 Domenico Roberti1†
Domenico Roberti1† Renata Conforti2†Teresa Giugliano2Barbara Brogna2Immacolata Tartaglione1Maddalena Casale1
Renata Conforti2†Teresa Giugliano2Barbara Brogna2Immacolata Tartaglione1Maddalena Casale1 Giulio Piluso2
Giulio Piluso2 Silverio Perrotta1*
Silverio Perrotta1*- 1Department of Woman, Child and General and Specialized Surgery, University of Campania “L. Vanvitelli”, Naples, Italy
- 2Department of Precision Medicine, University of Campania “L. Vanvitelli”, Naples, Italy
Diamond-Blackfan anemia (DBA) is a rare congenital erythroid aplasia with a highly heterogeneous genetic background; it usually occurs in infancy. Approximately 30–40% of patients have other associated congenital anomalies; in particular, facial anomalies, such as cleft palate, are part of about 10% of the DBA clinical presentations.
Pierre Robin sequence (PRS) is a heterogeneous condition, defined by the presence of the triad of glossoptosis, micrognathia and cleft palate; it occurs in 1/8500 to 1/14,000 births.
Klippel Feil (KF) syndrome is a complex of both osseous and visceral anomalies, characterized mainly by congenital development defects of the cervical spine.
We describe the case of a 22-years-old woman affected by DBA, carrying a de novo deletion about 500 Kb-long at 12q13.2-q13.3 that included RPS26 and, at least, others 25 flanking genes. The patient showed craniofacial anomalies due to PRS and suffered for KF deformities (type II). Computed Tomography study of cranio-cervical junction (CCJ) drew out severe bone malformations and congenital anomalies as atlanto-occipital assimilation (AOA), arcuate foramen and occipito-condylar hyperplasia. Foramen magnum was severely reduced. Atlanto-axial instability (AAI) was linked to atlanto-occipital assimilation, congenital vertebral fusion and occipito-condyle bone hyperplasia. Basilar invagination and platybasia were ruled out on CT and Magnetic Resonance Imaging (MRI) studies. Furthermore, the temporal Bone CT study showed anomalies of external auditory canals, absent mastoid pneumatization, chronic middle ear otitis and abnormal course of the facial nerve bones canal.
The described phenotype might be related to the peculiar deletion affecting the patient, highlighting that genes involved in the in the breakdown of extracellular matrix (MMP19), in cell cycle regulation (CDK2), vesicular trafficking (RAB5B), in ribonucleoprotein complexes formation (ZC3H10) and muscles function (MYL6 and MYL6B) could be potentially related to bone-developmental disorders. Moreover, it points out that multiple associated ribosomal deficits might play a role in DBA-related phenotypes, considering the simultaneous deletion of three of them in the index case (RPS26, PA2G4 and RPL41), and it confirms the association among SLC39A5 functional disruption and severe myopia.
This report highlights the need for a careful genetic evaluation and a detailed phenotype-genotype correlation in each complex malformative syndrome.
Background
Craniofacial disorders are highly variable developmental anomalies and may occur on their own or with other syndromes (Cielo and Marcus, 2015). Various syndromes, characterized by craniofacial disorders, are also associated with anomalies of the cranio-cervical region (Menezes and Vogel, 2008).
Diamond-Blackfan anemia (DBA) is a congenital erythroid aplasia, that is usually present in infancy as severe hypoplastic macrocytic anemia (Clinton and Gazda, 1993) and it has to be differentiated by diseases with similar onset such as Pearson syndrome (Tumino et al., 2011). It affects about 5 to 7 cases/million live births per year (Lipton et al., 2006). Most cases are sporadic, while approximately 10 to 25% are familial (Campagnoli et al., 2004; Quarello et al., 2012). It is usually associated with morphological abnormalities (Clinton and Gazda, 1993; Campagnoli et al., 2004).
Approximately 30–40% of patients with DBA suffer for congenital anomalies that may involve head, upper limb, heart and genitourinary systems (Vlachos et al., 2008; Boria et al., 2010). The craniofacial anomalies are the most common and characterized, mostly by hypertelorism and broad flat nasal bridge (Vlachos et al., 2008). The hand deformities include triphalangeal thumb and thenar muscle hypoplasia. There may also be weak radial pulse (Ball et al., 1996). Endocrine dysfunctions are common in DBA and, in order of frequency, these include: adrenal insufficiency, hypogonadism, hypothyroidism, growth hormone deficiency/resistance, diabetes mellitus and diabetes insipidus (Lahoti et al., 2016). Many affected children are below average height for their age, and may have delayed puberty (Ball et al., 1996).
Around 60% of patients with DBA have associated germline mutations (50%) or deletions (10%) in 15 ribosomal protein genes and, in rare cases, GATA1 (Choesmel et al., 2007; Boria et al., 2010; Mirabello et al., 2017). There are no clear genotype-phenotype correlations, with exception of patients with mutations in RPL5 and RPL11, which display a high frequency of developmental anomalies, especially cleft palates and triphalangeal thumbs (Gazda et al., 2012). The most common affected gene is RPS19 (Draptchinskaia et al., 1999; Campagnoli et al., 2004). Heterozygous mutations or deletions of RPS26 are relatively uncommon (Doherty et al., 2010; Quarello et al., 2012). It has been already described a patient with KFS associated with DBA due to a point mutation in RPS26 identified only by direct sequencing, missing analysis of the surrounding regions (Cmejla et al., 2011).
Pierre Robin sequence (PRS) is a rare etiologically nonspecific complex, defined by the clinical triad of glossoptosis, retro/micrognathia, and cleft or agenesis of the palate (Printzlau and Andersen, 2004; Butow et al., 2009).
Two forms of PRS have been reported in scientific literature: the syndromic PRS (S-PRS) and the non-syndromic PRS (N-PRS). The first one shows a prevalence of about 35%, compared with the 65% of the N-PRS (Printzlau and Andersen, 2004; Butow et al., 2009). The S-PRS, as in this case, can be associated with several different syndromes including Stickler Syndrome, Velo-cardio-Facial Syndrome, Fetal Alcohol Syndrome, mandibular Syndrome and trisomy 18.
Few studies have described the relation between KFS and PRS (Judge et al., 1999; Molnar et al., 2007; Al-Ani et al., 2009; Butow et al., 2009), and we have been able to find only 4 previous cases in literature until now.
Klippel-Feil (KF) syndrome is also a rare disease, that occurs from 1 in 40,000 to 42,000 newborns worldwide, with a slightly higher occurrence in females (Thomsen et al., 1997; Tracy et al., 2004). Symptoms of KFS are cervical vertebra fusion syndrome, KF deformity, and KF sequence disorder, characterized by abnormal fusion of two or more cervical vertebrae, which is present from birth with other osseous anomalies (Tracy et al., 2004; Samartzis et al., 2006; Conforti et al., 2012). The KFS has a heterogeneous clinical presentation and aetiology. Although the majority of cases are un-classified, four different genes have been described as causing this disease: two with an autosomal dominant transmission (GDF6 – KFS1 and GDF3 – KFS3) (Tassabehji et al., 2008; Ye et al., 2010) and two with an autosomal recessive one (MEOX1 – KFS2 and MYO18B – KFS4) (Mohamed et al., 2013; Alazami et al., 2015).
Case Presentation
Subject and Clinical Features
We describe the case of a 22 year-old woman (II2), known to be affected by PRS. She is the second-born of a mother who had had three term pregnancies (Figure S1). The two brothers are in good health, without signs of congenital abnormalities. The pregnancy was 38 weeks. After a natural childbirth, she weighed 2.1 kg and showed neonatal respiratory distress syndrome. She displayed a typical PRS (micrognathia, glossoptosis, cleft palate) and triphalangeal thumbs. She was also diagnosed with a congenital perimebranous ventricular defect, without haemodynamic effects.
On follow-up examinations, a neurodevelopmental delay was observed: she gained head control at 4–5 months, the ability to sit and to stand unassisted, respectively, at 11 months and at 13 months, and learned to walk, precariously, at 20 months. By this time, she gained a poor verbal language.
In her first months of life, she was hospitalized for the management of her congenital abnormalities; therefore, she was diagnosed with an inherited hyporigenerative anemia that required regular blood transfusion therapy throughout her life. She is currently transfused with four units of packed red blood cells per month.
Genetic characterization of the congenital anemia by multiplex ligation-dependent probe amplification (MLPA) assay led to the discovery of a de novo chromosomal deletion involving RPS26 (data not shown), allowing diagnosis of DBA (Doherty et al., 2010; Quarello et al., 2012).
The clinical examination on admission to our institute, when she was 21 years old, showed a peculiar face, skeletal abnormalities in a complex malformation syndrome and a mental deficiency. Dysmorphic facial features included, beside PRS, prominent nose bridge, low-set ears, bilateral external auditory meatus abnormalities, ocular asymmetry with buphthalmos, intermittent exotropia with left eye dominance, severe myopia, tooth decay and cavities. In addition, skeletal malformations consisted mainly of short stature, right-convex thoracic scoliosis with dorsal hump, hip dysmetria with heterometry of lower limbs, shortness and clinodactyly of fingers with hypoplasia of the distal phalanges of 1th fingers, cutis laxa with characteristic wrinkled palms and soles and multiple skin nevi. She also had several endocrinological alterations as primary amenorrhea, mild hyper-prolactinaemia and moderate familiar hyper-cholesterolaemia.
She showed trigeminal nerve palsy, bilateral mixed hearing loss, rhinolalia, dysarthria and acquired dysphagia for solid foods. She suffers by severe neck pain. Functional limitations in shoulder abduction were detected with lower limbs antigravity muscle weakness and decreased functional ability. Reflexes were normal, except for a bilateral indifferent plantar response.
She was properly informed about all the procedures and about the intent of anonymously publishing the data obtained; to confirm her acceptance, she signed informed consent forms agreeing to the procedures and to publication.
Radiological Features
The complex physical malformations required a neuro-radiological evaluation with Computed tomography (CT) study of the temporal bone, cranio-cervical junction (CCJ) and cervical spine.
Brain and spine Magnetic Resonance Imaging (MRI) studies were performed to evaluate neuro-radiological anomalies.
CCJ study focused on bone malformations, showing atlanto-occipital assimilation (AOA) caused by a complete fusion of the C1 anterior arch and lateral masses and by partial fusion of the posterior arch to the foramen magnum. Arcuate foramen was also observed on the left side (Figure 1A). The atlanto-dental interval (AADI: distance, on the sagittal plane, between the anterior assimilated C1 arch and the odontoid process) (Zong et al., 2017) appeared increased (10 mm) suggesting an atlanto-axial instability (AAI) (Figure 1A), defined for atlanto-dental interval >3 mm (Gamble and Rinsky, 1985; Ferreira and Botelho, 2015).
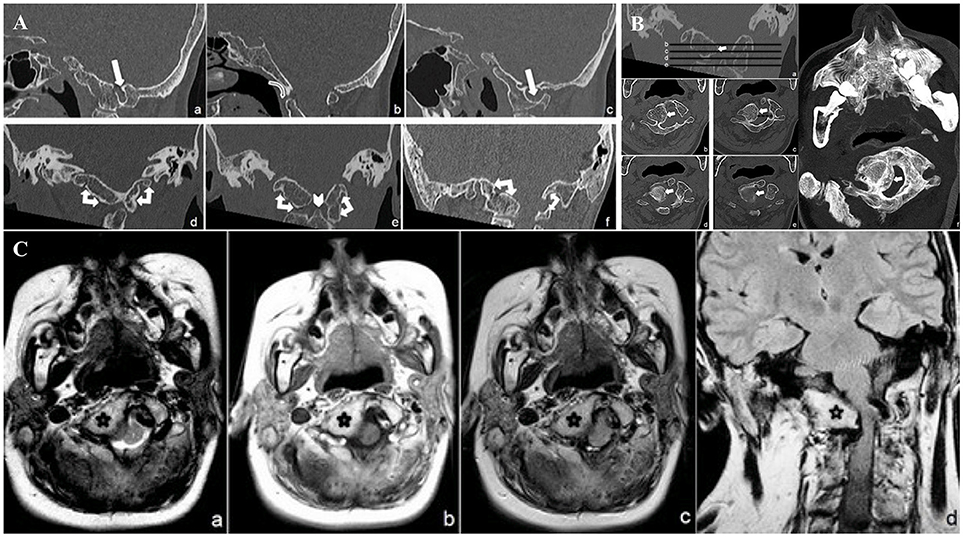
Figure 1. (A) CT minimum intensity projection (MIP) Sagittal (a: right parasagittal plane, b: midsagittal plane, c: left parasagittal plane) and Coronal (d: anterior atlanto-occipital complete fusion, e: lateral masses atlanto-occipital bilateral complete fusion, f: posterior atlanto-occipital partial fusion) CCJ reconstruction. Complete C 1 anterior arch and lateral masses fusion and partial fusion of the posterior arch to the occipital foramen (double-headed arrows). Foramen arcuate (white harrows). Note the “comma” appearance of complete anterior atlanto-occipital fusion (curved harrow) and the smaller odontoid process (arrowhead). (B) CT Coronal (a) and transverse minimum intensity projection (MIP) (b–e) and transverse shaded surface display (SSD) (f) reconstructions of narrowed foramen magnum due to right occipito-condylar hyperplasia (withe arrows). (C) MR transverse T2 (a), T1 (b), FLAIR (c) and coronal FLAIR (d) weighed sequences, show right ventrolateral compression of the lower medulla and upper cervical spinal cord (white arrow) at the level of narrowed foramen magnum due to CCJ complex malformation (black star).
The odontoid process was smaller (Figure 1A) than normal average (Zong et al., 2017). Sites of embryogenesis for the atlas' anterior arch and the atlantoaxial ligaments system are the same, so that the assimilation of the anterior arch of the atlas is often associated with AAI (Ferreira and Botelho, 2015). Very few authors already reported, as in this case, the association between PRS and AAI (Gamble and Rinsky, 1985; Molnar et al., 2007) due to AOA. The patient is affected as well by a right occipito-condylar hyperplasia, producing narrowing and malformation of both the foramen magnum and the upper cervical spine canal (Figure 1B). This hyperplasia causes the compression and displacement of the right side of the bulbo-medullary junction (Figure 1C). Basilar invagination (radiologically defined when the tip of the odontoid is located above the Chamberlin line) (Molnar et al., 2007) and platybasia (flattening of the skull base) were ruled out on both CT and MRI (Kisker et al., 1997; Zong et al., 2017).
Temporal bone CT study showed right hypoplasia and anomalous course of the external auditory channels, absence of pneumatization at the right mastoid, a chronic right ear otitis media, widening of the third part of the ipsilateral facial nerve bone canal, partial calcification of the left tympanic membrane and ossicular chain ankyloses (Figure 2A).
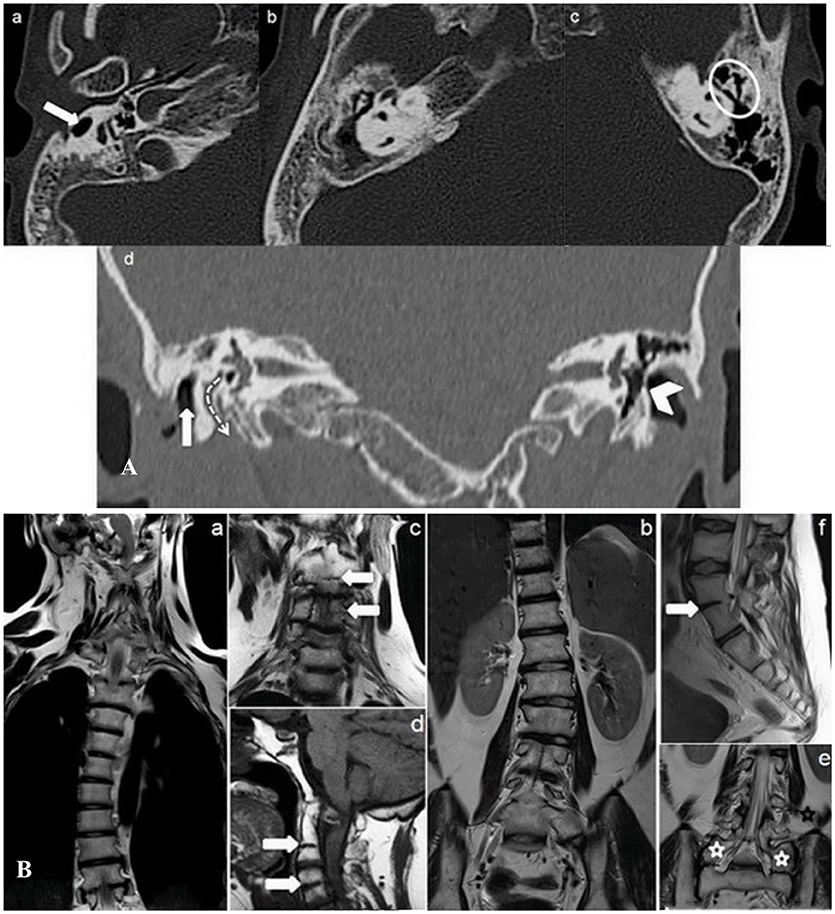
Figure 2. (A) Transverse (a–c) and Coronal (d) minimum intensity projection (MIP) CT temporal bone study. Right external auditory canal malformation (a) (white arrow); right chronic otitis media (b) (white star); ankyloses of left ossicular chain (c) (white circle); right widening of facial canal (third segment) (dotted arrow) and partial calcification of left tympanic membrane (arrowhead) (d). (B) MR Coronal T2 (a–c, e), Sagittal T1 (d), and Sagittal T2 (f) weighed sequences whole spine study. The study shows, besides rotoscoliosis (a, b), cervical (C 2–C 3, C 4–C 5) (d, f) and lumbar (L 4–L 5) vertebral fusion (white arrows), and L 5 bilateral sacralisation (e) (white stars) (KFS type II malformation).
Whole spinal cord MRI study added further information, showing not only rotoscoliosis, but also cervical (C2 – C3, C4 – C5) and lumbar (L4 – L5) vertebral fusion and bilateral L5 sacralisation (Figure 2B). The spine malformations led to a diagnosis of KFS type II (Samartzis et al., 2006), without malformations neither of the meninges nor of the nervous system.
Genetic Characterization
Dysmorphic features and malformations observed in the patient suggested to further investigate the initially identified genomic deletion affecting RPS26. We then carried out array-CGH analysis in the proband and her parents, confirming the presence of a wider de novo heterozygous microdeletion on chromosome 12q13.2-q13.3 (Figure 3). By Real-Time PCR, we further refined 5' and 3' breakpoint boundaries of the deletion, resulting in a minimal critical region of about 500 Kb in size (Figure 3 and Table S1). The microdeletion encompassed at least 26 coding genes (Table 1) and expression profiling by Taqman assay confirmed about 50% reduction of the expression for many of these genes (Table 1; data not shown). Some of them, in particular SARNP, DNAJC14, ORMDL2, DNAJC14, ANKRD52, COQ10A and CS, although heterozygously deleted, were not tested for expression (Table 1). At least three genes (MMP19, RPS26 and SLC39A5) already cause autosomal dominant diseases, while further 12 genes may be sensitive to haploinsufficiency, as they showed a pLI > 0.75 (Table 1). In addition, no similar deletions were annotated in Decipher (decipher.sanger.ac.uk) or have been previously described in literature.
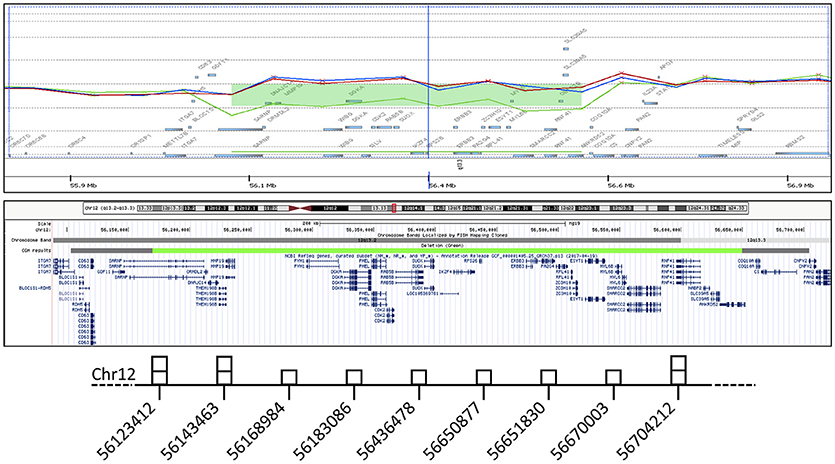
Figure 3. In the gene view (top), the probe distribution and signal intensity are shown for the index patient (green) and her parents (red and blue), with a green bar indicating the deletion detected with 8 probes. In the UCSC graphic view (middle), a similarly colored bar corresponds to the minimal aberration length, while the flanking grey bars indicate the 5' and 3' breakpoint boundaries. The results of genomic quantification by real-time PCR are schematically represented (bottom). The middle base-pair position of each amplicon along chromosome is surmounted by a square corresponding to the number of detected copies.
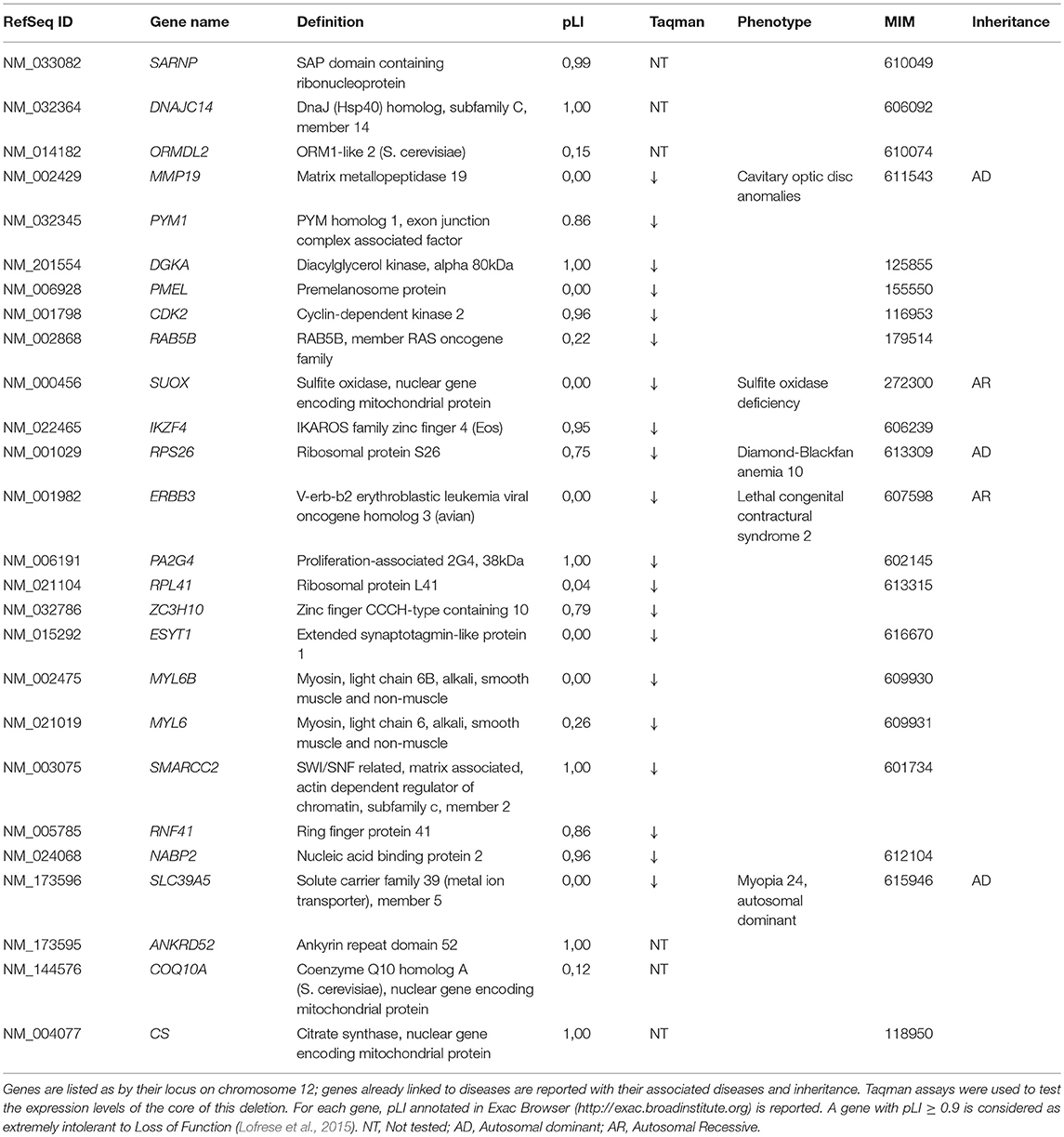
Table 1. List of genes deleted in the described patient, with results of Taqman assay and already associated inherited diseases.
Discussion
The index patient shows features at the boundaries of the three reported syndromes.
The hypoplastic mandible, the glossoptosis and the U-shaped cleft palate are strictly related to PRS and contribute to airway obstruction (Butow et al., 2009; Cielo and Marcus, 2015) as found in the described patient. In PRS, there is a 10.5% incidence rate of ear malformations (Breugem et al., 2016), which consist of multiple architectural anomalies involving the entire ear, including abnormal auricles, anomalies of the ossicular chain, abnormal stapes footplates and middle ear infection. Middle ear disturbances, as in the index case, are common and are mostly related to the cleft palate and associated to the Eustachian tube dysfunction (Sando et al., 1988; Yamaguchi et al., 1990; Robinson et al., 1993; Gruen et al., 2005). Chronic mucotympanum, chronic middle ear otitis, tympanic membrane retraction pockets and choleastotomas are the most frequent issues related to the middle ear (Gruen et al., 2005). Other anomalies include abnormal course of the facial nerve, as in the described case (Gruen et al., 2005; Rotondo et al., 2010), abnormal insertion of the tensor tympani tendon, ankylosis of the ossicules, and anomalous stapedial footplates, most of which can be related to anomalies of the branchial arches.
Typical elements of KFS are, instead, vertebral fusions, short and webbed neck, decreased range of motion in the cervical spine, deformed chest wall, high placed scapulae, abnormal curvature of the spine (scoliosis), raised scapula (Sprengel's scapula), rib defects, low hair line. The index case shows KF deformity with multiple fusions of non-contiguous cervical and lumbar vertebrae corresponded to type II of KFS, described by Samartzis et al. (Samartzis et al., 2006).
Radiological evaluation pointed out diffuse and severe CCJ bone malformations.
PRS and KFS could both display failure of segmentation between the fourth occipital sclerotome and the first cervical sclerotome resulting in AOA (Menezes and Vogel, 2008; Smoker and Khanna, 2008). The displacement of the assimilated atlas and the right occipito-condylar hyperplasia resulted, instead, in a narrowed foramen magnum, which was responsible for both severe compression of the underlying bulbo-medullary junction and AAI (Gamble and Rinsky, 1985; Yamaguchi et al., 1990; Molnar et al., 2007; Rotondo et al., 2010; Ferreira and Botelho, 2015; Zong et al., 2017). The described Occipito-condylar hyperplasia is an extremely rare congenital entity and only 3 other cases were previously described (Smoker and Khanna, 2008; Rojas et al., 2009; Lofrese et al., 2015). This bone malformation may be due to the excessive growth of the proatlas during the embryogenesis (Smoker and Khanna, 2008; Rojas et al., 2009; Lofrese et al., 2015). Upon birth, the displaced AOA and the overall shift in the head's balance could cause progressive growing of osteophytes, gradually constrain the bulb and the spinal cord. Therefore, neurological decline may occur due to the progressive degenerative ossification (Rojas et al., 2009; Lofrese et al., 2015; Shih et al., 2015). The ossification on the ligamentous interface and ligamentous holding forces may also increase axial instability (Gamble and Rinsky, 1985; Yamaguchi et al., 1990; Molnar et al., 2007; Rotondo et al., 2010; Ferreira and Botelho, 2015; Zong et al., 2017).
The patient's CCJ imbalance (due to skull-cervical spine malformation), coupled with the lack of stable articular joints, exposes her to a high risk of sudden neurological manifestations. It has been shown that common pathways exist between the neural cresta and the paraxial mesoderm embryogenesis during CCJ development (Menezes and Vogel, 2008).
Among genes deleted in the index case, three of them are involved in ribosomal biogenesis (RPS26, PA2G4, RPL41). Ribosomes catalyze for protein synthesis and their function impairment have been extensively involved in DBA pathogenesis. The deletion of this gene cluster could aggravate hematological features linked to DBA. Other genes in the deleted region are related to different pathways.
MMP19 is a gene related to autosomal dominant inherited congenital cavitary optic disc anomalies (Moore et al., 2000), encoding for a zinc-binding endopeptidases that degrades several components of the extracellular matrix. Zinc is an essential cofactor for hundreds of enzymes, thus being fundamental for hundreds of functions.
The identified deletion also included SLC39A5 that encodes for a protein showing structural characteristics of zinc transporters and it has been linked with an autosomal dominant severe form of myopia (Guo et al., 2014), as in the described patient.
Other genes involved in the deletion where expected to be extremely intolerant to Loss of Function (LoF) because of their pLI.
SAP domain containing ribonucleoprotein (SARNP) has a putative role in cell cycle progression acting as a single-stranded DNA binding protein (Fukuda et al., 2002).
DNAJC14 (DnaJ Heat Shock Protein Family (Hsp40) Member C14) regulates target proteins' export from the endoplasmic reticulum to the cell surface and it seems to be involved in protection from flavivirus infections (Yi et al., 2011). The exon junction complex (EJC) serves as a positional landmark for the intron exon structure of genes; the index patient has a deletion of PYM1, a key member of this complex, actin as a disassembly factor; it also associates with the 40S ribosomal subunit (Gehring et al., 2009). DGKA, instead, encodes for a Diacylglycerol kinase, ultimately removing it; it plays a role in intracellular signaling and phospholipid synthesis. Cell cycle control has been vaguely related to developmental syndromes (Tessadori et al., 2017). The index patient has an allelic deletion for CDK2, a key cell cycle related serine/threonine protein kinase. IKZF4 encodes a zinc-finger transcription factor required during early B cell development. The protein encoded by SMARCC2 displays helicase and ATPase activities, whose enzymatic disruption often leads to development disorders. Type 1 cytokine receptor signalling impairment has been, instead, linked previously to congenital Immunodeficiency, although their functions are redundant; NABP2 encodes for a Single-stranded DNA-binding protein, usually necessary for several DNA-related metabolic processes.
Ankyrin repeat domain 52 (ANKRD52) is a protein acting as a regulatory subunit of protein phosphatase 6 (PP6), involved in the recognition of phosphoprotein substrates (Watanabe et al., 2018). Citrate synthase is, instead, a protein codified by CS, acting as a Krebs tricarboxylic acid cycle enzyme catalazying the synthesis of citrate from oxaloacetate and acetyl coenzyme A (Hayward and Berendsen, 1998).
New studies focused on the role played by these genes, in developmental control and their regulatory functions and accurate craniometrical studies of CVJ anomalies, may offer early diagnosis, new therapeutic targets and specific treatment before neurological damage occurs.
This case-report points out that DBA, and mostly PRS and KF have a further genetic heterogeneity and it underlines the need for a careful genetic evaluation and a detailed phenotype-genotype correlation in each complex malformative syndrome.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Statement
We confirm that any aspect of the work covered in this manuscript that has involved the human patient has been approved by the ethical committee of our institution and has been conduct according to the The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans and to general Good clinical practice principles and respecting clinical expertise, patient values, and the best research evidence for the patient's care. The patient and her mother signed informed consents to undergo the procedures described. Moreover, a written informed consent has been obtained for publication in print and electronic form from the patient and her mother.
Author Contributions
DR takes care of the patient, performed the Taqman assays, coordinated the study evaluation and wrote the manuscript. RC performed the radiological evaluation and wrote the manuscript. TG performed the genetic assays. BB performed the analysis of the radiological data. IT and MC take care of the patient and performed the clinical evaluation. GP supervised the genetic analysis and wrote the manuscript. SP supervised the analysis and the manuscript preparation.
Funding
The authors declare that there have been no funding involved in the production of this case-report.
Consent for Publication
A written informed consent has been obtained for publication in print and electronic form from the patient and her mother.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank prof. Ugo Ramenghi for the initial BD anemia diagnosis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00549/full#supplementary-material
Abbreviations
AADI, Anterior atlanto-dental interval; AAI, Atlanto-axial instability; AOA, Atlanto-occipital assimilation; CCJ, Cranio-cervical junction; Cgh-Array, Comparative genomic hybridization Array; CT, Computed tomography; DBA, Diamond-Blackfan anemia; KF, Klippel Feil; MRI, Magnetic resonance imaging; PRS, Pierre Robin sequence.
References
Al-Ani, S. A., Rees, M., and De Chalain, T. M. (2009). Our experiences managing a patient with mandibular duplication and cervical spinal fusion. J. Craniofac. Surg. 20, 2118–2122. doi: 10.1097/SCS.0b013e3181bec654
Alazami, A. M., Kentab, A. Y., Faqeih, E., Mohamed, J. Y., Alkhalidi, H., Hijazi, H., et al. (2015). A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18B. J. Med. Genet. 52, 400–404. doi: 10.1136/jmedgenet-2014-102964
Ball, S. E., Mcguckin, C. P., Jenkins, G., and Gordon-Smith, E. C. (1996). Diamond-Blackfan anaemia in the U.K.: analysis of 80 cases from a 20-year birth cohort. Br. J. Haematol. 94, 645–653. doi: 10.1046/j.1365-2141.1996.d01-1839.x
Boria, I., Garelli, E., Gazda, H. T., Aspesi, A., Quarello, P., Pavesi, E., Ferrante, D., et al. (2010). The ribosomal basis of Diamond-Blackfan anemia: mutation and database update. Hum. Mutat. 31, 1269–1279. doi: 10.1002/humu.21383
Breugem, C. C., Evans, K. N., Poets, C. F., Suri, S., Picard, A., Filip, C., et al. (2016). Best practices for the diagnosis and evaluation of infants with robin sequence: a clinical consensus report. JAMA Pediatr. 170, 894–902. doi: 10.1001/jamapediatrics.2016.0796
Butow, K. W., Hoogendijk, C. F., and Zwahlen, R. A. (2009). Pierre Robin sequence: appearances and 25 years of experience with an innovative treatment protocol. J. Pediatr. Surg. 44, 2112–2118. doi: 10.1016/j.jpedsurg.2009.04.018
Campagnoli, M. F., Garelli, E., Quarello, P., Carando, A., Varotto, S., Nobili, B., et al. (2004). Molecular basis of Diamond-Blackfan anemia: new findings from the Italian registry and a review of the literature. Haematologica 89, 480–489. Available online at: http://www.haematologica.org/content/89/4/480.long
Choesmel, V., Bacqueville, D., Rouquette, J., Noaillac-Depeyre, J., Fribourg, S., Cretien, A., et al. (2007). Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood 109, 1275–1283. doi: 10.1182/blood-2006-07-038372
Cielo, C. M., and Marcus, C. L. (2015). Obstructive sleep apnoea in children with craniofacial syndromes. Paediatr. Respir. Rev. 16, 189–196. doi: 10.1016/j.prrv.2014.11.003
Clinton, C., and Gazda, H. T. (1993). “Diamond-Blackfan anemia,” in GeneReviews(R), eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H. Bean, T. D. Bird, N. Ledbetter, H. C. Mefford, R. J. H. Smith, and K. Stephens (Seattle, WA: University of Washington), 1993–2018.
Cmejla, R., Ludikova, B., Sukova, M., Blatny, J., and Pospisilova, D. (2011). Can mutations in the ribosomal protein S26 (RPS26) gene lead to Klippel-Feil syndrome in Diamond-Blackfan anemia patients? An update from the Czech Diamond-Blackfan anemia registry. Blood Cells Mol. Dis. 46, 300–301. doi: 10.1016/j.bcmd.2011.02.003
Conforti, R., Taglialatela, G., Rinaldi, F., Quaranta, E., Cirillo, M., and Paolisso, G. (2012). Cervical chordoma. A case report and differential diagnosis. Neuroradiol. J. 25, 185–187. doi: 10.1177/197140091202500205
Doherty, L., Sheen, M. R., Vlachos, A., Choesmel, V., O'donohue, M. F., Clinton, C., et al. (2010). Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 86, 222–228. doi: 10.1016/j.ajhg.2009.12.015
Draptchinskaia, N., Gustavsson, P., Andersson, B., Pettersson, M., Willig, T. N., Dianzani, I., et al. (1999). The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 21, 169–175. doi: 10.1038/5951
Ferreira, E. D., and Botelho, R. V. (2015). Atlas assimilation patterns in different types of adult craniocervical junction malformations. Spine 40, 1763–1768. doi: 10.1097/BRS.0000000000001045
Fukuda, S., Wu, D. W., Stark, K., and Pelus, L. M. (2002). Cloning and characterization of a proliferation-associated cytokine-inducible protein, CIP29. Biochem. Biophys. Res. Commun. 292, 593–600. doi: 10.1006/bbrc.2002.6680
Gamble, J. G., and Rinsky, L. A. (1985). Combined occipitoatlantoaxial hypermobility with anterior and posterior arch defects of the atlas in Pierre-Robin syndrome. J. Pediatr. Orthop. 5, 475–478. doi: 10.1097/01241398-198507000-00018
Gazda, H. T., Preti, M., Sheen, M. R., O'donohue, M. F., Vlachos, A., Davies, S. M., et al. (2012). Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum. Mutat. 33, 1037–1044. doi: 10.1002/humu.22081
Gehring, N. H., Lamprinaki, S., Hentze, M. W., and Kulozik, A. E. (2009). The hierarchy of exon-junction complex assembly by the spliceosome explains key features of mammalian nonsense-mediated mRNA decay. PLoS Biol. 7:e1000120. doi: 10.1371/journal.pbio.1000120
Gruen, P. M., Carranza, A., Karmody, C. S., and Bachor, E. (2005). Anomalies of the ear in the Pierre Robin triad. Ann. Otol. Rhinol. Laryngol. 114, 605–613. doi: 10.1177/000348940511400805
Guo, H., Jin, X., Zhu, T., Wang, T., Tong, P., Tian, L., et al. (2014). SLC39A5 mutations interfering with the BMP/TGF-beta pathway in non-syndromic high myopia. J. Med. Genet. 51, 518–525. doi: 10.1136/jmedgenet-2014-102351
Hayward, S., and Berendsen, H. J. (1998). Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30, 144–154.
Judge, B., Hamlar, D., and Rimell, F. L. (1999). Mandibular distraction osteogenesis in a neonate. Arch. Otolaryngol. Head Neck Surg. 125, 1029–1032. doi: 10.1001/archotol.125.9.1029
Kisker, C., Schindelin, H., Pacheco, A., Wehbi, W. A., Garrett, R. M., Rajagopalan, K. V., et al. (1997). Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell 91, 973–983. doi: 10.1016/S0092-8674(00)80488-2
Lahoti, A., Harris, Y. T., Speiser, P. W., Atsidaftos, E., Lipton, J. M., and Vlachos, A. (2016). Endocrine dysfunction in diamond-blackfan anemia (DBA): a report from the DBA registry (DBAR). Pediatr. Blood Cancer 63, 306–312. doi: 10.1002/pbc.25780
Lipton, J. M., Atsidaftos, E., Zyskind, I., and Vlachos, A. (2006). Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan anemia registry. Pediatr. Blood Cancer 46, 558–564. doi: 10.1002/pbc.20642
Lofrese, G., De Iure, F. F., Cappuccio, M., and Amendola, L. (2015). Occipital condyles congenital dislocation and condylus tertius: an unstable association revealing a new abnormality of the craniocervical junction. Spine 40, E992–E995. doi: 10.1097/BRS.0000000000000946
Menezes, A. H., and Vogel, T. W. (2008). Specific entities affecting the craniocervical region: syndromes affecting the craniocervical junction. Childs. Nerv. Syst. 24, 1155–1163. doi: 10.1007/s00381-008-0608-6
Mirabello, L., Khincha, P. P., Ellis, S. R., Giri, N., Brodie, S., Chandrasekharappa, S. C., et al. (2017). Novel and known ribosomal causes of Diamond-Blackfan anaemia identified through comprehensive genomic characterisation. J. Med. Genet. 54, 417–425. doi: 10.1136/jmedgenet-2016-104346
Mohamed, J. Y., Faqeih, E., Alsiddiky, A., Alshammari, M. J., Ibrahim, N. A., and Alkuraya, F. S. (2013). Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am. J. Hum. Genet. 92, 157–161. doi: 10.1016/j.ajhg.2012.11.016
Molnar, S., Szappanos, L., Kormendi, Z., and Veres, R. (2007). Occipitoatlantoaxial instability and congenital thoracic vertebral deformity in Pierre Robin sequence: a case report. Spine 32, E501–E504. doi: 10.1097/BRS.0b013e31811ea310
Moore, M., Salles, D., and Jampol, L. M. (2000). Progressive optic nerve cupping and neural rim decrease in a patient with bilateral autosomal dominant optic nerve colobomas. Am. J. Ophthalmol. 129, 517–520. doi: 10.1016/S0002-9394(99)00463-8
Printzlau, A., and Andersen, M. (2004). Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac. J. 41, 47–52. doi: 10.1597/02-055
Quarello, P., Garelli, E., Brusco, A., Carando, A., Mancini, C., Pappi, P., et al. (2012). High frequency of ribosomal protein gene deletions in Italian Diamond-Blackfan anemia patients detected by multiplex ligation-dependent probe amplification assay. Haematologica 97, 1813–1817. doi: 10.3324/haematol.2012.062281
Robinson, P. J., Lodge, S., Goligher, J., Bowley, N., and Grant, H. R. (1993). Secretory otitis media and mastoid air cell development. Int. J. Pediatr. Otorhinolaryngol. 25, 13–18. doi: 10.1016/0165-5876(93)90005-N
Rojas, C. A., Hayes, A., Bertozzi, J. C., Guidi, C., and Martinez, C. R. (2009). Evaluation of the C1-C2 articulation on MDCT in healthy children and young adults. AJR Am. J. Roentgenol. 193, 1388–1392. doi: 10.2214/AJR.09.2688
Rotondo, M., D'avanzo, R., Natale, M., Conforti, R., Pascale, M., and Scuotto, A. (2010). Post-traumatic peripheral facial nerve palsy: surgical and neuroradiological consideration in five cases of delayed onset. Acta Neurochir. 152, 1705–1709. doi: 10.1007/s00701-010-0747-x
Samartzis, D. D., Herman, J., Lubicky, J. P., and Shen, F. H. (2006). Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms. Spine 31, E798–E804. doi: 10.1097/01.brs.0000239222.36505.46
Sando, I., Shibahara, Y., Takagi, A., Takahara, T., and Yamaguchi, N. (1988). Frequency and localization of congenital anomalies of the middle and inner ears: a human temporal bone histopathological study. Int. J. Pediatr. Otorhinolaryngol. 16, 1–22. doi: 10.1016/0165-5876(88)90095-X
Shih, Y. T., Kao, T. H., Pan, H. C., Chen, H. T., and Tsou, H. K. (2015). The surgical treatment principles of atlantoaxial instability focusing on rheumatoid arthritis. Biomed Res. Int. 2015:518164. doi: 10.1155/2015/518164
Smoker, W. R., and Khanna, G. (2008). Imaging the craniocervical junction. Childs. Nerv. Syst. 24, 1123–1145. doi: 10.1007/s00381-008-0601-0
Tassabehji, M., Fang, Z. M., Hilton, E. N., Mcgaughran, J., Zhao, Z., De Bock, C. E., et al. (2008). Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum. Mutat. 29, 1017–1027. doi: 10.1002/humu.20741
Tessadori, F., Giltay, J. C., Hurst, J. A., Massink, M. P., Duran, K., Vos, H. R., et al. (2017). Germline mutations affecting the histone H4 core cause a developmental syndrome by altering DNA damage response and cell cycle control. Nat. Genet. 49, 1642–1646. doi: 10.1038/ng.3956
Thomsen, M. N., Schneider, U., Weber, M., Johannisson, R., and Niethard, F. U. (1997). Scoliosis and congenital anomalies associated with Klippel-Feil syndrome types I-III. Spine 22, 396–401. doi: 10.1097/00007632-199702150-00008
Tracy, M. R., Dormans, J. P., and Kusumi, K. (2004). Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin. Orthop. Relat. Res. 183–190. doi: 10.1097/01.blo.0000130267.49895.20
Tumino, M., Meli, C., Farruggia, P., La Spina, M., Faraci, M., Castana, C., et al. (2011). Clinical manifestations and management of four children with Pearson syndrome. Am. J. Med. Genet. A 155A, 3063–3066. doi: 10.1002/ajmg.a.34288
Vlachos, A., Ball, S., Dahl, N., Alter, B. P., Sheth, S., Ramenghi, U., et al. (2008). Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br. J. Haematol. 142, 859–876. doi: 10.1111/j.1365-2141.2008.07269.x
Watanabe, K., Umeda, T., Niwa, K., Naguro, I., and Ichijo, H. (2018). A PP6-ASK3 module coordinates the bidirectional cell volume regulation under osmotic stress. Cell Rep. 22, 2809–2817. doi: 10.1016/j.celrep.2018.02.045
Yamaguchi, N., Sando, I., Hashida, Y., Takahashi, H., and Matsune, S. (1990). Histologic study of eustachian tube cartilage with and without congenital anomalies: a preliminary study. Ann. Otol. Rhinol. Laryngol. 99, 984–987. doi: 10.1177/000348949009901210
Ye, M., Berry-Wynne, K. M., Asai-Coakwell, M., Sundaresan, P., Footz, T., French, C. R., et al. (2010). Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 19, 287–298. doi: 10.1093/hmg/ddp496
Yi, Z., Sperzel, L., Nurnberger, C., Bredenbeek, P. J., Lubick, K. J., Best, S. M., Stoyanov, C. T., et al. (2011). Identification and characterization of the host protein DNAJC14 as a broadly active flavivirus replication modulator. PLoS Pathog. 7:e1001255. doi: 10.1371/journal.ppat.1001255
Keywords: Diamond Blackfan anemia, Pierre Robin sequence, Klippel feil syndrome, musculoskeletal system development, craniofacial syndromes, craniocervical junction, craniometry
Citation: Roberti D, Conforti R, Giugliano T, Brogna B, Tartaglione I, Casale M, Piluso G and Perrotta S (2018) A Novel 12q13.2-q13.3 Microdeletion Syndrome With Combined Features of Diamond Blackfan Anemia, Pierre Robin Sequence and Klippel Feil Deformity. Front. Genet. 9:549. doi: 10.3389/fgene.2018.00549
Received: 01 August 2018; Accepted: 26 October 2018;
Published: 19 November 2018.
Edited by:
Prashant Kumar Verma, All India Institute of Medical Sciences, Rishikesh, IndiaReviewed by:
Maria Paola Lombardi, University of Amsterdam, NetherlandsEmanuele Angelucci, Ospedale Policlinico San Martino, Italy
Copyright © 2018 Roberti, Conforti, Giugliano, Brogna, Tartaglione, Casale, Piluso and Perrotta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silverio Perrotta, U2lsdmVyaW8uUGVycm90dGFAdW5pY2FtcGFuaWEuaXQ=
†These authors have contributed equally to this work