 Areerat Hnoonual
Areerat Hnoonual Potchanapond Graidist
Potchanapond Graidist Supika Kritsaneepaiboon4
Supika Kritsaneepaiboon4 Pornprot Limprasert
Pornprot Limprasert- 1Division of Human Genetics, Department of Pathology, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
- 2Department of Biomedical Sciences, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
- 3The Excellent Research Laboratory of Cancer Molecular Biology, Prince of Songkla University, Songkhla, Thailand
- 4Department of Radiology, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
- 5Faculty of Medicine, King Mongkut’s Institute of Technology Ladkrabang, Bangkok, Thailand
Autism spectrum disorder (ASD) is a highly heterogeneous neurodevelopmental disorder with many contributing risk genes and loci. To date, several intellectual disability (ID) susceptibility genes have frequently been identified in ASD. Here, whole exome sequencing was carried out on a proband with ASD and identified compound heterozygous mutations of the TRAPPC9, which plays a role in the neuronal NF-κB signaling pathway. These mutations consisted of a novel frameshift mutation (c.2415_2416insC, p.His806Profs∗9) and a rare splice site mutation (c.3349+1G>A) that were segregated from an unaffected father and unaffected mother, respectively. These two heterozygous mutations were also identified in the patient’s older brother with ID. Quantitative RT-PCR revealed a significant reduction of TRAPPC9 transcript in two siblings. This study first describes compound heterozygous mutations of the TRAPPC9 gene in two siblings with ASD and ID, which is notable as only homozygous mutations or compound heterozygous for copy number variations and rare variant in this gene have been reported to date and associated with autosomal recessive intellectual disability. The two siblings carrying compound heterozygous TRAPPC9 mutations presented with ID, developmental delay, microcephaly and brain abnormalities similarly to the clinical features found in almost cases with homozygous TRAPPC9 mutation in previous studies. Together this study provides evidence that clinical manifestations of TRAPPC9 mutations as seen in our patients with ID and autism may be broader than previous case reports have indicated.
Introduction
Autism spectrum disorder (ASD) is a highly heterogeneous neurodevelopmental disorder characterized by deficits in social communication and social interaction, and repetitive behavior and interests. However, the major genetic causes of ASD are still largely unknown. Approximately 70% of individuals with ASD are comorbid with intellectually disability (ID) (Yeargin-Allsopp et al., 2003; Chakrabarti and Fombonne, 2005; Christensen et al., 2016). Several studies have provided evidence that genes identified for ID, especially genes involved in neuronal pathways and brain development and functions, have frequently been implicated in ASD, supporting a shared similar genetic etiology between these disorders.
To date many genes have been associated with non-syndromic ID, including TRAPPC9 (MIM 611966) which plays a role in the neuronal NF-κB signaling pathway. Homozygous mutations, homozygous deletion, and compound heterozygous for copy number variations (CNVs)/rare variants in the TRAPPC9 gene have been implicated with significant contributions to autosomal recessive mental retardation 13 (MRT13, MIM 613192). Most previous reported cases with TRAPPC9 mutations presented with ID/developmental delay, microcephaly and brain abnormalities (Mir et al., 2009; Mochida et al., 2009; Philippe et al., 2009; Abou Jamra et al., 2011; Kakar et al., 2012; Marangi et al., 2013; Giorgio et al., 2016; Abbasi et al., 2017; Mortreux et al., 2018). However, to date, heterozygous mutation alone or compound heterozygous mutations in the TRAPPC9 gene have not been identified in patient with MRT13 or other associated diseases.
Recently, whole exome sequencing (WES) studies have led to the discovery of a rapidly increasing number of novel candidate genes and causative gene mutations associated with ASD and ID. Herein, we first report compound heterozygous mutations (c.2415_2416insC and c.3349+1G>A) of the TRAPPC9 gene in two Thai siblings with ASD and ID born to healthy and non-consanguineous parents through WES. Additionally, our findings and review of literature inform a broader understanding of the clinical features of patients with TRAPPC9 mutations.
Clinical Report
The two affected individuals were diagnosed according to the new structured interview for Thai children with ASD for research based on the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria (Hansakunachai et al., 2014). The affected proband (II:2), a 5-year-8-month female, was diagnosed with ASD from a previous study (Charalsawadi et al., 2014). She was the second child born to non-consanguineous healthy Thai parents. Her non-verbal IQ was 42 (moderate impairment) as evaluated by the Stanford-Binet Intelligence Scale : fifth edition, SB:V. Behavior and social evaluation using the Vineland Adaptive Behavior Scales (Interview edition survey form) showed that she had severe deficit in adaptive behaviors in all domains including communication, daily living skills and socialization. She started walking at the age of 24 months and to the time of our examination (5 years 8 months) had never developed speech. She presented with unilateral cleft lip at birth. The birth weight was 3,140 g (50th centile) and head circumference at birth was 34 cm (50th centile). After 3 years of age, she was found to have microcephaly (head circumference < 3rd centile). On examination, her height was 111 cm (50th centile) and weight was 24 kg (90th centile). Neither parent had a history of seizure and there was no family history of developmental delay. A brain MRI from 10 months of age is shown in Figures 1A–D. Standard karyotyping, CGG repeats of the FMR1 and MECP2 testings were all normal.
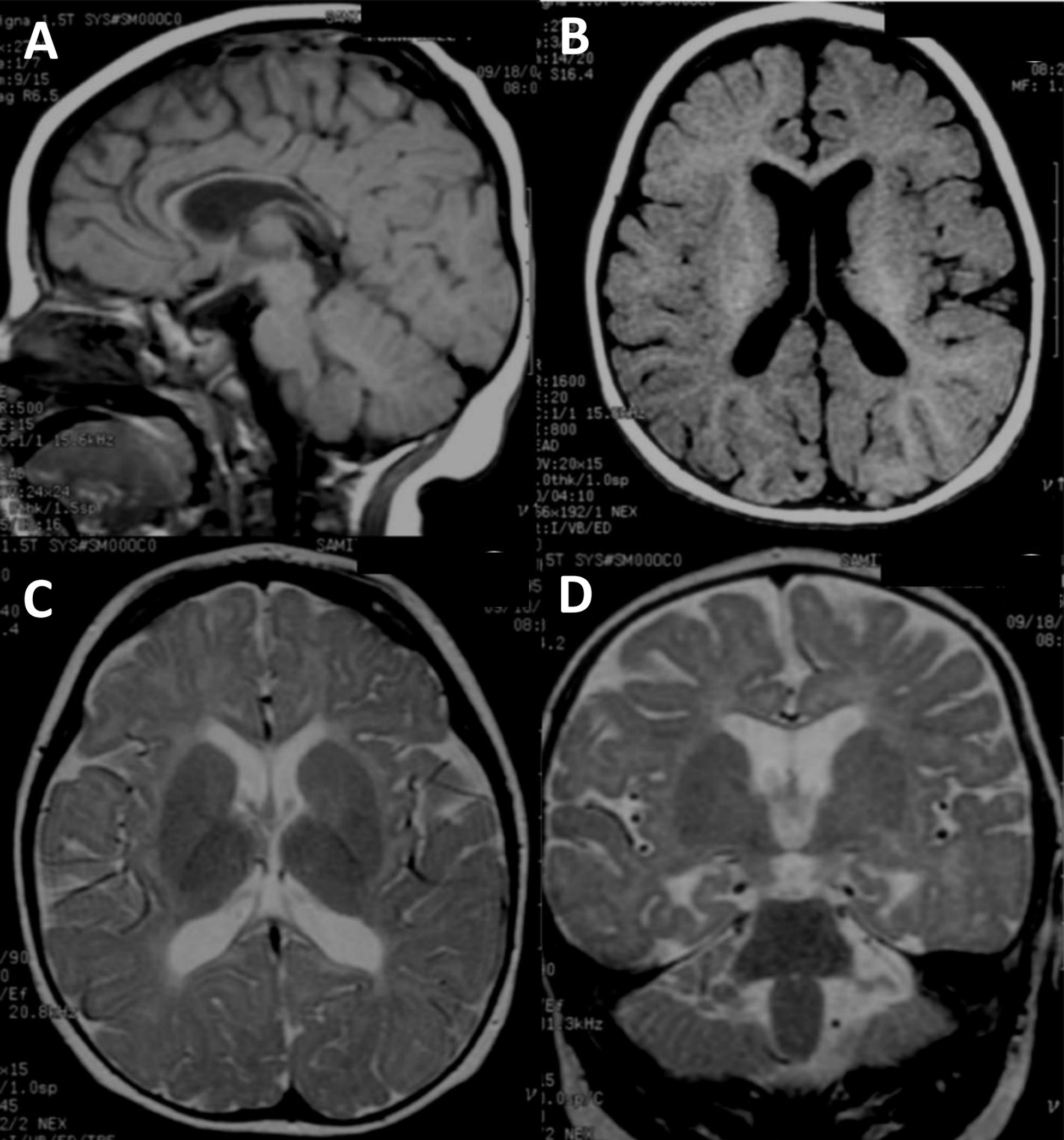
Figure 1. Brain MRI image of the ASD proband. (A–D) Brain MRI of the ASD proband at 10 months of age (II-2). (A) T1-weighted sagittal image shows fully formed but diffusely thin corpus callosum. The cerebellar vermis is normal. (B) T1-weighted axial image reveals normal gyral pattern of cerebral cortex and diminished cerebral white matter volume. (C) T2-weighted axial image demonstrates delayed myelination at anterior limbs of both internal capsules and diffuse white matter abnormalities seen as T2W hyperintensity at periventricular and subcortical white matter. (D) T2-weighted coronal image shows more obvious mild cerebral atrophy seen as enlarged CSF space and prominent temporal horns of both lateral ventricles. Diffuse white matter abnormalities at periventricular and subcortical white matter are noted.
The patient’s older brother (II:1), 11-year-5-month, was moderate ID (non-verbal IQ = 42, SB:V). Results from the Vineland Adaptive Behavior Scale of this patient also showed severe deficits in all adaptive behaviors domains. He started walking at 24 months and was able to speak one word at 36 months. His birth weight was 3,280 g (50th centile). At 11 years and 5 months of age, his height was 150 cm (75th centile) and his weight was 66 kg (>97th centile). He has also no history of seizure. His face presented repaired cleft lip and cupped ears. Birth head circumference was not available, but at 1 month was 35 cm (3rd centile). A brain MRI at 9 months of age showed a thin corpus callosum, delayed myelination and reduction in cerebral white matter. The gyral pattern of the cerebral cortex was also normal (figure not shown). Standard karyotyping and CGG repeats of the FMR1 studies were all normal. The pedigree for this family is shown in Figure 2.
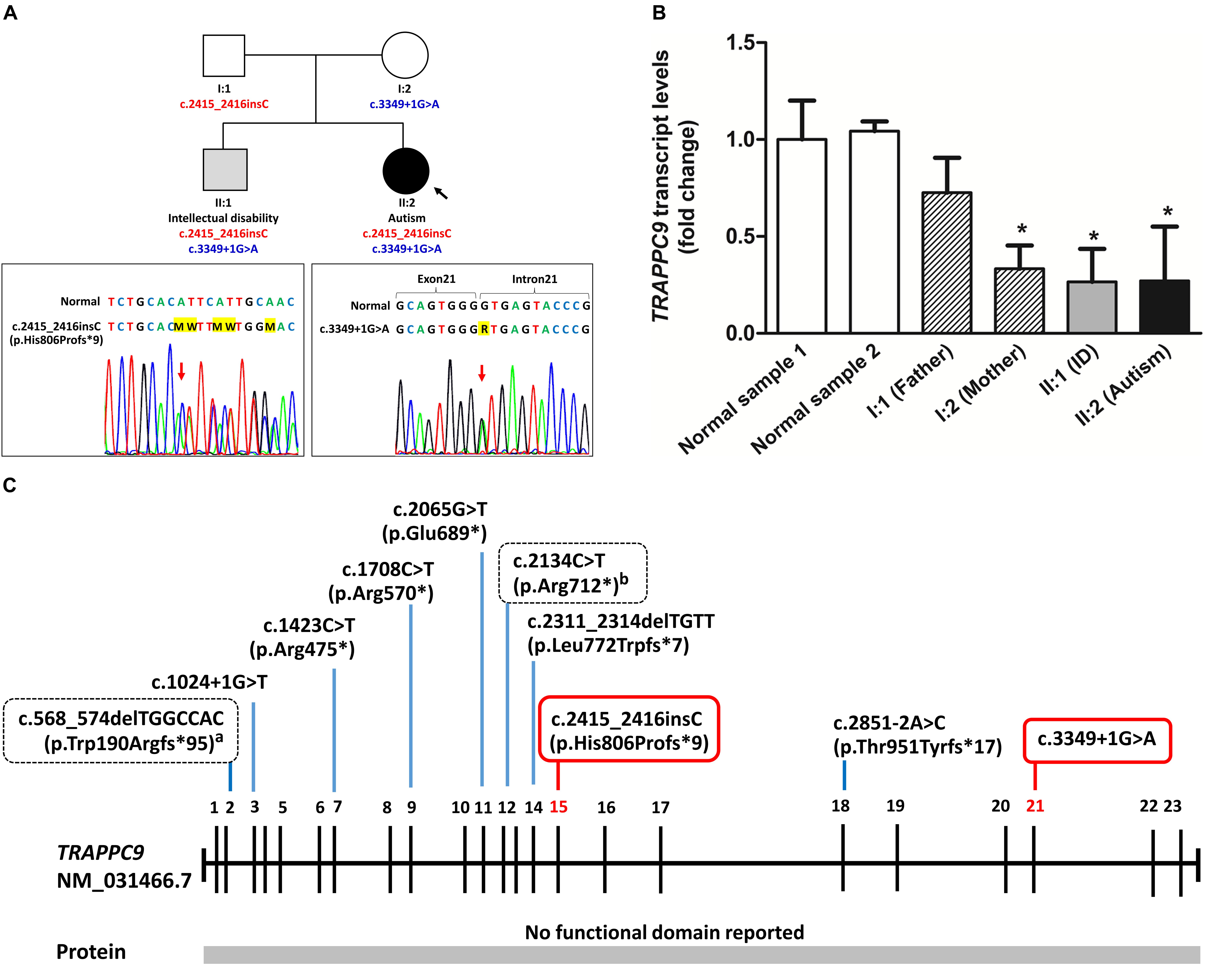
Figure 2. Family pedigree and molecular analysis of TRAPPC9 mutations. (A) Family pedigree and sequencing of splice site mutation (c.3349+1G>A) and insertion mutation (c.2415_2416insC, p.His806Profs∗9) of the TRAPPC9 gene. Unaffected individuals are shown in white. (B) Quantitative RT-PCR of TRAPPC9 mRNA expression representing fold change value calculated using the ΔΔCT method for a gene expression in family members with TRAPPC9 mutations compared to a normal control sample 1. Values are presented as the mean ± standard deviation (n = 3 independent experiments). ∗P < 0.05 as compared to the normal sample. (C) Summary of reported TRAPPC9 mutations. All mutations have previously been reported in homozygous changes except for the two heterozygous changes which are presented in box with dashed lines. Heterozygous mutations described in this study are presented in box with solid line. aPatient carries compound heterozygous for 119 kb duplication of 8q24.3 and a deletion variant (c.568_574delTGGCCAC, p.Trp190Argfs∗95); bPatient carries compound heterozygous for 189 kb deletion of 8q24.3 and a nonsense variant (c.2134C>T, p.Arg712∗).
We obtained written informed consent for genomic analysis of the patient and his family members and the parents provided written informed consent for the publication of the patient’s identifiable information. The study was approved by the Institutional Ethics Committee of the Faculty of Medicine, Prince of Songkla University (REC 48/364-006-3).
Materials and Methods
Single Nucleotide Polymorphism (SNP) Array
Single nucleotide polymorphism (SNP) array analysis was performed to identify clinically significant copy number variations (CNVs) in the proband with ASD (II:2) using the HumanCytoSNP-12 DNA Analysis BeadChip v2.1 kit (Illumina, San Diego, CA, United States), according to the manufacturer’s instructions. The data were then analyzed using BlueFuse Multi software v4.3 and GenomeStudio Data Analysis Software v. 2011.1 based on the reference human genome (hg19/GRCh37).
Whole Exome Sequencing (WES)
Whole exome sequencing was performed on the proband with ASD (II:2) using the SureSelect Human All Exon V4 Kit (Agilent Technologies, Santa Clara, CA, United States) and sequenced on an HiSeq2000 (Illumina) with 101-bp paired-end reads. Exome sequencing data processing, base calling and primary data analysis were performed using the Illumina Real-Time Analysis (RTA) version 1.12.4 and Illumina’s CASAVA pipeline 1.8.2 with default parameters. The paired-end reads were aligned to the reference human genome (hg19/GRCh37) using the Burrows-Wheeler Aligner (BWA). Variant calling was performed using SAMtools (Li et al., 2009) and sequence variants were annotated using ANNOVAR (Wang et al., 2010). The remaining variants were then filtered against our candidate ASD gene set (762 genes) from AutDB, SFARI, and TruSight Autism genes. Subsequently, to search more widely for the cause of disease in the patient, the variants were also filtered against a candidate ID gene set (1,912 genes) from literature reviews. Sanger sequencing was then performed to confirm sequence variants and to assess segregation in the family. The details and simplified diagram of WES analysis and variants filtering are shown in Supplementary Figure S1.
Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
To investigate the impact of the mutations on the transcript level of TRAPPC9, we performed a quantitative RT-PCR (qRT-PCR) on samples of proband and all available family members using SYBR Green Supermix (Bio-Rad) on a CFX96 Real-Time PCR detection system (Bio-Rad). Total RNA was extracted from fresh EDTA blood using TRIzol LS reagent (Invitrogen, Carlsbad, CA, United States) and cDNA was synthesized using the SuperScriptTM IV First-Strand cDNA synthesis system (Invitrogen) according to the manufacturer’s protocol. The primers used for the qRT-PCR have been described in a previous study (Mir et al., 2009; Supplementary Table S1). The relative quantification was calculated using a comparative CT method (ΔΔCT) and HPRT1 (Hypoxanthine phosphoribosyltransferase 1, MIM 308000) was used as the internal control housekeeping gene. The mRNA expression for the normal control sample 1 is set a value of 1.0, and mRNA levels of all family members are determined relative to this number.
Results
Single nucleotide polymorphism (SNP) array analysis was first performed on the proband with ASD (II:2), but no pathogenic copy number variations (CNVs) were detected. WES was then performed, which led to the identification of two heterozygous mutations of the TRAPPC9 gene. One mutation was an insertion (NM_031466.7: c.2415_2416insC) of exon 15, leading to a frameshift and premature stop codon (p.His806Profs∗9). This frameshift insertion mutation was segregated from the unaffected father. In addition, this frameshift mutation was absent from the 195 unrelated Thais from our in-house exome sequencing database as well as not being found in the public databases (the 1000 Genomes Project, the NHLBI-ESP 6500 exome, the ExAC database, dbSNP 137). The second mutation was a single donor splice site mutation (NM_031466.7: c.3349+1G>A,) after exon 21, which was segregated from the unaffected mother. This splice site mutation has been reported in one case of East Asian population in the 1000 Genomes Project (minor allele frequency = 0.0002) (rs539016732). Furthermore, these two heterozygous mutations in the TRAPPC9 gene were also identified using Sanger sequencing in the proband’s older brother with moderated ID (II:1, Figure 2A).
Quantitative RT-PCR results showed that the mRNA expression of TRAPPC9 was slightly decreased in the father (I:1) with the frameshift mutation (Fold Change = 0.725, P = 0.373), but the mRNA levels of TRAPPC9 were significantly decreased in the mother (I:2) with a rare splice site mutation (Fold Change = 0.333, P = 0.017), the older brother (II:1) (Fold Change = 0.266, P = 0.014) and the proband (II:2) (Fold Change = 0.271, P = 0.016) with compound heterozygous mutations compared to a normal control sample (normal sample 1) (Figure 2B). These results indicate that a splice site TRAPPC9 mutation clearly can affect transcription of the TRAPPC9 gene. RT-PCR followed by cDNA sequencing was then performed to investigate the consequences of the c.3349+1G>A splice site mutation on TRAPPC9 mRNA splicing. However, we did not observe aberrant form of the TRAPPC9 transcript in the proband, her brother and mother carrying a heterozygous splice site TRAPPC9 mutation. These results suggest that aberrant mRNA transcripts from a splice site TRAPPC9 mutation may sometimes be eliminated by nonsense-mediated mRNA decay (NMD) as discussed in previous studies (Mir et al., 2009; Philippe et al., 2009). However, further studies of the functional consequences of these two TRAPPC9 mutations are needed to elucidate more definitive conclusions.
Discussion
Trafficking Protein Particle Complex 9 (TRAPPC9, MIM 611966), also known as NIBP (NIK- and IKK-beta binding protein), contains 23 exons and encodes the NIBP protein which is mainly expressed in muscle, kidney, heart, placenta, and brain tissues, including postmitotic neurons of the cerebral cortex, hippocampus, and deep gray matter (Hu et al., 2005; Mochida et al., 2009). However, the function of the NIBP protein domains has not yet been determined. NIBP interacts directly with NIK and IKK-beta is involved in both the classical and alternative NF-κB signaling pathways (Hu et al., 2005) which may in turn be involved in several neuronal processes, including neuronal cells differentiation, synaptic plasticity and neurogenesis (Denis-Donini et al., 2008). Moreover, NF-κB signaling is also required for myelination in the central nervous system (Philippe et al., 2013; Blank and Prinz, 2014). A previous study found that patients with impaired NF-κB signaling presented with delayed myelination of the white matter (Philippe et al., 2013). These data support the possibility that TRAPPC9 mutations may lead to clinical manifestations in patients with ASD and ID through impairing neuronal NF-κB signaling.
To our knowledge, six homozygous mutations in the TRAPPC9 gene have been reported in patients with MRT13 in consanguineous families (Mir et al., 2009; Mochida et al., 2009; Philippe et al., 2009; Abou Jamra et al., 2011; Kakar et al., 2012; Marangi et al., 2013; Giorgio et al., 2016; Abbasi et al., 2017; Mortreux et al., 2018; Figure 2C and Supplementary Table S2). A nonsense TRAPPC9 mutation (c.1423C>T, p.Arg475∗) has been reported in families from four different ethnicities (Israeli Arab, Syrian, Pakistani and Egyptian) (Mir et al., 2009; Mochida et al., 2009; Abou Jamra et al., 2011; Giorgio et al., 2016; Abbasi et al., 2017). The other mutations reported to be found in the TRAPPC9 gene were nonsense mutation (c.1708C>T, p.Arg570∗) in a Tunisian family, nonsense mutation (c.2065G>T, p.Glu689∗) in a Pakistani family and a 4-bp frameshift deletion (c.2311_2314delTGTT, p.Leu772Trpfs∗7) in an Iranian family. In addition, splice site mutations (c.1024+1G>T and c.2851-2A>C) resulting in exon skipping with frameshift and premature truncation have been reported in a Pakistani and an Italian family, respectively. Recently, CNVs (deletion/duplication) on 8q24.3 encompassing the TRAPPC9 gene have been reported in patients with ID (Koifman et al., 2010; Mortreux et al., 2018) in two families. Compound heterozygous CNVs/rare variants in the TRAPPC9 gene including compound heterozygous for 119 kb duplication and a deletion variant (c.568_574delTGGCCAC, p.Trp190Argfs∗95), and compound heterozygous for 189 kb deletion and a nonsense variant (c.2134C>T, p.Arg712∗) have also been reported in patients with ID of non-consanguineous parents (Mortreux et al., 2018).
Reported clinical phenotypes associated with TRAPPC9 mutations include dysmorphic facial features, moderate to severe ID, developmental delay, microcephaly, obesity and brain MRI abnormalities, which match the clinical features of the patients in this study carrying compound heterozygous mutations of TRAPPC9 (Table 1). Of note, the clinical features including ID, developmental delay, microcephaly and brain abnormalities are observed in almost reported cases with TRAPPC9 mutations. Interestingly, all known patients with TRAPPC9 mutations from previous studies, including our patients, presented with brain abnormalities (thin corpus callosum, cerebral and cerebellar hypoplasia), supporting the important role of TRAPPC9 in brain development and functions. Patients in this study also show autistic feature and cleft lip which have been reported in very rare cases with TRAPPC9 mutations, suggesting a broader spectrum of clinical manifestations of TRAPPC9 mutations as seen in our patients, compared to previous case reports.
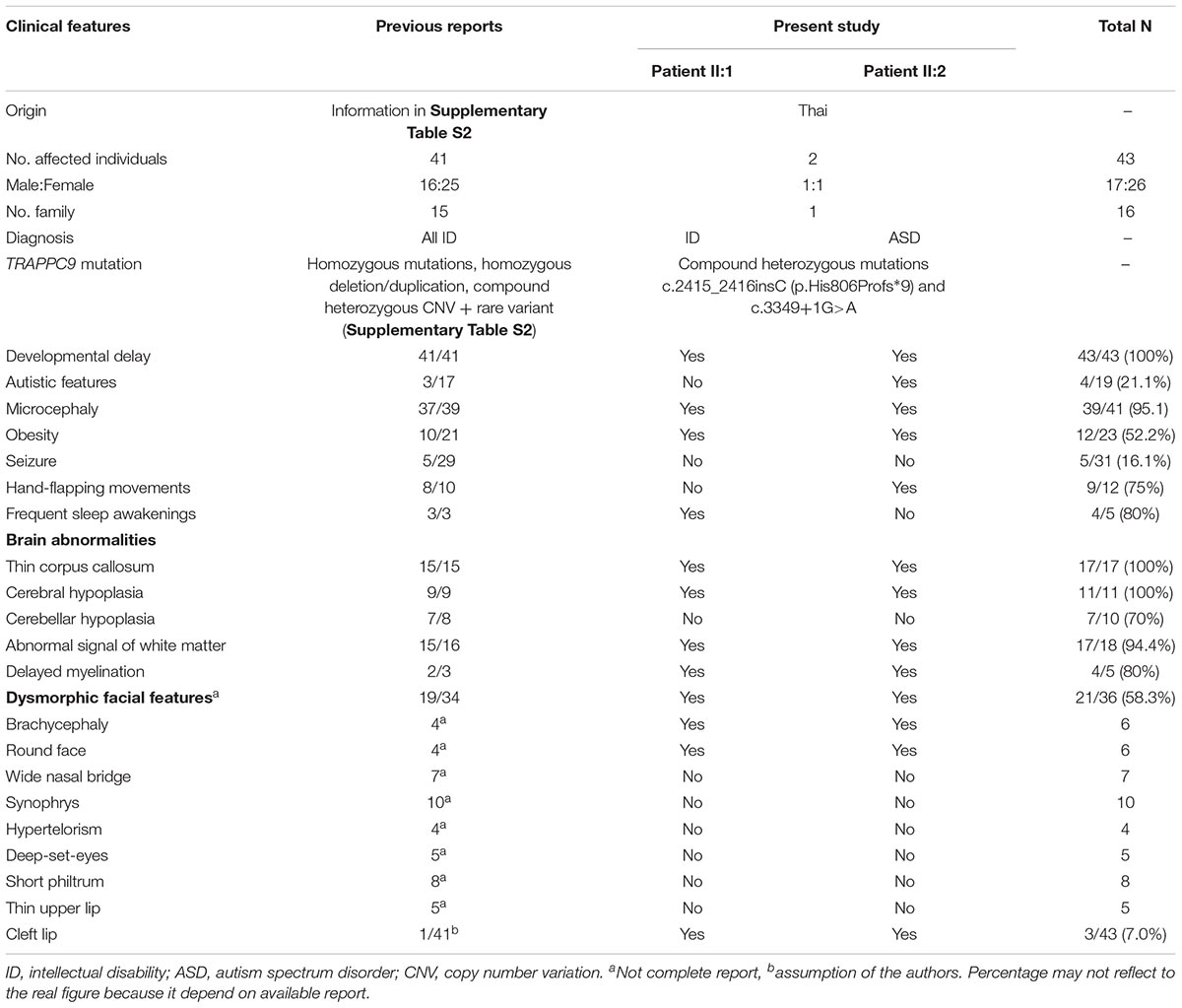
Table 1. Comparison of available clinical features between previous case reports with TRAPPC9 mutations (41 patients/15 families) and patients in this study.
Concluding Remarks
This study is the first to report the identification of novel compound heterozygous mutations of the TRAPPC9 gene in two Thai siblings with ASD and ID. The combination of these two TRAPPC9 mutations are most likely the cause of clinical features in the patients, especially ID, developmental delay, microcephaly and brain abnormalities. We identify the first case diagnosed with ASD carrying TRAPPC9 mutations. Thus, our findings suggest that TRAPPC9 mutations may constitute one of the genetic risk factors for ASD in the patient.
Author Contributions
PL and AH designed the study and wrote the manuscript. PL collected the patients’ clinical information. SK interpreted the brain MRIs of the patients. AH performed the analysis of WES data. AH and PG performed the molecular analysis and interpreted the results. All authors reviewed and approved the final manuscript.
Funding
This research was supported by grants from the Faculty of Medicine, Prince of Songkla University (48/364-006-3).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are thankful to the family for participation in this study, and to Dr. Tasanawat Sombuntham for providing the initial clinical information, Prof. Dr. Wasun Chantratita for providing the in-house Thai WES database, and Dr. Thanya Sripo and Ms. Oradawan Plong-On for technical support in qRT-PCR analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00061/full#supplementary-material
References
Abbasi, A. A., Blaesius, K., Hu, H., Latif, Z., Picker-Minh, S., Khan, M. N., et al. (2017). Identification of a novel homozygous TRAPPC9 gene mutation causing non-syndromic intellectual disability, speech disorder, and secondary microcephaly. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 839–845. doi: 10.1002/ajmg.b.32602
Abou Jamra, R., Wohlfart, S., Zweier, M., Uebe, S., Priebe, L., Ekici, A., et al. (2011). Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet. 19, 1161–1166. doi: 10.1038/ejhg.2011.98
Blank, T., and Prinz, M. (2014). NF-kappaB signaling regulates myelination in the CNS. Front. Mol. Neurosci. 7:47. doi: 10.3389/fnmol.2014.00047
Chakrabarti, S., and Fombonne, E. (2005). Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am. J. Psychiatry 162, 1133–1141. doi: 10.1176/appi.ajp.162.6.1133
Charalsawadi, C., Maisrikhaw, W., Praphanphoj, V., Wirojanan, J., Hansakunachai, T., Roongpraiwan, R., et al. (2014). A case with a ring chromosome 13 in a cohort of 203 children with non-syndromic autism and review of the cytogenetic literature. Cytogenet. Genome Res. 144, 1–8. doi: 10.1159/000365909
Christensen, D. L., Baio, J., Van Naarden Braun, K., Bilder, D., Charles, J., Constantino, J. N., et al. (2016). Prevalence and characteristics of autism spectrum disorder among children aged 8 years–autism and developmental disabilities monitoring network, 11 sites. United States, 2012. MMWR Surveill. Summ. 65, 1–23. doi: 10.15585/mmwr.ss6503a1
Denis-Donini, S., Dellarole, A., Crociara, P., Francese, M. T., Bortolotto, V., Quadrato, G., et al. (2008). Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J. Neurosci. 28, 3911–3919. doi: 10.1523/jneurosci.0148-08.2008
Giorgio, E., Ciolfi, A., Biamino, E., Caputo, V., Di Gregorio, E., Belligni, E. F., et al. (2016). Whole exome sequencing is necessary to clarify ID/DD cases with de novo copy number variants of uncertain significance: two proof-of-concept examples. Am. J. Med. Genet. A 170, 1772–1779. doi: 10.1002/ajmg.a.37649
Hansakunachai, T., Roongpraiwan, R., Sombuntham, T., Limprasert, P., and Ruangdaraganon, N. (2014). A new structured interview for children with autism spectrum disorder based on the DSM-IV. J. Med. Assoc. Thai. 97(Suppl. 8), S7–S14.
Hu, W. H., Pendergast, J. S., Mo, X. M., Brambilla, R., Bracchi-Ricard, V., Li, F., et al. (2005). NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J. Biol. Chem. 280, 29233–29241. doi: 10.1074/jbc.M501670200
Kakar, N., Goebel, I., Daud, S., Nurnberg, G., Agha, N., Ahmad, A., et al. (2012). A homozygous splice site mutation in TRAPPC9 causes intellectual disability and microcephaly. Eur. J. Med. Genet. 55, 727–731. doi: 10.1016/j.ejmg.2012.08.010
Koifman, A., Feigenbaum, A., Bi, W., Shaffer, L. G., Rosenfeld, J., Blaser, S., et al. (2010). A homozygous deletion of 8q24.3 including the NIBP gene associated with severe developmental delay, dysgenesis of the corpus callosum, and dysmorphic facial features. Am. J. Med. Genet. A 152a, 1268–1272. doi: 10.1002/ajmg.a.33319
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Marangi, G., Leuzzi, V., Manti, F., Lattante, S., Orteschi, D., Pecile, V., et al. (2013). TRAPPC9-related autosomal recessive intellectual disability: report of a new mutation and clinical phenotype. Eur. J. Hum. Genet. 21, 229–232. doi: 10.1038/ejhg.2012.79
Mir, A., Kaufman, L., Noor, A., Motazacker, M. M., Jamil, T., Azam, M., et al. (2009). Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am. J. Hum. Genet. 85, 909–915. doi: 10.1016/j.ajhg.2009.11.009
Mochida, G. H., Mahajnah, M., Hill, A. D., Basel-Vanagaite, L., Gleason, D., Hill, R. S., et al. (2009). A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am. J. Hum. Genet. 85, 897–902. doi: 10.1016/j.ajhg.2009.10.027
Mortreux, J., Busa, T., Germain, D. P., Nadeau, G., Puechberty, J., Coubes, C., et al. (2018). The role of CNVs in the etiology of rare autosomal recessive disorders: the example of TRAPPC9-associated intellectual disability. Eur. J. Hum. Genet. 26, 143–148. doi: 10.1038/s41431-017-0018-x
Philippe, O., Rio, M., Carioux, A., Plaza, J. M., Guigue, P., Molinari, F., et al. (2009). Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am. J. Hum. Genet. 85, 903–908. doi: 10.1016/j.ajhg.2009.11.007
Philippe, O., Rio, M., Malan, V., Van Esch, H., Baujat, G., Bahi-Buisson, N., et al. (2013). NF-kappaB signalling requirement for brain myelin formation is shown by genotype/MRI phenotype correlations in patients with Xq28 duplications. Eur. J. Hum. Genet. 21, 195–199. doi: 10.1038/ejhg.2012.140
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Keywords: autism, ASD, intellectual disability, whole exome sequencing, TRAPPC9
Citation: Hnoonual A, Graidist P, Kritsaneepaiboon S and Limprasert P (2019) Novel Compound Heterozygous Mutations in the TRAPPC9 Gene in Two Siblings With Autism and Intellectual Disability. Front. Genet. 10:61. doi: 10.3389/fgene.2019.00061
Received: 12 July 2018; Accepted: 24 January 2019;
Published: 11 February 2019.
Edited by:
Swarkar Sharma, Shri Mata Vaishno Devi University, IndiaReviewed by:
Muhammad Jawad Hassan, National University of Medical Sciences (NUMS), PakistanIbtessam Ramzi Hussein, King Abdulaziz University, Saudi Arabia
Anjana Munshi, Central University of Punjab, India
Copyright © 2019 Hnoonual, Graidist, Kritsaneepaiboon and Limprasert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pornprot Limprasert, cG9ybnByb3QubGlAa21pdGwuYWMudGg=