 Melissa Bizzarri1
Melissa Bizzarri1 Stefano Cassanelli1
Stefano Cassanelli1 Laura Bartolini1
Laura Bartolini1 Leszek P. Pryszcz2
Leszek P. Pryszcz2 Michala Dušková3
Michala Dušková3 Hana Sychrová3
Hana Sychrová3 Lisa Solieri1*
Lisa Solieri1*- 1Department of Life Sciences, University of Modena and Reggio Emilia, Reggio Emilia, Italy
- 2Laboratory of Zebrafish Developmental Genomics, International Institute of Molecular and Cell Biology, Warsaw, Poland
- 3Department of Membrane Transport, Institute of Physiology, Czech Academy of Sciences, Prague, Czechia
The pre-whole genome duplication (WGD) Zygosaccharomyces clade comprises several allodiploid strain/species with industrially interesting traits. The salt-tolerant yeast ATCC42981 is a sterile and allodiploid strain which contains two subgenomes, one of them resembling the haploid parental species Z. rouxii. Recently, different mating-type-like (MTL) loci repertoires were reported for ATCC42981 and the Japanese strain JCM22060, which are considered two stocks of the same strain. MTL reconstruction by direct sequencing approach is challenging due to gene redundancy, structure complexities, and allodiploid nature of ATCC42981. Here, DBG2OLC and MaSuRCA hybrid de novo assemblies of ONT and Illumina reads were combined with in vitro long PCR to definitively solve these incongruences. ATCC42981 exhibits several chimeric MTL loci resulting from reciprocal translocation between parental haplotypes and retains two MATa/MATα expression loci, in contrast to MATα in JCM22060. Consistently to these reconstructions, JCM22060, but not ATCC42981, undergoes mating and meiosis. To ascertain whether the damage of one allele at the MAT locus regains the complete sexual cycle in ATCC42981, we removed the MATα expressed locus by gene deletion. The resulting MATa/- hemizygous mutants did not show any evidence of sporulation, as well as of self- and out-crossing fertility, probably because incomplete silencing at the chimeric HMLα cassette masks the loss of heterozygosity at the MAT locus. We also found that MATα deletion switched off a2 transcription, an activator of a-specific genes in pre-WGD species. These findings suggest that regulatory scheme of cell identity needs to be further investigated in Z. rouxii protoploid yeast.
Introduction
Polyploidization, a state resulting from doubling of a genome within a species (autopolyploidy) or the merging between different species (allopolyploidy) (Campbell et al., 2016), is an important evolutionary force which shapes eukaryotic genomes (Albertin and Marullo, 2012), triggers speciation, and can result in phenotypic changes driving adaptation (Ohno, 1970). A whole-genome duplication (WGD) event occurred approximately 100–200 Mya in the common ancestor of six yeast genera in the family Saccharomycetaceae, including Saccharomyces cerevisiae (as reviewed by Wolfe et al., 2015). WGD was recently proposed to be a direct consequence of an ancient hybridization between two ancestral species (Marcet-Houben and Gabaldón, 2015), followed by genome doubling of initially sterile hybrid to regain fertility, i.e., the ability to undergo meiosis and produce viable spore (Wolfe, 2015).
Different mechanisms can contribute to hybrid infertility, such as chromosomal missegregation caused by meiosis I non-disjunction (Boynton et al., 2018), chromosomal rearrangements (Liti et al., 2006; Rajeh et al., 2018), and Dobzhansky–Muller gene incompatibilities either between nuclear genes (Bizzarri et al., 2016) or between mitochondrial and nuclear genes (Lee et al., 2008). Specialized loci, called the mating-type (MAT)-like (MTL) cassettes, regulate mating between haploid cells with opposite MATa and MATα idiomorphs, as well as meiosis in diploid a/α cells. In diplontic yeast S. cerevisiae MAT locus on chromosome III contains either the a1 or the α1 and α2 genes in Ya and Yα segments, respectively, surrounded by X and Z regions at the left and right sides. In haploid α cells, α1 activates the α-specific genes (αsgs), while α2 represses a cohort of a-specific genes (asgs), which a cells transcribe by default (Haber, 2012). Finally, diploid a/α cells are meiosis but not mating-competent, because the a1-α2 heterodimer positively regulates IME1 (Inducer of Meiosis) gene expression and represses the transcription of RME1, a haploid-specific gene (hsg) that inhibits entry into meiosis, and of other hsgs required for mating responses. S. cerevisiae cells also have extra copies of MAT genes at the HMRa and HMLα loci located close to telomeres of chromosome III and silenced by a combination of the Sir1–4 proteins (Hickman et al., 2011). These extra copies serve as donors during the mating-type switching which enables MATa cells to convert into MATα cells, or vice versa, and to mate each other. This autodiploidization event is triggered by a site-specific endonuclease called HO which induces double-strand break at Z region of the MAT locus. In Saccharomyces interspecies hybrids, experimental deletion of one MAT locus or elimination of the entire chromosome carrying one MAT locus yielded fertile allotetraploids (Greig et al., 2002; Pfliegler et al., 2012; Karanyicz et al., 2017). More recently, the MAT locus damage was proposed to be the most plausible evolutionary route which enables natural interspecies hybrids of the Zygosaccharomyces bailii complex to rescue mating and meiosis (Ortiz-Merino et al., 2017; Braun-Galleani et al., 2018).
In the Saccharomycetaceae lineage, Z. rouxii stands on the crossroad where different and relevant evolutionary events take their way (Dujon and Louis, 2017). This evolutionary route involves ancient allopolyploidization between two parental lineages, one of which was close to Z. rouxii and Torulaspora delbrueckii (ZT) clade (Marcet-Houben and Gabaldón, 2015). Z. rouxii represents the early branching species before WGD that recruits HO from a LAGLIDADG intein to catalyze the first step of mating-type switching (Fabre et al., 2005). Furthermore, Z. rouxii exhibits the triplication of MTL loci, which is a genomic landmark of the Saccharomycetaceae family, but, in contrast to S. cerevisiae, it lacks of MAT-HMR linkage. Whereas the route of αsg regulation appears to be conserved, the regulatory circuit of asgs has been extensively rewired across the Saccharomycotina clade. Instead of the negative regulatory circuit widespread in post-WGD species, several pre-WGD species activate asgs by an HMG-domain protein (a2) that is encoded by MATa (Tsong et al., 2003). Conventionally, Z. rouxii displays haplontic life style, where heterothallic haploid cells with opposite mating-type mate each other or, alternatively, homothallic haploid cells switch mating-type and subsequently undergo mating between mother and daughter cells. In both cases, the transient diploid zygote should sporulate to restore the haploid state. Alternatively, stable allodiploid strains arose from mating between divergent haploid parents. One parental haplotype (called T-subgenome) resembles Z. rouxii and was 15% different from the other parental haplotype (called P-subgenome) (Gordon and Wolfe, 2008; Bizzarri et al., 2016, 2018; Watanabe et al., 2017).
Both haploid and allodiploid strains show highly variable gene arrangements around MTL, suggesting that these loci are recombination hotspot during error-prone mating-type switching events (Watanabe et al., 2013; Solieri et al., 2014). Structural rearrangements are so rampant in these regions that different stock cultures of the same haploid (Watanabe et al., 2013) or allodiploid (Bizzarri et al., 2016; Watanabe et al., 2017) strains can display distinct MTL repertoires. For instance, differences in MTL loci were recently found between two sub-cultures of the allodiploid strain ATCC42981. In our previous work, we found 7 MTL loci in in-house stock of ATCC42981 (termed ATCC42981_R for convenience) (Bizzarri et al., 2016), while Watanabe et al. (2017) detected 6 MTL loci in strain JCM22060, the Japanese stock of ATCC42981. Ectopic recombination between MTL-flanking regions from divergent parental haplotypes yields chimeric arrangements hardly to resolve both by targeted long PCR approaches (Bizzarri et al., 2016) and by genome sequencing technologies based on short reads (Watanabe et al., 2017).
In 2014, the MinION sequencing device (Oxford Nanopore Technology, ONT) was released and initially exploited to sequence and assemble PCR products or microbial genomes (Jain et al., 2016). Recent improvements in protein pore (a laboratory-evolved Escherichia coli CsgG mutant named R9.4), library preparation techniques (1D ligation and 1D rapid), sequencing speed (450 bases/s), and control software enabled the usage of Nanopore sequence data, in combination with other sequencing technologies, for assembling eukaryotic genomes including yeasts, nematodes and human (Istace et al., 2017; Jansen et al., 2017; Yue et al., 2017; Jain et al., 2018). The main advantage of ONT is that reads can reach tens of kilobases (Jain et al., 2016), making more easy to resolve repeat regions and to detect structural variation. Recently, the genome of allodiploid strain ATCC42981_R was sequenced and assembled through a de novo hybrid strategy which combined MinION long and Illumina short reads (Bizzarri et al., 2018).
Here, we took advantage from the newly released genome of ATCC42981_R (Bizzarri et al., 2018), in order to resolve incongruences in the highly dynamic MTL loci. Furthermore, we deleted the expressed MATαP locus in ATCC42981_R to test whether the loss of MAT heterozygosity can induce genome doubling and rescue fertility in allodiploid cells of the ZT clade.
Materials and Methods
Strains, Plasmids, and Culture Conditions
Yeast strains and plasmids used in this study are listed in Table 1. Yeast cells were routinely propagated at 28°C in YPD (1% yeast extract, 2% peptone, 2% glucose) medium with 1.5% agar when necessary. Stock cultures were stored at -80°C with glycerol at final concentration of 25% (v/v) for long-term preservation. For sporulation and mating assays, MEA (5% malt extract, 2% agar) with and without 6% NaCl and YM (0.3% yeast extract, 0.5% peptone, 0.3% malt extract, 1% dextrose, 1.5% agar) media were used. Z. parabailii strain G21C was used as control for conjugated asci formation after growth on MEA medium. When required, YPD medium was supplemented with G418 (100 mg mL-1; MP Biomedicals, Germany) to the final concentration of 200 μg mL-1.
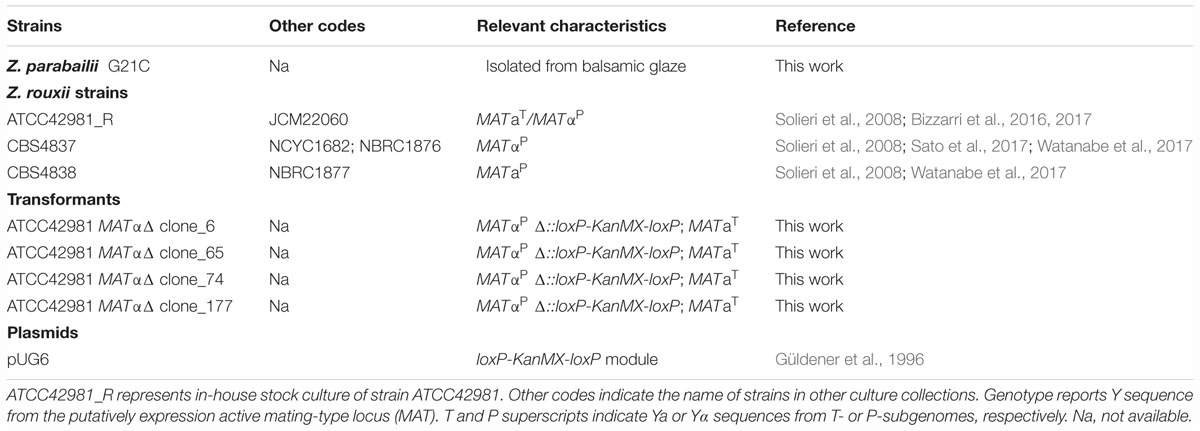
Table 1. Yeast strains and plasmid used in this work.
DNA Manipulations
DNA manipulations were performed according to standard protocols (Sambrook et al., 1989). Genomic DNA from yeast cells was isolated according to Hoffman and Winston (1987), while plasmid DNA from E. coli was isolated using the GenEluteTM Plasmid Miniprep Kit (Sigma). DNA quantity and quality were evaluated electrophoretically and spectrophotometrically using a NanoDrop ND-1000 device (Thermo Scientific, Waltham, MA, United States). ZymocleanTM Gel DNA Recovery and DNA Clean & ConcentratorTM-5 Kits (Zymo Research, Orange, CA, United States) were used for the isolation of DNA fragments from agarose gels and for PCR amplicons purification, respectively. Long PCR amplifications were carried out with rTAQ DNA polymerase (Takara Bio, Shiga, Japan) according to manufacturer’s instructions. For colony PCR 1 μl of DNA extracted with lithium acetate-SDS method (Lõoke et al., 2011) was amplified with DreamTaq polymerase (Thermo Scientific, Waltham, MA, United States) according to the manufacturer’s instructions in 20 μl reaction volume. All PCR amplifications were carried out in a T100 Thermal cycler (Bio-Rad, Hercules, CA, United States). All primers used in this study are listed in Supplementary Table S1.
Genome Re-assembly
Hybrid assembly of ATCC42981_R genome from Oxford Nanopore and Illumina reads was released to the European Nucleotide Archive under accession number PRJEB26771 (Bizzarri et al., 2018). In the deposited assembly Platanus contigs were scaffolded into 33 scaffolds with corrected MinION reads using DBG2OLC (Ye et al., 2016). These scaffolds were submitted to two-step polishing with long reads using Racon v1.2.0 (Vaser et al., 2017) and with short reads using Pilon v1.22 (Walker et al., 2014), and, finally, reduced using Redundans v.014 (Pryszcz and Gabaldón, 2016). Here, both long and short reads were assembled jointly with the alternative assembly algorithm Maryland Super-Read Celera Assembler v.3.2.2 (MaSuRCA) (Zimin et al., 2017) with default settings. Gene identification and annotation were carried out through the Yeast Genome Annotation Pipeline (YGAP)1 without frameshift correction (Proux-Wéra et al., 2012). MaSuRCA assembly completeness was assessed by Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.2 (Simão et al., 2015) using saccharomycetales_odb9 data set.
MTL Loci Search and Sanger-Based Validation
Search for MTL loci on scaffolds generated by DBG2OLC and MaSuRCA hybrid assemblies was carried out with a custom BLAST server built using the Sequenceserver software package (Priyam et al., 2015). Ya and Yα sequences and MTL flanking genes from the haploid reference genome of Z. rouxii CBS732T (Souciet et al., 2009) were used as queries.
The in silico MTL arrangements were in vitro validated by PCR and Sanger sequencing. Specific primer sets were built on MTL-flanking regions outside the X and Z regions (Supplementary Table S1). For putatively active MATαP cassette, walking strategy was adopted to cover ∼1 Kb downstream and upstream Yα (Wang et al., 2011). According Watanabe et al. (2017), MTL and flanking genes were marked with T and P superscripts when they shared >99% identity with Z. rouxii CBS732T or with P-subgenome from allodiploid NBRC110957 (Watanabe et al., 2017), respectively. N superscript was used to identify gene variants divergent from both T and P counterparts (identity lower than 99%). The 5′ MTL-flanking gene ZYRO0F18524g was named as CHA1L for brevity. Sequences were aligned with Clustal Omega (Sievers and Higgins, 2014) and viewed using Jalview (Waterhouse et al., 2009). Neighbor-joining (NJ) tree was built using MEGA v.6 software (Tamura et al., 2013).
Deletion Cassettes Construction and Yeast Transformation
Deletion of the active MATα locus from P-subgenome (abbreviated as MATαP) was performed with the reusable loxP-kanMX-loxP cassette as described previously (Güldener et al., 1996). The MATα1/2cp2-kanMX-F-80nt and MATα1/2cp2-kanMX-R-80nt primers contained ∼80 bp homology sequences outside the X ad Z regions of MATαP locus, respectively, and were used to amplify the kanMX deletion cassette from pUG6. After purification, the resulting PCR product was used to transform Z. rouxii cells by electroporation with a modified protocol from Pribylova and Sychrova (2003). Briefly, cells were grown (28°C; 180 rpm) in 80 ml of YPD medium supplemented with 300 mM NaCl until the exponential phase (corresponding to OD600nm of 0.7–0.8). After washing with ddH2O, cells were resuspended into 16 ml of TE buffer (Tris-hydrochloride buffer, pH 8.0, containing 1.0 mM EDTA) supplemented with 25 mM dithiothreitol and 20 mM LiAc, and incubated at 30°C for 30 min with gently shaking. Cells were centrifuged at 6,000 g for 5 min at 4°C, and washed twice by resuspension in 20 mL of ice-cold 1 M sorbitol. Finally, cells were washed in 5 ml of ice-cold 1M sorbitol and resuspended in 800 μl of ice-cold 1 M sorbitol. One hundred microliter of competent cell suspension were transferred into a pre-chilled 2-mm electroporation cuvette (Molecular Bioproducts Inc., San Diego, CA, United States) and 1 μg of loxP-kanMX-loxP deletion cassette was added before the electroporation at 2250 V/cm for 5 ms (Eporator, Eppendorf, Germany). Immediately after electroporation, 100 μl of ice-cold 1 M sorbitol were added to electroporation mixture. Before plating on selective YPDA medium supplemented with G418, the transformation mixtures were incubated for 2 h in 5 ml of YPD at 30°C. In G418R clones, targeted gene disruption was confirmed by full-length, 5′-, and 3′-end diagnostic PCRs (Supplementary Figure S1).
RNA Extraction, cDNA Synthesis and RT-PCR
RNA was extracted from ATCC42981 wild type and deletion mutants cultured in YPD and harvested at stationary phase, as previously reported (Solieri et al., 2016). RNAs were reverse transcribed using 0.5 μM oligo (dT) and RevertAid H Minus Reverse Transcriptase (Thermo Scientific, Waltham, MA, United States) according to the manufacturer’s instructions. cDNAs (25 ng) were amplified using DreamTaq polymerase with primers specific for different variants of MATa1, MATα1, and MATα2 genes, as well as for T and P variants of asgs AGA2, STE2, and STE6 (Supplementary Table S1).
Results
Inventory of ATCC42981_R MTL Cassettes
To unambiguously characterize MTL loci in our stock culture, we exploited the new available ATCC42981_R draft genome (Bizzarri et al., 2018). This draft genome relies on the hybrid DBG2OLC assembly of MinION ultra-long and Illumina MiSeq short reads to resolve high heterozygosity and span repetitive regions, which represent the greatest technical challenges during the assembly of complex non-haploid genomes (Treangen and Salzberg, 2012; Del Angel et al., 2018).
Custom BLAST searches using Sequenceserver identified six scaffolds harboring 8 MTL loci (2 MTLαT, 4 MTLαP, and 2 MTLa) mainly at the scaffold edge (Table 2). As this pattern matched only partially either with our previous results (Bizzarri et al., 2016) or with the JCM22060 set of MTL loci (Watanabe et al., 2017), we took into account the possibility of misassembled segments, mainly considering that reference P-type genome is not available. Misassemblies could be more burdensome at the MTL loci which contain the long non-tandem repeated X and Z sequences enriched in homopolymeric stretches. To circumvent these caveats, we validated the MTL cassettes found in DBG2OLC assembly in silico by using the alternative assembler MaSuRCA, as well as in vitro by direct PCR and Sanger sequencing. With appropriate caution, agreement between these assemblies – which are completely independent in assembly algorithms – and among assemblies and Sanger sequencing can confirm the integrity of MTL cassettes.
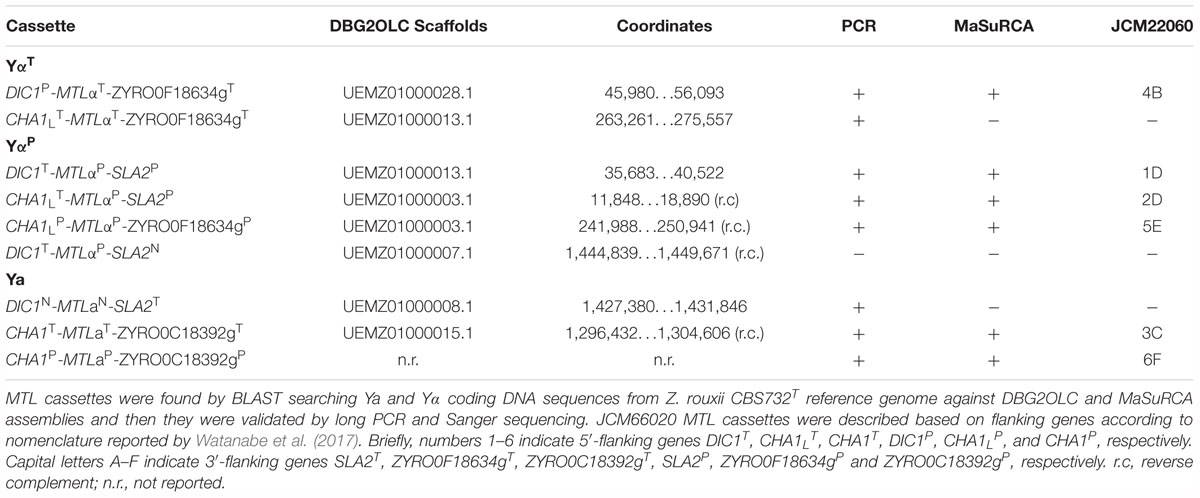
Table 2. Overview of the MTL cassettes confirmed by hybrid de novo genome assemblies and PCR approach.
MaSuRCA assembly resulted in an assembled genome size of 21.09 Mb distributed across 59 scaffolds with N50 of 1.34 Mb (Table 3). In our previous analysis, 10,524 predicted genes were estimated by Exonerate (Slater and Birney, 2005; Bizzarri et al., 2018). Here, gene number was re-calculated for both DBG2OLC and MaSuRCA assemblies using YGAP software. Based on this analysis, DBG2OLC and MaSuRCA displayed roughly the same number of predicted genes (Table 3). Single-copy orthologs analysis by BUSCO 3.0 revealed a high degree of completeness in both assemblies (>98.0%), even if MaSuRCA retrieved more duplicated orthologs than DBG2OLC.
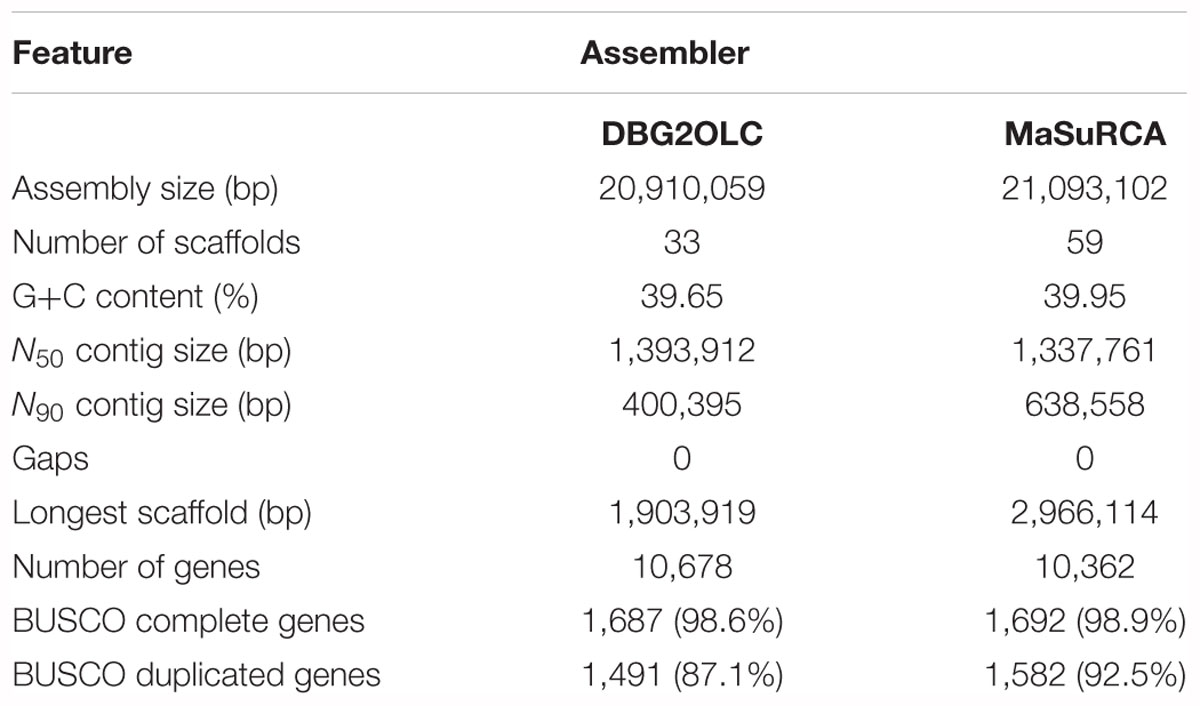
Table 3. Assembly metrics and annotation completeness obtained by using BUSCO universal fungal genes (saccharomycetales_odb9) data set.
MaSuRCA validated five out of eight DBG2OLC MTL cassettes, while one was MaSuRCA assembly specific (Table 2 and Supplementary Table S2). All six MaSuRCA cassettes were consistent with JCM22060. Like in DBG2OLC, MaSuRCA-derived MTL cassettes especially laid at the scaffold edges, confirming difficulties in scaffolding over repeated X and Z sequences shared by multiple and partially divergent MTL-flanking regions. Figure 1 showed that direct in vitro PCR validated eight MTL arrangements. Moreover, MaSuRCA consensus sequences were often more consistent with Sanger sequencing compared with DBG2OLC. Probably, this discrepancy resulted from a more aggressive DBG2OLC approach enabled to reduce the genome fragmentation, but at the price of local assembling accuracy.
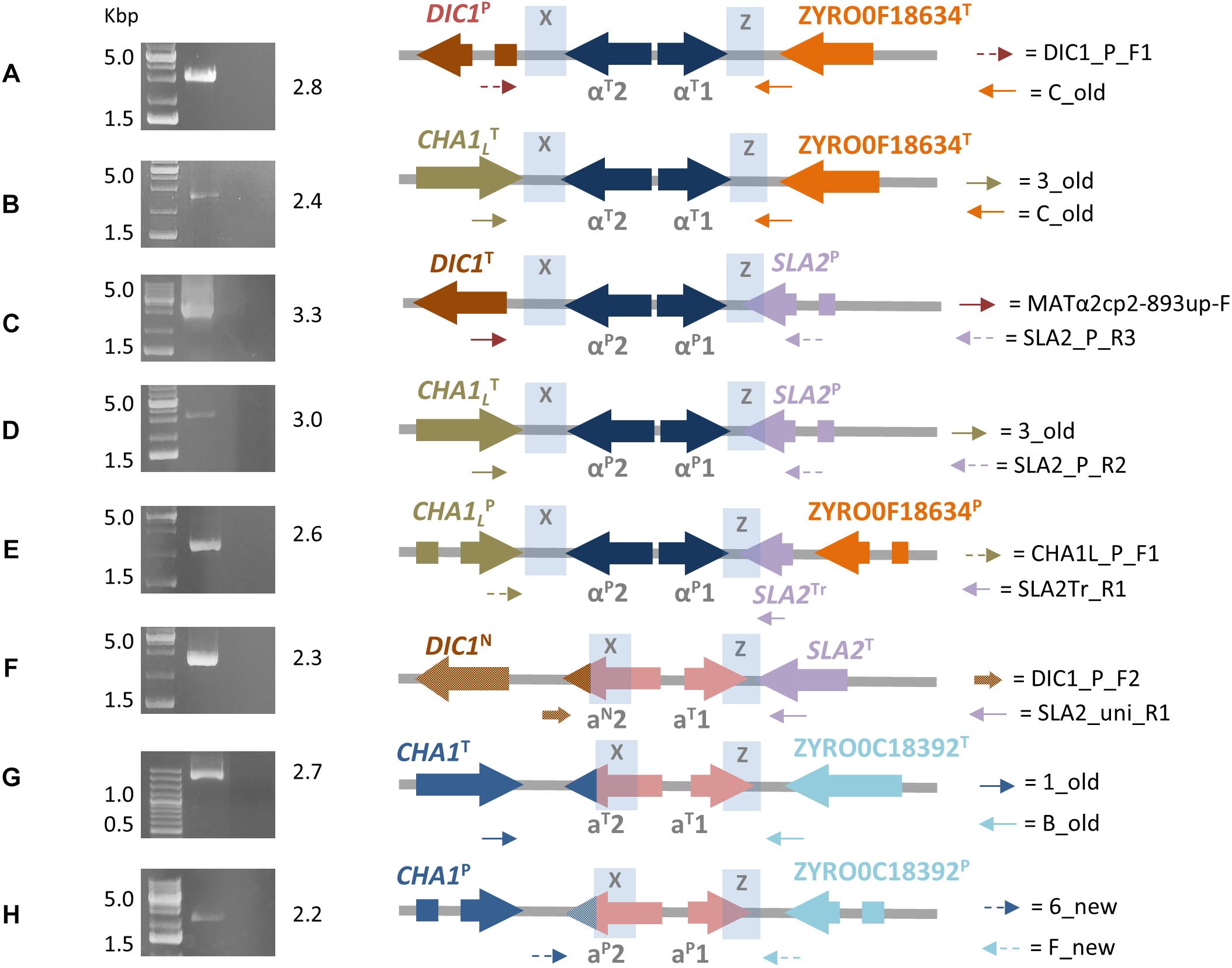
Figure 1. Final ATCC42981_R MAT-like cassettes organization resulting from DBG2OLC and MaSuRCA assemblies. Picture depicts full-length PCR results (left) and the corresponding inferred MTL arrangements (right). Genes from T- and P-subgenomes are marked with T and P superscripts, respectively. SLA2 truncated variant is marked with Tr superscript, while DIC1 and MATa2 new variants with N superscript. Blue shading denotes the X and Z regions. Dot arrows indicate P-variants of MAT flanking genes, while full arrows stand for T-variants. Variable MATa2 3′-end tags are indicated as colored tips. Capital letters from A to H refer to primer pairs listed in Supplementary Table S1. Numbers on the right of gel electrophoresis images indicate PCR product length in Kbp.
MTLαP Cassettes
Congruently with our previous data (Bizzarri et al., 2016), the DBG2OLC and MaSuRCA assemblies supported the cassettes DIC1T-MTLαP-SLA2P and CHA1LT-MTLαP-SLA2P (Table 2). PCR approach confirmed these arrangements (Figure 1). Pairwise comparisons showed that DIC1T and CHA1LT were 100% identical to the Z. rouxii CBS732T counterparts. In cassette DIC1T-MTLαP-SLA2P, the 3′-flanking gene SLA2P diverged from CBS732T counterpart (83.65% identity), and resembled SLA2 found in allodiploid NBRC110957 and NBRC1876 (99.58% identity) (Sato et al., 2017; Watanabe et al., 2017). In CHA1LT-MTLαP-SLA2P cassette, DBG2OLC assembly reported mismatches compared to SLA2P in NBRC110957 (93.12% identity), which were not supported by MaSuRCA. Sanger sequencing confirmed the accuracy of MaSuRCA assembling (Supplementary Figure S2).
According to the model of T- and P-subgenomes, DIC1T-MTLαP-SLA2P and CHA1LT-MTLαP-SLA2P should be chimeric cassettes arisen from rearrangements involving the X regions. NBRC110957 also contains the DIC1T-MTLaP-SLA2P chimeric arrangement (Watanabe et al., 2017; Supplementary Table S2), suggesting that recombination is frequent upstream the Y sequence. Recombinant sites at the MAT locus were also documented in several Saccharomyces lager yeasts (Bond et al., 2004; Hewitt et al., 2014). Breakpoints frequently occurred at the right of the MAT locus resulting in hybrid S. cerevisiae–S. eubayanus chromosomes III. These chromosomes contain S. eubayanus sequences in the W region and S. cerevisiae in the Y region hitch-hiking downstream genes or vice versa (Monerawela and Bond, 2017). In lager yeast Ws34/70 a possible location for the recombination event is a 9-bp insertion in the S. eubayanus X region compared to S. cerevisiae. We found a similar indel between X regions of ATCC42981_R DIC1 variants (Supplementary Figure S3), confirming that X region could represent a specific ‘fragile’ chromosomal location susceptible to double strand breakage (DSB).
Novel sets of P-subgenome-specific primers confirmed an additional MTLαP locus (CHA1LP-MTLαP-ZYRO0F18634gP) which escaped our previous reconstruction (Bizzarri et al., 2016) (Figure 1). Based on Watanabe et al. (2017), this locus should be a cryptic HML cassette, which did not affect the true cell identity. This cassette had a truncated SLA2 sequence downstream the Z region, confirming DNA erosion on the right side of MAT locus (Gordon et al., 2011). Interestingly, in both DBG2OLC and MaSuRCA assemblies this cassette is linked to CHA1LT-MTLαP-SLA2P on the same scaffold (Supplementary Figure S4).
MTLαT Cassettes
DBG2OLC and MaSuRCA assemblies failed to congruently reconstruct MTLαT loci (Table 2). DBG2OLC scaffold UEMZ01000013.1 contains CHA1LT-MTLαT-ZYRO0F18634gT linked to the chimeric cassette DIC1T-MTLαP-SLA2P, while another MTLαT locus (DIC1P-MTLαT-ZYRO0F18634gT) lies on the scaffold UEMZ01000028.1. MaSuRCA assembly reported only the DIC1P-MTLαT-ZYRO0F18634gT cassette. Moreover, MTL cassette linkage differed between DBG2OLC and MaSuRCA: DIC1T-MTLαP-SLA2P was linked to CHA1LT-MTLαT-ZYRO0F18634gT in DBG2OLC, while it was linked to CHA1LT-MTLαP-SLA2P and CHA1LP-MTLαP-ZYRO0F18634gP in MaSuRCA (Supplementary Figure S4). PCR approach supported both MTLαT cassettes from DBG2OLC assembly (Figure 1), while scaffold comparison suggests that MaSuRCA collapsed the CHA1LT flanking regions into a single locus (Supplementary Figure S4).
MTLa Cassettes
Blast search against the DBG2OLC assembly revealed two MTLa cassettes (Table 2). The arrangement CHA1T-MTLaT-ZYRO0C18392gT was also supported by MaSuRCA and PCR approach, and was congruent with our previous reconstruction (Bizzarri et al., 2016) and with JCM22060 (Watanabe et al., 2017) (Supplementary Table S2).
The second MTLa locus resolved by DBG2OLC, DIC1N-MTLaN-SLA2T, contained aT1 and a novel aN2 gene variant (indicated with N superscript) which was 97.99% identical to MATa2 from NBRC110957 DIC1P-MTLaT-ZYRO0C18392T cassette (Figure 2). PCR approach demonstrated that this cassette really exists in ATCC42981_R genome, even if it was missing both in MaSuRCA assembly and in JCM22060 (Figure 1). Like in case of SLA2P from CHA1LT-MTLαP-SLA2P, DBG2OLC MATa2 sequence showed some indels in homopolymeric stretches compared to the Sanger-sequence data (98.54% pairwise identity), resulting in a prematurely interrupted ORF (data not shown). The neighbor genes at the 5′ and 3′ sides were a novel DIC1 variant (named DIC1N) and the SLA2T gene, respectively. Noteworthy, the DIC1-MAT-SLA2 arrangement is retained around the transcriptionally active MAT loci in almost all the pre-WGD species (Gordon et al., 2011). Therefore DIC1N-MTLaN-SLA2T cassette could be a good candidate to be the active MATa cassette in ATCC42981_R.
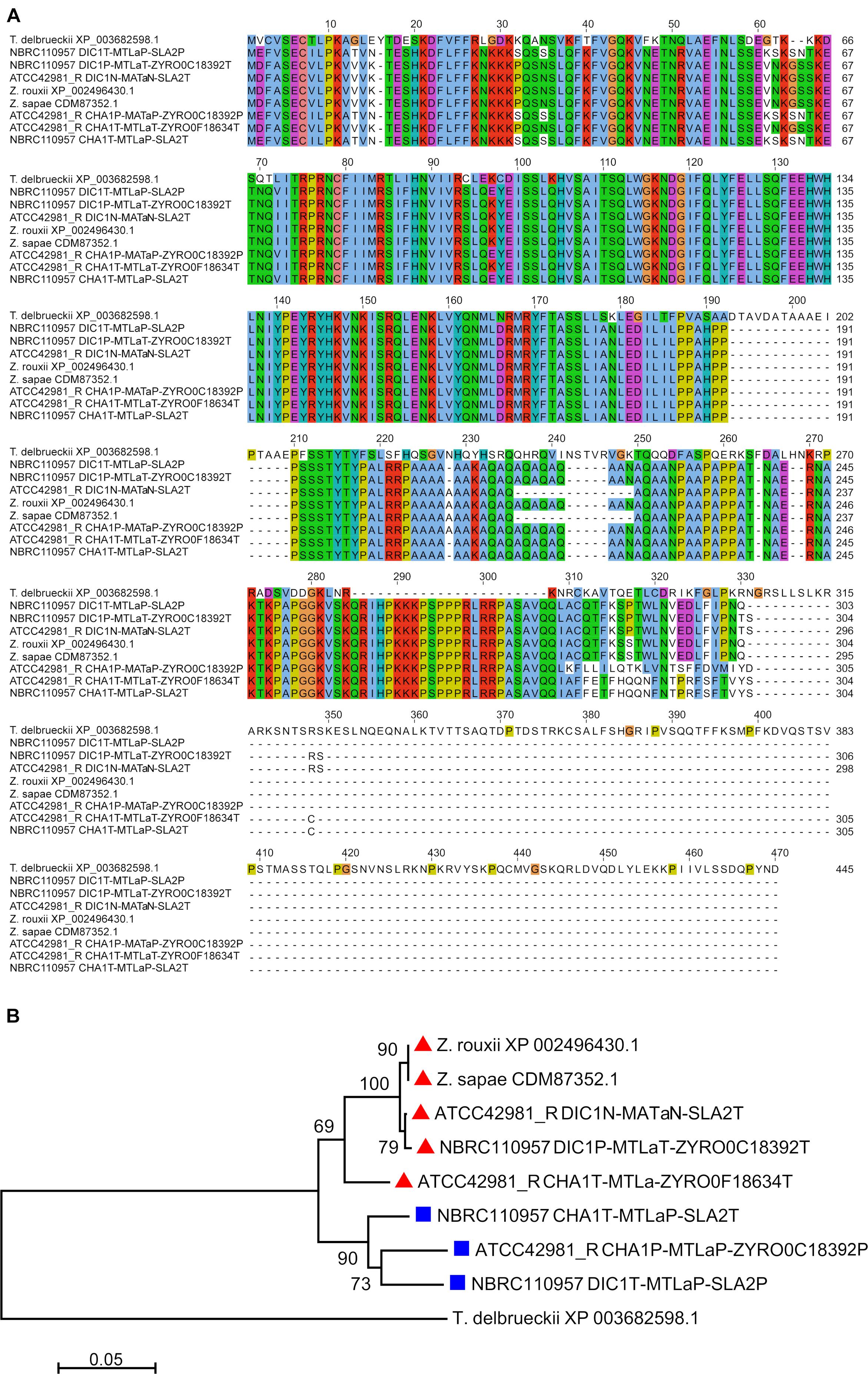
Figure 2. Multiple sequence alignment and phylogenetic analysis of MATa2 proteins. (A) Depicts the alignment involving 9 MATa2 amino acid sequences. The amino acid identities were colored according to Clustal Omega color scheme (Sievers and Higgins, 2014). In (B) dendrogram was inferred using the Neighbor-Joining method. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches, when ≥50%. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. All positions containing gaps and missing data were eliminated. Red triangles and blue squares marked T and P variants.
Finally, PCR approach with haplotype P-specific primers identified a third MTLa locus (CHA1P-MTLaP-ZYRO0C18392gP) which was present in JCM22060 and in MaSuRCA assembly (Table 2). Blast search for CHA1P gene revealed that DBG2OLC assembler did not extend scaffold UEMZ01000005.1 beyond this gene.
Reconstruction of MTL Structure
Analysis of regions around MTL loci assisted us to reconstruct the putative MTL structure in ATCC42981_R. NBRC1130T culture retains ancestral MTL arrangement compared with CBS732T (Watanabe et al., 2013) and was used as reference strain. In this strain, chromosome C contains MAT and HML loci flanked by sets of genes which were also conserved around ATCC42981_R MTL cassettes (Supplementary Figure S5). In particular, MAT locus was flanked on the left by PEX2 and CBP1 and on the right by SUI1 and CWC25, while HML cassette was flanked by VAC17 at the left side and by FET4 and COS12 at the right side (Figure 3). Blast analysis indicated that DBG2OLC scaffold UEMZ01000008.1 was almost collinear to NBRC1130T chromosome C in the first 1,427,380 bp. Genes upstream and downstream the MATaN cassette were P and T-type, respectively. Congruently, MATaN cassette retained the synteny with PEX2P and CBP1P at 5′- and SUI1T and CWC25T at 3′-end. However, 3′-end side was interrupted at RAD50T. Scaffold UEMZ01000003.1 (rc) linked CHA1LT-MTLαP-SLA2P and CHA1LP-MTLαP-ZYRO0F18634gP cassettes (Figure 3). Reciprocal translocation between chromosomes C from T and P haplotypes led to a similar arrangement in CBS4837 (Watanabe et al., 2017). As result, in CBS4837 the MATαP expression cassette is linked to CHA1LT-MTLαP-SLA2P and CHA1LP-MTLαP-ZYRO0F18634gP. In ATCC42981_R, flanking gene analysis also supported a linkage between MATaN and CHA1LT-MTLαP-SLA2P/CHA1LP-MTLαP-ZYRO0F18634gP cassettes, suggesting that scaffolds UEMZ01000008.1 and UEMZ01000013.1 contributed to the chimeric chromosome C. Like in CBS4837 (Watanabe et al., 2017), this chromosome C could arise from a reciprocal translocation between two ancestral T and P chromosomes C.
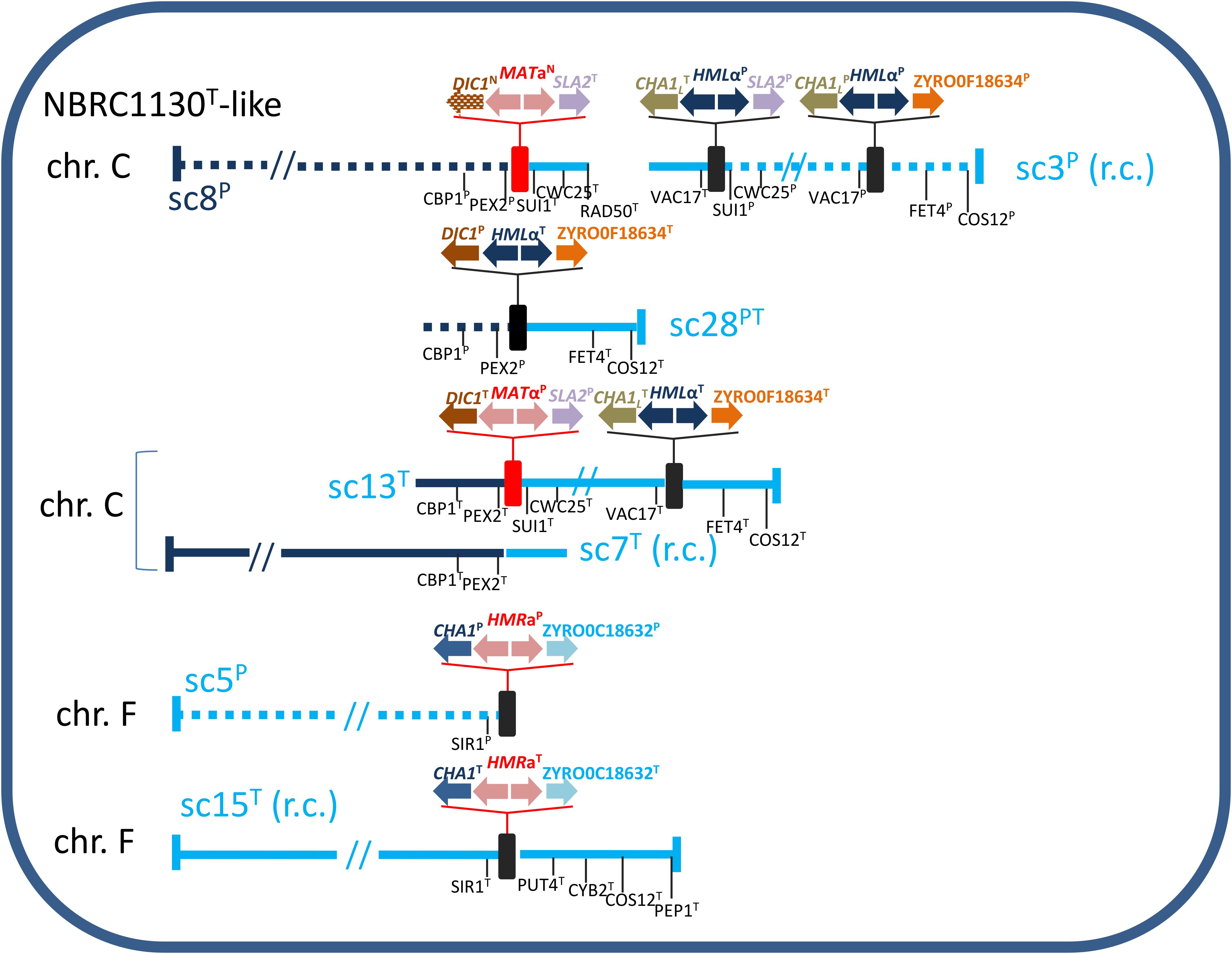
Figure 3. Inferred gene organization around the MTL loci in ATCC42981_R. Scaffold (sc) numbers referred to the DBG2OLC genome assembly deposited in European Nucleotide Archive under accession number PRJEB26771 (Bizzarri et al., 2018); for brevity each scaffold is identified by the last number of ENA code (i.e., UEMZ01000028.1 in short sc28). Solid and dotted lines referred to T- and P-subgenomes, respectively. Genes from T- and P-subgenomes are marked with T and P superscripts, respectively, while DIC1 and MATa2 new variants with N superscript. Red and black rectangles defined MAT and HML/HMR loci, respectively. Scaffold lengths are not in scale. r.c., reverse complement.
Scaffold UEMZ01000028.1 was chimeric with P-type (PEX2 and CBP1) and T-type (FET4 and COS12) genes upstream and downstream the cassette DIC1P-MTLαT-ZYRO0F18634gT, respectively (Figure 3). The loss of gene block between MAT and HML cassettes suggested that a deletion between MAT and HML cassettes led to this arrangement, similar to that described in strain NBRC0686 (Watanabe et al., 2013; Supplementary Figure S5). Alternatively, in CBS4837 a similar arrangement resulted from reciprocal translocation leading to chimeric chromosome C (Watanabe et al., 2017).
DBG2OLC scaffold UEMZ01000013.1 exhibited T-type flanking genes around DIC1T-MTLαP-SLA2P and CHA1LT-MTLαT-ZYRO0F18634gT. Overlapping region with scaffold UEMZ01000007.1 suggested that scaffolds UEMZ01000013.1 and UEMZ01000007.1 could contribute to the T-type chromosome C in ATCC42981_R (Figure 3).
NBRC1130T strain has the HMRa locus on chromosome F. SIR1 and a set of genes including PUT4, CYB2, COS12, and PEP1 are upstream and downstream to HMRa, respectively (Supplementary Figure S5). ATCC42981_R DBG2OLC assembly exhibited two scaffolds retaining this synteny, namely 5 and 15 (rc). Scaffold UEMZ01000005.1 contained P-type genes, including SIR1P (Figure 3). Unfortunately, DBG2OLC assembler interrupted this scaffold after CHA1P. However, MaSuRCA assembly retained PUT4P, CYB2P, COS12P, and PEP1P downstream of HMRaP, suggesting that ATCC42981_R has a P-type chromosome F collinear to NBRC1130 chromosome F. Syntenic relationships and Blast analysis supported scaffold UEMZ01000015.1 as the T-type version of NBRC1130T chromosome F (Supplementary Figure S5).
Disclosing the True Cell Identity
Watanabe et al. (2017) identified two MTL patterns: strains with pattern A, such as NBRC110957, exhibit two active MAT loci, namely DIC1T-MATP-SLA2P and CHA1T-MTLP-SLA2T, while strains with pattern B have DIC1T-MATP-SLA2P as active MAT locus, even if they also actively transcribed genes from CHA1LT-MTLP-SLA2P. JCM66020 belongs to this last group, exhibits a MATαP idiomorph and, congruently, mates only the tester strain a (CBS4838). Conversely, ATCC42981_R displays another pattern of putatively active MAT loci, namely, DIC1T-MATαP-SLA2P and DIC1N-MATaN-SLA2T, in addition to the CHA1LT-MTLP-SLA2P cassette. RT-PCR analysis confirmed that αP1, αP2, aN2 and aT1 genes were expressed, while aP1 gene encoded by CHA1P-MTLP-ZYRO0C18392gP cassette was silent (Figure 4). Interestingly, aT1-specific RT-PCR resulted in two PCR amplicons compatible with alternative spliced intronic sequence.
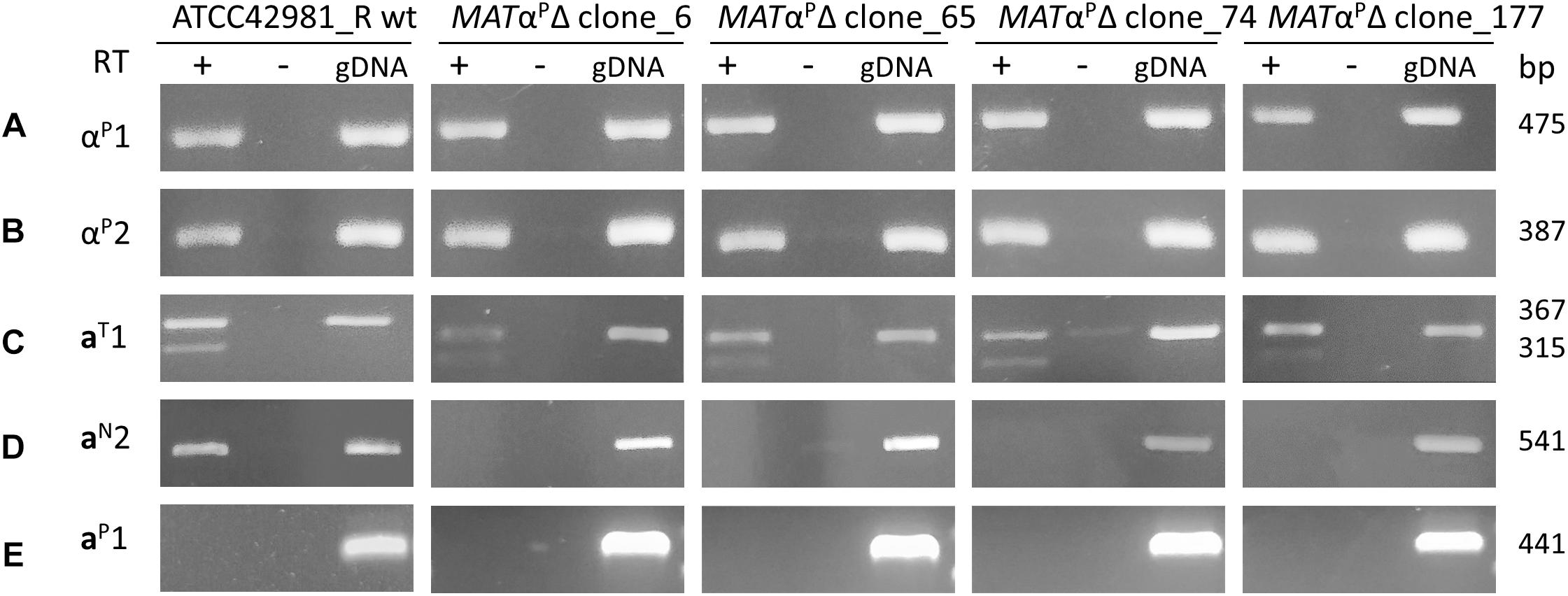
Figure 4. Gene expression at the MAT loci in ATCC42981_R wild type and MATαPΔ deletion mutants. cDNA was amplified from total RNA extracted from stationary growing cells. Plus or minus indicates with or without reverse transcriptase in cDNA synthesis reaction, respectively. Gene variants from T- and P-subgenomes are indicated with T and P superscripts, while divergent MATa2 gene variant from DIC1N-MATaN-SLA2T cassette with N superscript. Capital letters from A to E refer to primer sets listed in Supplementary Table S1. wt, wild type; gDNA, genomic DNA.
Genome comparison with other pre-WGD yeasts indicates that HMLα silent cassettes are generally 5′-flanked by CHA1L (Gordon et al., 2011). Conversely, strains with pattern B actively transcribed MTL genes from CHA1LT-MTL-SLA2P cassette without that these transcripts affect cell identity (Watanabe et al., 2017). This is evident for strain CBS4837, where genes encoding opposite αP and aP idiomorphs are both expressed by DIC1T-MATP-SLA2P and CHA1LT-MTLP-SLA2P cassettes, respectively. In JCM22060 (encoding αP genes at both these loci), outcross experiment with CBS4837 and gamete segregation support that cell identity was determined by DIC1T-MATP-SLA2P cassette. To establish which cassette contributes to cell identity in ATCC42981_R, we deleted αP idiomorph genes by replacing the entire segment including αP1, αP2 encoding genes and the intergenic region from DIC1T-MATαP-SLA2P with loxP-kanMX-loxP module. From approximatively 300 screened colonies we obtained four G418R clones. PCR genotyping showed that these clones are MATαPΔ deletants containing loxP-kanMX-loxP surrounded by DIC1T and SLA2P instead of MATαP locus (Supplementary Figure S1).
Gene deletion of DIC1T-MATαP-SLA2P cassette should abolish the heterozygosity at the MATa/α active loci and results in an allodiploid partially resembling a haploid cell with a mating-type. Conversely, ATCC42981_R MATαPΔ still showed αP1 and αP2 gene expression (Figure 4). These mRNAs could be only transcribed by the not completely silenced cassettes CHA1LT-MTLαP-SLA2P or by CHA1LP-MTLαP-ZYRO0F18634gP.
Since allodiploid lacking one MAT active locus should behave like haploid with opposite mating-type, we expected to detect both a1 and a2 transcripts in ATCC42981_R MATαPΔ mutants. In some haploid pre-WGD species, a2 gene encodes a transcription activator of asgs, while a1 should not affect asgs in a cells (Tsong et al., 2003, 2006; Baker et al., 2012). Unexpectedly, RT-PCR showed that MATαP deletion switched off a2 but not a1 gene expression (Figure 4). By contrast, ATCC42981_R wild type both transcribed a1 and a2 genes. Preliminary end-point RT-PCRs showed that the asgs AGA2, STE6, and STE2 are transcriptionally active in both wild type and MATαPΔ cells (data not shown).
Mating and Sporulation Competence Assays
To test whether the MATαP deletion rescues the mating competence in ATCC42981_R, we carried out self- and out-cross fertility assays of the wild type strain and the MATαPΔ transformants as monoculture or in mixture with CBS4837 (α) or CBS4838 (a) mating testers, respectively. If MATαPΔ transformants behave as homothallic haploids, they should produce shmoo and conjugated asci as monoculture, while, if they are like heterothallic haploids, they should mate and sporulate in mixture either with CBS4837 or CBS4838. We used three media reported in literature to promote zygote formation and conjugated asci of Zygosaccharomyces cells, as proved for Z. parabailii G21C (Figure 5). In particular, 5-6% NaCl addition was reported to increase sporulation occurrence (Mori and Onishi, 1967). Like the wild type strain, MATαPΔ mutants did not show any evidence of conjugative bridge and/or conjugative asci either as monoculture or in mixture with the mating testers (Figure 5). The composition of three test media did not affect the inability to mate or to undergo meiosis. Overall, these evidences indicate that the deletion of active MATαP locus did not make ATCC42981_R cells phenotypically heterothallic or homothallic haploids.
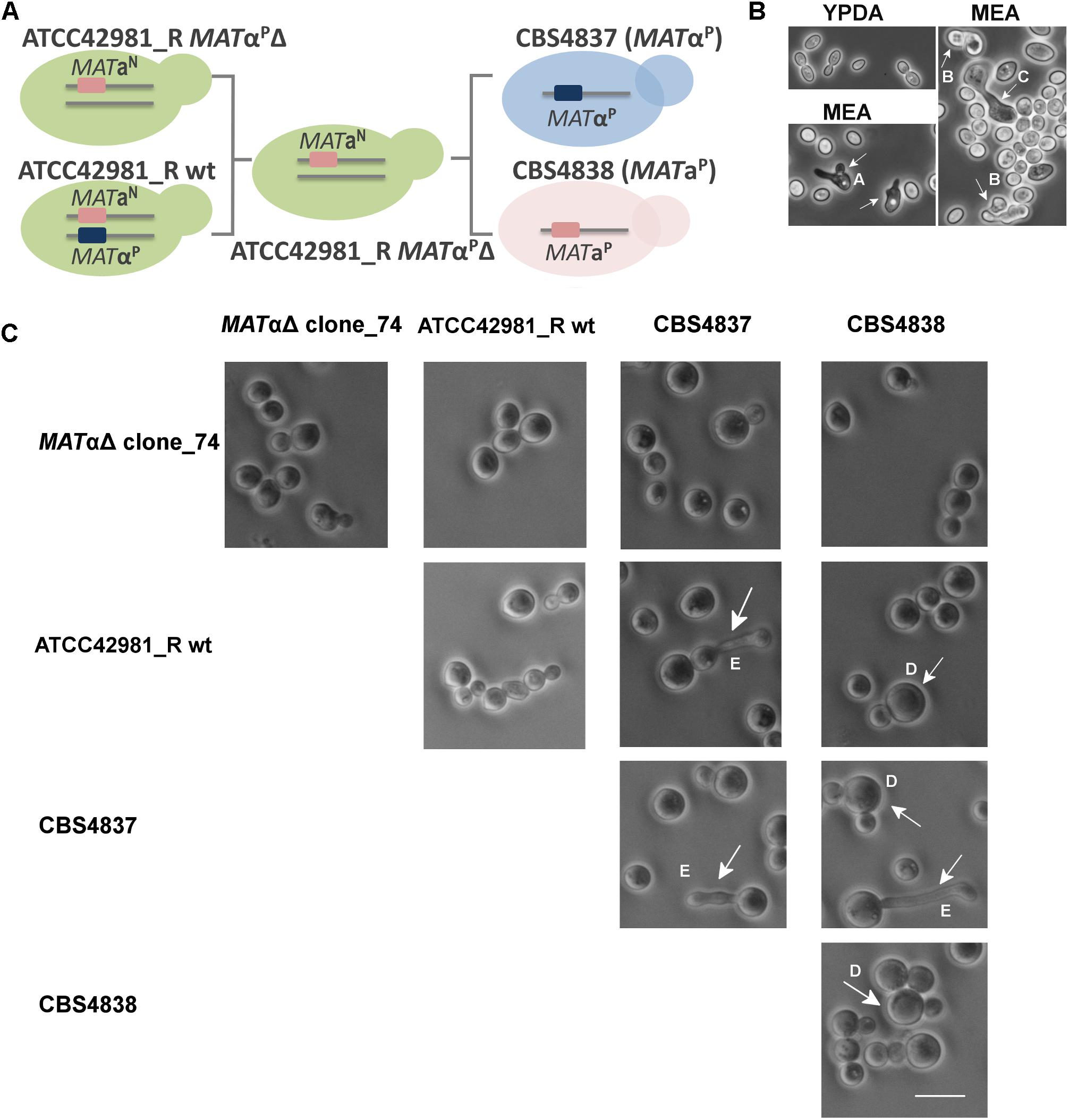
Figure 5. Self- and out-cross fertility assays of ATCC42981_R wild type and MATαPΔ deletion mutant. (A) Shows the scheme of self- and out-cross fertility assays involving ATCC42981_R wild type and MATαΔ clone_74, while (B) depicts representative phase contrast microscopic images of control strain Zygosaccharomyces parabailii G21C grown in YPDA and MEA media. (C) Shows selected phase contrast microscopic images of representative ATCC42981_R MATαΔ clone_74 grown as monoculture and in mixture with ATCC42981_R wild type, CBS4837 (α) and CBS4838 (a). Phase contrast microscopic images of ATCC42981_R wild type, CBS4837 and CBS4838 monocultures were reported for comparative purposes. The scale bar represents 11 μm and was reported in only one picture for brevity. Capital letters from A to E indicate abortive shmoo, conjugated asci with ascospores, conjugative bridge, giant cells, and abnormal conjugative tube, respectively. All cells were photographed after 7 days on incubation on MEA medium at 27°C. wt, wild type.
Discussion
Our study is the first to combine the Nanopore whole-genome sequencing to conventional PCR-based methods in order to survey MTL loci in a Z. rouxii allodiploid genome. This yeast is particularly prone to outbreeding and provides a particularly appealing platform to study genome re-shaping after the merger of two parental subgenomes. Recombination and introgression between subgenomes have been rampant in hybrid yeasts, resulting in loss of heterozygosity and gradual genome reduction (Sipiczki, 2008). In Z. rouxii MTL loci markedly contribute to this genomic plasticity (Watanabe et al., 2013; Solieri et al., 2014). As consequence, this species frequently undergoes chromosomal translocations at the MTL loci, which make hard the understanding of true cell identity by simple MTL genotyping. For example, haploid Z. rouxii strain CBS732T switched mating-type at the CHA1-MAT-SLA2 locus (Bizzarri et al., 2018), suggesting that CHA1 gene flanks the actively transcribed MAT locus instead of DIC1. Several assortments of different flanking gene variants and distinct idiomorph encoding genes make challenging and laborious to resolve the complex genetic MTL architecture by PCR targeted approaches. For these reasons, we generated a high-quality genome assembly in order to dissect complex rearrangements at the MTL loci that were not fully resolvable from the earlier survey based only on long-range PCR amplification (Bizzarri et al., 2016). One of the major advantages of the ONT is the possibility of sequencing very long DNA fragments, which span the entire MTL cassettes. This strategy assures to accurately reconstruct gene order around different MTLs. On the other hand, using noisy ultra-long reads for self-correction and assembling of highly heterozygous genomes can affect the consensus sequence accuracy and the parental haplotypes sorting. In case of ATCC42981_R, distinguishing between homeologous sequences is further challenging as only the Z. rouxii parental genome is available to guide homeologous scaffold assembly. Error rate made necessary to polish MinION reads with Illumina-derived reads, resulting into DBG2OLC-driven hybrid de novo genome assembly (Bizzarri et al., 2018). However, our result showed that a single “best assembler” does not exist to resolve highly heterozygous and highly repeated MTL regions. DBG2OLC assembly suffers from poor performance in certain sequence contexts, such as in regions with low coverage or regions that contain short repeats. Besides, the new assembly generated with MaSuRCA showed higher sequencing accuracy compared to DBG2OLC, but loses some MTL cassettes. As bottom-end validation step, PCR approach was used to discard artificial MTL arrangements arisen from flawed contig assemblies. This strategy resolves controversies over MTL loci in ATCC42981_R genome derived from the analysis of the Japanese stock JCM22060 (Watanabe et al., 2017).
Reconstruction of MTL structure indicates that ATCC42981_R resembles CBS4837 for the exception of an additional scaffold containing DIC1T-MTLαP-SLA2P linked to CHA1LT-MTLαT-ZYRO0F18634gT (Figure 3). This assessment was congruent with previous PFGE-Southern blotting which showed two signals for MATα-specific probe (Bizzarri et al., 2016). The most significant difference between ATCC42981_R and JCM22060 is that ATCC42981_R harbors the transcriptionally active MATaN cassette in addition to the expected MATαP. Differently from Z. parabailii (Ortiz-Merino et al., 2017), MATaN cassette of ATCC42981_R contains MATa1 gene. This means that Z. rouxii retains the ancestral regulatory circuit based on a1–α2 heterodimer as diploid cell sensor (Booth et al., 2010). Watanabe et al. (2017) showed that strain JCM22060, which contains only MATαP, mates the tester strain a in a medium containing Shoyu-koji extract. By contrast, we did not find any evidence of meiosis or mating in ATCC42981_R (Bizzarri et al., 2016), when grown on the media reported in literature to promote Z. rouxii mating and sporulation (James and Stratford, 2011). Watanabe et al. (2017) argued that difference in medium composition could account for the phenotypic discrepancy between ATCC42981_R and the sister stock JCM22060. As the Shoyu-koji extract is difficult to gain in western countries, we cannot rule out this hypothesis. Otherwise, heterozygosity at the MAT locus could significantly contribute to the allodiploid infertility. In particular, the hybrid heterodimer with divergent a1 and α2 subunits brings the cell in an ‘haploid-diploid intermediate’ functional state which hamper both the meiosis commitment and the responsiveness to mating stimuli (Bizzarri et al., 2016).
In Saccharomyces clade, experimental deletion of one MAT locus leads to allotetraploids suitable to undergo meiosis (Greig et al., 2002; Pfliegler et al., 2012). Similarly, Z. parabailii and Z. pseudobailii hybrid strains ATCC60483 and MT15 were recently supposed to be fertile due to the accidental breakage of 1 of the 2 homeologous copies of the MAT locus (Ortiz-Merino et al., 2017; Braun-Galleani et al., 2018). A prediction of this model is that artificial deletion of one MAT locus in Zygosaccharomyces cells should override the arrest in mating commitment. In our model, ATCC42981_R cells did not behave as haploids with idiomorph a, when the MATαP locus was deleted. This suggests that mechanism underpinning the cell identity in Z. rouxii hybrids could be different from those involved in cell identity regulation of the sister species Z. parabailii and Z. pseudobailii.
Gene deletion of transcriptionally active MATαP locus did not rescue the ability to produce conjugated asci in ATCC42981_R, while the persistence of α1 and α2 transcripts suggests that HMLα silencing was leaky in ATCC42981_R. Consequently, αP genes either from CHA1LT-MTLαP-SLA2P or CHA1LP-MTLαP-ZYRO0F18634gP are transcriptionally active in MATαPΔ mutants. Strain NBRC110957, which does not have the CHA1LT-MTLαP-SLA2P cassette, uses CHA1LP-MTLαP-ZYRO0F18634gP as donor during switching from aP to αP (Watanabe et al., 2017). This suggests that CHA1LP-MTLαP-ZYRO0F18634gP cassette is most likely silenced and that αP could be expressed by the CHA1LT-MTLαP-SLA2P in ATCC42981_R. Congruently, strain CBS4837 actively transcribed genes from CHA1LT-MTLaP-SLA2P cassette. These findings make less probable the alternative hypothesis that MATαP deletion induces HMLα cassette de-silencing. Abnormal expression of cryptic HMR/HML loci has been described in Vanderwaltozyma polyspora, the Z. rouxii closest relative that branched after WGD (Roberts and Van der Walt, 1959). Consequently, V. polyspora haploid cells behave as a/α diploid and appear mating-incompetent for many generations only to subsequently restore silencing. Significantly, V. polyspora lacks of Sir1 histone deacetilase, which mediates the HM loci silencing in S. cerevisiae together with the SIR complex (Sir2/Sir3/Sir4). In S. cerevisiae failure to recruit Sir1 is thought to account for the instability of subtelomeric silencing relative to HM loci (Chien et al., 1993). Like V. polyspora, Candida glabrata is another species close to Z. rouxii, which lacks of a SIR1 ortholog (Gabaldón et al., 2013). A defective silencing system leads to the expression of MATa genes in C. glabrata MATα cells (Muller et al., 2008) and makes HML more prone to HO cleavage at the Y/Z junctions (Boisnard et al., 2015). Z. rouxii has the archetypal member of the SIR1 family, KOS3 (Kin of Sir1 3) (Gallagher et al., 2009). In pre-WGD species Torulaspora delbrueckii KOS3 located ∼1 kb away from HMR and plays a key role in HML/HMR silencing (Ellahi and Rine, 2016). Strikingly, in ATCC42981_R we also found two KOS3 copies, KOS3T and KOS3P, upstream of HMRaT and HMRaP loci, respectively. In addition, Sir1 and the components of SIR complex have been reported to rapidly evolve in the Saccharomycetaceae family. This could potentially jeopardize the efficiency of the silencing machinery in interspecific hybrids. For example, Sir1, Sir4 and the cis-acting silencer sequences are incompatible in S. cerevisiae × S. uvarum hybrids (Zill et al., 2010, 2012). In ATCC42981_R, heterochromatin formation across silent loci could be less effective due to the incompatibility in the silencing machinery between the T- and P-subgenomes. Watanabe et al. (2017) suggest that chimeric MTL cassettes could display epigenetic expression control when only E silencer sequence is maintained around MTL locus. This could produce allodiploid single cells which undergo epigenetic silencing at one of MAT loci and restore fertility. In ATCC42981_R two DIC1-MAT-SLA2 cassettes assure active transcription of opposite idiomorphs, while the presence of E silencer only at the right side of HMLαP locus could unlock the silencing and mask the loss of heterozygosity at the MAT locus induced by MATα locus deletion.
Strikingly, the depletion of αP1 and αP2 genes switched off the a2 but not the a1 gene transcription. Moreover in both deleted and wild type strains two a1 alternative spliced isoforms are present, one of them compatible with the retention of first intron. In S. cerevisiae exon–intron structure is conserved and the retention of first intron resulted in a functional a1 transcriptional factor that prevents mating (Ner and Smith, 1989). Since α1 activates the αsgs in the ancestral circuit of yeast cell identity (Baker et al., 2011), we rule out the possibility that α1 is involved in a2 gene repression. In S. cerevisiae, α2 represses asgs by binding asgs cis-regulatory sequences cooperatively with a MADS-box transcription regulator, Mcm1 (Tsong et al., 2003). Z. rouxii, which branched from the S. cerevisiae lineage prior to the loss of a2 gene, should maintain both the a2 activation and the α2 repression of asgs (Tsong et al., 2006; Baker et al., 2012). In Lachancea kluyveri haploid cells, α2 deletion induces the transcription of the asgs AGA1 and AGA2, while a2 deletion decreases the asgs transcript levels (Baker et al., 2012). However, to the best of our knowledge, no evidence has been provided until now about the consequences of α2 gene deletion in diploid cells which retain a2 gene. As a1 is still expressed in Z. rouxii MATαΔ/MATa hemizygous cells, we speculate that a2 silencing could be a promoter-driven event directly or indirectly regulated by α2. Furthermore, in our MATαΔ/MATa model, the asgs were expressed even when a2 was switched off by the MATα2 deletion, suggesting the existence of a different asgs regulatory network in the ATCC42981_R hybrid compared to Z. rouxii.
Conclusion
This study revised the pattern of MTL loci in allodiploid strain ATCC42981_R. By taking advantage from ONT technology, we captured a novel MATa cassette which did not correspond to the expected T and P counterparts, providing preliminary evidences that a third haplotype contributes to this genome. The differences between ATCC42981_R and JCM22060 support that MTLs are a root source of genetic variation, leading to novel chimeric MTL cassettes, different cell identities and, consequently, distinct phenotypic behaviors. While further researches are required to investigate mechanisms responsible of this extensive MTL reshaping, our results confirm that these yeast stocks are genetically unstable (Watanabe et al., 2013; Bizzarri et al., 2018). We also demonstrated how HMR/HML silencing is crucial to establish the cell identity, as leakage in HML silencing prevents allodiploid MATαPΔ cells to behave like haploids. How allodiploid cell modulates a2 expression via α2 transcriptional factor represents an unexplored regulatory circuit that has to be investigated in future.
Data Availability
The whole genome sequence datasets generated for this study can be found under the NCBI BioProject number PRJEB26771.
Author Contributions
SC and LS contributed conception and design of the study. MB conducted the experiments described in this study. LB contributed to in vitro PCR validation and asg expression. HS and MD contributed to deletion mutant construction. SC and LP performed bioinformatic analysis of the whole genome sequence data. LS wrote the manuscript. SC and MB contributed to draft revision. All authors read and approved the final manuscript.
Funding
LS was partially supported by the Italian Ministry of Education, University and Research (MIUR), within the framework of the Italian National Grant for Fundamental Research (FFABR 2017). The work of HS group was supported by the Ministry of Education, Youth and Sports of CR (MEYS) within the LQ1604 National Sustainability Program II (Project BIOCEV-FAR) and by the project “BIOCEV” (CZ.1.05/1.1.00/02.0109).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Prof. Paolo Giudici for his valuable comments and to Marcello Benevelli for help in mating assays.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00137/full#supplementary-material
Footnotes
References
Albertin, W., and Marullo, P. (2012). Polyploidy in fungi: evolution after whole-genome duplication. Proc. R. Soc. B. Biol. Sci. 279, 2497–2509. doi: 10.1098/rspb.2012.0434
Baker, C. R., Booth, L. N., Sorrells, T. R., and Johnson, A. D. (2012). Protein modularity, cooperative binding, and hybrid regulatory states underlie transcriptional network diversification. Cell 151, 80–95. doi: 10.1016/j.cell.2012.08.018
Baker, C. R., Tuch, B. B., and Johnson, A. D. (2011). Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc. Natl. Acad. Sci. U.S.A. 108, 7493–7498. doi: 10.1073/pnas.1019177108
Bizzarri, M., Cassanelli, S., Pryszcz, L. P., Gawor, J., Gromadka, R., and Solieri, L. (2018). Draft genome sequences of the highly halotolerant strain Zygosaccharomyces rouxii ATCC 42981 and the novel allodiploid strain Zygosaccharomyces sapae ATB301T obtained using the MinION platform. Microbiol. Res. Announc. 7:e874-18. doi: 10.1128/MRA.00874-18
Bizzarri, M., Cassanelli, S., and Solieri, L. (2017). Mating-type switching in CBS 732T derived subcultures unveils potential genetic and phenotypic novelties in haploid Zygosaccharomyces rouxii. FEMS Microbiol. Lett. 365, 1–8. doi: 10.1093/femsle/fnx263
Bizzarri, M., Giudici, P., Cassanelli, S., and Solieri, L. (2016). Chimeric sex-determining chromosomal regions and dysregulation of cell-type identity in a sterile Zygosaccharomyces allodiploid yeast. PLoS One 11:e0152558. doi: 10.1371/journal.pone.0152558
Boisnard, S., Li, Y. Z., Arnaise, S., Sequeira, G., Raffoux, X., Enache-Angoulvant, A., et al. (2015). Efficient mating-type switching in Candida glabrata induces cell death. PLoS One 10:e0140990. doi: 10.1371/journal.pone.0140990
Bond, U., Neal, C., Donnelly, D., and James, T. C. (2004). Aneuploidy and copy number breakpoints in the genome of lager yeasts mapped by microarray hybridisation. Curr. Genet. 45, 360–370. doi: 10.1007/s00294-004-0504-x
Booth, L. N., Tuch, B. B., and Johnson, A. D. (2010). Intercalation of a new tier of transcription regulation into an ancient circuit. Nature 468, 959–963. doi: 10.1038/nature09560
Boynton, P. J., Janzen, T., and Greig, D. (2018). Modeling the contributions of chromosome segregation errors and aneuploidy to Saccharomyces hybrid sterility. Yeast 35, 85–98. doi: 10.1002/yea.3282
Braun-Galleani, S., Ortiz-Merino, R. A., Wu, Q., Xu, Y., and Wolfe, K. H. (2018). Zygosaccharomyces pseudobailii, another yeast interspecies hybrid that regained fertility by damaging one of its MAT loci. FEMS Yeast Res. 18:foy079. doi: 10.1093/femsyr/foy079
Campbell, M. A., Ganley, A. R., Gabaldon, T., and Cox, M. P. (2016). The case of the missing ancient fungal polyploids. Am. Nat. 188, 602–614. doi: 10.1086/688763
Chien, C. T., Buck, S., Sternglanz, R., and Shore, D. (1993). Targeting of SIR1 protein establishes transcriptional silencing at HM loci and telomeres in yeast. Cell 75, 531–534. doi: 10.1016/0092-8674(93)90387-6
Del Angel, D. V., Hjerde, E., Sterck, L., Capella-Gutierrez, S., Notredame, C., Pettersson, O. V., et al. (2018). Ten steps to get started in genome assembly and annotation. F1000Research 7:ELIXIR-148. doi: 10.12688/f1000research.13598.1
Dujon, B. A., and Louis, E. J. (2017). Genome diversity and evolution in the budding yeasts (Saccharomycotina). Genetics 206, 717–750. doi: 10.1534/genetics.116.199216
Ellahi, A., and Rine, J. (2016). Evolution and functional trajectory of Sir1 in gene silencing. Mol. Cell Biol. 36, 1164–1179. doi: 10.1128/MCB.01013-15
Fabre, E., Muller, H., Therizols, P., Lafontaine, I., Dujon, B., and Fairhead, C. (2005). Comparative genomics in hemiascomycete yeasts: evolution of sex, silencing, and subtelomeres. Mol. Biol. Evol. 22, 856–873. doi: 10.1093/molbev/msi070
Gabaldón, T., Martin, T., Marcet-Houben, M., Durrens, P., Bolotin-Fukuhara, M., Lespinet, O., et al. (2013). Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14:623. doi: 10.1186/1471-2164-14-623
Gallagher, J. E. G., Babiarz, J. E., Teytelman, L., Wolfe, K. H., and Rine, J. (2009). Elaboration, diversification and regulation of the Sir1 family of silencing proteins in Saccharomyces. Genetics 181, 1477–1491. doi: 10.1534/genetics.108.099663
Gordon, J. L., Armisén, D., Proux-Wéra, E., ÓhÉigeartaigh, S. S., Byrne, K. P., and Wolfe, K. H. (2011). Evolutionary erosion of yeast sex chromosomes by mating-type switching accidents. Proc. Natl. Acad. Sci. U.S.A. 108, 20024–20029. doi: 10.1073/pnas.1112808108
Gordon, J. L., and Wolfe, K. H. (2008). Recent allopolyploid origin of Zygosaccharomyces rouxii strain ATCC 42981. Yeast 25, 449–456. doi: 10.1002/yea.1598
Greig, D., Borts, R. H., Louis, E. J., and Travisano, M. (2002). Epistasis and hybrid sterility in Saccharomyces. Proc. Biol. Sci. 269, 1167–1171. doi: 10.1098/rspb.2002.1989
Güldener, U., Heck, S., Fiedler, T., Beinhauer, J., and Hegemann, J. H. (1996). A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24, 2519–2524. doi: 10.1093/nar/24.13.2519
Haber, J. E. (2012). Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191, 33–64. doi: 10.1534/genetics.111.134577
Hewitt, S. K., Donaldson, I. J., Lovell, S. C., and Delneri, D. (2014). Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS One 9:e92203. doi: 10.1371/journal.pone.0092203
Hickman, M. A., Froyd, C. A., and Rusche, L. N. (2011). Reinventing heterochromatin in budding yeasts: Sir2 and the origin recognition complex take center stage. Eukaryot. Cell 10, 1183–1192. doi: 10.1128/EC.05123-11
Hoffman, C. S. and Winston, F. (1987). A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 51, 267–272. doi: 10.1016/0378-1119(87)90131-4
Istace, B., Friedrich, A., d’Agata, L., Faye, S., Payen, E., Beluche, O., et al. (2017). De novo assembly and population genomic survey of natural yeast isolates with the oxford nanopore MinION sequencer. Gigascience 6, 1–13. doi: 10.1093/gigascience/giw018
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., and Sasani, T. A. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. doi: 10.1038/nbt.4060
Jain, M., Olsen, H. E., Paten, B., and Akeson, M. (2016). The oxford nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17:239. doi: 10.1186/s13059-016-1103-0
James, S. A., and Stratford, M. (2011). “Zygosaccharomyces Barker (1901),” in The Yeasts, a Taxonomic Study, eds C. P. Kurtzman, J. W. Fell, and T. Boekhout (London: Elsevier), 937–947.
Jansen, H. J., Liem, M., Jong-Raadsen, S. A., Dufour, S., Weltzien, F. A., Swinkels, W., et al. (2017). Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 7:7213. doi: 10.1038/s41598-017-07650-6
Karanyicz, E., Antunovics, Z., Kallai, Z., and Sipiczki, M. (2017). Non-introgressive genome chimerisation by malsegregation in autodiploidised allotetraploids during meiosis of Saccharomyces kudriavzevii x Saccharomyces uvarum hybrids. Appl. Microbiol. Biotechnol. 2017, 4617–4633. doi: 10.1007/s00253-017-8274-9
Lee, H., Chou, J., Cheong, L., Chang, N. H., Yang, S. Y., and Leu, J. Y. (2008). Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 135, 1065–1073. doi: 10.1016/j.cell.2008.10.047
Liti, G., Barton, D. B. H., and Louis, E. J. (2006). Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174, 839–850. doi: 10.1534/genetics.106.062166
Lõoke, M., Kristjuhan, K., and Kristjuhan, A. (2011). Extraction of genomic dna from yeasts for pcr-based applications. BioTechniques 50, 325–328. doi: 10.2144/000113672
Marcet-Houben, M., and Gabaldón, T. (2015). Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker’s yeast lineage. PLoS Biol. 13:e1002220. doi: 10.1371/journal.pbio.1002220
Monerawela, C., and Bond, U. (2017). Recombination sites on hybrid chromosomes in Saccharomyces pastorianus share common sequence motifs and define a complex evolutionary relationship between group I and II lager yeasts. FEMS Yeast Res. 17:fox047. doi: 10.1093/femsyr/fox047
Mori, H., and Onishi, H. (1967). Diploid hybridization in a heterothallic haploid yeast, Saccharomyces rouxii. Appl. Microbiol. 15, 928–934.
Muller, H., Hennequin, C., Gallaud, J., Dujon, B., and Fairhead, C. (2008). The asexual yeast Candida glabrata maintains distinct a and α haploid mating-types. Eukaryot. Cell 7, 848–858. doi: 10.1128/EC.00456-07
Ner, S. S., and Smith, M. (1989). Role of intron splicing in the function of the MATa1 gene of Saccharomyces cerevisiae. Mol. Cell. Biol. 9, 4613–4620. doi: 10.1128/mcb.9.11.4613
Ohno, S. (1970). Evolution by Gene Duplication. New York, NY: Springer-Verlag. doi: 10.1007/978-3-642-86659-3
Ortiz-Merino, R. A., Kuanyshev, N., Braun-Galleani, S., Byrne, K. P., Porro, D., and Branduardi, P. (2017). Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol. 15:e2002128. doi: 10.1371/journal.pbio.2002128
Pfliegler, P. W., Antunovics, Z., and Sipiczki, M. (2012). Double sterility barrier between Saccharomyces species and its breakdown in allopolyploid hybrids by chromosome loss. FEMS Yeast Res. 12, 703–718. doi: 10.1111/j.1567-1364.2012.00820.x
Pribylova, L., and Sychrova, H. (2003). Efficient transformation of the osmotolerant yeast Zygosaccharomyces rouxii by electroporation. J. Microbiol. Methods 55, 481–484. doi: 10.1016/S0167-7012(03)00197-0
Priyam, A., Woodcroft, B. J., Rai, V., Munagala, A., Moghul, I., Ter, F., et al. (2015). Sequenceserver: a modern graphical user interface for custom BLAST databases. bioRxiv [Preprint]. doi: 10.1101/033142
Proux-Wéra, E., Armisén, D., Byrne, K. P., and Wolfe, K. H. (2012). A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach. BMC Bioinformatics 13:237. doi: 10.1186/1471-2105-13-237
Pryszcz, L., and Gabaldón, T. (2016). Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44:e113. doi: 10.1093/nar/gkw294
Rajeh, A., Lv, J., and Lin, Z. (2018). Heterogeneous rates of genome rearrangement contributed to the disparity of species richness in Ascomycota. BMC Genomics 19:282. doi: 10.1186/s12864-018-4683-0
Roberts, C., and Van der Walt, J. P. (1959). The life cycle of Kluyveromyces polysporus. C. R. Trav. Lab. Carlsberg Chim. 31, 129–148.
Sambrook, J., Maniatis, T., and Fritsch, E. (1989). Molecular Cloning: A Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory.
Sato, A., Matsushima, K., Oshima, K., Hattori, M., and Koyama, Y. (2017). Draft genome sequencing of the highly halotolerant and allopolyploid yeast Zygosaccharomyces rouxii NBRC 1876. Genome Announc. 5:e1610-16. doi: 10.1128/genomeA.01610-16
Sievers, F., and Higgins, D. G. (2014). “Clustal omega, accurate alignment of very large numbers of sequences,” in Multiple Sequence Alignment Methods, ed. D. J. Russell (Totowa, NJ: Humana Press). doi: 10.1007/978-1-62703-646-7_6
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv35
Sipiczki, M. (2008). Interspecies hybridization and recombination in Saccharomyces wine yeasts. FEMS Yeast Res. 8, 996–1007. doi: 10.1111/j.1567-1364.2008.00369.x
Slater, G. S. C., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31
Solieri, L., Cassanelli, S., Croce, M. A., and Giudici, P. (2008). Genome size and ploidy level: new insights for elucidating relationships in Zygosaccharomyces species. Fungal Genet. Biol. 45, 1582–1590. doi: 10.1016/j.fgb.2008.10.001
Solieri, L., Dakal, T. C., Giudici, P., and Cassanelli, S. (2014). Sex-determination system in the diploid yeast Zygosaccharomyces sapae. G3 4, 1011–1025. doi: 10.1534/g3.114.010405
Solieri, L., Vezzani, V., Cassanelli, S., Dakal, T. C., Pazzini, J., and Giudici, P. (2016). Differential hypersaline stress response in Zygosaccharomyces rouxii complex yeasts: a physiological and transcriptional study. FEMS Yeast Res. 16:fow063. doi: 10.1093/femsyr/fow063
Souciet, J. L., Dujon, B., and Gaillardin, C. (2009). Comparative genomics of protoploid Saccharomycetaceae. Genome Res. 19, 1696–1709. doi: 10.1101/gr.091546.109
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Treangen, T. J., and Salzberg, S. L. (2012). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13:36. doi: 10.1038/nrg3117
Tsong, A. E., Miller, M. G., Raisner, R. M., and Johnson, A. D. (2003). Evolution of a combinatorial transcriptional circuit: a case study in yeasts. Cell 115, 389–399. doi: 10.1016/S0092-8674(03)00885-7
Tsong, A. E., Tuch, B. B., Li, H., and Johnson, A. D. (2006). Evolution of alternative transcriptional circuits with identical logic. Nature 443, 415–420. doi: 10.1038/nature05099
Vaser, R., Sovic′, I., Nagarajan, N., and Šikic′, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, Z., Ye, S., Li, J., Zheng, B., Bao, M., and Ning, G. (2011). Fusion primer and nested integrated PCR (FPNI-PCR): a new high-efficiency strategy for rapid chromosome walking or flanking sequence cloning. BMC Biotechnol. 11:109. doi: 10.1186/1472-6750-11-109
Watanabe, J., Uehara, K., and Mogi, Y. (2013). Diversity of mating-type chromosome structures in the yeast Zygosaccharomyces rouxii caused by ectopic exchanges between MAT-Like loci. PLoS One 8:e62121. doi: 10.1371/journal.pone.0062121
Watanabe, J., Uehara, K., Mogi, Y., and Tsukioka, Y. (2017). Mechanism for restoration of fertility in hybrid Zygosaccharomyces rouxii generated by interspecies hybridization. Appl. Environ. Microbiol. 83:AEM.1187-17. doi: 10.1128/AEM.01187-17
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Wolfe, K. H. (2015). Origin of the yeast whole-genome duplication. PLoS Biol. 13:e1002221. doi: 10.1371/journal.pbio.1002221
Wolfe, K. H., Armisen, D., Proux-Wera, E., OhEigeartaigh, S. S., Azam, H., Gordon, J. L., et al. (2015). Clade- and species-specific features of genome evolution in the Saccharomycetaceae. FEMS Yeast Res. 15:fov035. doi: 10.1093/femsyr/fov035
Ye, C., Hill, C. M., Wu, S., Ruan, J., and Ma, Z. S. (2016). DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 6:31900. doi: 10.1038/srep31900
Yue, J. X., Li, J., Aigrain, L., Hallin, J., Persson, K., Oliver, K., et al. (2017). Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat. Genet. 49, 913–924. doi: 10.1093/bioinformatics/bty614
Zill, O. A., Scannell, D., Teytelman, L., and Rine, J. (2010). Co-evolution of transcriptional silencing proteins and the DNA elements specifying their assembly. PLoS Biol. 8:e1000550. doi: 10.1371/journal.pbio.1000550
Zill, O. A., Scannell, D. R., Kuei, J., Sadhu, M., and Rine, J. (2012). Evolutionary analysis of heterochromatin protein compatibility by interspecies complementation in Saccharomyces. Genetics 192, 1001–1014. doi: 10.1534/genetics.112.141549
Keywords: mating-type, MinION, sexual cycle, Zygosaccharomyces, chimeric loci, interspecies hybridization, yeast
Citation: Bizzarri M, Cassanelli S, Bartolini L, Pryszcz LP, Dušková M, Sychrová H and Solieri L (2019) Interplay of Chimeric Mating-Type Loci Impairs Fertility Rescue and Accounts for Intra-Strain Variability in Zygosaccharomyces rouxii Interspecies Hybrid ATCC42981. Front. Genet. 10:137. doi: 10.3389/fgene.2019.00137
Received: 01 October 2018; Accepted: 11 February 2019;
Published: 01 March 2019.
Edited by:
Isabel Sá-Correia, University of Lisbon, PortugalReviewed by:
Paola Branduardi, Università degli Studi di Milano-Bicocca, ItalyGeraldine Butler, University College Dublin, Ireland
Copyright © 2019 Bizzarri, Cassanelli, Bartolini, Pryszcz, Dušková, Sychrová and Solieri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Solieri, bGlzYS5zb2xpZXJpQHVuaW1vcmUuaXQ=