 Tianjiao Li1
Tianjiao Li1 Sandra Martins2
Sandra Martins2 Yun Peng1Puzhi Wang1Xiaocan Hou1
Yun Peng1Puzhi Wang1Xiaocan Hou1 Zhao Chen1Chunrong Wang1Zhaoli Tang1Rong Qiu3Chao Chen4Zhengmao Hu4Kun Xia4
Zhao Chen1Chunrong Wang1Zhaoli Tang1Rong Qiu3Chao Chen4Zhengmao Hu4Kun Xia4 Beisha Tang1,4,5,6,7,8,9Jorge Sequeiros10
Beisha Tang1,4,5,6,7,8,9Jorge Sequeiros10 Hong Jiang1,4,6*
Hong Jiang1,4,6*- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2IPATIMUP – Institute of Molecular Pathology and Immunology of the University of Porto, Instituto de Investigação e Inovação em Saúde (i3S), Porto, Portugal
- 3School of Information Science and Engineering, Central South University, Changsha, China
- 4Laboratory of Medical Genetics, Central South University, Changsha, China
- 5National Clinical Research Center for Geriatric Diseases, Xiangya Hospital, Central South University, Changsha, China
- 6Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 7Parkinson’s Disease Center of Beijing Institute for Brain Disorders, Beijing, China
- 8Collaborative Innovation Center for Brain Science, Shanghai, China
- 9Collaborative Innovation Center for Genetics and Development, Shanghai, China
- 10IBMC – Institute for Molecular and Cell Biology, i3S – Instituto de Investigação e Inovação em Saúde, ICBAS (Instituto de Ciências Biomédicas Abel Salazar), University of Porto, Porto, Portugal
Machado-Joseph disease (MJD, also known as spinocerebellar ataxia 3 or SCA3) is the most common dominant ataxia worldwide, with an overall average prevalence of 1–5/100,000. To this date, two major ancestral lineages have been found throughout the world. In China, the relative frequency of MJD among the SCAs reaches as high as 63%, however, little is known about its mutational origin in this country. We analyzed 50 families with MJD patients in two or more generations to study the hypothesis that new mutational events have occurred in this population. Haplotypes based on 20 SNPs have shown new genetic backgrounds segregating with MJD mutations in our cohort from China. We found the “Joseph-derived” lineage (Joseph lineage with a G variant in rs56268847) to be very common among Chinese MJD patients. Moreover, we estimated the time for the origin of this MJD SNP background based on STR diversity flanking the (CAG)n of ATXN3. It was surprising to find that the Chinese MJD population originated from 8,000 to 17,000 years ago, far earlier than the previous literature reports, which will be an important evidence to explain the origin, spread and founder effects of MJD.
Introduction
Machado-Joseph disease (MJD, OMIM#109150), also known as spinocerebellar ataxia type 3 (SCA3), is one of the polyQ diseases. It is a rare autosomal dominantly inherited neurodegenerative disease that causes progressive cerebellar ataxia, resulting in lacks of muscle control and coordination of the upper and lower extremities, with symptoms including dysarthria, dysphagia, pyramidal signs, progressive external ophthalmoplegia, and dystonia (Bettencourt and Lima, 2011; Costa Mdo and Paulson, 2012; Wang et al., 2015; Paulson et al., 2017). This autosomal dominant disease is caused by an elongated polyglutamine stretch encoded by more than 51 CAG repeats on the ATXN3 allele (Rüb et al., 2013; Ding et al., 2016; Chen et al., 2017; Long et al., 2018). MJD is spread worldwide, but it has a higher relative frequency among the SCAs in Portugal (57.8%) (Vale et al., 2010), Brazil (59.6%) (de Castilhos et al., 2014), Japan (43%) (Takano et al., 1998), and Germany (42%) (Schöls et al., 1997). Our previous study has shown that mainland China has a high prevalence of MJD with a relative frequency of 62.6% (Chen et al., 2018).
This disease was first reported in an extended family of Portuguese-Azorean ancestry, in 1972 (Nakano et al., 1972), and was believed to be very frequent in Portugal due to founder effects. Also, Japan, Brazil and France all had founder effects of MJD (Marie-Françoise Chesselet, 2001). Furthermore, a linkage disequilibrium analysis was carried out with three single nucleotide polymorphisms (SNPs, A669TG/G669TG, C987GG/G987GG, and TAA1118/TAC1118) and five short tandem repeats (STRs, D14S1015, D14S995, D14S973, D14S1016, and D14S977), in 249 families of various ethnic backgrounds. Four different SNP haplotypes were identified segregating with MJD expansions: A-C-A, A-G-A, G-G-A, and G-G-C (Gaspar et al., 2001). Following this discovery, a worldwide study of extended haplotypes was performed in 264 MJD families, and two major ancestral lineages were confirmed: the GTGGCA background or the Machado lineage, probably originated in Portugal; and the TTACAC or Joseph lineage, observed in 19 countries, including Japan (Martins et al., 2007). To determine the occurrence of new mutation events and clarify the spread of ancestral MJD lineages in different populations, genetic distances were determined using a total of 20 SNPs and 4 microsatellites for the purpose of haplotypes identification (Martins et al., 2012), however, little information on MJD haplotypes is available in China, although the frequency of MJD is very high in this Asian population.
In this study, to determine the origin and estimate the age of MJD mutational event(s) in China, we performed a haplotype analysis of 20 SNPs and 7 microsatellites in 50 Chinese MJD families.
Materials and Methods
Patients and Controls
We analyzed 50 families with MJD, from Southern China, including 109 patients, and 105 healthy individuals. All participants agreed with our request for collection of peripheral blood samples and signed an informed consent form. The study was approved by Ethics Committee of Xiangya Hospital of Central South University in China. Genomic DNA was isolated using a standard protocol. Each family has at least two generations and has at least four individuals. Healthy spouses served as the control group. In one of the families studied, a patient homozygous for an MJD expansion was analyzed together with both parents and his sister.
Genotyping and Haplotype Analysis
We analyzed 20 SNPs located upstream and downstream the (CAG)n, including the previously analyzed six core SNPs and variants within a 4 kb area flanking the repeat (Figure 1; Martins et al., 2006; Costa et al., 2019). PCR amplification reactions were done with TSE 101 Golden Star T6 Super PCR Mix (1.1×). The location of all loci and Primers for amplification are listed in the Supplementary Table S1. Genotyping of SNPs was performed through Sanger sequencing. We inferred allelic phases by segregation for most of analyzed families; some haplotypes were reconstructed by PHASE software version 2.1.11 (only haplotypes with probabilities greater than 0.6 were used for subsequent analyses). We compared the distribution of SNPs in the Chinese MJD population with the two ancestral lineages described previously, and selected four different SNPs among these to test for Hardy-Weinberg equilibrium and Chi-square test (X2). In this test, 49 healthy spouses of probands served as controls. We first distinguished the haplotypes of the six core SNPs and conducted a Fisher’s exact test. Basically, the formula δ = (Fd-Fc)/ (1-Fc) is used to calculate the approximate risk of population attribution and provide evidence of LD. Then, we extended the haplotype to 20 SNPs for further analysis (Devlin and Risch, 1995; Gaspar et al., 2001).
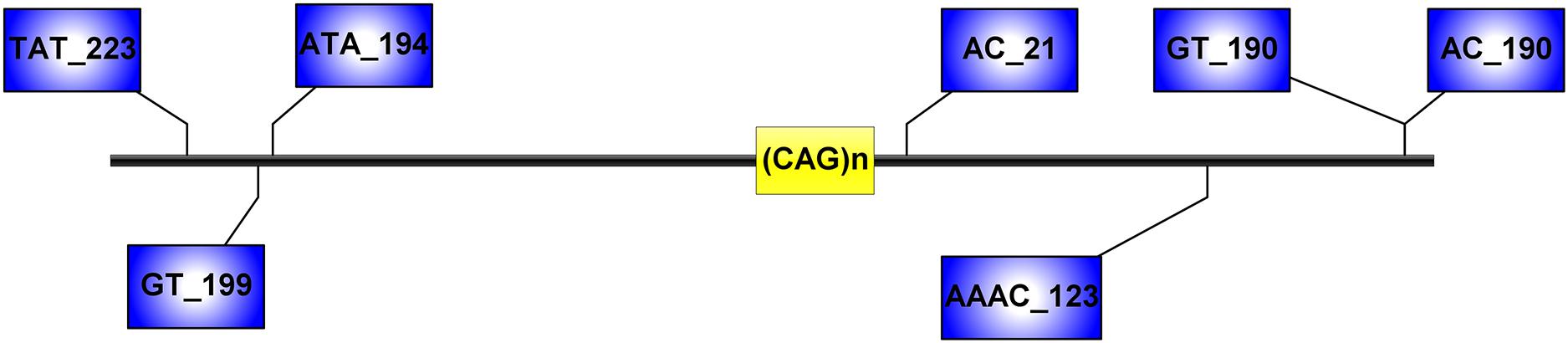
Figure 1. Microsatellite loci flanking the (CAG)n of ATXN3, analyzed in this study. Distances from the (CAG)n, included in the STR name, are expressed in kilobases.
STR-Based Haplotype Analysis and Age Estimation
Capillary electrophoresis was used to detect the seven STRs (TAT_223)n, (GT_199)n, (ATA_194)n, (AC_21)n, (AAAC)n, (GT)n, and (AC_190)n (Figure 1), and haplotypes reconstructed by segregation and with the PHASE software version 2.1.1 (only haplotypes with probabilities greater than 0.6 were used for subsequent analyses). We determine ancestral haplotypes and steps of mutation by STR-based haplotype from the most common haplotype based on 20 SNPs and assess the age of mutation events. This formula 𝜀 = 1-[(1-c) (1-μ)] is used to calculate the probability of change for each generation, where c represents the recombination rate and μ represents the mutation rate. The multiplication of 𝜀 and t yields the average of mutations and reorganizations (λ), where t is the number of generations (Martins et al., 2007).
Results
SNP Haplotyping of Chinese MJD Families
We identified 13 disease-associated haplotypes in our cohort of Chinese MJD families. We compared these haplotypes with the Joseph lineage and indicated their differences (Table 1). It is worthy of mentioning that, for four SNPs, we found new alleles segregating with expanded alleles, not previously associated to Machado or Joseph ancestral MJD lineages (Table 2). The G allele occurs in the rs56268847 of the pathogenic chromosomes in the 28 families, exceeding 50% of the total fifty families involved in the study. Regarding SNPs rs16999141, rs10467857 and rs77086230, alleles never found in phase with expanded MJD alleles were here observed in families 21, 31 and 15, respectively.
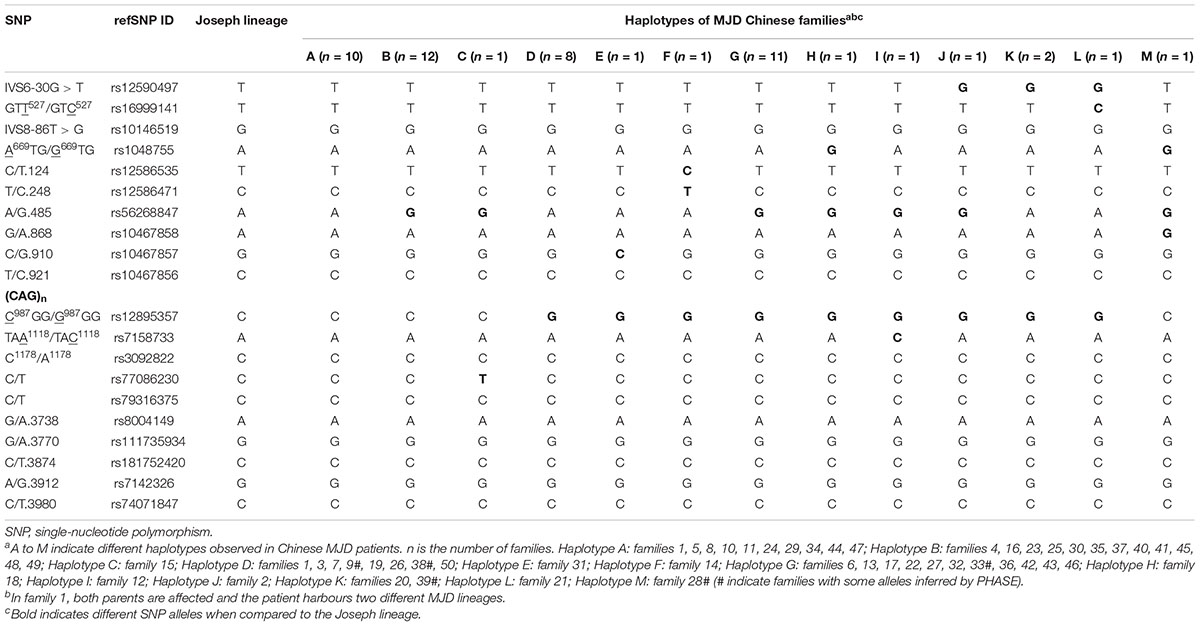
Table 1. SNP-based haplotypes of MJD Chinese families in comparison to Joseph MJD lineage.
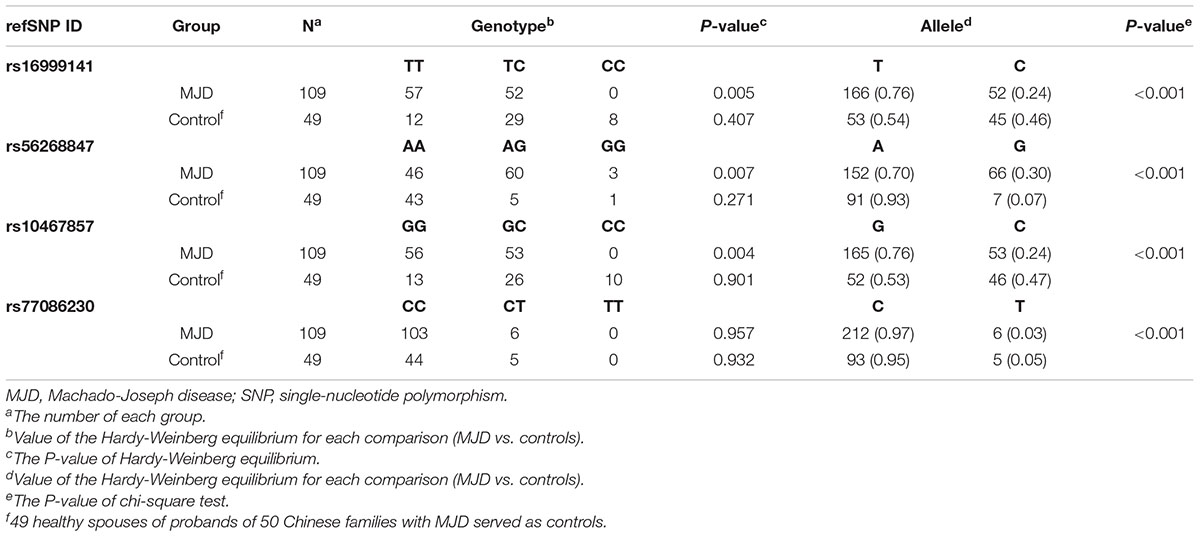
Table 2. Allele frequency of 4 different SNPs analyzed by Hardy-Weinberg equilibrium and chi-square test.
Hardy-Weinberg Equilibrium and Chi-Square Test for the 4 of SNPs
Hardy-Weinberg equilibrium and chi-square test were carried out to examine the SNP variations. The control group consisting of 49 spouses confirmed by the Hardy-Weinberg equilibrium that satisfy the genetic equilibrium in the four heterogeneous SNPs. In rs16999141, no genotype CC was found in MJD patients, 57 of 109 patients shared TT, and the rest were TC. For SNP rs56268847, significant variation from AA, AG to GG was detected with the p-value of the Hardy-Weinberg equilibrium at this position is 0.007. More specifically, 46 out of 109 SCA3/MJD patients had a genotype of AA, while 60 patients had AG, and by contrast, only three patients had GG. Different distribution for SNP rs10467857 was detected. Of the 109 identical patients, 56 are genotyped as GG and 53 as GC (p-value of the Hardy-Weinberg equilibrium 0.004), respectively. Similar situation was found in the SNP rs77086230 in a way that Only CC and CT were discovered with a predominance of CC over CT. The p-value of Hardy-Weinberg equilibrium of this SNP in SCA3/MJD group and three SNPs in the control group are more significant than 0.05. Moreover, all p-values used chi-square tests for three SNPs are less than 0.001 (Table 2).
Analysis of Haplotypes Based on Six SNPs
Through linkage analysis of six SNPs such as IVS6-30G > T, GTT527/GTC527, A669TG/G669TG, C987GG/G987GG, TAA1118/TAC1118, and C1178/A1178, we found that Chinese MJD patients only shared 7 haplotypes, while healthy people shared 21 haplotypes (Table 3). Among them, T-T-A-G-C-C only appears in the MJD patients. The remaining 6 haplotypes were confirmed statistically significant by chi-square test, and all the p-values were less than 0.001.
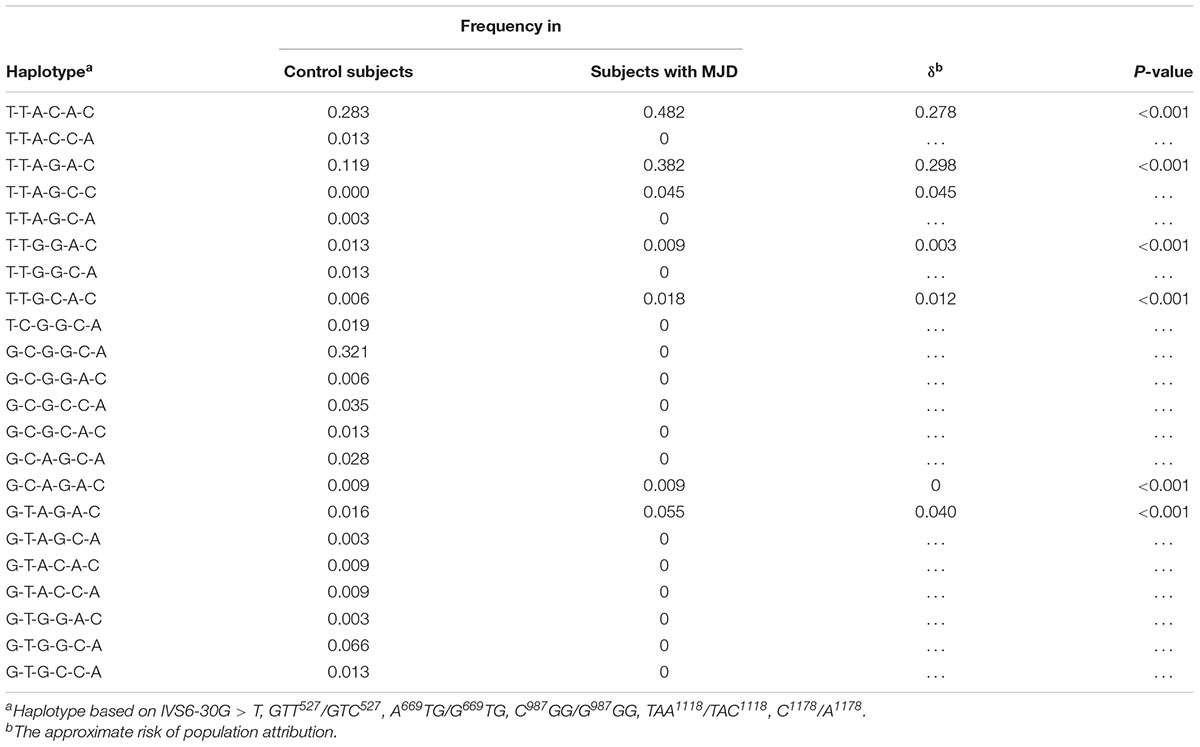
Table 3. Overall linkage disequilibrium analysis of Chinese families for intragenic haplotypes based on 6 SNPs.
Age Estimation
We performed STR analysis and age estimation on four major haplotypes A, B, D, and G based on 20 SNPs, and inferred the Phylogenetic networks of the four haplotypes (Figure 2). Surprisingly, MJD haplotypes A and D seemed to be present in the Chinese population as remote as 16,335 ± 1,966 and 11,837 ± 1,871 years, respectively; whereas introduction of haplotypes B and G probably occurred simultaneously, a few million years later, 9,272 ± 1,352 and 9,254 ± 1,411 years ago, respectively (Table 4).
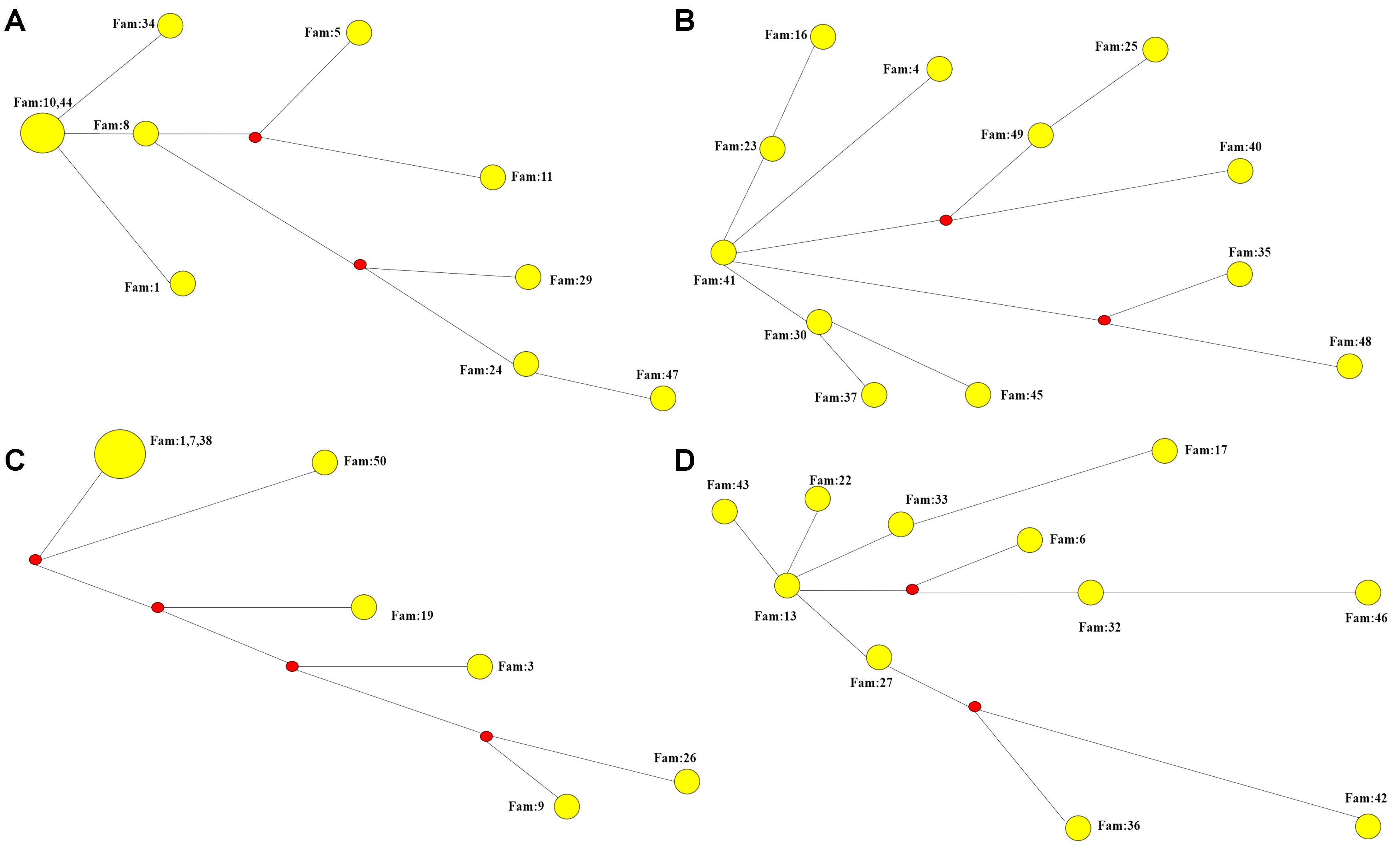
Figure 2. Phylogenetic networks of the four major haplotypes based on 7 microsatellite loci. (A) Phylogenetic network of haplotype A. (B) Phylogenetic network of haplotype B. (C) Phylogenetic network of haplotype D. (D) Phylogenetic network of haplotype G.
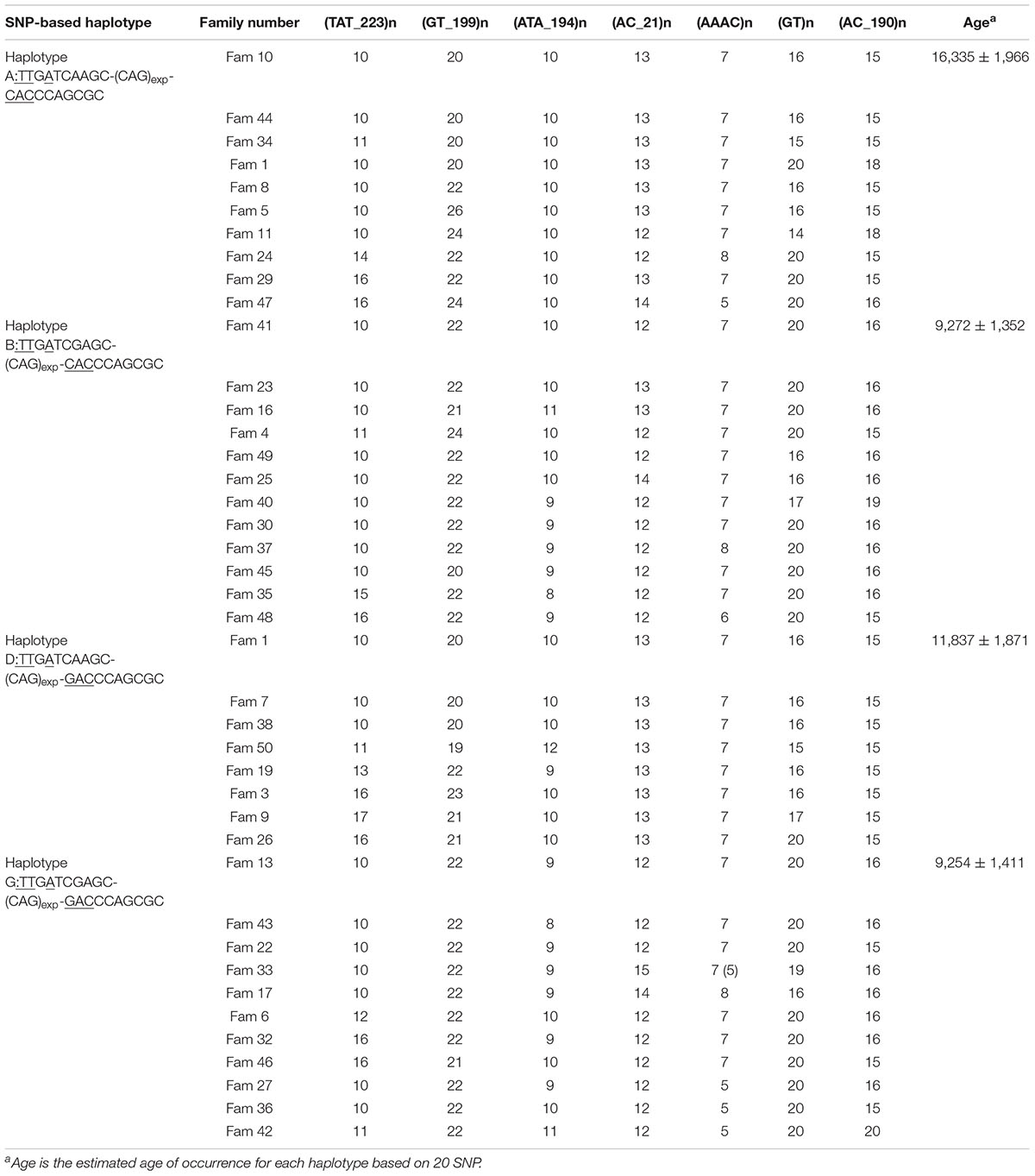
Table 4. Haplotypes and age estimation with 7 STR flanking the (CAG)n at the ATXN3, from families sharing the four most common SNP-based haplotypes.
Discussion
For the selection of participants, we require that the proband must have more than one children or proband’s family must bear more than two generations. The stringency of the selection criteria for the participants made only 5 of the families inferred as haplotypes by software, suggesting the vast majority of families inferred haplotypes through the pedigree structure to make our results more accurate and unquestionable.
The rs56268847 found in Asian SCA3/MJD patients was not statistically significant in the previous report (Martins et al., 2012), most probably due to the small sample size and different national backgrounds. Pathogenic chromosomes of 28 probands in 50 families carry G allele with a frequency of 0.56. The p-value of the Hardy-Weinberg equilibrium is <0.05 in the SCA3/MJD group, indicating that this allele does not reach a genetic equilibrium. However, the difference between the control group confirmed to achieve the genetic balance after the same test and Chi-square test, and the SCA3/MJD group is statistically significant. Thus, it is reasonable to speculate that the difference might be primarily caused by the disease. Furthermore, since the G allele at rs56268847 has been observed to segregate with the expanded allele in more than half of our Chinese MJD families, this suggests that a point mutation at this SNP must have occurred early in the introduction of MJD in China or, even, in other Asian countries.
Additionally, for the first time we detected base variations (new base addition) in three SNPs rs16999141, rs10467857, and rs77086230 in the Chinese SCA3/MJD patients compared to the previously reported two ancestral lineages. Both ancestral lineages are reported as T on rs16999141, and we detected T/C. While G is reported in the ancestral lineage SNP rs10467857, we detected G/C instead of G only. Similarly, C/T is found in the Chinese SCA3/MJD patients other than C only as reported in the ancestral lineage SNP rs77086230. As for the significance in our new discovery of the new base variation in 3 sites, although so far only one family were examined for each SNP, two test methods have confirmed the significant difference. To be more conclusive to say that the new base variations occurred relatively recently, tests of more families are required.
Previous study showed that the A-G-A haplotype for the SNPs rs1048755, rs12895357, and rs7158733 in the 249 of families, exist in MJD patient (Gaspar et al., 2001), while they were not statistically significant most probably due to the different backgrounds. More convincingly, the p-value obtained by Fisher’s exact test is less than 0.001. Thus, we believe the significant difference in the frequency of A-G-A haplotype between the SCA3/MJD and the control group.
The first studies on the epidemiology of MJD were based on families described before the gene ATXN3 was known (Sequeiros and Coutinho, 1993). Later, two major MJD haplotypes have been identified (Gaspar et al., 2001; Martins et al., 2007); here, in addition to the worldwide spread Joseph lineage, we found three other major haplotypes that differ from Joseph at (1) rs56268847 (lineage B, 12 families), (2) rs12895357 (lineage D, 8 families), and (3) both rs56268847 and rs12895357 (lineage G, 11 families). Interestingly, lineage B has been previously described among Australian aborigines, probably introduced in this population via Asia (Martins et al., 2012). Independent mutational origins do not necessarily underlie these four MJD SNP-haplotypes since recurrent mutations on the 2 SNPs may have occurred: backgrounds B and D may have evolved from the Joseph lineage by recurrence at rs56268847 and rs12895357, respectively; recombination could be a possible explanation for the origin of SNP backgrounds G, although the short distance between the two SNPs and the deleterious (CAG)n makes it unlikely. Other MJD backgrounds, phylogenetically more distant from the Joseph lineage, were found in single MJD families; de novo expansions may be on the origin of some of them, but a larger cohort should confirm that their low frequency is explained by a recent event or genetic drift.
This discovery is of importance to clarify the prevalence of SCA3/MJD and could become indispensable evidence that supports the founder effect in this disease. More importantly, this finding could help decipher the genetic basis of the SCA3/MJD by further study on the haplotypes.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Author Contributions
TL completed the collection of samples, analysis of data, and writing of the manuscript. SM, JS, ZH, KX, BT, and HJ completed the design of the experiments. YP, PW, XH, ZC, CW, and ZT assisted in the collection of samples. RQ and CC contributed to the analysis of the data.
Funding
This work was supported by the National Key Research and Development Program of China (Nos. 2016YFC0905100 and 2016YFC0901504 to HJ), the National Natural Science Foundation of China (Nos. 81771231 and 81471156 to HJ), the Clinical Research Funds of Xiangya Hospital (2014L03 to HJ), the Clinical and rehabilitation fund of Peking University Weiming Biotech Group (No. xywm2015I10 to HJ), Youth Foundation of Xiangya Hospital (No. 2017Q03 to ZC), and Independent Exploration and Innovation Project of Graduate Students of Central South University (No. 1053320170177 to TL). Moreover, SM is funded by the project IF/00930/2013 from FCT. This article is a result of the project NORTE-01-0145-FEDER-000008, supported by Norte Portugal Regional Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, and through the European Regional Development Fund (ERDF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely appreciate all patients and their families that participated in the study voluntarily and supported this study. Moreover, we hope to thank all those who gave this study help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00740/full#supplementary-material
Footnotes
References
Bettencourt, C., and Lima, M. (2011). Machado-joseph disease: from first descriptions to new perspectives. Orphanet J. Rare Dis. 6:35. doi: 10.1186/1750-1172-6-35
Chen, Z., Wang, C., Zheng, C., Long, Z., Cao, L., Li, X., et al. (2017). Ubiquitin-related network underlain by (CAG)n loci modulate age at onset in Machado-Joseph disease. Brain 140:e25. doi: 10.1093/brain/awx028
Chen, Z., Wang, P., Wang, C., Peng, Y., Hou, X., Zhou, X., et al. (2018). Updated frequency analysis of spinocerebellar ataxia in China. Brain 141:e22. doi: 10.1093/brain/awy016
Costa, I. P. D., Almeida, B. C., Sequeiros, J., Amorim, A., and Martins, S. (2019). A pipeline to assess disease-associated haplotypes in repeat expansion disorders: the example of MJD/SCA3 Locus. Front. Genet. 10:38. doi: 10.3389/fgene.2019.00038
Costa Mdo, C., and Paulson, H. L. (2012). Toward understanding machado-joseph disease. Prog. Neurobiol. 97, 239–257. doi: 10.1016/j.pneurobio.2011.11.006
de Castilhos, R. M., Furtado, G. V., Gheno, T. C., Schaeffer, P., Russo, A., Barsottini, O., et al. (2014). Spinocerebellar ataxias in Brazil–frequencies and modulating effects of related genes. Cerebellum 13, 17–28. doi: 10.1007/s12311-013-0510-y
Devlin, B., and Risch, N. (1995). A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29, 311–322. doi: 10.1006/geno.1995.9003
Ding, D., Li, K., Wang, C., Chen, Z., Long, Z., Peng, Y., et al. (2016). ATXN2 polymorphism modulates age at onset in Machado-Joseph disease. Brain 139:e59. doi: 10.1093/brain/aww176
Gaspar, C., Lopes-Cendes, I., Hayes, S., Goto, J., Arvidsson, K., Dias, A., et al. (2001). Ancestral origins of the Machado-Joseph disease mutation: a worldwide haplotype study. Am. J. Hum. Genet. 68, 523–528. doi: 10.1086/318184
Long, Z., Li, T., Chen, Z., Peng, Y., Wang, C., Hou, X., et al. (2018). Cerebellar lncRNA expression profile analysis of SCA3/MJD Mice. Int. J. Genom. 2018, 1–6. doi: 10.1155/2018/5383517
Marie-Françoise Chesselet, M. D. (2001). Molecular Mechanisms of Neurodegenerative Diseases. New York, NY: Humana Press.
Martins, S., Calafell, F., Gaspar, C., Wong, V. C., Silveira, I., Nicholson, G. A., et al. (2007). Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch. Neurol. 64, 1502–1508. doi: 10.1001/archneur.64.10.1502
Martins, S., Calafell, F., Wong, V. C., Sequeiros, J., and Amorim, A. (2006). A multistep mutation mechanism drives the evolution of the CAG repeat at MJD/SCA3 locus. Eur. J. Hum. Genet. 14, 932–940. doi: 10.1038/sj.ejhg.5201643
Martins, S., Soong, B. W., Wong, V. C., Giunti, P., Stevanin, G., Ranum, L. P., et al. (2012). Mutational origin of machado-joseph disease in the australian aboriginal communities of groote eylandt and yirrkala. Arch. Neurol. 69, 746–751. doi: 10.1001/archneurol.2011.2504
Nakano, K. K., Dawson, D. M., and Spence, A. (1972). Machado disease: a hereditary ataxia in portuguese emigrants to massachusetts. Neurology 22, 49–49. doi: 10.1212/wnl.22.1.49
Paulson, H. L., Shakkottai, V. G., Clark, H. B., and Orr, H. T. (2017). Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat. Rev. Neurosci. 18, 613–626. doi: 10.1038/nrn.2017.92
Rüb, U., Schöls, L., Paulson, H., Auburger, G., Kermer, P., Jen, J. C., et al. (2013). Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 104, 38–66. doi: 10.1016/j.pneurobio.2013.01.001
Schöls, L., Amoiridis, G., Büttner, T., Przuntek, H., Epplen, J. T., and Riess, O. (1997). Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann. Neurol. 42, 924–932. doi: 10.1002/ana.410420615
Sequeiros, J., and Coutinho, P. (1993). Epidemiology and clinical aspects of Machado-Joseph disease. Adv. Neurol. 61, 139–153.
Takano, H., Cancel, G., Ikeuchi, T., Lorenzetti, D., Mawad, R., and Stevanin, G. (1998). Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and Caucasian populations. Am. J. Hum. Genet. 63, 1060–1066. doi: 10.1086/302067
Vale, J., Bugalho, P., Silveira, I., Sequeiros, J., Guimaraes, J., and Coutinho, P. (2010). Autosomal dominant cerebellar ataxia: frequency analysis and clinical characterization of 45 families from portugal. Eur. J. Neurol. 17, 124–128. doi: 10.1111/j.1468-1331.2009.02757.x
Wang, C., Chen, Z., Yang, F., Jiao, B., Peng, H., Shi, Y., et al. (2015). Analysis of the GGGGCC repeat expansions of the C9orf72 Gene in SCA3/MJD Patients from China. PLoS One 10:e0130336. doi: 10.1371/journal.pone.0130336
Keywords: spinocerebellar ataxia type 3, SCA3, Machado-Joseph disease, founder effect, haplotype, mutational origins
Citation: Li T, Martins S, Peng Y, Wang P, Hou X, Chen Z, Wang C, Tang Z, Qiu R, Chen C, Hu Z, Xia K, Tang B, Sequeiros J and Jiang H (2019) Is the High Frequency of Machado-Joseph Disease in China Due to New Mutational Origins? Front. Genet. 9:740. doi: 10.3389/fgene.2018.00740
Received: 24 June 2018; Accepted: 22 December 2018;
Published: 20 February 2019.
Edited by:
James J. Cai, Texas A&M University, United StatesReviewed by:
Tina T. Hu, Princeton University, United StatesSajid Ali, University of Agriculture, Peshawar, Pakistan
Copyright © 2019 Li, Martins, Peng, Wang, Hou, Chen, Wang, Tang, Qiu, Chen, Hu, Xia, Tang, Sequeiros and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Jiang, amlhbmdob25nNzM4NjhAMTI2LmNvbQ==