Multidrug resistance transporters Snq2p and Pdr5p mediate caffeine efflux in Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 79, 1103–1110. doi: 10.1080/09168451.2015.1010476
van Leeuwen, J. S., Ünlü, B., Vermeulen, N. P. E., and Vos, J. C. (2012). Differential involvement of mitochondrial dysfunction, cytochrome P450 activity, and active transport in the toxicity of structurally related NSAIDs. Toxicol. In Vitro 26, 197–205. doi: 10.1016/j.tiv.2011.11.013
Van Meer, G., Voelker, D. R., and Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. doi: 10.1038/nrm2330
Ververidis, P., Davrazou, F., Diallinas, G., Georgakopoulos, D., Kanellis, A. K., and Panopoulos, N. (2001). A novel putative reductase (Cpd1p) and the multidrug exporter Snq2p are involved in resistance to cercosporin and other singlet oxygen-generating photosensitizers in Saccharomyces cerevisiae. Curr. Genet. 39, 127–136. doi: 10.1007/s002940100189
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M., and Barton, G. J. (2009). Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Wehrschütz-Sigl, E., Jungwirth, H., Bergler, H., and Högenauer, G. (2004). The transporters Pdr5p and Snq2p mediate diazaborine resistance and are under the control of the gain-of-function allele PDR1-12. Eur. J. Biochem. 271, 1145–1152. doi: 10.1111/j.1432-1033.2004.04018.x
Whelan, S., and Goldman, N. (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699. doi: 10.1093/oxfordjournals.molbev.a003851
Keywords: ABC transporters, PDR18 and SNQ2, phylogenetic and genomic neighborhood analyzes, comparative genomics and evolution, multidrug resistance
Citation: Godinho CP, Dias PJ, Ponçot E and Sá-Correia I (2018) The Paralogous Genes PDR18 and SNQ2, Encoding Multidrug Resistance ABC Transporters, Derive From a Recent Duplication Event, PDR18 Being Specific to the Saccharomyces Genus. Front. Genet. 9:476. doi: 10.3389/fgene.2018.00476
Received: 26 June 2018; Accepted: 26 September 2018;
Published: 15 October 2018.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Adam Michael Reitzel, University of North Carolina at Charlotte, United StatesKenneth Wolfe, University College Dublin, Ireland
Copyright © 2018 Godinho, Dias, Ponçot and Sá-Correia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Sá-Correia, aXNhY29ycmVpYUB0ZWNuaWNvLnVsaXNib2EucHQ=
†These authors have contributed equally to this work