 Muhammad Younus1,2
Muhammad Younus1,2 Farooq Ahmad2Erum Malik3
Farooq Ahmad2Erum Malik3 Muhammad Bilal2Mehran Kausar2,4
Muhammad Bilal2Mehran Kausar2,4 Safdar Abbas2Shabnam Shaheen5Mohib Ullah Kakar6Majid Alfadhel7,8
Safdar Abbas2Shabnam Shaheen5Mohib Ullah Kakar6Majid Alfadhel7,8 Muhammad Umair2,8*
Muhammad Umair2,8*- 1State Key Laboratory of Membrane Biology, Beijing Key Laboratory of Cardiometabolic Molecular Medicine, Institute of Molecular Medicine, Peking-Tsinghua Center for Life Sciences, PKU-IDG/McGovern Institute for Brain Research, Peking University, Beijing, China
- 2Department of Biochemistry, Faculty of Biological Sciences, Quaid-i-Azam University, Islamabad, Pakistan
- 3Department of Biochemistry, Shah Abdul Latif University Khairpur, Khairpur, Pakistan
- 4DMLS Department, The University of Lahore, Islamabad Campus, Islamabad, Pakistan
- 5Department of Higher Education, Government Girls Degree College Serai Naurang (Lakki Marwat), Peshawar, Pakistan
- 6School of Life Sciences, Beijing Institute of Technology, Beijing, China
- 7Division of Genetics, Department of Pediatrics, King Abdulaziz Medical City, Riyadh, Saudi Arabia
- 8Medical Genomics Research Department, King Abdullah International Medical Research Center (KAIMRC), King Saud Bin Abdulaziz University for Health Sciences, Ministry of National Guard–Health Affairs (MNGHA), Riyadh, Saudi Arabia
Background: Limb-girdle muscular dystrophy (LGMD) is an increasingly heterogeneous category of inherited muscle diseases, mainly affecting the muscles of shoulder areas and the hip, segregating in both autosomal recessive and dominant manner. To-date, thirty-one loci have been identified for LGMD including seven autosomal dominant (LGMD type 1) and twenty four autosomal recessive (LGMD type 2) inherited loci.
Methodology/Laboratory Examination: The present report describes a consanguineous family segregating LGMD2F in an autosomal recessive pattern. The affected individual is an 11-year-old boy having two brothers and a sister. Direct targeted next generation sequencing was performed for the single affected individual (VI-1) followed by Sanger sequencing.
Results: Targeted next generation sequencing revealed a novel homozygous nonsense mutation (c.289C>T; p.Arg97∗) in the exon 3 of the delta-sarcoglycan (SGCD) gene, that introduces a premature stop codon (TCA), resulting in a nonsense mediated decay or a truncated protein product.
Discussion and Conclusion: This is the first report of LGMD2F caused by an SGCD variant in a Pakistani population. The mutation identified in the present investigation extends the body of evidence implicating the gene SGCD in causing LGMD2F and might help in genetic counseling, which is more important to deliver the risk of carrier or affected in the future pregnancies.
Introduction
Limb-girdle muscular dystrophies (LGMDs) are a group of an inherited muscle disorder, which is heterogeneous in nature, typically affecting the pelvic girdle (hips) and voluntary muscles of the shoulders. LGMDs primarily affects the proximal limb muscles however, other muscles like the respiratory and cardiac muscles also may get degenerated (Nigro and Savarese, 2014).
Limb-girdle muscular dystrophies can be broadly classified into two classes namely autosomal dominant form known as limb-girdle muscular dystrophies type 1 (LGMD1) and autosomal recessive form known as limb girdle muscular dystropies type 2 (LGMD2). LGMD1 can be further sub-classified into seven subtypes whereas LGMD2 into twenty four subtypes (Thompson and Straub, 2016).
Sarcoglycanopathies in humans are rare genetic disorders with an incidence of 1/178,000 live births (Fanin et al., 1997). Sarcoglycanopathies such as alpha (α), beta (β), gamma (γ), and delta (δ) sarcoglycans are a subset of recessive, severe form of LGMDs (LGMD2C-F) that present in early childhood (Magri et al., 2014). These encode single-pass transmembrane glycoproteins (α-, β-, δ-, γ-sarcoglycans), making up the tetrameric sarcoglycan-sarcospan complex (SGC). Sarcoglycans are basically N-glycosylated transmembrane proteins forming a hetero-tetrameric complex, which is mostly linked to the dystrophin–dystroglycan complex (Sandonà and Betto, 2009). SGC is critical for maintaining sarcolemmal stability and pathogenic sequence variants in the SGCA, SGCB, SGCD, or SGCG might result in non-assembly of the SGC (Shi et al., 2004; Tarakci and Berger, 2016).
The delta sarcoglycan (δ) gene (SGCD) is the largest LGMD gene spanning a genomic region of 433 kb on chromosome 5q33.3 and the fully functional transcript has a total of 9 exons. The second (2nd) intron of δ-sarcoglycan is one of the largest intron in human genome having a size of 164 kb. Both gamma (γ) and delta (δ) sarcoglycan can have a size of 35 kDa and are homologous to each other (Dickens et al., 2002).
Here, we characterized a consanguineous Pakistani family with LGMD2F caused by a novel homozygous nonsense mutation (c.289C>T; p.Arg97∗) in the exon 3 of the SGCD gene (NM_001128209.1).
Case Presentation
The present study describes a single affected boy (IV-1; 11 years) that belongs to a consanguineous Pakistani family (Figure 1A). The affected individual (IV-1) has three normal siblings (two boys (IV-2; 10 years; IV-4; 7 years) and a girl (IV-5; 1.5 years). The proband (IV-1) presented here, was the first son of consanguineous Pakistani parents and had a birth weight of 3.5 kg (50th centile). His first symptoms presented at 3–4 years of age when his parents noted different walking style and less weight gain. He started sitting without assistance at 8 months and walking at 15 months of age. He started to run at 2.5 years of age, never jumped or hopped and suffered frequent 2–3 falls per day. The affected boy had difficulty in walking and climbing stairs since 3 years of age. He has relatively normal extensor muscles of the fingers, wrist, toes flexors and hip abductors, with severe weakness of other muscles (Figures 1B–E). The cardiac review suggested mild cardiac hypertrophy, muscular weakness, normal peripheral pulse and no murmurs were observed. Venus pressure was normal and signs of heart failure were not observed. He was diagnosed having LGMD on the basis of the observed phenotypes, and molecular diagnosis. Detailed clinical findings observed in the proband (IV-1) have been described in the Supplementary Table 1.
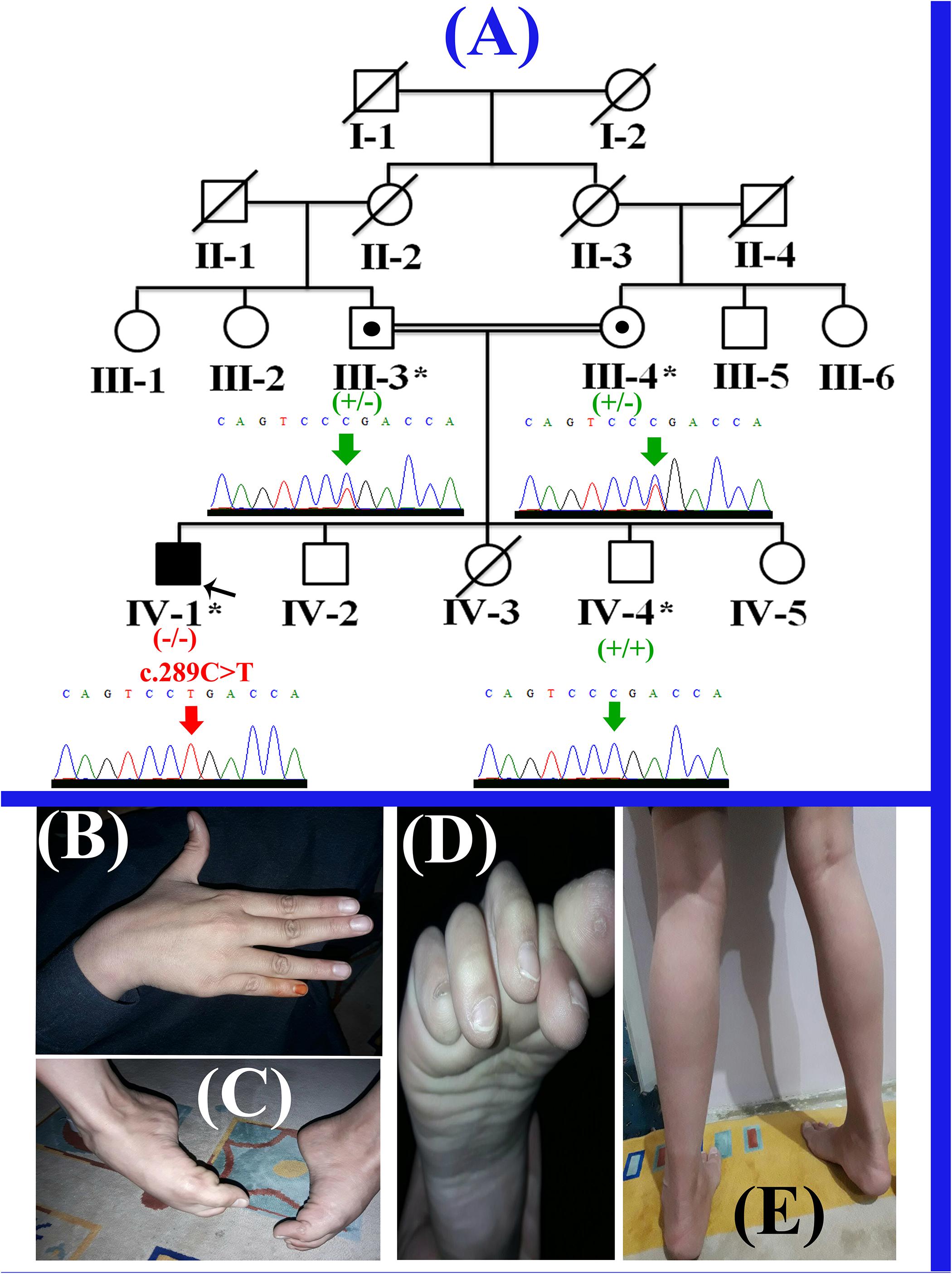
Figure 1. (A) Pedigree of the present family, segregating autosomal recessive form of LGMD2F. Clear circles and squares represent normal females and males, respectively. Black symbol represents the affected individual (proband). Double lines indicate consanguineous union. Individual numbers labeled with asterisks, indicate those who were available for the present study. (B) Affected individual’s (IV-1) hand showing contractures. (C,D) Right foot of the affected individual (IV-1) showing fixed flection deformity and stiffness of the toes. (E) Feet showing the weakness in the gastrocnemius muscle and the inability to stand without support.
Echocardiography
Echocardiography showed situs solitus, normal atrial and ventricular arrangements. The cardiac chambers were not enlarged. The inferior vena cava (IVC) was not dilated and there was a pulsatile descending aorta. Views of the atrial septum were limited and there was suspicion of a tiny patent foramen ovale (PFO) with a left to right shunting. The mitral valve was normal with no significant regurgitation or stenosis. The tricuspid valve was also normal with no signs of stenosis or regurgitation and no ventricular septal defect was observed. The aortic valve and the pulmonary valve were normal with only trivial pulmonary regurgitation and the left ventricle functioned normally. The mitral valve inflow Doppler demonstrated a wave of 0.47 m/s and an E wave of 0.67 m/s. The E/A ratio was 1.42 and the mitral valve acceleration time was 164 ms. Tissue Doppler indices of the left ventricular free wall demonstrated a normal pattern of E/A, and S waves, while the S wave velocity was around 10 cm/sec. The right ventricular function was normal with a Tricuspid annular plane systolic excursion (TAPSE) of 15.5 mm, while the Isovolumic relaxation time (IVRT) of the left ventricle was 83 ms and E/e prime ratios did not suggest that left atrial pressures were significantly elevated. Left ventricular systolic function was normal. The left ventricular end diastolic diameter was observed 24 mm having fractional shortening of 33%. In the short axis view prolapse of the mitral valve was observed, not causing any significant regurgitation. The coronary arteries were normal. There were confluent pulmonary arteries with no patent ductus arteriosus, normal left sided arch and no pericardial effusion. Furthermore, ECG which did not reveal any significant signs of arrhythmia or ischaemia. The affected individual (IV-1) muscle weakness and lack of mobility limit his physical capabilities but he has not experienced any significant signs of exercise incompetence, although he suffered from episodes of syncope. A muscle biopsy of the affected individual was not available. Parents of the affected individual were healthy and showed no abnormality.
DNA Extraction and Targeted Next Generation Sequencing
The present study was performed following the declaration of Helsinki protocols, and written informed consent for conducting the study and for publication of photographs was obtained from the parents. Peripheral blood samples were obtained from four individuals including three unaffected (III-3, III-4, IV-4) and single affected (IV-1) individual in an EDTA containing Vacutainer tubes (BD, Franklin Lakes, NJ, United States). Extraction of genomic DNA was performed using GenEluteTM Blood Genomic DNA Kit (Sigma-Aldrich, MO, United States) and quantified using Nanodrop-1000 spectrophotometer (Thermal Scientific, Wilmington, MA, United States).
Selective analysis of 31 genes associated with LGMD was performed using SureSelect focused Exome +1 capture (Agilent Technologies, CA, United States). Sequencing was performed using NextSeq 500 platform (Illumina, San Diego, CA, United States) using Burrows-Wheeler Aligner (BWA v 0.7.5), and all reads were aligned against human assembly hg19 (GRCh37). Variant calling (Free bayes), variant annotation (Alamut-batch) was performed using in-house pipeline, with a minimum sensitivity of 99.9%. Since the pedigree clearly depicted an autosomal recessive inheritance of the phenotype (Figure 1A), therefore to identify a putative pathogenic homozygous recessive variants, we first searched for the homozygous variants in the 24 known LGMD genes (Supplementary Table 2). To search for disease causing variants, targeted NGS and screening was performed as described previously (Umair et al., 2017a). Variants screening was performed based on autosomal recessive inheritance pattern for the LGMD phenotype, rare variants and OMIM list of LGMD genes. We focused only on pathogenic, disease causing non-synonymous (NS) variants, causing nonsense, missense, splice acceptor/donor site, short frame-shift coding deletions (indel) or insertions and large deletions/duplications. Considering LGMD as a rare disorder, it is highly unlikely that the disease causing variant could be present in homozygous form in general population, therefore the data was cross-matched with online public databases. Following step-by-step filtering process for screening homozygous and compound heterozygous variants in the known genes causing LGMDs, a nonsense mutation (c.289C>T; p.Arg97∗) was identified in the exon 3 of the SGCD gene, located on chromosome 5q33.2-q33.3 (Figures 2A,B).
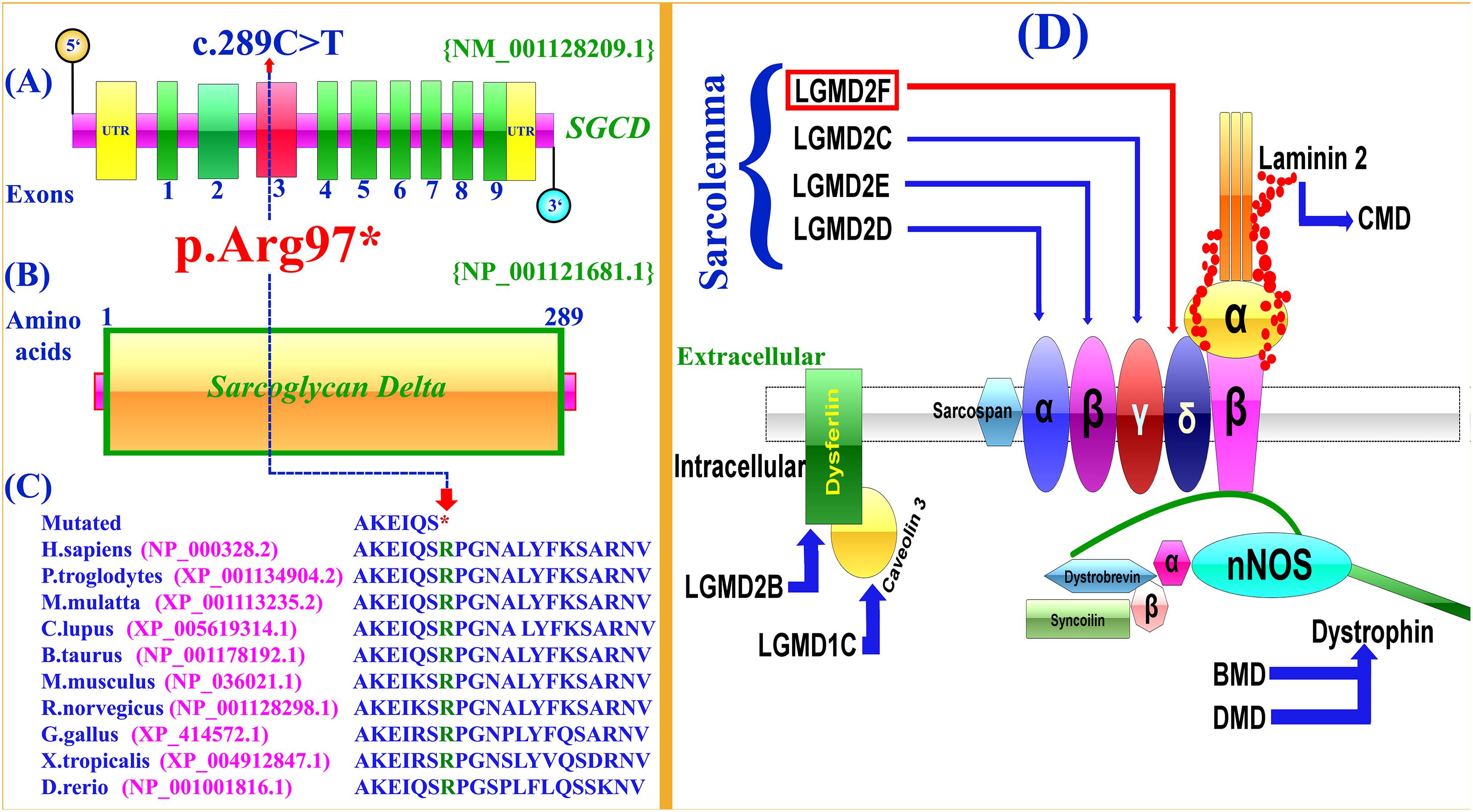
Figure 2. Schematic representation of exons and protein product of SGCD gene. (A,B) Arrows show the position the mutation (c.289C>T; p.Arg97∗) identified in the present study. (C) Conservation of the Arg (R) 97 amino acid across different species. Notably, the nonsense mutation (c.298C>T) results in a premature stop codon (TCA). (D) Schematic representation of the sarcoglycan complex. The intracellular and extracellular regions of sarcoglycans are separated by a cell membrane. Different sarcoglycans, α, β, γ, δ, dysferlin, caveolin 3, dystrophin and laminin are shown and associated disorders are also shown using arrows.
In Silico Analysis
The variant (c.289C>T) was not found in homozygous state in the dbSNP1, 1000 Genomes2, EVS3, genomAD4, and Exome Aggregation Consortium ExAC5. In the ExAC database containing more than 60,000 human exome data, the identified SGCD variant (c.289C>T) was found in heterozygous state with an allelic frequency of 0.000153 in European (Finnish) population.
The identified variant was predicted to be deleterious using different online available bioinformatics tools such as Mutation Taster6, Sorting Intolerant From Tolerant (SIFT)7, and Polymorphism Phenotyping V2 (PolyPhen-2)8. The variant is also highly conserved across multiple species (Figure 2C). To exclude polymorphic nature of the variant, it was cross checked within different online variant databases, 160 in-house exome (Pakistan) from unrelated Pakistani individuals, and 185 ethnically matched controls.
Sanger Sequencing Validation
The homozygous sequence variant (c.289C>T; p.Arg97∗), identified in the present study was verified using Sanger sequencing in all the available family members and the variant perfectly segregated with the disease phenotype (Figure 1A). Genomic sequence of the SGCD gene was retrieved from the University of California Santa Cruz (UCSC) genome database browser9. Primer sequences for PCR amplification of the exon 3 were designed using Primer3 software10. Primers sequences used for the present study include (Forward primer-F1: GGAATGGGAAACCTGAGG; Reverse primer-R1: AGCTGGGTAGCAGGGGTTG). Sanger sequencing procedure has been described earlier (Umair et al., 2016). Mutation was identified via BIOEDIT sequence alignment editor version 6.0.7 (Ibis Biosciences Inc., Carlsbad, CA, United States).
Discussion
In the present study, we performed genetic and molecular analysis on a consanguineous family having a single affected individual (IV-1), segregating LGMD2F in an autosomal recessive manner. Consanguineous marriages are preferred in a county like Pakistan due to its socioeconomic and cultural obligations (Umair et al., 2018). The coefficient of inbreeding is increased by consanguinity, which increases the presence of variant in a homozygous state. Pakistani population displays rare genetic ailments due to higher coefficient of inbreeding. In such a situation, WES is an efficient, fast and economical strategy for detection of pathogenic disease-causing variants (Umair et al., 2017b). LGMD phenotypes observed in the affected individual were similar to those reported previously (Dinçer et al., 2000). Cardiomyopathy is not a core component of sarcoglycanopathies, still there have been reports of several patients without cardiac involvement and mutations in the SGCD gene have also been reported to cause autosomal-dominant dilated cardiomyopathy (DCM) independently from LGMD phenotypes (Tsubata et al., 2000).
LGMD2F is the fourth sarcoglycanopathy (LGMD2C to LGMD2F) type, characterized when the causative mutation in the δ-SG gene was identified for the first time in Brazilian LGMD patients with a Duchenne muscular dystrophy (DMD) type features linked to chromosome 5q33-34. Since then, a large number of mutations causing both LGMD2F and δ-SG-associated cardiomyopathy have been described (Leiden Muscular Dystrophy database11).
In the present investigation, targeted next-generation sequencing revealed a novel homozygous nonsense mutation (c.289C>T; p.Arg97∗), segregating with the disease phenotype within the family. To-date only twelve mutations have been identified in the SGCD gene associated with LGMD including four nonsense, five missense, two small deletions and one gross deletion (Supplementary Table 3). In silico analysis, revealed that the variant (c.289C>T; p.Arg97∗) might result in truncated SGCD protein lacking 192 conserved amino acids and most likely degraded by nonsense mediated mRNA decay (NMD).
Six sarcoglycans, α, β, γ, δ, η and ζ have been identified so far. Mutations in the genes for α, β, γ, and δ-sarcoglycan causes heterogeneous group of autosomal recessive limb-girdle muscular dystrophies (LGMD2C-F) (Bönnemann et al., 1995; Noguchi et al., 1995). The sarcoglycan deficient LGMDs are characterized by progressive weakness of the pelvic and shoulder girdle musculature with symptoms ranging from mild to highly severe (Zatz et al., 2003). Patients with LGMD2C-F, in particular with β-sarcoglycan deficient (LGMD2E) and δ-sarcoglycan-deficient (LGMD2F), often develop a progressive and potentially fatal DCM in association with muscular dystrophy (Politano et al., 2001).
The sarcoglycans are part of the dystrophin-glycoprotein complex (DGC), an oligomeric complex spanning the plasma membrane of skeletal and cardiac muscle fibers. Sarcoglycans plays a key role in the stability of cytoskeleton of muscle membrane. In the plasma membrane of muscle, they form a tetrameric complex that stabilizes the association of dystrophin with α and β dystroglycans (Figure 2D).
SGCD mutations causing LGMD2F and its clinical presentation are largely similar among the four sarcoglycanopathies (Nigro and Savarese, 2014). It is assumed that integrity of the sarcoglycan complex is crucial for maintaining the mechanical and non-mechanical linkage between the subsarcolemmal cytoskeleton and the extracellular matrix (Ozawa et al., 2005; Heydemann and McNally, 2007). Sgcd knockout mice also showed both muscular dystrophy (Loss of vascular smooth muscle SG-SSPN complex) and cardiomyopathy (focal areas of necrosis as the histological hallmark in cardiac and skeletal muscle) features (Coral-Vazquez et al., 1999).
In conclusion, we have reported a consanguineous Pakistani family having a novel SGCD variant causing LGMD2F. It is evident that δ-SG pathogenesis is complex and multifactorial which might require combinatorial therapeutic strategies. The clinical and molecular manifestation of such a heterogeneous disease is required timely, that might also involve different genetic modifiers. Still, the primary cure through gene or cell therapy may be beneficial for the patient in the future. This molecular finding increases the mutational spectrum of SGCD related pathogenesis and also provides proper genetic counseling to the affected family.
Author Contributions
MY and MU drafted the manuscript. MY, FA, MB, SA, MK, SS, and EM collected the samples and clinical data, analyzed the data, and performed the experiments. SA, EM, and MUK analyzed the genomic data. MF, MA edited the manuscript. MU and MA conceived and designed the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We highly appreciate the cooperation and participation of the family members in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00727/full#supplementary-material
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/SNP/
- ^ http://www.1000genomes.org
- ^ http://evs.gs.washington.edu/EVS/
- ^ http://gnomad.broadinstitute.org/
- ^ http://exac.broadinstitute.org/
- ^ http://www.mutationtaster.org/
- ^ https://sift.bii.a-star.edu.sg/
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://genome.ucsc.edu/cgi-bin/hgGateway
- ^ http://bioinfo.ut.ee/primer3-0.4.0/
- ^ http://www.dmd.nl/
References
Bönnemann, C. G., Modi, R., Noguchi, S., Mizuno, Y., Yoshida, M., Gussoni, E., et al. (1995). Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat. Genet. 11, 266–273. doi: 10.1038/ng1195-266
Coral-Vazquez, R., Cohn, R. D., Moore, S. A., Hill, J. A., Weiss, R. M., Davisson, R. L., et al. (1999). Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 98, 465–474. doi: 10.1016/S0092-8674(00)81975-3
Dickens, N. J., Beatson, S., and Ponting, C. P. (2002). Cadherin-like domains in alpha-dystroglycan, alpha/epsilon-sarcoglycan and yeast and bacterial proteins. Curr. Biol. 12, R197–R199. doi: 10.1016/S0960-9822(02)00748-0
Dinçer, P., Bönnemann, C. G., Erdir Aker, O., Akçoren, Z., Nigro, V., Kunkel, L. M., et al. (2000). A homozygous nonsense mutation in delta-sarcoglycan exon 3 in a case of LGMD2F. Neuromuscul. Disord. 10, 247–250. doi: 10.1016/S0960-8966(00)00100-0
Fanin, M., Duggan, D. J., Mostacciuolo, M. L., Martinello, F., Freda, M. P., Sorarù, G., et al. (1997). Genetic epidemiology of muscular dystrophies resulting from sarcoglycan gene mutations. J. Med. Genet. 34, 973–977. doi: 10.1136/jmg.34.12.973
Heydemann, A., and McNally, E. M. (2007). Consequences of disrupting the dystrophin-sarcoglycan complex in cardiac and skeletal myopathy. Trends Cardiovasc. Med. 17, 55–59. doi: 10.1016/j.tcm.2006.12.002
Magri, F., Brajkovic, S., Brusa, R., Comi, G. P., and Govoni, A. (2014). Revised genetic classification of limb girdle muscular dystrophies. Curr. Mol. Med. 14, 934–943. doi: 10.2174/1566524014666141010130244
Nigro, V., and Savarese, M. (2014). Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 33, 1–12.
Noguchi, S., McNally, E. M., Ben Othmane, K., Hagiwara, Y., Mizuno, Y., Yoshida, M., et al. (1995). Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science 270, 819–822. doi: 10.1126/science.270.5237.819
Ozawa, E., Mizuno, Y., Hagiwara, Y., Sasaoka, T., and Yoshida, M. (2005). Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 32, 563–576. doi: 10.1002/mus.20349
Politano, L., Nigro, V., Passamano, L., Petretta, V., Comi, L. I., Papparella, S., et al. (2001). Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul. Disord. 11, 178–185. doi: 10.1016/S0960-8966(00)00174-7
Sandonà, D., and Betto, R. (2009). Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev. Mol. Med. 11:e28. doi: 10.1017/S1462399409001203
Shi, W., Chen, Z., Schottenfeld, J., Stahl, R. C., Chan, Y. M., and Kunkel, L. M. (2004). Specific assembly pathway of sarcoglycans is dependent on beta- and deltasarcoglycan. Muscle Nerve 29, 409–419. doi: 10.1002/mus.10566
Tarakci, H., and Berger, J. (2016). The sarcoglycan complex in skeletal muscle. Front. Biosci. 21:744–756. doi: 10.2741/4418
Thompson, R., and Straub, V. (2016). Limb-girdle muscular dystrophies – international collaborations for translational research. Nat. Rev. Neurol. 12, 294–309. doi: 10.1038/nrneurol.2016.35
Tsubata, S., Bowles, K. R., Vatta, M., Zintz, C., Titus, J., Muhonen, L., et al. (2000). Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Invest. 106, 655–662. doi: 10.1172/JCI9224
Umair, M., Ahmad, F., and Ullah, A. (2018). Whole exome sequencing as a diagnostic tool for genetic disorders in Pakistan. Pak. J. Med. Res. 57, 90–91.
Umair, M., Alhaddad, B., Rafique, A., Jan, A., Haack, T. B., Graf, E., et al. (2017a). Exome sequencing revealed a novel homozygous splice site variant in WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr. Res. 82, 753–758. doi: 10.1038/pr.2017.149
Umair, M., Shah, K., Alhaddad, B., Haack, T. B., Graf, E., Strom, T. M., et al. (2017b). Exome sequencing revealed a splice site variant in the IQCE gene underlying post-axial polydactyly type A restricted to lower limb. Eur. J. Hum. Genet. 25, 960–965. doi: 10.1038/ejhg.2017.83
Umair, M., Hassan, A., Jan, A., Ahmad, F., Imran, M., Samman, M. I., et al. (2016). Homozygous sequence variants in the FKBP10 gene underlie osteogenesis imperfecta in consanguineous families. J. Hum. Genet. 61, 207–213. doi: 10.1038/jhg.2015.129
Keywords: limb-girdle muscular dystrophies, LGMD, targeted NGS, SGCD, LGMD2F, homozygous variant, nonsense mutation
Citation: Younus M, Ahmad F, Malik E, Bilal M, Kausar M, Abbas S, Shaheen S, Kakar MU, Alfadhel M and Umair M (2019) SGCD Homozygous Nonsense Mutation (p.Arg97∗) Causing Limb-Girdle Muscular Dystrophy Type 2F (LGMD2F) in a Consanguineous Family, a Case Report. Front. Genet. 9:727. doi: 10.3389/fgene.2018.00727
Received: 12 July 2018; Accepted: 22 December 2018;
Published: 23 January 2019.
Edited by:
Noor Ahmad Shaik, King Abdulaziz University, Saudi ArabiaReviewed by:
Vishnu Hosur, The Jackson Laboratory, United StatesPreetha Jayasheela Shetty, Gulf Medical University, United Arab Emirates
Copyright © 2019 Younus, Ahmad, Malik, Bilal, Kausar, Abbas, Shaheen, Kakar, Alfadhel and Umair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Umair, a2h1Z29vNHVAeWFob28uY29t; dW1haXJtdUBuZ2hhLm1lZC5zYQ==