 Sofia Santos Costa1
Sofia Santos Costa1 Benjamin Sobkowiak2
Benjamin Sobkowiak2 Ricardo Parreira1
Ricardo Parreira1 Jonathan D. Edgeworth3
Jonathan D. Edgeworth3 Miguel Viveiros1
Miguel Viveiros1 Taane G. Clark2,4*
Taane G. Clark2,4* Isabel Couto1*
Isabel Couto1*- 1Global Health and Tropical Medicine, Instituto de Higiene e Medicina Tropical, Universidade NOVA de Lisboa, Lisbon, Portugal
- 2Faculty of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, London, United Kingdom
- 3Department of Infectious Diseases, Centre for Clinical Infection and Diagnostics Research, Guy’s and St Thomas’ NHS Foundation Trust, King’s College London, London, United Kingdom
- 4Faculty of Epidemiology and Population Health, London School of Hygiene and Tropical Medicine, London, United Kingdom
NorA is the best studied efflux system of Staphylococcus aureus and therefore frequently used as a model for investigating efflux-mediated resistance in this pathogen. NorA activity is associated with resistance to fluoroquinolones, several antiseptics and disinfectants and several reports have pointed out the role of efflux systems, including NorA, as a first-line response to antimicrobials in S. aureus. Genetic diversity studies of the gene norA have described three alleles; norAI, norAII and norAIII. However, the epidemiology of these alleles and their impact on NorA activity remains unclear. Additionally, increasing studies do not account for norA variability when establishing relations between resistance phenotypes and norA presence or reported absence, which actually corresponds, as we now demonstrate, to different norA alleles. In the present study we assessed the variability of the norA gene present in the genome of over 1,000 S. aureus isolates, corresponding to 112 S. aureus strains with whole genome sequences publicly available; 917 MRSA strains sourced from a London-based study and nine MRSA isolates collected in a major Hospital in Lisbon, Portugal. Our analyses show that norA is part of the core genome of S. aureus. It also suggests that occurrence of norA variants reflects the population structure of this major pathogen. Overall, this work highlights the ubiquitous nature of norA in S. aureus which must be taken into account when studying the role played by this important determinant on S. aureus resistance to antimicrobials.
Introduction
Staphylococcus aureus is one of the major human pathogens in the hospital and community settings, causing a wide array of clinical manifestations, from mild skin infections to life-threatening systemic infections (Chambers and DeLeo, 2009; Tong et al., 2015). Development and acquisition of resistance to antibiotics and other antimicrobials is of paramount importance in S. aureus, as exemplified by the common occurrence and dissemination of strains displaying a phenotype of multidrug resistance (MDR), including methicillin-resistant Staphylococcus aureus (MRSA) strains (Chambers and DeLeo, 2009). MRSA are spread worldwide and are a public health threat, ranked amongst the major nosocomial pathogens (Lee et al., 2018; Tacconelli et al., 2018). MRSA epidemiology has revealed the local or global dissemination of a few number of clones, namely the lineages in clonal complexes CC1, CC5, CC8, CC22, CC30, CC45, CC59, and CC80 (Lee et al., 2018).
In recent years, several studies have supported drug efflux as a player in the emergence of resistance toward antibiotics and other antimicrobials in S. aureus (DeMarco et al., 2007; Furi et al., 2013; Kwak et al., 2013; Costa et al., 2015). Of particular interest are multidrug efflux pumps (MDR EPs), which extrude a wide range of chemically dissimilar antimicrobials, being frequently associated with MDR phenotypes in bacteria (Piddock, 2006; Poole, 2007). In S. aureus, more than twenty putative MDR EPs are encoded in the chromosome (Schindler et al., 2015), of which several have already been characterized (Costa et al., 2013b). Among these, NorA is the most well studied, being frequently used as a model for studying efflux-mediated resistance in this pathogen.
NorA is a 388 aminoacid protein with 12 transmembrane segments (TMS) that belongs to the Major Facilitator Superfamily (MFS) of secondary transporters. This MDR EP uses the proton motive force to extrude from the cell fluoroquinolones, ethidium bromide, quaternary ammonium compounds, and other antimicrobials (Yoshida et al., 1990; Kaatz et al., 1993; Neyfakh et al., 1993). Several reports have associated NorA activity to low-level resistance to fluoroquinolones and reduced susceptibility to biocides (DeMarco et al., 2007; Huet et al., 2008; Kosmidis et al., 2010; Costa et al., 2011, 2015; Furi et al., 2013). Other studies support a broader substrate range for NorA, including siderophores (Deng et al., 2012) and fusaric acid (Marchi et al., 2015).
The NorA encoding gene, norA, was first identified in the chromosome of a fluoroquinolone-resistant S. aureus isolate, collected in 1986 at a Japanese hospital (Ubukata et al., 1989). Early studies have shown that genetic diversity of this gene can be captured by three alleles, differing up to 10% at the level of the nucleotide sequence and 5% in the polypeptide sequence; norAI (Yoshida et al., 1990), norAII (also described as norA23) (Noguchi et al., 2004) and norAIII (also described as norA1199) (Kaatz et al., 1993). The occurrence of norA variants is also strengthened by a recent study by Brooks et al. (2018) that refers to the variability of this gene in a set of over 150 S. aureus strains. Although some studies have been conducted to ascertain the impact of this genetic diversity on the efflux activity of NorA (Schmitz et al., 1998; Sierra et al., 2000; Noguchi et al., 2004), this effect remains unclear.
Despite this early characterization, there are still contradictory reports in literature on the role of NorA in S. aureus efflux-mediated antimicrobial resistance. Several studies have reported on the putative absence of norA, most probably due to failure to amplify this gene with primers directed to only one of the possible norA alleles. The present study aims at clarifying some of these aspects, by demonstrating that the norA gene is part of the core genome of S. aureus, reflecting on the genetic variability of the gene, its distribution amongst S. aureus clonal lineages, including both methicillin-resistant and -susceptible strains and possible impact on NorA function.
Materials and Methods
Study Datasets
Four sets of nucleotide sequences of the norA structural gene and the corresponding polypeptide sequences were used in this study, comprising (i) the sequences of the three norA alleles described to date in literature; (ii) the norA sequences from 112 S. aureus whole genome sequences retrieved from the GenBank database; (iii) the norA sequences from 917 MRSA strains sourced from a London-based study (Auguet et al., 2018) (ENA accession PRJEB11177); (iv) the norA sequences of nine MRSA isolates collected in a major Hospital in Lisbon, Portugal, representative of the circulating norA alleles in that hospital at the time (Costa et al., 2011, 2016), which were deposited in GenBank.
A detailed list of all the sequences comprised in this study and respective accession numbers can be found in Supplementary Information S1. The Lisbon MRSA isolates were previously characterized for their susceptibility toward fluoroquinolones and biocides and presence of mutations in the quinolone-resistance determining region of grlA/gyrA genes (Costa et al., 2011, 2013a) – Supplementary Information S1. Information on the remaining S. aureus strains was gathered from the GenBank database and relevant published papers.
Whenever sequence types (ST) were not provided, in silico MLST was performed using the MLST 1.8 (Larsen et al., 2012) and SRST2 (Inouye et al., 2014) softwares. The datasets were used to construct a pan-genome (set of all genes within a given species), and a core genome (set of genes found in all strains of that species) for S. aureus (Tettelin et al., 2005; van Tonder et al., 2014).
Sequence Analysis of norA and MLST Alleles
De novo assemblies were performed for all London samples using Velvet (Zerbino and Birney, 2008) and VelvetOptimiser (Zerbino, 2010). Resulting contig FASTA files and FASTA files of the S. aureus samples obtained from GenBank were annotated using Prokka (Seemann, 2014). The pan-genome of these samples was then constructed with Roary (Page et al., 2015), and the sequences of the norA and MLST genes (arcC, aroE, glpF, gmk, pta, tpi, and yqiL) isolated using custom R scripts (R Core Team, 2016).
For the set of the nine Portuguese MRSA clinical isolates, norA was amplified by PCR using three pairs of primers (Table 1). PCR reaction mixtures were prepared in 0.05 mL containing 2.5 U Taq Polymerase (Thermo Scientific, Waltham, MA, United States); 1X Taq buffer (Thermo Scientific); 30 pmol of each primer (Invitrogen, Carlsbad, CA, United States); 0.2 mM dNTPs (GE Healthcare, Chicago, IL, United States); 1.75 mM MgCl2 (Thermo Scientific). The amplification conditions were the following: initial DNA denaturation step at 95°C for 3 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 52°C [norA(a)], 45°C [norA(b)] or 50°C [norA(c)] for 1 min, and extension at 72°C for 1 min, followed by a final extension step at 72°C for 5 min. The PCR products were sequenced using the same set of primers and the sequences analyzed and assembled to make up the entire fragment using the software MAFFT v. 61 and BioEdit v. 7.0.9.02.
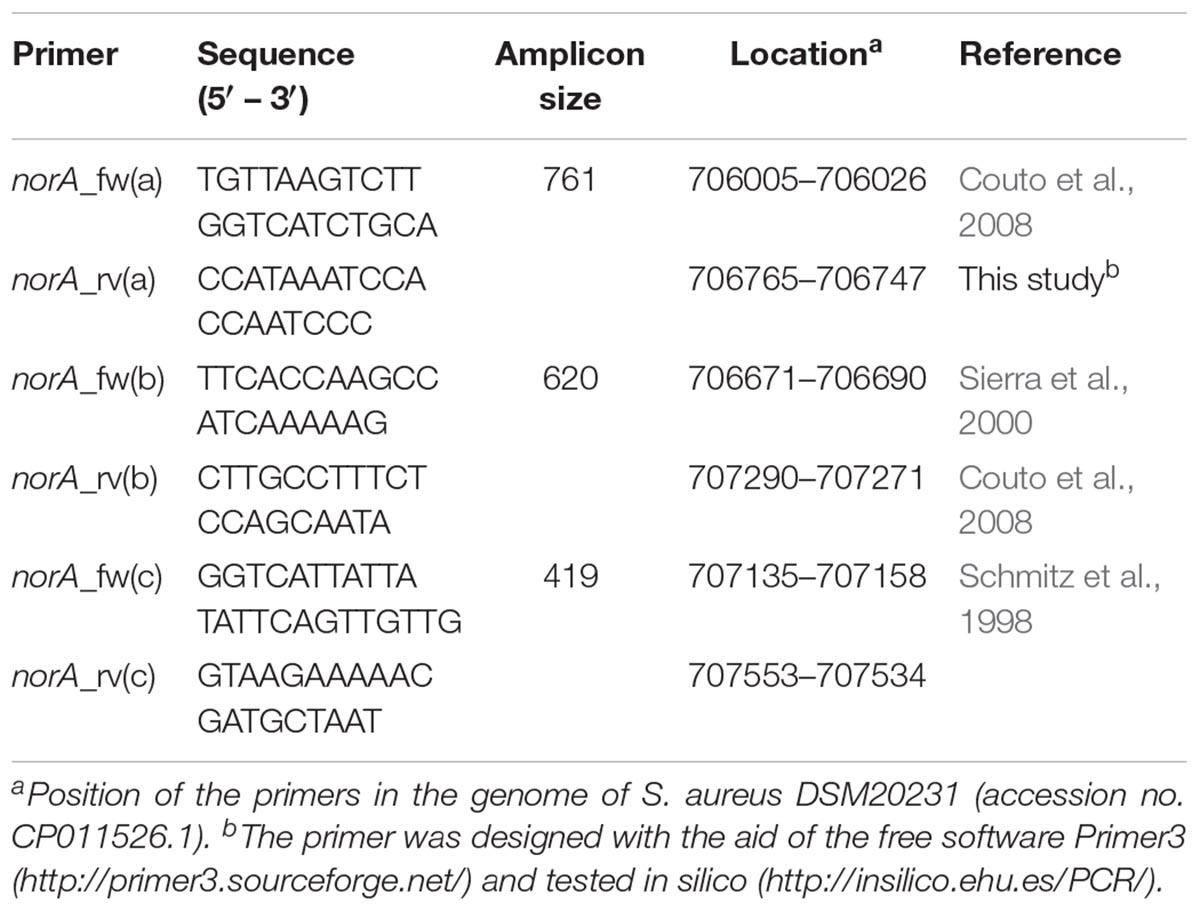
Table 1. Primers used to sequence the norA promoter and coding region of Staphylococcus aureus clinical strains.
The norA and MLST gene sequences from all four datasets were aligned using custom R scripts. Samples that shared identical norA and MLST sequences were grouped together and representative sequences used for phylogenetic reconstruction. Maximum-likelihood (ML) tree construction was performed on the aligned norA and concatenated MLST gene sequences separately using RAxML (Stamatakis, 2014) with a GTRGAMMA evolutionary model. The resulting phylogenetic trees were displayed and annotated using FigTree3.
Analysis of the Impact of norA Variability on NorA Activity
The tridimensional structure of the NorA efflux pump was predicted via the in silico platform PHYRE2 (Protein Homology/analogY Recognition Engine v2.04) (Kelley et al., 2015) based on the nucleotide sequences of the three norA variants described in literature. A mutational analysis based on the predicted tridimensional structure was conducted with the Phyre2 Investigator tools, using the SuSPect algorithm (Yates et al., 2014). This algorithm produces a table of scores from 0 to 100 according to predicted deleteriousness (0 = neutral to 100 = deleterious). A score of 50 is recommended as a cut-off between neutral and deleterious variants, with extreme scores being more confident predictions (Yates et al., 2014). In this work, a score of ≥75 was used as cut-off value.
Results and Discussion
norA Is Part of the Core Genome of S. aureus
Our study sought to establish if norA is part of the core genome of S. aureus. This is a relevant question since a significant part of the studies on efflux-mediated resistance in this pathogen focus mainly on the activity of NorA and some studies have reported the absence of the norA gene in several S. aureus strains (Monecke and Enricht, 2005; Vali et al., 2008; Conceição et al., 2015; Hasanvand et al., 2015; Liu et al., 2015; Ammar et al., 2016; Taheri et al., 2016; Antiabong et al., 2017; Hassanzadeh et al., 2017; Hadadi et al., 2018; Kernberger-Fischer et al., 2018). This reported absence could be explained by the genetic diversity of the gene and a consequent primer failure during norA PCR screening. To test if norA is part of the core genome of S. aureus, we constructed the pan-genome from the 1029 S. aureus isolates whole genome sequence datasets (112 GenBank; 917 London S. aureus isolates). The core genome of S. aureus consists of 1551 genes (found in ≥99% of individuals with 95% BLAST identity; Figure 1A) with the genome of each individual isolate containing a median of 2196 genes (with a range of 1955 to 2469), marginally lower than the number of genes found in S. aureus reference strains (Li et al., 2011). Rarefaction curves were calculated and determined that the number of samples used was adequate to accurately reconstruct the pan-genome of the population; 90% of the total gene diversity was present when considering just 49.5% (514/1029) of the samples and there was a stabilization of the core genome size after 24.9% (256/1029) of the total samples (Figure 1B). The norA gene was found to be present in all London and GenBank samples and thus is part of the core genome, with each sample containing a single gene variant. Additionally, we performed a Blastn search using as query the nucleotide sequence of the norAI allele (GenBank accession no. D90119.1) against all S. aureus genome sequences (complete, scaffold or contig formats) deposited in GenBank5, corresponding to more than 8.000 (circa 8.150) sequences. All the Blastn searched sequences retrieved hits with ≥90% identity to norAI allele. This result supports the notion that norA is a S. aureus core gene, occurring in all S. aureus genomes. The finding that norA is part of S. aureus core genome and thus is present in all S. aureus strains, implies that reporting the detection of this gene is not sufficient to make a direct association with a particular resistance phenotype. To make such a correlation, one must carry out expression analysis of the norA gene as well as of other efflux pump genes, as it has been shown that S. aureus strains can display different efflux pump gene expression patterns (DeMarco et al., 2007; Kosmidis et al., 2010, 2012), even under pressure of the same antimicrobial (Huet et al., 2008; Costa et al., 2011, 2015).
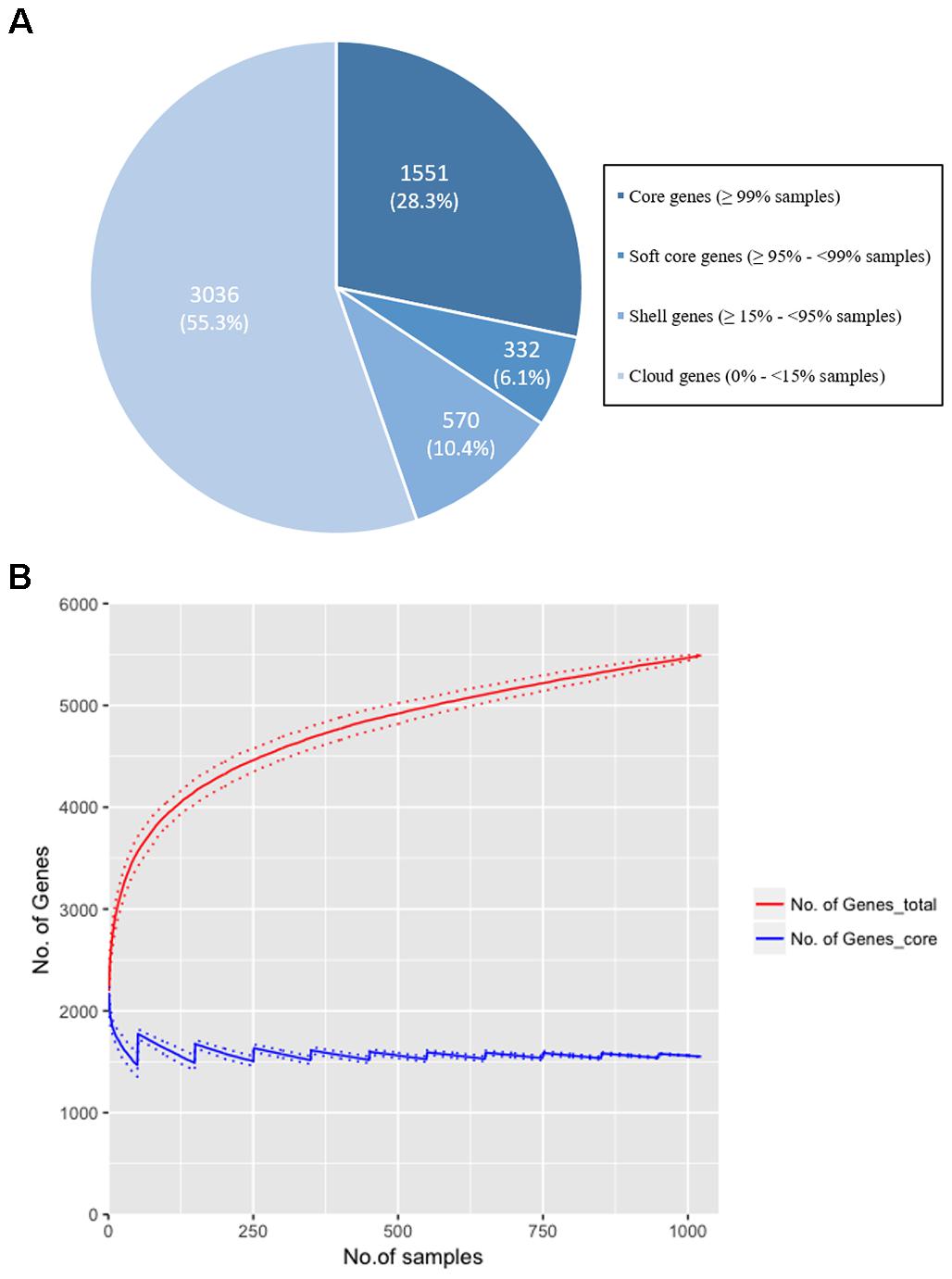
Figure 1. (A) The distribution of genes in the pan-genome of the London and GenBank S. aureus isolates (n = 1029). A total of 5,489 genes were identified with 1,551 (28.25%) genes found in more than 99% of strains. The majority of genes (55.3%) were identified as ‘cloud’ genes, found only in a small number of samples (<15%), demonstrating the diversity of the S. aureus genome. (B) Rarefaction curves for total (red line) and core (blue line) genes identified when increasing the sample size to reconstruct the pan-genome. Random sampling of samples was conducted 100 times and the mean size of the core genome and number of total genes [and standard deviation (dotted lines)] is shown as the sample size is increased by 1 to the total sample size (n = 1029). 90% of the total genes are identified after 514 samples (49.5% of total samples), and there is a plateau in size of the core genome (±5% of the final genome size) at sample 256 (24.9% of total samples). The rarefaction curve confirms that the gene diversity of the strains has been adequately captured with the number of samples included in the analysis.
Genetic Diversity of the norA Gene
One of the goals of this study was to determine the distribution of the norA variants amongst the several S. aureus clonal lineages. A phylogenetic analysis was performed with the three norA alleles and norA nucleotide sequences retrieved from complete S. aureus genomes available in GenBank and from the Portuguese and London-based S. aureus collections (Figure 2). As expected, a certain degree of genetic variability was revealed by the maximum-likelihood tree obtained, despite this gene being relatively conserved. Figure 3 shows the distribution of the pairwise genetic distance within norA across the samples used to construct the phylogenetic tree, with a maximum of 143 SNP differences between samples (12.3% of the total gene length of 1164 bp).
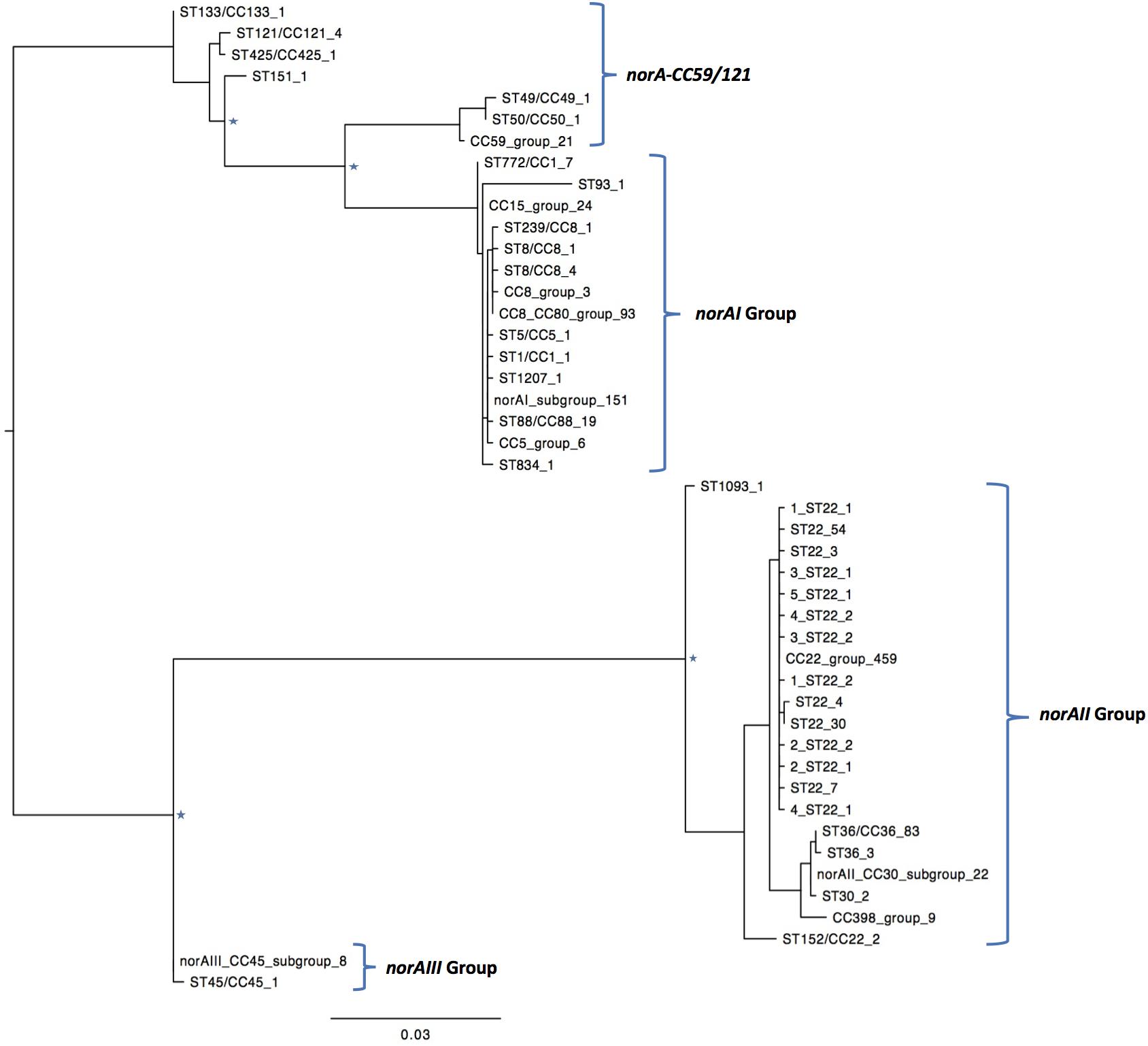
Figure 2. Maximum likelihood phylogeny of the norA sequences analyzed in this study. Identical norA sequences among all samples used in this study have been collapsed and represented by a single label, which denotes a representative ST/CC of that group. The number of sequences is denoted by the last number of the tip label. Four major groups are found, clustering with the norA alleles described in literature, norAI (D90119.1), norAII (AB019536.1), norAIII (M97169.1), and the allelic variant norA-CC59/121. The norAI associated group is the most diverse, including samples belonging to CC1, CC5, CC8, CC15, CC80, and a wide range of sequence types. The norAII associated group contains samples belonging to clonal complexes CC22, CC30, CC36, and CC398. Major nodes supported with bootstrap values above 75% are denoted with a star.
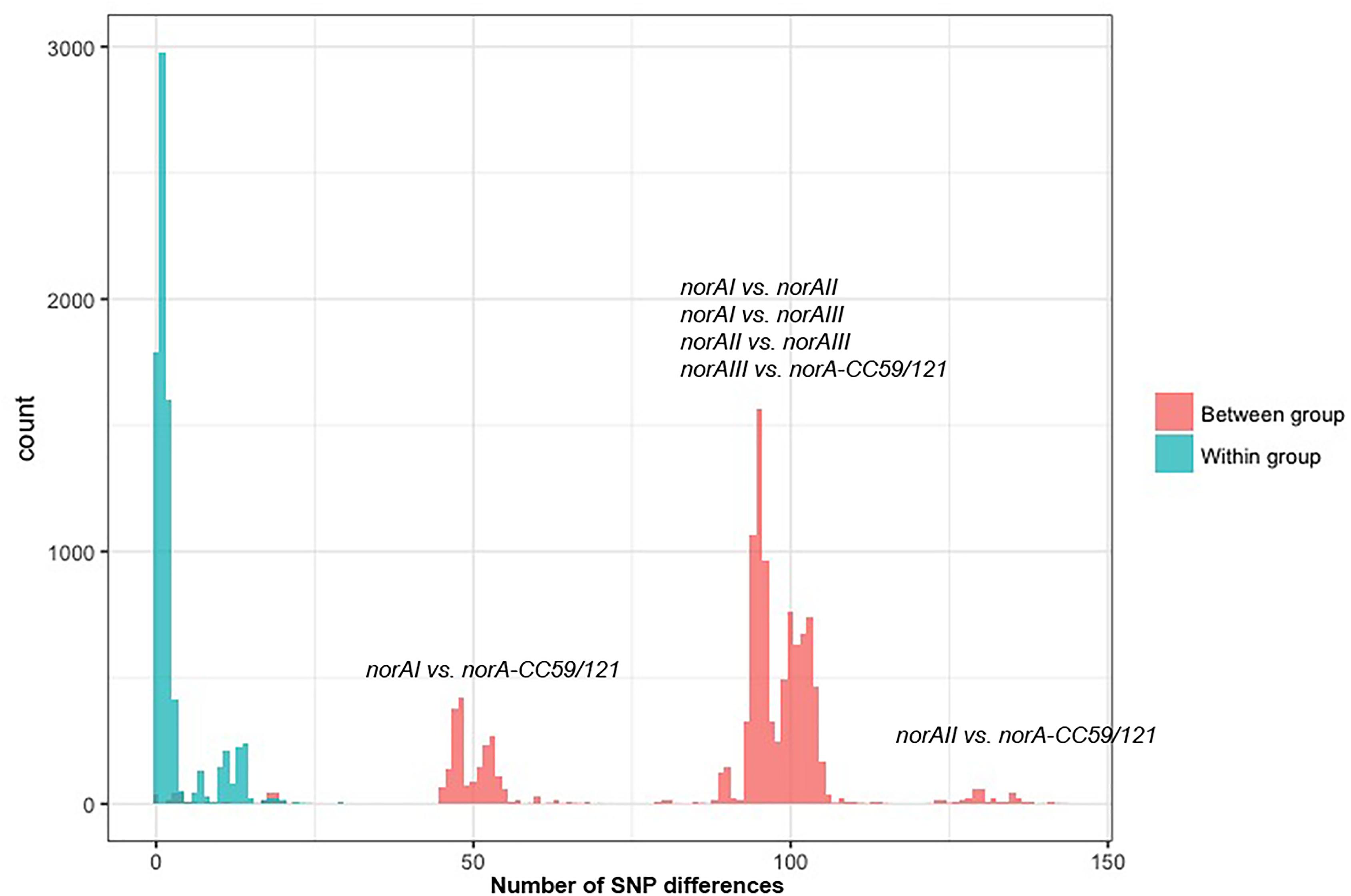
Figure 3. Distribution of the pairwise SNP distance within norA sequences used in this study. The norA sequences include norAI (D90119.1), norAII (AB019536.1) and norAIII (M97169.1), the nine Portuguese isolates, 112 complete assemblies from GenBank, and 75 representative sequences of London isolates. The figure is annotated to show the pairwise SNP distance between isolates both within (blue) and between (red) each of the four major norA groups described in the main text: norAI group, norAII group, norAIII group, and CC121/CC59 group.
A global analysis showed a clear correlation between occurrence of norA alleles and specific S. aureus clonal lineages. The norAI allele was mainly associated with genetic backgrounds of the clonal complexes CC1, CC5, CC8, and CC80 or closely related lineages. On the other hand, norAII and close variants appear to be restricted to strains belonging to CC30, CC398, and CC22 (Figure 2). The norAIII allele is only associated with the S. aureus lineage CC45 and novel but related STs.
Among the sequences retrieved from GenBank, the norAI allele is the most frequent (76 out of 112 strains) and the norAII allele the second most frequent allele (21 out of 112 strains), whereas norAIII occurs in just two CC45 strains. We also detected 13 more divergent variants, most closely related to the norAI allele, that were associated with other lineages, such as CC59 and CC121 (Figures 2, 3). Although these findings may be biased as the dataset analyzed reflects the current epidemiologically S. aureus relevant clones, with a frequency of ∼70% MRSA strains (78 out of 112 genomes), it clearly shows that norAI and related variants are the most prevalent alleles across lineages, as suggested by earlier studies (Schmitz et al., 1998; Sierra et al., 2000; Noguchi et al., 2004). Interestingly, a sample isolated in Brazil in 2010 (FCFHV36) carried a norAI type variant of the norA gene that included a frameshift mutation in codon 129. This mutation caused a premature stop codon at codon number 147, resulting in potential pseudogenization of norA in this isolate.
Regarding the London isolates, we found the norAII allele to be the most common allele, identified in 671/917 samples, due to the high number of CC22 strains in the study samples (539/917). As with the GenBank samples, the norAIII allele was the least common variant found in the London isolates, with only the three CC45 and four novel ST strains carrying closely related variants. The norAI allele was found in 220/917 strains consisting of the broadest range of lineages, comprising 21 different STs. Additionally, one ST22 sample possessed a norA variant with a frameshift mutation at codon 365 that results in a premature stop codon at codon number 367 and, again, possible pseudogenization.
Nine MRSA isolates, representative of a collection of 53 S. aureus clinical isolates isolated at a major Portuguese Hospital and previously characterized for efflux activity and clonal lineage (Costa et al., 2011, 2013a, 2015, 2016) were added to the study to ascertain their norA allele. The full sequence of the norA gene was determined for these isolates and compared with norA alleles from major clonal lineages (Figure 4). Both the norAI and norAII alleles were found amongst the Portuguese isolates and their distribution reflects the findings for the other datasets analyzed. Eight isolates from clonal lineages CC5, CC8, and CC88 carried norAI and a single isolate from clonal lineage CC22 harboured the norAII allele (Figure 4). Combining information on norA allele, clonal lineage and previous characterization of these isolates (Costa et al., 2011) showed no correlation between norA allele, norA expression levels, efflux capacity, or antimicrobial resistance levels (Supplementary Information S1).
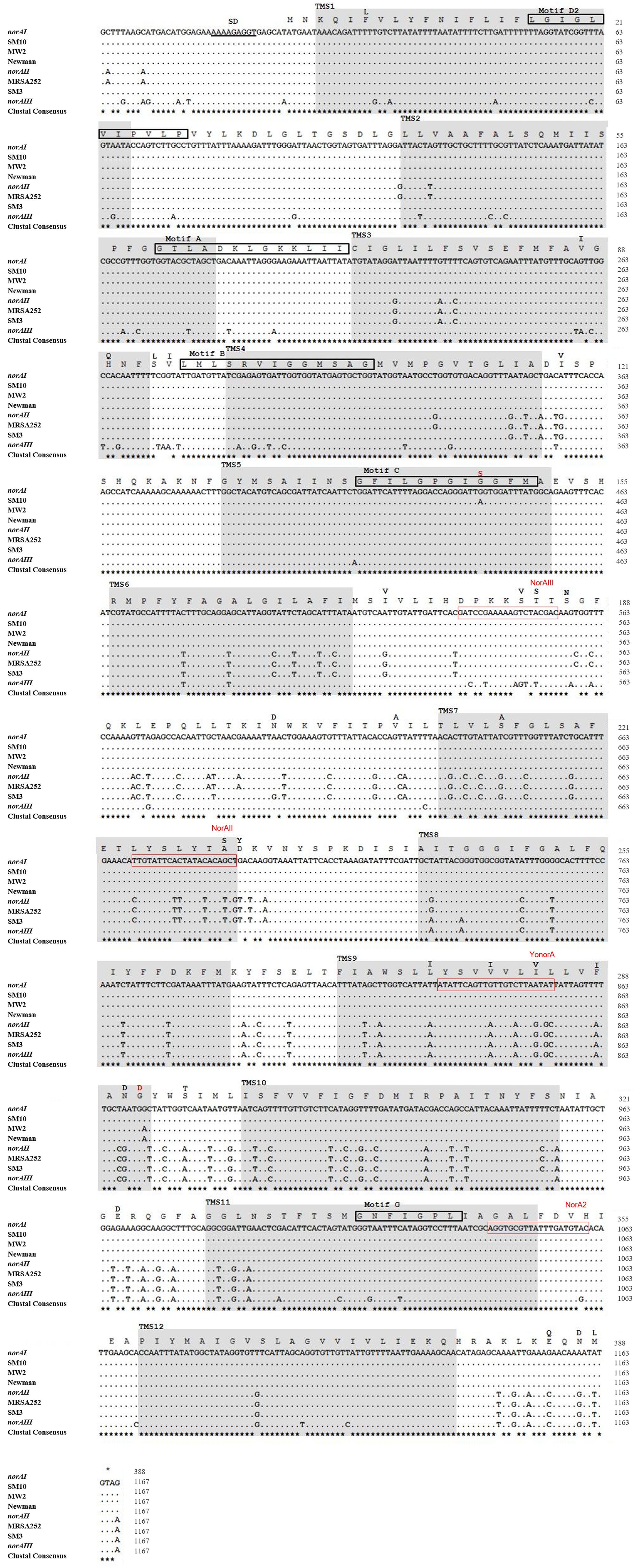
Figure 4. Multiple alignment of norA sequences of the three norA alleles, from selected Portuguese MRSA clinical isolates (SM) and from strains of representative clonal lineages. The MRSA strains displayed were selected to represent the main clones found, namely SM10 – CC5; SM3 – CC22; MW2 – CC1; Newman – CC8; MRSA252 – CC30. The Shine-Dalgarno (SD) sequence is underlined in black. The shaded regions correspond to the putative TMS of NorA as suggested by Paulsen et al. (1996). The aminoacid regions inside the boxes are relative to the conserved motifs described for MFS transporters with 12 TMS. The black residues shown above the main polypeptide sequence represent the variations present in the various NorA polypeptide sequences relatively to the NorAI variant. The red residues represent alterations found among the SM collection of Portuguese MRSA clinical isolates. The red boxes indicate the primers described in Table 2 for distinction of norA alleles.
A wet lab PCR-based strategic approach is proposed for detection and differentiation of the several norA alleles occurring in S. aureus. The norAI and norAII alleles can be easily distinguished using two sets of specific primers (Table 2 and Figure 4). To amplify and differentiate norAIII from the norA allele associated with CC59/CC121, an approach consisting in PCR amplification followed by restriction of the PCR product with HindIII can be applied (Table 2 and Figure 4).
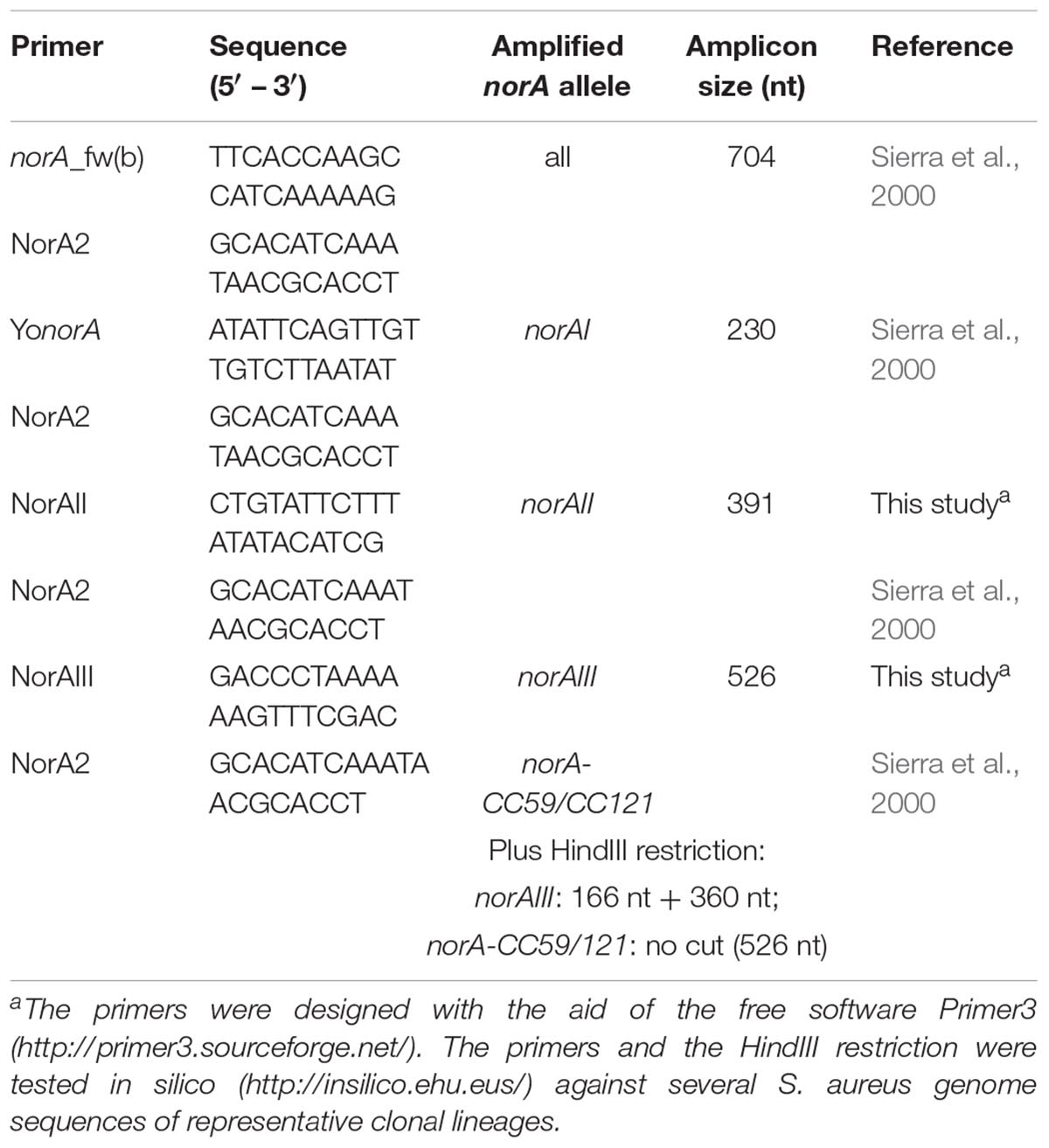
Table 2. List of primers proposed to amplify and differentiate the norA alleles by conventional PCR.
Impact of Genetic Variability on NorA Function
A mutational analysis was performed in silico via the PHYRE2 platform to construct a NorA structural model, using the SuSPect algorithm to predict the effect of each residue substitution on NorA function. This analysis is limited because of the lack of available structural data for bacterial MFS pumps, mostly limited to substrate-specific transporters. Nevertheless, the polypeptide sequences encoded by the three norAI alleles were used to predict a tri-dimensional structure of NorA. All three NorA variants showed the highest identity with the putative MFS transporter YajR of Escherichia coli (Jiang et al., 2013). A second NorA in silico model, based on the structure of glycerol-3-phosphate MFS transporter from E. coli, described by Bhaskar et al. (2016) was taken into account for comparison purposes. The SuSPect-derived mutational analysis revealed 42 residues for which particular mutations could potentially impair NorA activity (Supplementary Information S2). Of these, residues Pro110, Pro158, Pro311, and Gly326 were particularly susceptible to mutations.
Analysis of the polypeptide sequence corresponding to the three norA variants revealed a total of 43 amino acid alterations. Figure 5 summarizes these alterations, their location in NorA and their distribution amongst S. aureus lineages. There was no overlap between these alterations and the residues identified by SuSPect. We also compared the S. aureus NorAI variant with the NorA efflux pump encoded in the Staphylococcus epidermidis ATCC 12228 chromosome, which presents the same substrate profile (Costa et al., 2018; Supplementary Information S2). The differences encountered between the two polypeptide sequences correspond to residues that were not highlighted as pivotal for the protein activity by SuSPect. These results suggest conservation of NorA function in these two main staphylococcal species.
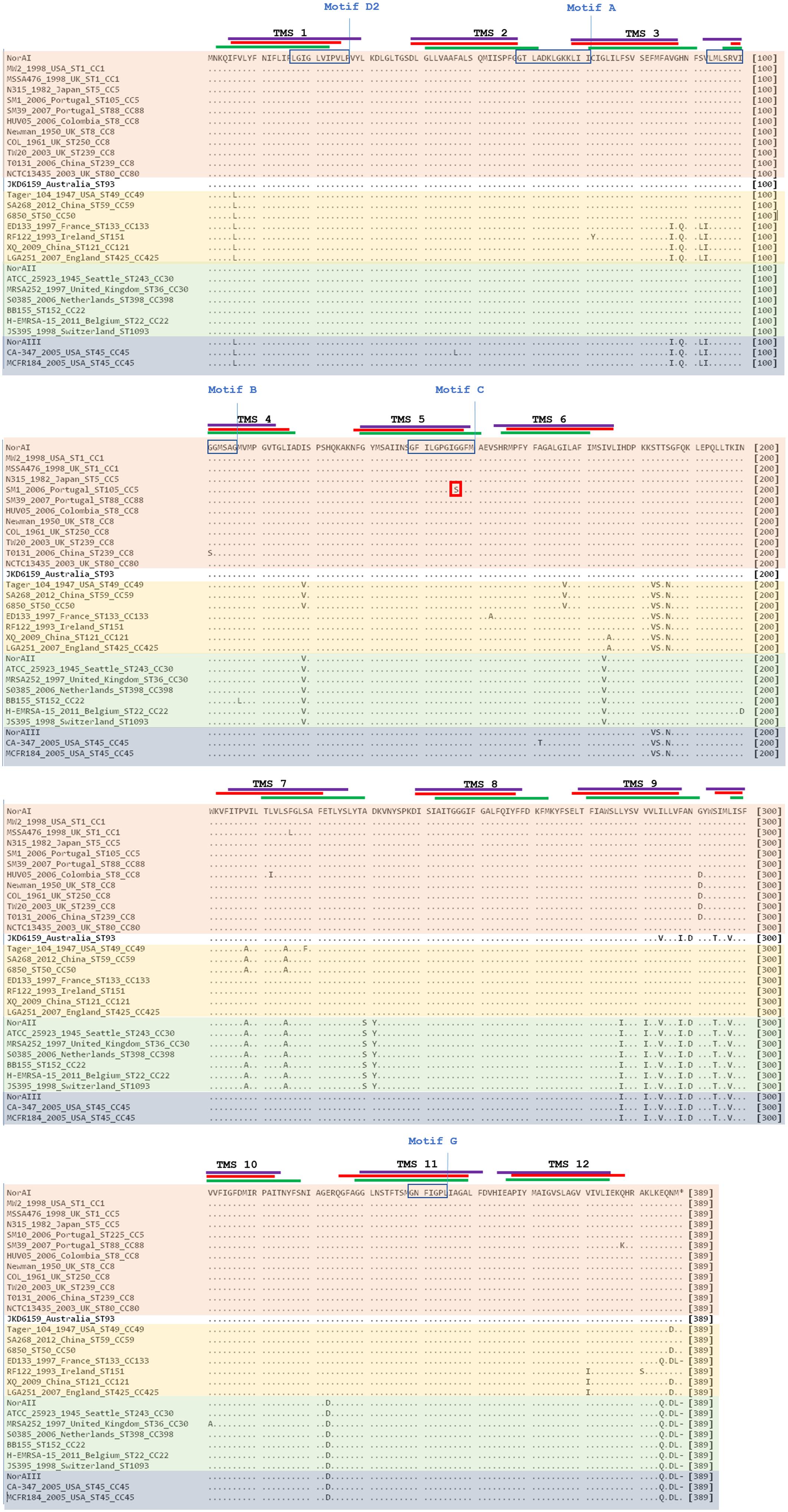
Figure 5. Multiple alignment of NorA polypeptide sequences derived from alleles norAI (Yoshida et al., 1990), norAII (Noguchi et al., 2004), and norAIII (Kaatz et al., 1993) and from the norA alleles from selected S. aureus strains. The MRSA strains displayed were selected to represent the main S. aureus clonal lineages found for the strains with genomes publicly available and for the Portuguese strains (SM10 – CC5; SM39 – CC88). Sequences are grouped according to the respective norA allele (orange – norAI; green – norAII; blue - norAIII; yellow – norA-CC59/121). The lines correspond to the transmembrane segments (TMS) of NorA, as predicted by Paulsen et al. (1996) (green), by the HMMTOP method as presented in Bhaskhar et al. (2016) (red) and as predicted by the Phyre2-based NorA model (purple). The aminoacid regions inside the boxes are relative to the conserved motifs described for MFS transporters with 12 TMS (Paulsen et al., 1996). The red box corresponds to the amino acid substitution likely to affect NorA function.
One alteration, Gly147Ser located within putative TMS 5, may affect NorA function (Figure 4). This alteration was detected in all four strains belonging to ST105 (three of which are of Portuguese origin) and in one of the strains belonging to ST225 (Portuguese origin) (Supplementary Information S2), but was absent from strains from other CC5 lineages, such as ST5. Residue 147 is predicted to be located within the conserved motif C (gxxxGPxiGGxl) (Paulsen et al., 1996) on the TMS 5, facing a water-filled channel. This motif is conserved among MFS drug-H+ antiporters and has been implicated in the binding of the substrate, with the glycines being essential for that function (Ginn et al., 2000; Tamura et al., 2001). In fact, in a mutagenesis study in S. aureus efflux pump TetA(K), also a 12-TMS member of the MFS family, the mutation Gly147Ser was responsible for the loss of 80% of the activity of that efflux pump (Ginn et al., 2000). Although Tet transporters and NorA are distinct in their substrate specificity, these pumps share abundance of glycines in TMS 5, a trait that has been postulated to confer conformational plasticity to EPs (Ginn et al., 2000). Thus, the mutation Gly147Ser may have a deleterious effect on NorA activity. Additional studies are necessary to fully ascertain the impact of this alteration in the efflux capacity of NorA. The same applies to the full understanding of possible correlations between a given norA allele and corresponding NorA efflux capacity, probably requiring carefully controlled genetic systems because of overlapping substrates between many staphylococcal multidrug efflux systems and the redundancy in their response to antimicrobials (Costa et al., 2013b; Schindler and Kaatz, 2016).
Conclusion
In this study we provide evidence that the efflux pump gene norA is part of core genome of S. aureus. This gene presents some genetic variability which may impair its detection by conventional laboratorial techniques, such as PCR, due to potential primer mismatch. Strikingly, a correlation was observed between the several norA alleles and specific S. aureus lineages, suggesting that the occurrence of norA variants reflects the population structure of this major pathogen.
Data Availability
The sequences analyzed in this study are available in GenBank and their accession numbers detailed in Supplementary Information S1, except for WGS data for the London-based strains that are available from the European Nucleotide Archive database under accession number PRJEB11177.
Author Contributions
IC, SC, and TC conceived and designed the study. SC performed the wet lab norA analysis and the in silico mutational analysis. BS conducted the pan-genome analysis. SC and BS performed the phylogenetic analysis. JE provided genomic data on the London-based MRSA strains. RP, JE, MV, TC, and IC contributed to the analysis of these data. SC, BS, TC, and IC wrote the first draft of the manuscript. All authors have reviewed the final version of the manuscript.
Funding
This work was partially supported by Fundação para a Ciência e a Tecnologia (FCT, Portugal), through funds to GHTM – UID/Multi/04413/2013. SC was supported by grant SFRH/BPD/97508/2013 from FCT, Portugal. TC was funded by the Medical Research Council United Kingdom (Grant Nos. MR/K000551/1, MR/M01360X/1, MR/N010469/1, and MR/R020973/1) and BBSRC United Kingdom (BB/R013063/1). BS was funded by the Medical Research Council United Kingdom (Grant No. MR/N010469/1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The MRC eMedLab computing resource was used for bioinformatics and statistical analysis. The authors deeply acknowledge Professor José Melo-Cristino for providing the Portuguese S. aureus strains analyzed in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00710/full#supplementary-material
Footnotes
- ^ mafft.cbrc.jp/alignment/software/
- ^ https://bioedit.software.informer.com/
- ^ http://tree.bio.ed.ac.uk/software/figtree/
- ^ http://www.sbg.bio.ic.ac.uk/phyre2
- ^ www.ncbi.nlm.nih.gov/genome/genomes/154
References
Ammar, A. M., Attia, A. M., Abd El-Hamid, M. I., El-Shorbagy, I. M., and Abd El-Kader, S. A. (2016). Genetic basis of resistance waves among methicillin resistant Staphylococcus aureus isolates recovered from milk and meat products in Egypt. Cell Mol. Biol. 62, 7–15.
Antiabong, J. F., Kock, M. M., Bellea, N. M., and Ehlers, M. M. (2017). Diversity of multidrug efflux genes and phenotypic evaluation of the in vitro resistance dynamics of clinical Staphylococcus aureus isolates using methicillin; a model β-lactam. Open Microbiol. J. 11, 132–141. doi: 10.2174/1874285801711010132
Auguet, O. T., Stabler, R. A., Betley, J., Preston, M. D., Dhaliwal, M., Gaunt, M., et al. (2018). Frequent undetected MRSA ward-based transmission linked to patient sharing between hospitals. Clin. Infect. Dis. 66, 840–848. doi: 10.1093/cid/cix901
Bhaskar, B. V., Babu, T. M., Reddy, N. V., and Rajendra, W. (2016). Homology modeling, molecular dynamics, and virtual screening of NorA efflux pump inhibitors of Staphylococcus aureus. Drug Des. Dev. Ther. 10, 3237–3252. doi: 10.2147/DDDT.S113556
Brooks, L. E., Ul-Hasan, S., Chan, B. K., and Sistrom, M. J. (2018). Quantifying the evolutionary conservation of genes encoding multidrug efflux pumps in the ESKAPE pathogens to identify antimicrobial drug targets. mSystems 3:e00024-18. doi: 10.1128/mSystems.00024-18
Chambers, H. F., and DeLeo, F. R. (2009). Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641. doi: 10.1038/nrmicro2200
Conceição, T., Coelho, C., de Lencastre, H., and Aires-de-Sousa, M. (2015). High prevalence of biocide resistance determinants in Staphylococcus aureus isolates from three african countries. Antimicrob. Agents Chemother. 60, 678–681. doi: 10.1128/AAC.02140-15
Costa, S. S., Falcão, C., Viveiros, M., Machado, D., Martins, M., Melo-Cristino, J., et al. (2011). Exploring the contribution of efflux on the resistance to fluoroquinolones in clinical isolates of Staphylococcus aureus. BMC Microbiol. 11:e241. doi: 10.1186/1471-2180-11-241
Costa, S. S., Palma, C., Kadlec, K., Fessler, A. T., Viveiros, M., Melo-Cristino, J., et al. (2016). Plasmid-borne antimicrobial resistance of Staphylococcus aureus isolated in a Hospital in Lisbon. Portugal. Microb. Drug Resist. 22, 617–626. doi: 10.1089/mdr.2015.0352
Costa, S. S., Junqueira, E., Palma, C., Viveiros, M., Melo-Cristino, J., Amaral, L., et al. (2013a). Resistance to antimicrobials mediated by efflux pumps in Staphylococcus aureus. Antibiotics 2, 83–99. doi: 10.3390/antibiotics2010083
Costa, S. S., Viveiros, M., Amaral, L., and Couto, I. (2013b). Multidrug efflux pumps in Staphylococcus aureus: an update. Open Microbiol. J. 7, 59–71. doi: 10.2174/1874285801307010059
Costa, S. S., Viveiros, M., Pomba, C., and Couto, I. (2018). Active antimicrobial efflux in Staphylococcus epidermidis: building up of resistance to fluoroquinolones and biocides in a major opportunistic pathogen. J. Antimicrob. Chemother. 73, 320–324. doi: 10.1093/jac/dkx400
Costa, S. S., Viveiros, M., Rosato, A. E., Melo-Cristino, J., and Couto, I. (2015). Impact of efflux in the development of multidrug resistance phenotypes in Staphylococcus aureus. BMC Microbiol. 15:232. doi: 10.1186/s12866-015-0572–578
Couto, I., Costa, S. S., Viveiros, M., Martins, M., and Amaral, L. (2008). Efflux-mediated response of Staphylococcus aureus exposed to ethidium bromide. J. Antimicrob. Chemother. 62, 504–513. doi: 10.1093/jac/dkn217
DeMarco, C. E., Cushing, L. A., Frempong-Manso, E., Seo, S. M., Jaravaza, T. A. A., and Kaatz, G. W. (2007). Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 51, 3235–3239. doi: 10.1128/AAC.00430-07
Deng, X., Ji, Q., Liang, H., Missiakas, D., Lan, L., and He, C. (2012). Expression of multidrug resistance efflux pump gene norA is iron responsive in Staphylococcus aureus. J. Bacteriol. 194, 1753–1762. doi: 10.1128/JB.06582-11
Furi, L., Ciusa, M. L., Knight, S., Di Lorenzo, V., Tocci, N., Cirasola, D., et al. (2013). Evaluation of reduced susceptibility to quaternary ammonium compounds and bisbiguanidines in clinical isolates and laboratory-generated mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 57, 3488–3497. doi: 10.1128/AAC.00498-13
Ginn, S. L., Brown, M. H., and Skurray, R. A. (2000). The TetA(K) tetracycline/H+ antiporter from Staphylococcus aureus: mutagenesis and functional analysis of motif C. J. Bacteriol. 182, 1492–1498. doi: 10.1128/JB.182.6.1492-1498.2000
Hadadi, M., Heidari, H., Ebrahim-Saraie, H. S., and Motamedifar, M. (2018). Molecular characterization of vancomycin, mupirocin and antiseptic resistant Staphylococcus aureus strains. Mediterr. J. Hematol. Infect. Dis. 10:e2018053. doi: 10.4084/MJHID.2018.053
Hasanvand, A., Ghafourian, S., Taherikalani, M., Jalilian, F. A., Sadeghifard, N., and Pakzad, I. (2015). Antiseptic resistance in methicillin sensitive and methicillin resistant Staphylococcus aureus isolates from some major hospitals, Iran. Recent Pat. Antiinfect. Drug Discov. 10, 105–112. doi: 10.2174/1574891X10666150623093259
Hassanzadeh, S., Mashhadi, R., Yousefi, M., Askari, E., Saniei, M., and Pourmand, M. R. (2017). Frequency of efflux pump genes mediating ciprofloxacin and antiseptic resistance in methicillin-resistant Staphylococcus aureus isolates. Microb. Pathog. 111, 71–74. doi: 10.1016/j.micpath.2017.08.026
Huet, A. A., Raygada, J. L., Mendiratta, K., Seo, S. M., and Kaatz, G. W. (2008). Multidrug efflux pump overexpression in Staphylococcus aureus after single and multiple in vitro exposures to biocides and dyes. Microbiology 154, 3144–3153. doi: 10.1099/mic.0.2008/021188-0
Inouye, M., Dashnow, H., Raven, L.-A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Jiang, D., Zhao, Y., Wang, X., Fan, J., Heng, J., Liu, X., et al. (2013). Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. U.S.A. 110, 14664–14669. doi: 10.1073/pnas.1308127110
Kaatz, G. W., Seo, S. M., and Ruble, C. A. (1993). Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 37, 1086–1094. doi: 10.1128/AAC.37.5.1086
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kernberger-Fischer, I. A., Krischek, C., Strommenger, B., Fiegen, U., Beyerbach, M., Kreienbrock, L., et al. (2018). Susceptibility of methicillin-resistant and -susceptible Staphylococcus aureus isolates of various clonal lineages from Germany to eight biocides. Appl. Environ. Microbiol. 84:, e799–e718. doi: 10.1128/AEM.00799-18
Kosmidis, C., DeMarco, C. E., Frempong-Manso, E., Seo, S. M., and Kaatz, G. W. (2010). In silico genetic correlations of multidrug efflux pump gene expression in Staphylococcus aureus. Int. J. Antimicrob. Agents 36, 222–229. doi: 10.1016/j.ijantimicag.2010.05.015
Kosmidis, C., Schindler, B. D., Jacinto, P. L., Patel, D., Bains, K., Seo, S. M., et al. (2012). Expression of multidrug resistance efflux pump genes in clinical and environmental isolates of Staphylococcus aureus. Int. J. Antimicrob. Agents 40, 204–209. doi: 10.1016/j.ijantimicag.2012.04.014
Kwak, Y. G., Truong-Bolduc, Q. C., Kim, H. B., Song, K. H., Kim, E. S., and Hooper, D. C. (2013). Association of norB overexpression and fluoroquinolone resistance in clinical isolates of Staphylococcus aureus from Korea. J. Antimicrob. Chemother. 68, 2766–2772. doi: 10.1093/jac/dkt286
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total genome sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Lee, A. S., de Lencastre, H., Garau, J., Kluytmans, J., Malhotra-Kumar, S., Peschel, A., et al. (2018). Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 4:18033. doi: 10.1038/nrdp.2018.33
Li, Y., Cao, B., Zhang, Y., Zhgou, J., Yang, B., and Wang, L. (2011). Complete genome sequence of Staphylococcus aureus T0131, an ST239-MRSA-SCCmec type III clone isolated in China. J. Bacteriol. 193, 3411–3412. doi: 10.1128/JB.05135-11
Liu, Q., Zhao, H., Han, L., Shu, W., Wu, Q., and Ni, Y. (2015). Frequency of biocide-resistant genes and susceptibility to chlorhexidine in high-level mupirocin-resistant, methicillin-resistant Staphylococcus aureus (MuH MRSA). Diagn. Microbiol. Infect. Dis. 82, 278–283. doi: 10.1016/j.diagmicrobio.2015.03.023
Marchi, E., Furi, L., Arioli, S., Morrissey, I., Di Lorenzo, V., Mora, D., et al. (2015). Novel insight into antimicrobial resistance and sensitivity phenotypes associated to qac and norA genotypes in Staphylococcus aureus. Microbiol. Res. 170, 184–194. doi: 10.1016/j.micres.2014.07.001
Monecke, S., and Enricht, R. (2005). Rapid genotyping of methicillin-resistant Staphylococcus aureus (MRSA) isolates using miniaturized oligonucleotide arrays. Clin. Microbiol. Infect. 11, 825–833. doi: 10.1111/j.1469-0691.2005.01243.x
Neyfakh, A. A., Borsch, C. M., and Kaatz, G. W. (1993). Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob. Agents Chemother. 37, 128–129. doi: 10.1128/AAC.37.1.128
Noguchi, N., Okada, H., Narui, K., and Sasatsu, M. (2004). Comparison of the nucleotide sequence and expression of norA genes and microbial susceptibility in 21 strains of Staphylococcus aureus. Microb. Drug Resist. 10, 197–203. doi: 10.1089/mdr.2004.10.197
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paulsen, I. T., Brown, M. H., and Skurray, R. A. (1996). Proton-dependent multidrug efflux systems. Microbiol. Rev. 60, 575–608.
Piddock, L. J. V. (2006). Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 19, 382–402. doi: 10.1128/CMR.19.2.382-402.2006
Poole, K. (2007). Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 39, 162–176. doi: 10.1080/07853890701195262
R Core Team (2016). R: A Language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
Schindler, B. D., Frempong-Manso, E., DeMarco, C. E., Kosmidis, C., Matta, V., Seo, S. M., et al. (2015). Analyses of multidrug efflux pump-like proteins encoded on the Staphylococcus aureus chromosome. Antimicrob. Agents Chemother. 59, 747–748. doi: 10.1128/AAC.04678-14
Schindler, B. D., and Kaatz, G. W. (2016). Multidrug efflux pumps of Gram-positive bacteria. Drug Resist. Updat. 27, 1–13. doi: 10.1016/j.drup.2016.04.003
Schmitz, F. J., Hertel, B., Hofmann, B., Scheuring, S., Verhoef, J., Fluit, A. C., et al. (1998). Relationship between mutations in the coding and promoter regions of the norA genes in 42 unrelated clinical isolates of Staphylococcus aureus and the MICs of norfloxacin for these strains. J. Antimicrob. Chemother. 42, 561–563. doi: 10.1093/jac/42.4.561
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sierra, J. M., Ruiz, J., de Anta, M. T. J., and Vila, J. (2000). Prevalence of two different genes encoding NorA in 23 clinical strains of Staphylococcus aureus. J. Antimicrob. Chemother. 46, 145–146. doi: 10.1093/jac/46.1.145
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., Monnet, D. L., et al. (2018). Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. doi: 10.1016/S1473-3099(17)30753-3
Taheri, N., Ardebili, A., Amouzandeh-Nobaveh, A., and Ghaznavi-Rad, E. (2016). Frequency of antiseptic resistance among Staphylococcus aureus and coagulase-negative Staphylococci isolated from a university hospital in central Iran. Oman Med. J. 31, 426–432. doi: 10.5001/omj.2016.86
Tamura, N., Konishi, S., Iwaki, S., Kimura-Someya, T., Nada, S., and Yamaguchi, A. (2001). Complete cysteine-scanning mutagenesis and site-directed chemical modification of the Tn10-encoded metal-tetracycline/H+ antiporter. J. Biol. Chem. 276, 20330–20339. doi: 10.1074/jbc.M007993200
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tong, S. Y. C., Davis, J. S., Eichenberger, E., Holland, T. L., and Fowler, V. G. Jr. (2015). Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661. doi: 10.1128/CMR.00134-14
Ubukata, K., Itoh-Yamashita, N., and Konno, M. (1989). Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 33, 1535–1539. doi: 10.1128/AAC.33.9.1535
Vali, L., Davies, S. E., Lai, L. L. G., Dave, J., and Amyes, S. G. B. (2008). Frequency of biocide resistance genes, antibiotic resistance and the effect of chlorhexidine exposure on clinical methicillin-resistant Staphylococcus aureus isolates. J. Antimicrob. Chemother. 61, 524–532. doi: 10.1093/jac/dkm520
van Tonder, A. J., Mistry, S., Bray, J. E., Hill, D. M., Cody, A. J., Farmer, C. L., et al. (2014). Defining the estimated core genome of bacterial populations using a Bayesian decision model. PLoS Comput. Biol. 10:e1003788. doi: 10.1371/journal.pcbi.1003788
Yates, C. M., Filippis, I., Kelley, L. A., and Sternberg, M. J. (2014). SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features. J. Mol. Biol. 426, 2692–2701. doi: 10.1016/j.jmb.2014.04.026
Yoshida, H., Bogaki, M., Nakamura, S., Ubukata, K., and Konno, M. (1990). Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J. Bacteriol. 172, 6942–6949. doi: 10.1128/jb.172.12.6942-6949.1990
Zerbino, D. R. (2010). Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinformatics Chapter 11:Unit 11.5. doi: 10.1002/0471250953.bi1105s31
Keywords: Staphylococcus aureus, norA, alleles, variability, efflux
Citation: Costa SS, Sobkowiak B, Parreira R, Edgeworth JD, Viveiros M, Clark TG and Couto I (2019) Genetic Diversity of norA, Coding for a Main Efflux Pump of Staphylococcus aureus. Front. Genet. 9:710. doi: 10.3389/fgene.2018.00710
Received: 28 August 2018; Accepted: 18 December 2018;
Published: 09 January 2019.
Edited by:
Silvia Buroni, University of Pavia, ItalyReviewed by:
Maria José Saavedra, Universidade de Trás os Montes e Alto Douro, PortugalTeruo Kuroda, Hiroshima University, Japan
Copyright © 2019 Costa, Sobkowiak, Parreira, Edgeworth, Viveiros, Clark and Couto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taane G. Clark, dGFhbmUuY2xhcmtAbHNodG0uYWMudWs= Isabel Couto, aWNvdXRvQGlobXQudW5sLnB0