 Caihua Zhang1†Wenchun Luo1†
Caihua Zhang1†Wenchun Luo1† Yanda Li2Xu Zhang1Xiaotao Bai1Zhimin Niu1Xiao Zhang1
Yanda Li2Xu Zhang1Xiaotao Bai1Zhimin Niu1Xiao Zhang1 Zhijun Li3
Zhijun Li3 Dongshi Wan1*
Dongshi Wan1*- 1State Key Laboratory of Grassland Agro-Ecosystem, School of Life Sciences, Lanzhou University, Lanzhou, China
- 2Computer Science and Engineering Department, University of California, San Diego, La Jolla, CA, United States
- 3Xinjiang Production & Construction Corps, Key Laboratory of Protection and Utilization of Biological Resources in Tarim Basin, College of Life Sciences, Tarim University, Xinjiang, China
As a major abiotic stress, soil salinity limits seed germination and plant growth, development and production. Seed germination is highly related not only to the seedlings survival rate but also subsequent vegetative growth. Populus euphratica and P. pruinosa are closely related species that show a distinguished adaptability to salinity stress. In this study, we performed an integrative transcriptome analyses of three seed germination phases from P. euphratica and P. pruinosa under salt stress. A two-dimensional data set of this study provides a comprehensive view of the dynamic biochemical processes that underpin seed germination and salt tolerance. Our analysis identified 12831 differentially expressed genes (DEGs) for seed germination processes and 8071 DEGs for salt tolerance in the two species. Furthermore, we identified the expression profiles and main pathways in each growth phase. For seed germination, a large number of DEGs, including those involved in energy production and hormonal regulation pathways, were transiently and specifically induced in the late phase. In the comparison of salt tolerance between the two species, the flavonoid and brassinosteroid pathways were significantly enriched. More specifically, in the flavonoid pathway, FLS and F3′5′H exhibited significant differential expression. In the brassinosteroid pathway, the expression levels of DWF4, BR6OX2 and ROT3 were notably higher in P. pruinosa than in P. euphratica. Our results describe transcript dynamics and highlight secondary metabolite pathways involved in the response to salt stress during the seed germination of two desert poplars.
Introduction
Soil salinization is caused by many factors and conditions, such as unsuitable irrigation practices, irrigation with salinized water and seasonal effects (Ottow et al., 2005; Annunziata et al., 2017). As one of the most prominent abiotic stresses, salinity stress is considered the greatest threat to crop production and environmental conservation (Ottow et al., 2005; Arbona et al., 2013). Salinity stress leads to osmotic and ionic stress, which reduces cell and tissue expansion, and to ion excesses that changes the osmotic potential of plant cells and induce nutritional imbalances (Munns, 2002), sequentially affecting plant growth, development and survival (Carillo et al., 2019). To solve the serious problem of soil salinization, various efforts have been made; these efforts mainly concentrate on enhancing the salt resistance of economically important salt-sensitive plants through traditional breeding and biotechnological approaches or the use of plants that naturally display high salt tolerance (Flowers, 2004).
Populus euphratica and its sister species P. pruinosa are naturally distributed in China’s western desert region; due to their extraordinary adaptability to desert environments (Chen et al., 2002; Hukin et al., 2005), both species are also called desert poplars. The distinguished adaptability of these species provides beneficial ecological effects in northwest China. Currently, both poplars are considered important genetic resources in tree breeding and in research elucidating physiological and molecular mechanisms involving stress tolerance in trees (Tuskan et al., 2006; Wullschleger et al., 2013). As the genome data of P. euphratica becomes available (Ma et al., 2013), the resistance mechanism of both poplars have been revealed at multiple levels, e.g., a phylogenetic analysis shows that the two species diverged approximately 1–2 million years ago (Wang J. et al., 2011), and ancient polymorphisms contributed to their genomic divergence (Ma et al., 2018). In addition to leaf morphology and leaf trichome differences (Ma et al., 2016), both poplars occupy different ecological habitats. P. pruinosa prefers desert areas with high ground water levels, while P. euphratica can grow in desert areas where the groundwater levels are low (Ottow et al., 2005). These differences between the sister poplars result from differences in genetic mechanisms, such as the adaptive evolution of genes (Ma et al., 2013; Zhang et al., 2014) and gene expression divergences among orthologs (Qiu et al., 2011; Zhang et al., 2013).
Seed germination is constrained significantly by soil salinity (Kaya et al., 2003). Soil salinity creates an osmotic potential around the outside of seeds, resulting in decreased water uptake during germination and an increase in the excessive uptake of ions, which causes the toxic effects of Na+ and Cl- ions to seeds (Murillo-Amador et al., 2002; Khajeh-Hosseini et al., 2003). Therefore, salt stress can inhibit or delay seed germination (Almansouri et al., 2001). However, studies focusing on the genetic mechanism of seed germination under salt stress are limited.
Seed germination begins with imbibition and ends with the embryonic axis breaking through the seed coat (Bewley et al., 2012). Seed germination includes three phases. In phase I, the seed begins to expand, with a rapidly increasing water content. Then, the seed enters a plateau phase (phase II), in which the water uptake remains at a stable level. In phase III, the water uptake increases rapidly. Phase III ceases as the embryonic axis breaks through the seed coat, upon which seed germination is complete (Bewley, 1997). Energy production and respiration play important roles in the seed germination process. In the early stage, anaerobic respiration provides the main energy source, and then respiratory activity increases with oxygen uptake. Subsequently, plant hormones, such as gibberellins (GA), abscisic acid (ABA), brassinosteroids (BRs), ethylene, auxins, and cytokinins, are widely involved in determining the physiological state of a seed and regulating the germination process (Kucera et al., 2005; Holdsworth et al., 2008; Müller et al., 2009; North et al., 2010). Furthermore, numerous complex networks, including those related to gene expression and regulation commanded by various transcription factors (Chen et al., 2002), ion transporting processes, such as NHX (Na+/H+ antiporter), SOS (salt overly sensitive) (Zhu, 2001), and HKT (high-affinity K+ transporter) (Ren et al., 2005) processes, and secondary metabolism are all involved in the response to salt stress (Bewley, 1982, 1997; Bewley and Black, 1984; Biligetu et al., 2011). Recently, transcriptomic analyses of several poplar species under various stresses have been extensively conducted (Chen et al., 2002, 2012; Brinker et al., 2010; Janz et al., 2010; Qiu et al., 2011; Wang J. et al., 2011; Ma et al., 2013; Ziemann et al., 2013). These data provide us with a basic understanding of seed germination. However, detailed transcriptomic dynamics and physiological mechanisms under salt stress during seed germination have not yet been revealed. Such an exploration might be useful to identify the genes that improve poplar salt tolerance by biotechnological manipulation. Moreover, most genes associated with seed germination are poorly understood due to the complexity of the germination process.
Here, we present a comprehensive transcriptome study encompassing the whole process of seed germination for two species under salt stress, which provides a valuable gene resource for genetic manipulation in poplar breeding.
Materials and Methods
Plant Materials and Growth Conditions
We collected three replicate samples of the seeds of the two studied species from a total of 18 trees in the Tarim Basin (Xinjiang, China) and stored the seeds at 4°C. For germination, vigorous seeds were imbibed in distilled water (control), 0.2%, 0.4%, 0.6%, 0.8%, 1.0%, 1.2%, 1.4%, and 1.6% NaCl, and then germinated on wet filter paper in 9 cm diameter Petri dishes in a plant growth incubator (21°C 200 μmol m-2s-1, 16 h: 8 h light/dark photoperiod). The germination rate was measured using the Chinese national standard test (GB2772-1999). Each sample contained 50 seeds and had three replicates (Wang et al., 2013). The germinating seeds were scanned and photographed using a stereo microscope (Nikon SM Z1500, Japan) to record their morphology. The moisture content of the seed samples was measured in seeds oven dried at 75°C to a constant weight. The moisture content [g (g FW)-1] was calculated as [(FW-DW)/FW].
The percentage of seeds with two cotyledons turning green or with emerging radicles (>1 mm) was considered the germination rate (Wang Y. et al., 2011). For the germination percentage, counts were made until no additional germination was observed for 72 h (Bradford, 1990). To elucidate the threshold salinity for the two species under the salt treatments, we measured relative indexes, including GR, RGP, GT, GI, K, and RSH (Imit et al., 2015). For RNA isolation, the seeds were imbibed in 1.0% NaCl (to expose them to salt stress) and then removed after 4, 12, 24, 48, and 72 h for RNA preparation. The control samples were collected from dry seeds (0 h). We rapidly transferred all the samples to storage at -80°C before RNA extraction.
Reactive Oxygen Species (ROS) Level and Enzyme Activity Determination
For germination, seeds were imbibed in 0%, 0.4%, 0.8% and 1.0% NaCl as described above for 24 h. The levels of ROS, superoxide dismutase (SOD) and catalase (CAT) were measured using the standard protocol for the toolkit from Suzhou Comin Biotechnology.
Determination of RNA Extraction and Quality
Using the CTAB procedure, we extracted and purified total RNA three times from each of the sample set (Chang et al., 1993). The A260/A280 ratios of all the RNA samples ranged from 1.9 to 2.0. We examined the integrity of all RNA samples by the Agilent 2100 Bioanalyzer, and all the RNA integrity number (RIN) values ranged from 7 to 10.
cDNA Library Construction and RNA Sequencing
Construction of the cDNA library and RNA sequencing were performed by BIOMARKER (Beijing, China) using the Illumina (San Diego, CA, United States) Genome Analyzer platform in accordance with the manufacturer’s protocols. Paired-end sequencing was performed using a HiSeq 2500 (Illumina) platform with a read length of 125 bp.
Initial Mapping of Reads
We trimmed reads by removing adapter sequences, reads with too many (> 5%) unknown base calls (N), low-complexity sequences, and low-quality bases (i.e., sequences for which > 65% of the bases had a quality score ≤ 7). HISAT2 (Kim et al., 2015) was used to align all reads of the two species to the P. euphratica genome (Ma et al., 2013). Because the intrinsic divergence between the species could result in poor mapping, we did not map RNA-seq reads from the two species onto their own genomes. Next, StringTie (Pertea et al., 2015) created multiple isoforms of genes and estimated the gene expression levels (FPKM) (Trapnell et al., 2010) during assembly. To reduce the effects of background transcription, genes with FPKM ≥ 1 were used for the subsequent analysis. We calculated the Pearson correlation coefficient between biological replicates with R software using the expression data. The Pearson correlation calculated by R was used to evaluate repeatability between biological replicates.
Analysis of DEGs
We applied Ballgown (Frazee et al., 2015) to determine which transcripts were differentially expressed between two or more experiments, confirming their significance with an F-test. Ballgown allows both time-course and fixed-condition differential expression analyses. Therefore, two methods were employed to identify DEGs: (1) time as the main variable and species as the covariate; (2) species as the main variable and time as the covariate.
Hierarchical Clustering and Gene Co-expression Analysis
Using normalized log2 (FPKM+1) values, hierarchical clustering was completed with the pvclust package. Based on the normalized FPKM values, K-means clustering was performed by the K-Means/K-Medians Support Module (KMS) embedded in MEV 4.91.
Gene Functional Enrichment and qRT-PCR Analysis
GO and KEGG enrichment analyses of the two differently expressed transcript data sets were performed using a modified Chi-square test and Fisher’s exact test in R (p-value < 0.01 and false discovery rate < 0.05). Transcription levels of genes were quantified with a MX3005P Real-Time PCR Detection System (Agilent) based on the 2(-delta C(T)) method (Livak and Schmittgen, 2001). The experiment was performed in a 20 μL volume reaction system containing 10 μL 2 × SYBR Premix ExTaq (TaKaRa) with the intercalating dye SYBR Green. All primers were designed using PRIMER5.0 software and are listed in Supplementary Table S4.
Results
Physiological and Morphological Changes During Seed Germination
To evaluate the effect of salt stress on seed germination, the progress of seed germination has traditionally been divided into three phases based on seed water uptake during imbibition (Nonogaki et al., 2010). The first phase (phase I) occurs within the period of 0 h–36 h; the plateau phase (phase II) occurs within the period of 36 h–64 h; and phase III is continuous for 64 h–120 h during the transition to seedling growth (Figure 1A). We investigated the relationship between the germination rate and NaCl concentration. Seeds exhibiting high germination rates were selected and cultured in distilled water with a gradient of NaCl concentrations (0%, 0.2%, 0.4%, 0.6%, 0.8%, 1.0%, 1.2%, 1.4%, and 1.6%) (Figure 1B). With increasing NaCl concentration, seed germination was significantly inhibited. At different NaCl concentrations, the seed germination rates of P. pruinosa were higher than those of P. euphratica. The relative germination percentage of the two species exceeded 80% in the 0.4% NaCl solution, whereas the value approached zero in 2.4% NaCl. We hypothesized that when the relative germination percentages were 75%, 50%, and 25%, the corresponding salt concentrations could be considered suitable, critical and limiting for seed germination, respectively. In our study, for P. euphratica, the suitable, critical and limiting values were 0.602%, 1.161%, and 1.72%, respectively, whereas for P. pruinosa, these values were 0.599%, 1.179%, and 1.759%, respectively, suggesting that the threshold salinity for the two species differed. We also measured the germination index, salt tolerance index, relative salt harm rate and germination energy (Figure 1B). The average germination percentage, subordinate function values, and threshold salinity for P. pruinosa were higher than those for P. euphratica. Based on the results, a 1.0% NaCl concentration was selected for the subsequent salt treatment. The seed phenotypes of the two species were observed at four time points (Figure 1C). In the controls, the radicle emergence was completed within 12 h, and the hypocotyl and cotyledons emerged from the seed coat by 24 h. The cotyledons started to open by 36 h and opened fully and turned green by 48 h. In contrast, under the salt treatment, the seeds were still in the imbibition stage at 12 h, the radicle emergence stage was completed by 24 h, and the subsequent stages were all delayed by 12 h.
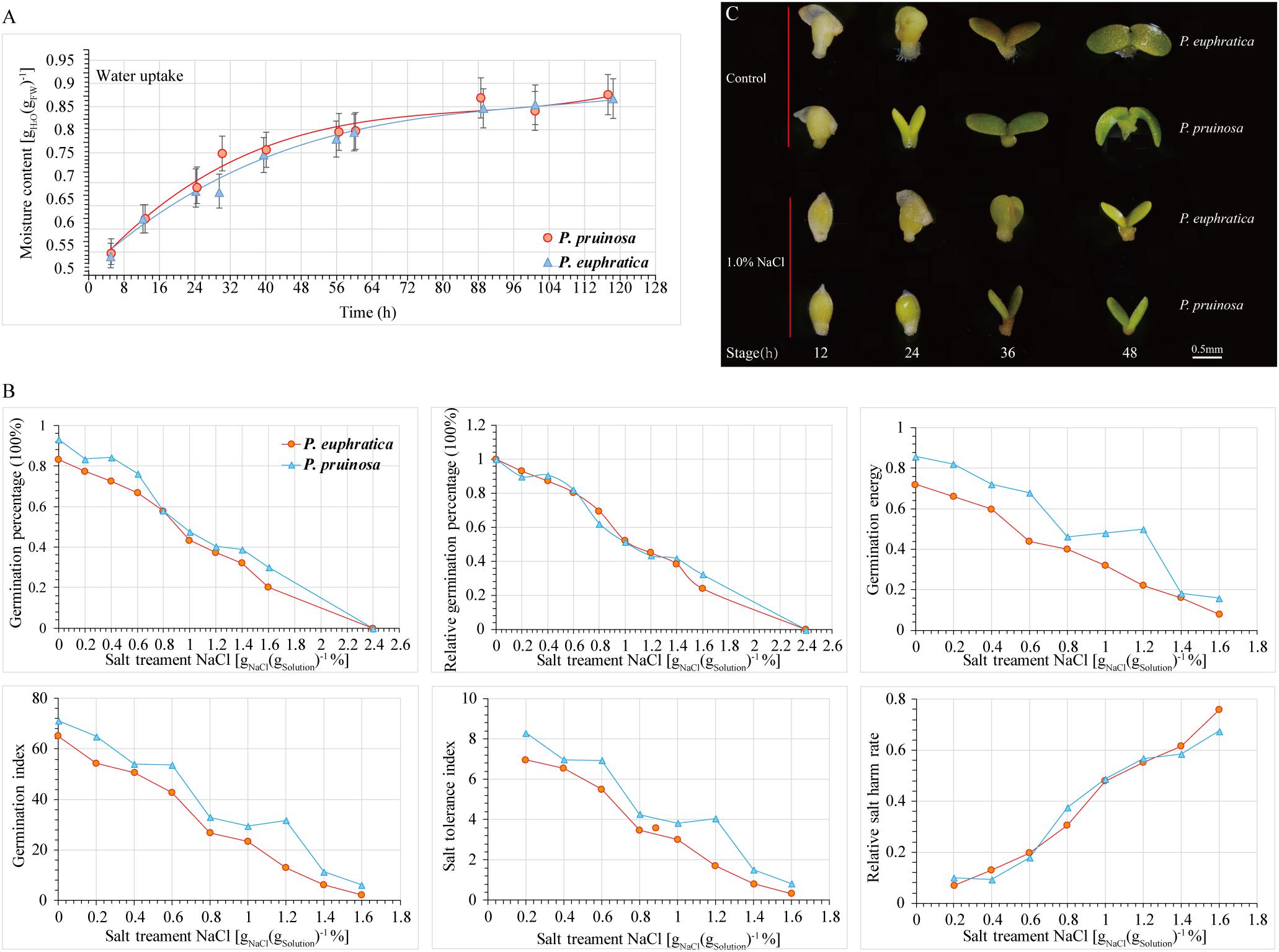
Figure 1. Comparison of physiological indices for P. euphratica and P. pruinosa seeds under salt stress treatment. (A) Time series of changes in moisture content of P. euphratica and P. pruinosa seeds during germination. (B) Effect of different NaCl concentrations on 6 physiological indexes of P. euphratica and P. pruinosa. (C) Morphological changes in P. euphratica and P. pruinosa seeds from 12 h to 48 h under salt stress and control. Bar = 0.5 mm.
RNA-Seq and Mapping of Illumina-Solexa Sequencing Reads
To systematically investigate the transcriptome dynamics of the two species’ seeds during germination under salt stress, we obtained 36 transcriptome samples. After removing low-quality sequences and trimming adapter sequences, 3–6 GB 125-bp paired-end clean reads were generated from each library (Supplementary Table S1). Approximately 80% of the reads matched the genome (Supplementary Table S2). All the genes and transcripts were reassembled (Table 1).
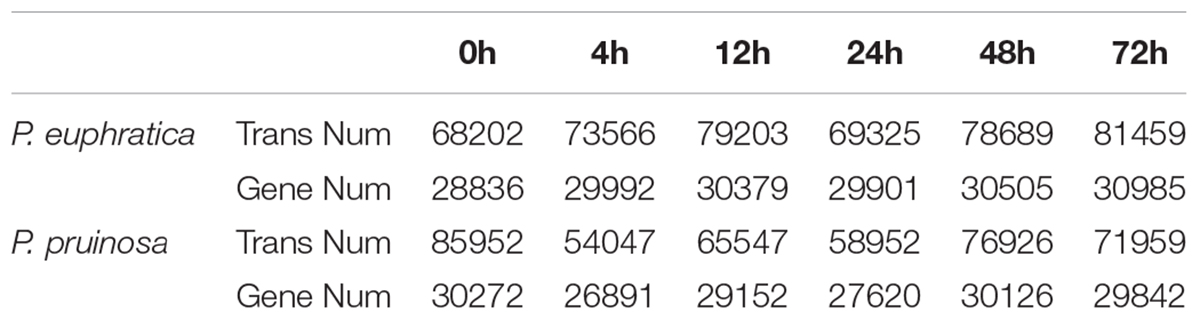
Table 1. Numbers of assembled genes and transcriptions.
In the detection of minor differential gene expression between time points and the two species, we used three biological replicates (Supplementary Figures S1, S2) to assess our data quality. The results showed that the expression values of biological replicates from the same samples were highly correlated (average R2 > 0.8). Among the genes, FPKM values exceeding 73% ranged from 1 to 100 at each time point (Supplementary Figure S3A). We used the average RPKM of the biological replicates as the expression quantity. To examine the divergence in gene expression between the two species under salt stress in more detail, we performed a hierarchical clustering analysis for all the expressed genes from P. euphratica and P. pruinosa at each time point using bootstrapping (Supplementary Figure S3B). The correlation dendrogram in Supplementary Figure S3B shows that samples collected at 0, 4 and 12 h clustered together, while those collected at 24, 48 and 72 h clustered into another group. This result indicates that one set of genes was activated during the early stress and germination stages, while there was another set of genes that was differentially expressed after 48 h. Therefore, based on a Spearman correlation analysis, the germinating seed samples from 0, 4 and 12 h were in the early phase, the seeds in the sample from 24 h were in the middle phase, and the seeds from 48 h to 72 h were in the late phase of the germination process.
Identification of DEGs, Temporal Expression Trends and GO Functional Enrichment
To identify global transcriptional changes that occurred during seed germination under salt stress, we confirmed the two data sets of DEGs, including 12831 DEGs and 19004 differentially expressed transcripts (DETs) for seed germination processes, and 8071 DEGs and 19000 DETs for salt tolerance, of two species. The DEGs were grouped into ten clusters (designated K1–K10) (Supplementary Figure S4) to examine the temporal expression trends of seed germination processes. To better understand the functions of the DEGs and obtain a view of functional transitions across time during seed germination in the two species, GO category enrichment analysis was performed (Supplementary Figure S5) to identify important events (biophysical, biochemical, and cellular processes) during seed germination.
According to the cluster analysis results, all the clusters of P. pruinosa and P. euphratica could be divided into early (0–12 h), middle (24 h), and late (48–72 h) phases (Supplementary Figure S3B). The early phase (represented by clusters K1 to K4) was strongly expressed at 0–12 h and gradually downregulated between 12 and 72 h in the two species. Based on the GO enrichment results, genes related to “adenyl nucleotide binding,” “adenyl ribonucleotide binding,” “purine ribonucleoside binding,” and “purine nucleoside binding” were increasingly expressed after imbibition (Supplementary Figure S5). Second, some genes associated with “structural molecule activity,” “structural constituent of cytoskeleton,” “intracellular non-membrane-bounded organelle,” “non-membrane-bounded organelle” and “cellular structure restoration” were enriched (Supplementary Figure S5). In addition, some genes associated with “ATP binding” were enriched (Supplementary Figure S5).
Genes in cluster K5 were highly expressed at 0 to 24 h and downregulated from 48 to 72 h. In the middle phase, the enriched genes included genes associated with “catalytic activity,” “mitochondrial part,” “nutrient reservoir activity,” “electron transport chain,” and “respiratory electron transport chain” (Supplementary Figure S5). Each of the five co-expression modules of the two species could be roughly categorized in the late (K6 to K10) phase. Transcripts of these modules were significantly upregulated during at least the last two time points. Many genes of this stage were typified by the enriched functions of “catabolic process,” “generation of precursor metabolites and energy,” “lipid metabolic process,” “carbohydrate metabolic process,” “hydrolase activity,” and “catalytic activity” (Supplementary Figure S5). Moreover, some upregulated genes of this stage were associated with “cellular nitrogen compound biosynthetic process” and “NAD binding” (Supplementary Figure S5).
Functional Regulatory Network Analysis (KEGG Pathway Enrichment) of Seed Germination Process DEGs
To further elucidate the seed germination process DEGs associated with biochemical pathways, we performed a KEGG pathway enrichment analysis. A total of 3847 out of 12831 DEGs enriched 328 pathways, and 58 pathways were significantly (p-value ≤ 0.01) overrepresented during seed germination (Supplementary Figure S6).
The early phase was exemplified by an observed statistically significant enrichment of “ribosome,” “proteasome,” and “protein processing in endoplasmic reticulum” pathways (Supplementary Figure S6). The middle phase exhibited the enrichment of “flavonoid biosynthesis,” “oxidative phosphorylation,” “ribosome,” “proteasome” and “spliceosome” pathways (Supplementary Figure S6). While many genes related to the metabolism of free amino acids were enriched in phase III (Supplementary Figure S6), most of the major pathways were enriched in the late phase, including “carbon metabolism,” “glycolysis/gluconeogenesis,” “starch and sucrose metabolism,” “oxidative phosphorylation,” “photosynthesis,” “porphyrin and chlorophyll metabolism,” and “carotenoid biosynthesis” (Supplementary Figure S6). “Oxidative phosphorylation” provides ATP for other metabolism pathways, such as mitochondrial repair and differentiation (Weitbrecht et al., 2011). The glyoxylate pathway contains a key step in the conversion of fatty acids to sucrose (Pritchard et al., 2002).
DEGs Related to Energy Production for Seed Germination Processes
During the preliminary phase, due to the inactivation of photosynthesis, the degradation of storage needed for energy production via processes such as glycolysis, the glyoxylate cycle, and the tricarboxylic acid (TCA) cycle, largely determines germination vigor. We defined the relative functional categories to be “carbon metabolism,” “glyoxylate and dicarboxylate metabolism,” “glycolysis/gluconeogenesis,” and “starch and sucrose metabolism.” Then, we identified the four major energy production processes, i.e., fermentation, the TCA cycle, glyoxylate and glycolysis, representing significantly overrepresented functional pathways, and we examined the expression patterns of the related DEGs (Figure 2). Here, ten gene families participating in the TCA cycle were differentially expressed over time in the two species. With respect to glycolysis, numerous gene families were upregulated, such as GALM, PFK, FBP, ALDO, GAPDH, and PK. In anaerobic respiration, three related gene families, PDC, ADH, and LDH, were all upregulated in the two species.
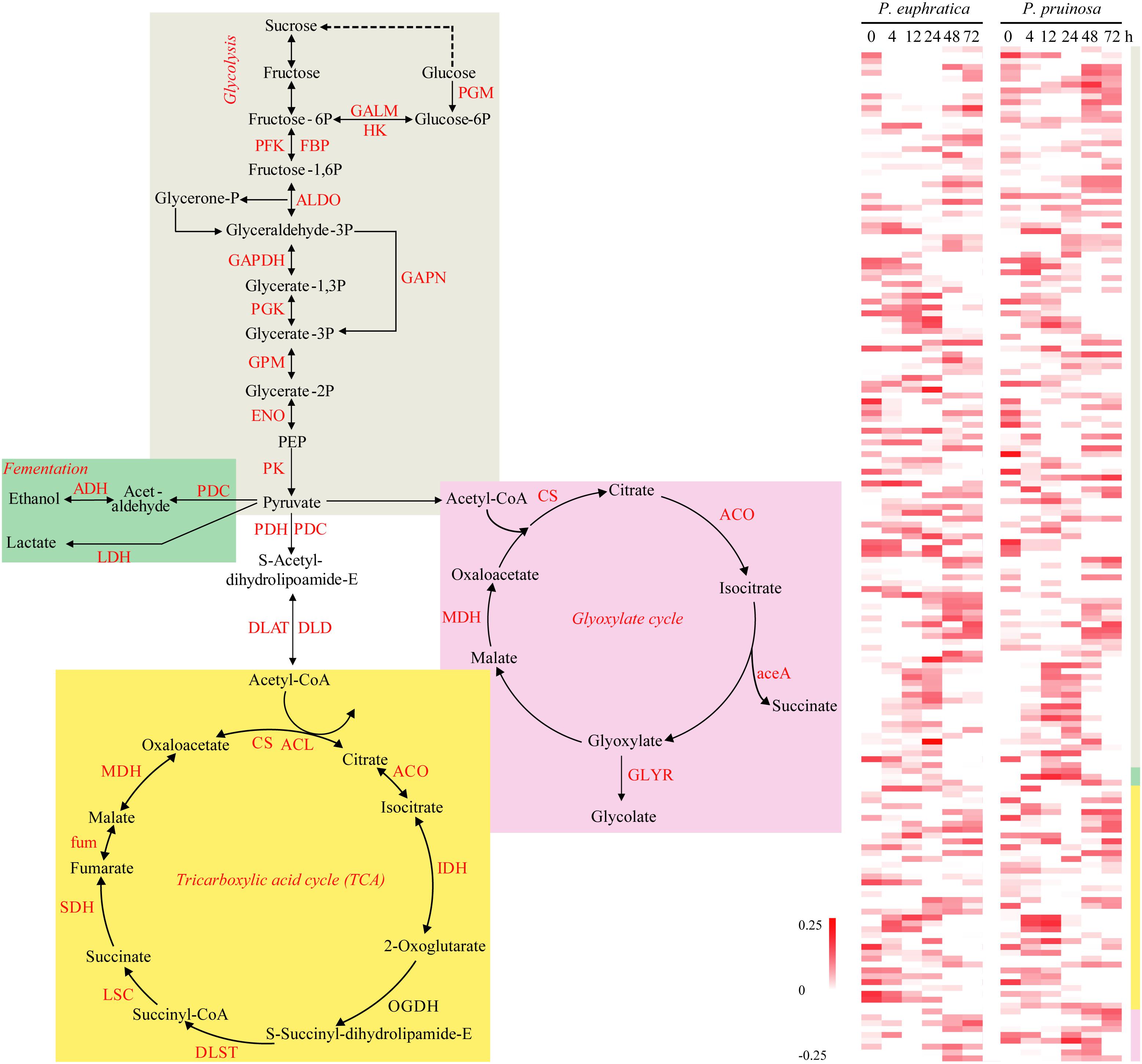
Figure 2. The energy production during seed germination and its possible relationship to early transcriptome changes in P. euphratica and P. pruinosa. Glycolysis (gray region), TCA cycle (yellow region) and fermentation respiration (green region) are the common pathways associated with ATP production. Glyoxylate (pink region) is also an important energy source for the germinating seed via lipid metabolism. The colors behind the heat map represent expression patterns of the counterpart genes in the four pathways. For details of abbreviations, see Supplementary Table S3.
Hormonal Regulation of Seed Germination in P. euphratica and P. pruinosa Under Salt Stress
In our study, 100 genes associated with “plant hormone signal transduction” were differentially expressed over time in the two species. We identified the key hormone signal transduction genes and further compared the expression profiles of the multi-step signaling pathways of ABA, GA and ethylene (Figure 3 and Supplementary Figure S7). Genes related to ABA signal transduction, e.g., PYL/PYR1, the negative regulator PP2C and the positive regulator SnRK2, exhibited similar expression patterns in the two poplars. The expression level of PP2C was high at 0 and 4 h but decreased after 12 h. In the GA signaling pathway, DEGs exhibited different regulatory expression patterns between the species during germination under salt stress. Specifically, the DELLA protein expression was upregulated from 0 to 12 h in P. euphratica but was continuously high level in P. pruinosa. GID1 was strongly upregulated during the middle and late phases of seed germination, while the expression of specific genes differed between the two species. Furthermore, most GA signal transcription-related genes were upregulated in the middle and late phases. We also identified the genes involved in ethylene signaling, as shown Figure 3. The expression pattern analysis indicated that most of the DEGs exhibited similar expression patterns in the two species for ETR and EIN3 (Supplementary Figure S7). CTR expression was upregulated in the late phase, while ETR was highly expressed after the early phase of germination in the two species.
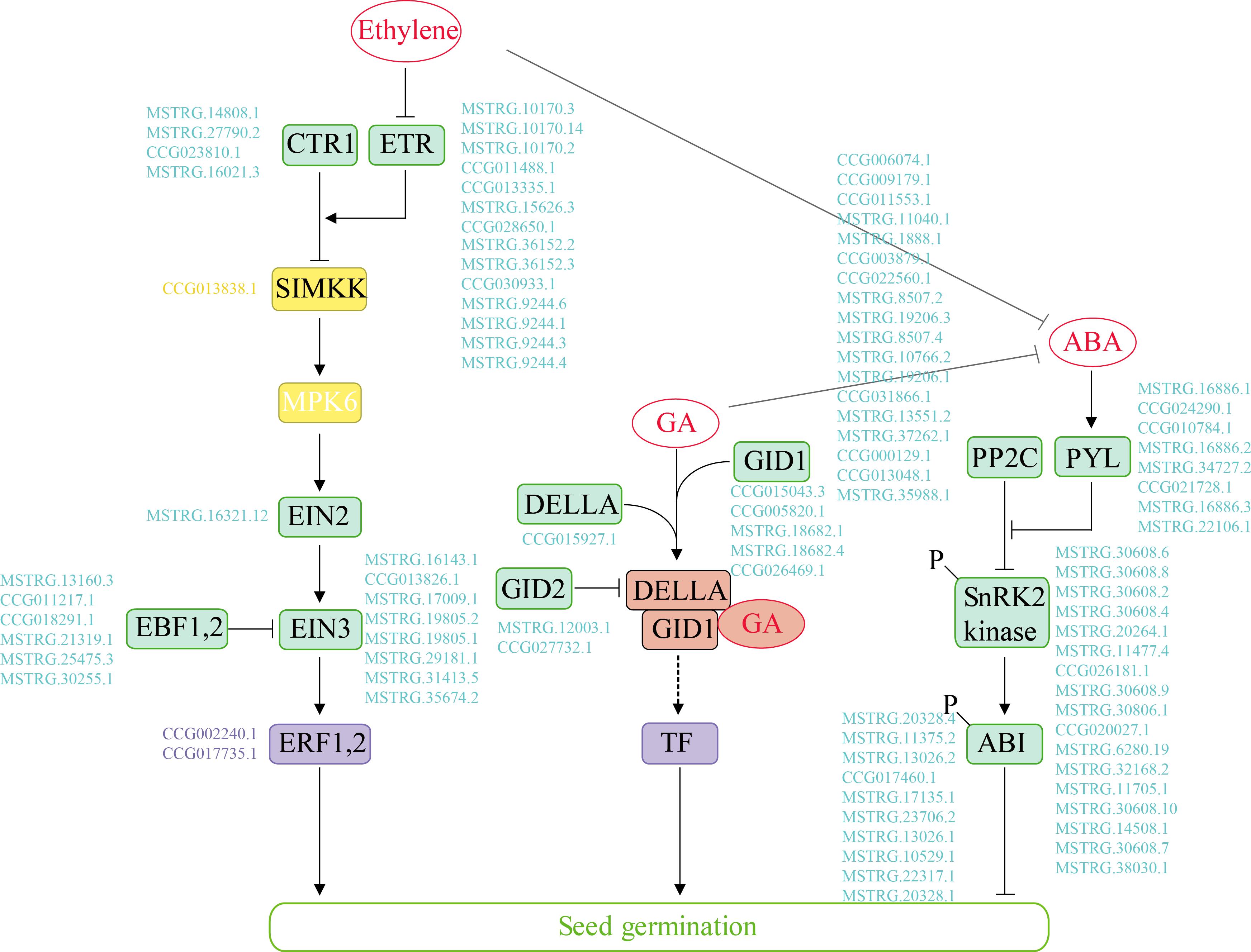
Figure 3. The Ethylene, GA and ABA major metabolic pathways in the germinating seeds of P. euphratica and P. pruinosa under salt stress. Promotion or inhibition is indicated by thick arrows and blocks, respectively. Enzymes and genes are shown. A MAPK cascade (yellow box) consisted of SIMKK and MPK6. Examples of transcription factors (purple box) and other regulators (green box) contributing to these processes are presented. Genes are indicated with blue letters. The GA-GID1-DELLA complex (red box) is recognized by an F-box protein (GID2) for ubiquitination in the SCF complex (included in the dotted arrow). For details of abbreviations, see Supplementary Table S3.
Transcription Factors and Genes Involved in Salt Responses During Seed Germination
Numerous transcription factors that regulate the response to salt stress in desert poplars have been identified (Trapnell et al., 2010). Here, a total of 1582 and 1573 expressed transcripts were categorized as transcription factors in P. euphratica and P. pruinosa, respectively (Figure 4A). In total, 1480 transcription factors were expressed in both species (Figure 4B). Relatively few genes displayed species-specific expression. MYBs, bZIPs, WRKY, and ERF, as key response factors to abiotic stresses, were all induced by salt stress (Figure 4B), and the changes in their expression dynamics may reveal their critical functions in response to salt stress (Yamaguchi-Shinozaki and Shinozaki, 2005). Furthermore, some proteins regulating Na+/H+ transport and controlling ion homeostasis, such as NHXs, SOS1, SOS2, SOS3, and HKTs, were induced by salt stress (Figure 4). These results confirm that the genes related to ion transport and chloride channels play vital roles in maintaining and re-establishing homeostasis in the cytoplasm (Hasegawa et al., 2000; Wang et al., 2008; Sun et al., 2009; Ye et al., 2009; Qiu et al., 2011). BCH1 and ZEP, which are involved in the biosynthesis of ABA, were highly upregulated in salt-stressed samples in the two species (Figure 4B). In addition, the expression of BADH and GolS, which are involved in critical solute biosynthesis processes that help plants maintain high osmotic pressure under salt stress. (Taji et al., 2002; Bartels and Sunkar, 2005), was induced by the salt treatment. Nevertheless, the expression patterns of genes responding to salt stress in P. euphratica were consistent with those in P. pruinosa, indicating there is extensive transcriptional consistency in the two species with respect to their responses to salt stress.
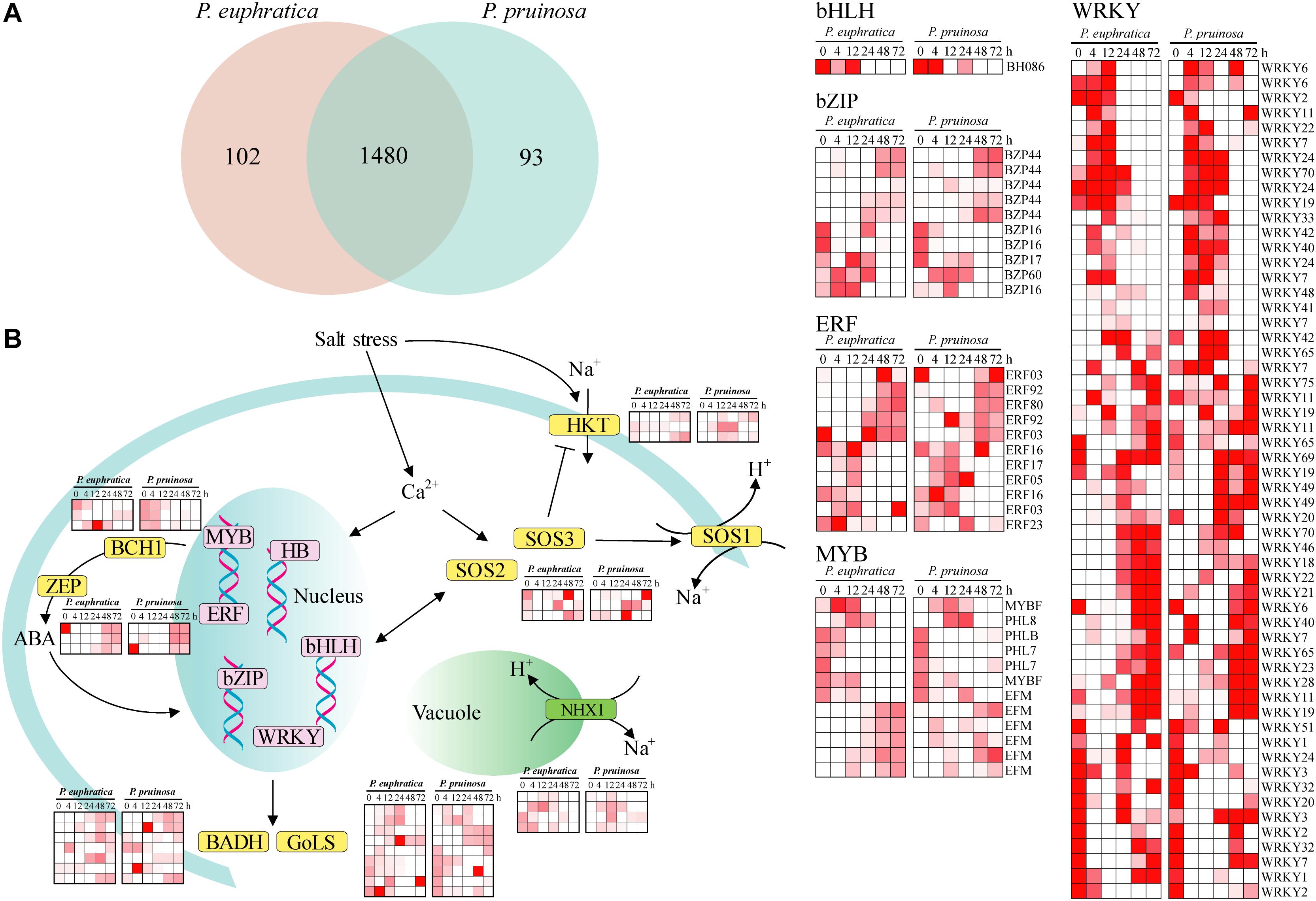
Figure 4. The current known components and relationships of transcription factors related to salt response in P. euphratica and P. pruinosa. (A) Venn diagram showing overlaps between the transcription factors of P. euphratica and P. pruinosa. (B) The expanded and positively selected genes in the salt response pathways of P. euphratica and P. pruinosa (yellow). Expression of the differently expressed transcription factors of P. euphratica and P. pruinosa (pink). The heatmap was generated from hierarchical cluster analysis of genes.
GO Functional Enrichment Between the Two Species Over the Time Series
The temporal expression trends of DEGs between the two species during germination were obviously different, suggesting that the two desert poplars might have evolved different gene expression patterns to adapt to different salty desert habitats. To obtain a better view of the functional differences between the species over the course of germination, GO enrichment analysis was employed, comparing the two species in two phases (the middle phase had only one DEG) (Supplementary Figure S8). The results indicated that in the early phase, 2766 DEGs were mainly enriched, and these DEGs were associated with the functional classifications “ribosomes,” “amide biosynthetic process,” “cellular macromolecule biosynthetic process,” “protein activity,” and “ATP binding.” In the late phase, 5305 enriched DEGs were associated with “response to stress,” “response to oxidative stress,” “response to abiotic stress,” “response to stimulus,” “ATP metabolic process,” “photosystem,” “photosynthesis,” “growth,” “developmental process,” “ion binding,” “calcium ion binding,” and “oxidoreductase activity.”
KEGG Functional Enrichment in the Two Species Over the Time Series
To further elucidate the different enriched biochemical pathways, DEGs of the two species were mapped into 352 pathways, 11 of which were significantly (p-value ≤ 0.01) enriched, including “flavonoid biosynthesis,” “stilbenoid, diarylheptanoid and gingerol biosynthesis,” “brassinosteroid biosynthesis,” “phenylpropanoid biosynthesis,” “diterpenoid biosynthesis,” and “monoterpenoid biosynthesis” (Supplementary Figure S9). The results indicate that many antioxidants, antioxidases and secondary metabolites are involved in the adaptation to salt stresses by these two species (Burritt and Mackenzie, 2003). The second metabolite in the flavonoid pathway plays vital roles in stress protection, but the biosynthesis of this metabolite is regulated by key enzymes (Winkel-Shirley, 2002). In this study, PAL was induced at 12 h in P. euphratica seeds and at 48 h in P. pruinosa seeds (Figure 5). CHS, whose five gene copies had different expression patterns between the two species, initiated flavonoid biosynthesis. Furthermore, the FLS expression in P. pruinosa was higher than that in P. euphratica. Specifically, FLS was highly expressed in the early phase in P. euphratica and was significantly and highly expressed during the seed germination process. In addition, the expression levels of F3′5′H and CHS in P. pruinosa were significantly higher than those in P. euphratica (Figure 5). F3H converts naringenin to dihydrokaempferol which is further converted to kaempferol and quercetin by FLS. The duplication of FLS may allow the ability to diversify the types and amounts of flavonols produced in different tissues and under different stresses(Winkel-Shirley, 2002).
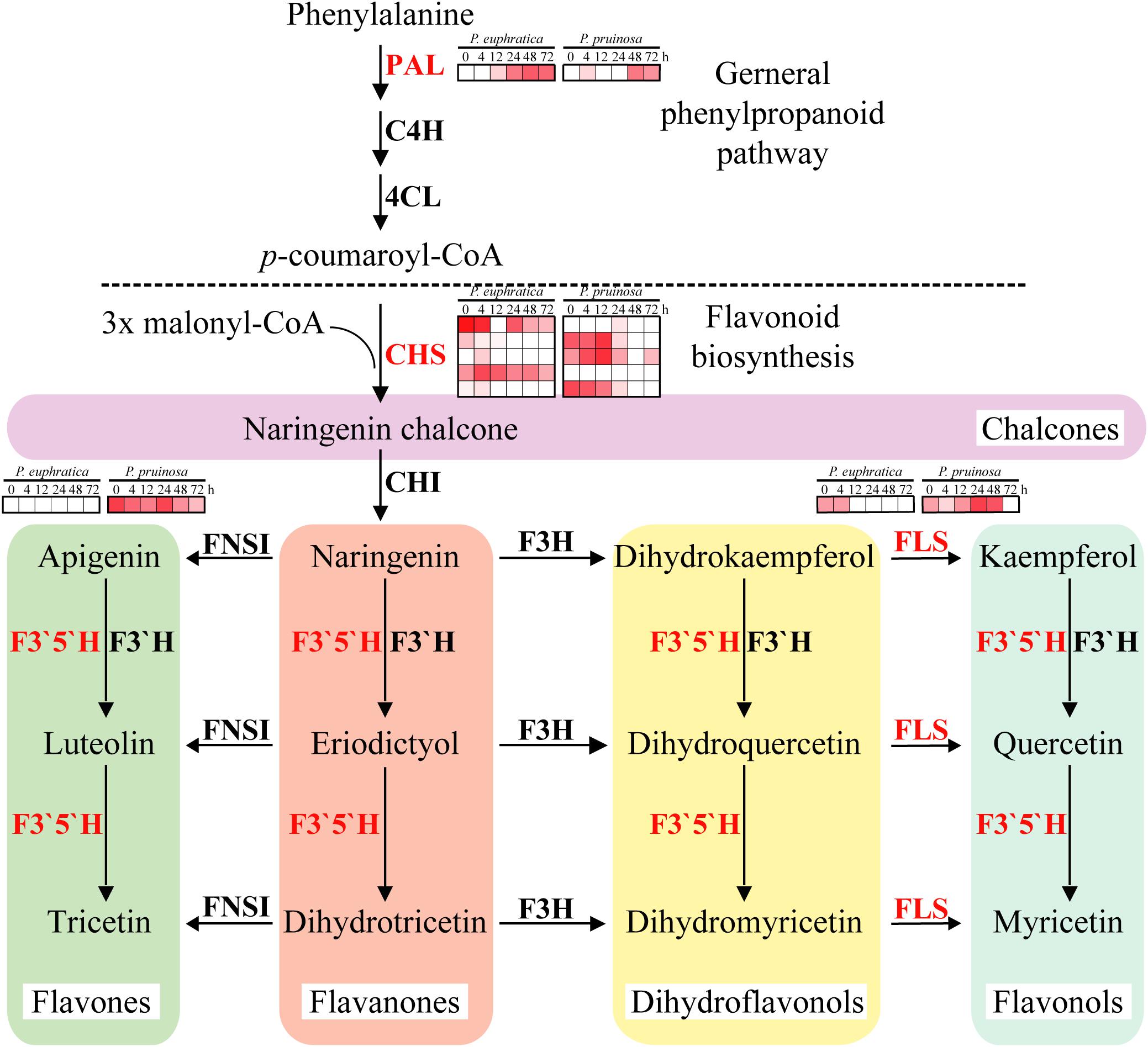
Figure 5. Regulatory network of flavonoid biosynthesis underlying the co-regulated DEGs in P. euphratica and P. pruinosa. For details of abbreviations, see Supplementary Table S3.
Brassinosteroids are involved in an extensive range of effects, such as cell division, cell expansion, xylem differentiation and seed germination, in plants (Kagale et al., 2007). In the two species, six gene families related to brassinosteroid metabolism were enriched, including DET2, DWF4, BR6OX1, BRox2, ROT3 and BAS1. Among them, DET2 and BAS1 were highly expressed in P. euphratica and exhibited relatively low expression in P. pruinosa, while ROT3 was highly expressed in P. pruinosa but was not detected in P. euphratica (Figure 6). Moreover, there were two copies of both DWF4 and BR6OX2, and each copy exhibited a different expression pattern between the two poplars.
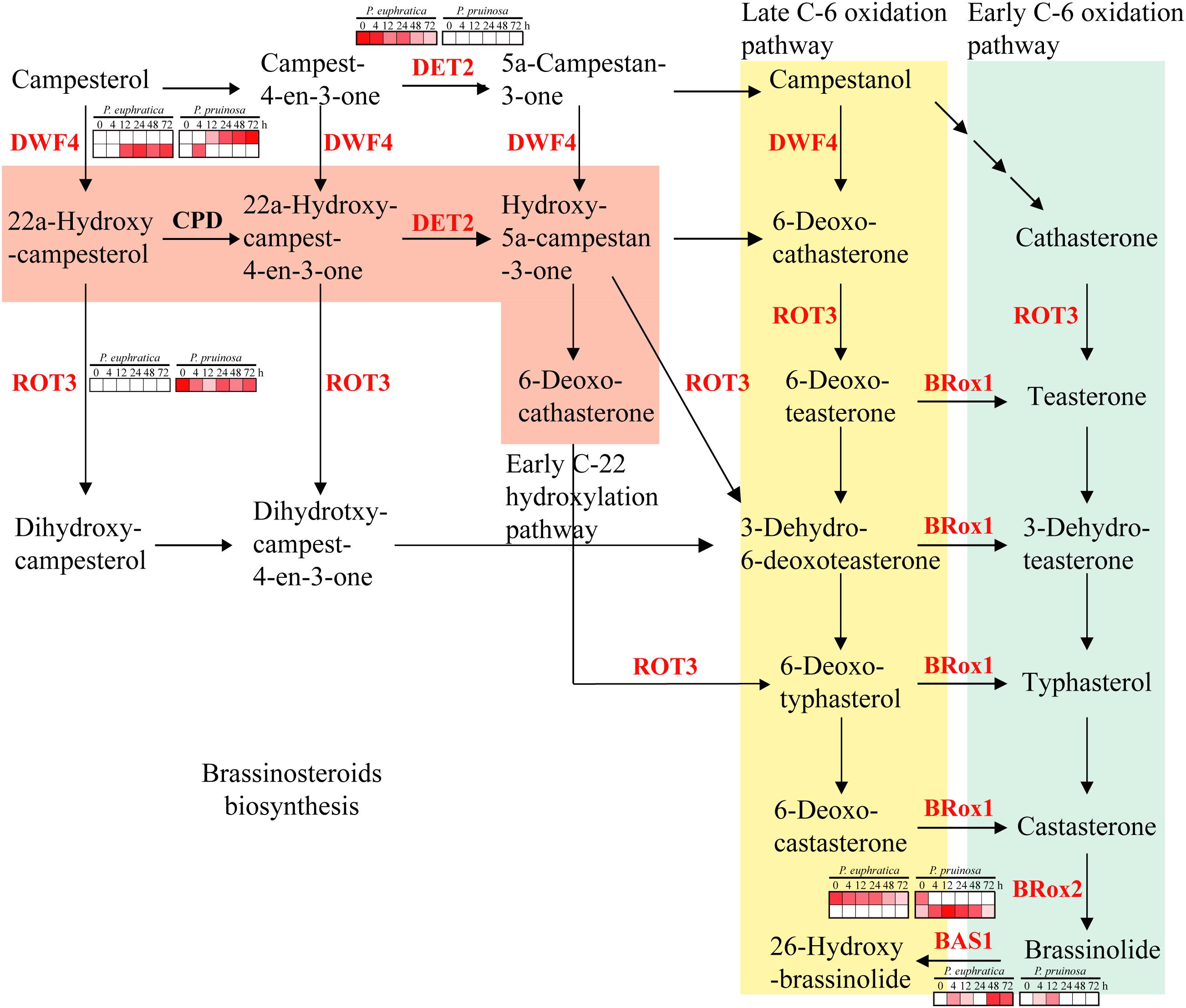
Figure 6. Regulatory network of brassinosteroid biosynthesis underlying the co-regulated DEGs in P. euphratica and P. pruinosa. For details of abbreviations, see Supplementary Table S3.
ROS Level and Enzyme Activity Determination
We measured ROS levels and related enzyme activities. The quantification assay indicated that more hydrogen peroxide accumulated in P. euphratica than in P. pruinosa under the various salt conditions, especially in 1.0% NaCl, where the levels were approximately 2-fold higher in P. euphratica than in P. pruinosa (Figure 7). Therefore, SOD activities were significantly higher in P. pruinosa than in P. euphratica after treatment with 0.4% NaCl solution. The CAT activities in the treatment with 1.0% NaCl solution demonstrated a similar pattern.
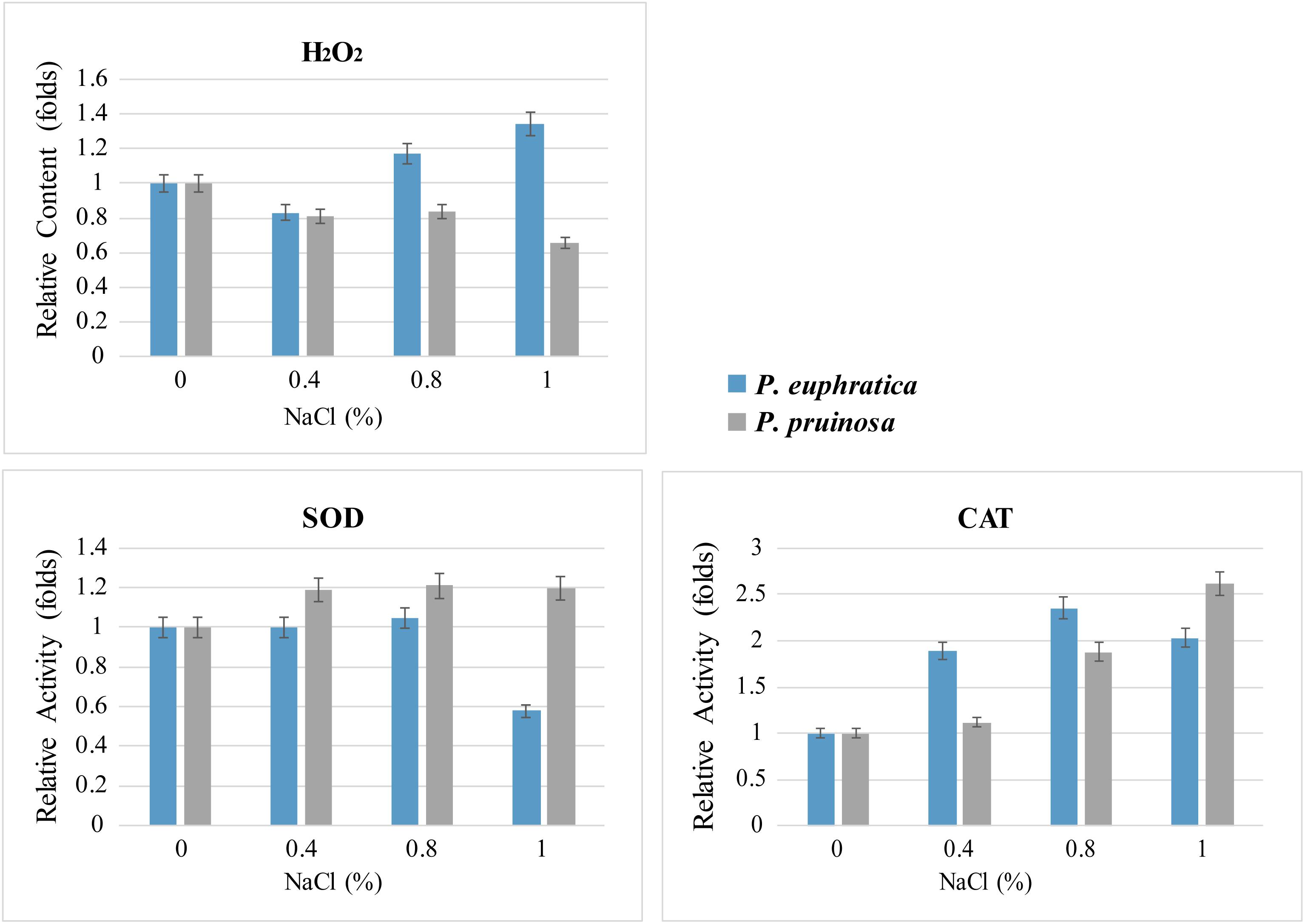
Figure 7. Quantitative comparison of superoxide contents and antioxidant enzyme activities (SOD and CAT) for the two species exposed to salt stress. The changes in content and activity were analyzed with different salt treatments.
Verification of Expression Patterns by qRT-PCR
To validate the RNA-seq results, qRT-PCR analysis was conducted at different time points during seed germination in the two species (Figure 8). The results of genes studied by the RT-PCR analysis, including those in the plant hormone signal transduction (PYL and GID1), flavonoid biosynthesis (PAL) and brassinosteroid biosynthesis (ROT3 and DWF4) pathways, were all similar to the RNA-seq results.
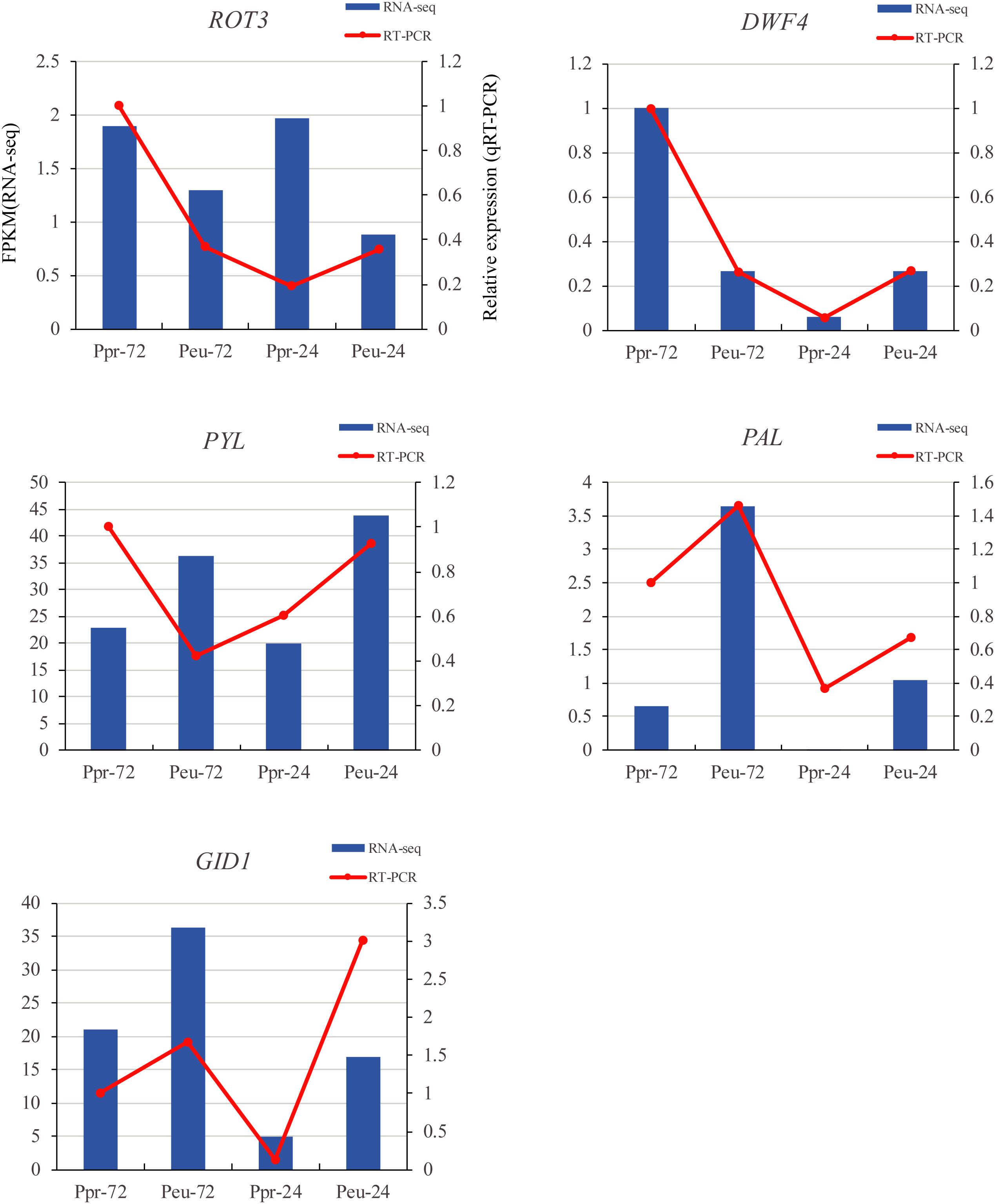
Figure 8. qRT-PCR verification of five selected DEGs. We compared the RNA-seq data (blue bar) with qRT-PCR data (red lines). And we indicated the normalized expression level (FPKM) of RNA-seq on the y-axis to the left. The relative qRT-PCR expression level is shown on the y-axis to the right. CYC063 was set as the internal control. Both methods agree, showing similar gene expression trends.
Discussion
P. pruinosa Showed a Higher Salt Tolerance Than P. euphratica at the Three Seed Germination Stages
Populus euphratica and P. pruinosa diverged from a recent common ancestor between 1 and 2 million years ago (Wang J. et al., 2011) and exhibited different ecological adaptations to desert habitats. In this context, the two desert poplars have evolved different genetic strategies (Ma et al., 2013; Zhang et al., 2013). However, it is not known whether these genetic variations also underlie differences in seed germination.
In the present study, the rate of seed germination in P. pruinosa faster than that in P. euphratica during seed germination (Figure 1C). Based on the seed moisture content, the seed germination time courses for the two species, upon the transfer of seeds to water, can be divided into three phases, which agree with the three classical phases of seed germination (Nonogaki et al., 2010; Figure 1A). We also investigated the relationship between the germination rate and NaCl concentration. The average germination percentage, subordinate function values, and threshold salinity of P. pruinosa were higher than those of P. euphratica. Based on transcriptome analysis, approximately 80% of the reads matched with the genome, and the number of mapped genes in each library was 52%–70%, indicating that most genes were expressed in the seeds of the two species under salt stress. The correlation dendrogram is consistent with the separate phases classified by seed water uptake (Supplementary Figure S3B), suggesting that, in two stages (early and late phases), the sister species evolved divergent regulatory and metabolic pathways associated with seed germination in different salt habitats.
Biochemical Processes of Poplar Seeds Are Regulated by Highly Coordinated Transcript Dynamics
The expression data in the three seed germination phases showed a high reproducibility in both species, and each phase was clearly distinguished by expression dynamics. The DEGs induced early in seed germination (early phase) appeared to be associated with the repair of genetic materials, the cellular structure and the resumption of energy metabolism. During seed germination, the free amino acids involved in protein synthesis are provided by storage protein degradation induced by osmopriming in the first hours of imbibition (Wang et al., 2012). Accordingly, proteases are newly synthesized and accumulate during imbibition (Yang et al., 2007). Therefore, we speculate that in P. euphratica and P. pruinosa, amino acid biosynthesis genes are expressed after 48 h of the seed germination process, and their products provide for the synthesis and metabolism of de novo proteins in the growing embryo (Joosen et al., 2013). Thus, the stored proteins in seeds act not only as important sources of amino acids but also as a source of energy (Angelovici et al., 2011). The middle phase was associated with the active nutrient reservoir, amino acid metabolism and catalytic activity. In this stage, producing a redox state is likely a primary function of the fast recovery of cellular metabolism at the beginning of imbibition (Rosental et al., 2014). The functions of the enriched genes not only produce energy but also promote the activity of essential enzymes to support the completion of germination (Van Dongen et al., 2011). Moreover, flavonoids can induce a delay in the germination rate and play important roles in protection against diverse stresses (D’Auria and Gershenzon, 2005). Most flavonoid genes in this study were enhanced in P. pruinosa in the middle phase, indicating that the “flavonoid biosynthesis” pathway might lead to a difference in the seed germination rate between P. pruinosa and P. euphratica.
Reserves used for the germination of seeds are primarily stored in the form of starch, lipids and proteins in the embryo or endosperm (Yang et al., 2009). Proteins related to hydrolase activity contribute to starch and protein degradation (Rosental et al., 2014), while catalytic activity proteins may increase enzyme activities or provide the energy required during seed germination. Nitrogen-containing compounds release seeds from dormancy, presumably leading to the oxidation of NADPH and therefore providing an increased carbon flow through the glycolytic and oxidative pentose phosphate pathways (PPP) (Roberts, 1964; Hendricks and Taylorson, 1974; Roberts and Lord, 1979; Cohn et al., 1983; Hilhorst and Karssen, 1989). NADP, as a coenzyme of glucose-6-phosphate dehydrogenase, plays a key role in linking the glycolysis pathway and PPP (Nonogaki et al., 2010). Here, we investigated the DEGs involved in fermentation, the TCA cycle, glyoxylate and glycolysis during seed germination. We found that energy production mainly occurred in the later phase and increased gradually. Interestingly, many genes of key enzymes in the TCA cycle were expressed during the early phase, which would lead to the accumulation of many key enzymes during early germination (Weitbrecht et al., 2011). In the late phase, some of the enriched genes were linked to “the photosystem II oxygen evolving complex,” “photosystem,” and “photosynthesis,” suggesting that the seeds had already started to photosynthesize, contributing to the energy supply and powering the productivity of the seed (Ruuska et al., 2004; Goffman et al., 2005; Allen et al., 2009). Meanwhile, “glyoxylate metabolism” activity was enriched in the last phase of germination, which suggests that lipid metabolism is also an important energy source for seed germination. The results demonstrated that activation of energy metabolism during early germination is necessary for seed germination, however, energy production is more complex in the late phase than in earlier phases. In addition to energy metabolism, the expression of genes associated with detoxification were also involved in responses to salt stress in the two desert poplars (Zhu, 2001); these genes included genes associated with “glutathione metabolism,” “flavonoid biosynthesis” and “cytochrome P450,” which all are tightly correlated with seed tolerance to salt stress (Alscher, 1989). These observations suggest that P. euphratica and P. pruinosa quickly establish energetic and developmental balances under salt stress. Germination is actuated by a large number of cellular processes, such as transcription, translation, repair mechanisms, responses to various stresses, organelle reassembly and cellular structure reconstruction. All the processes are supported by metabolism for energy generation. Together, the above results indicate that the transition of primary biochemical processes over time during the seed germination of the two studied poplars is produced partly by highly coordinated transcript dynamics. As the two desert poplars have adapted to different salty desert habitats, these species may have developed different genetic pathways under salt stress during seed germination.
Hormonal Regulation Contributes to the Difference in Seed Germination Phases
Some genes were enriched in the “plant hormone signal transduction” functional category, which is key for physiological state determination and the regulation of seed germination, especially the GA-ABA balance (Meyer et al., 2009). ABA positively regulates the induction of dormancy and negatively regulates germination. Here, the genes related to ABA signal transduction exhibited similar expression patterns in the two poplars. For example, PYL/PYR1, which are considered ABA receptors, exhibited upregulated expression during the first stage, suggesting that the ABA content of the dry seeds was high and decreased during imbibition (Preston et al., 2009). In addition, the negative regulator PP2C has been found to be a major core component of ABA signaling; its expression level was high at 0 and 4 h but decreased after 12 h (Fujii and Zhu, 2009; Umezawa et al., 2009).
GAs play an important role in the promotion of germination and the release of dormancy (Kucera et al., 2005) by stimulating ABA degradation. Here, many DEGs associated with the GA signaling pathway exhibited different expression patterns during germination under salt stress. Specifically, DELLA proteins belonging to the GRAS family were negatively regulated in the GA signaling pathway (Sun and Gubler, 2004) and upregulated from 0 to 12 h in P. euphratica, while they were continuously expressed at high levels in P. pruinosa. GID1, coding a soluble GA receptor, was strongly upregulated during the middle and late phases of seed germination. The GA protein can interact with DELLA when bioactive GAs are present (Ueguchi-Tanaka et al., 2007). Furthermore, most GA signaling transcription-related genes were upregulated in the middle and late phases, which corresponds to the results of a previous study showing that the GA content increased during germination in seeds during phase II.
Ethylene is implicated in the promotion of germination in many species. Here, we identified the DEGs involved in ethylene signaling in seed germination. We found that most of these DEGs, alongside ETR and EIN3, exhibited similar expression patterns in the two species (Supplementary Figure S7). In the absence of ethylene, ETR1 activates CTR1, which negatively regulates downstream signaling components and is inactive in the presence of ethylene. CTR expression was upregulated in the late phase of germination in the two species, while ETR was highly expressed after the early phase of germination. These proteins are regulated by ethylene levels during seed germination by the inactivation of a MAPK cascade comprising SIMKK and MPK6, which are positive regulators of the ethylene response pathway (Ouaked et al., 2003). EIN3 and EIN3-LIKE proteins bind to the promoter of the ERF1 (ethylene responsive factor 1) gene and thereby confer a hierarchy of transcription factors involved in ethylene signaling (Lee and Kim, 2003). Most importantly, the expression patterns of DEGs related to the ethylene pathway were different between P. euphratica and P. pruinosa, indicating that ETR and EIN3 distinctly regulate ethylene signal transcription pathways during seed germination.
Overall, GAs increase and counteract ABA inhibition in the early and late phases of germination (North et al., 2010). Ethylene counteracts ABA inhibition by interfering with ABA signaling during the late phase of germination, while the ABA content is regulated by an equilibrium between the biosynthesis and catabolism of ABA (Nambara and Marion-Poll, 2005). Thus, many of the DEGs exhibited analogous expression patterns in the two species in the models for GA, ABA and ethylene in response to salinity stress but exhibited completely different expression patterns during seed germination.
The Fine Regulation of the Synthesis of Flavonoids and Brassinosteroids in Desert Poplars Contributes to Their Environmental Adaptation
Flavonoids have an extensive range of biological functions, including protecting plants under various stresses (Winkel-Shirley, 2002). Flavonoids are synthesized by the phenylpropanoid pathway and found in most seeds and grains; the major types of flavonoids in seeds are flavonols, anthocyanins, phlobaphenes, isoflavones and proanthocyanidins (Lepiniec et al., 2006). Several genes that encode key enzymes in the flavonoid biosynthetic pathway were expressed differently between the seeds of P. euphratica and P. pruinosa under salt stress (Figure 5). We suggest that the phenylpropanoid pathway, especially the flavonoid metabolism pathway, is widely involved in protection from salt stress in both desert poplars. In general, salt stress is often accompanied by an oxidative burst in plants. In this study, the hydrogen peroxide (H2O2) accumulation in P. euphratica was 2-fold higher than that in P. pruinosa under the various salt conditions, especially in 1.0% NaCl (Figure 7), suggesting that salt treatment might induce oxidative stress in the seeds of P. euphratica. The unavoidable accumulation of H2O2 and scavenging pathways activity should be maintained in balance, where H2O2 could either perform a signaling role or reach a nontoxic level in plants under salt stress conditions. To alleviate and eliminate highly reactive oxygen species, plants have evolved a battery of antioxidative mechanisms, and the antioxidant defense system includes hydrophilic and hydrophobic antioxidants and enzymes such as SOD and CAT (Shalata and Tal, 1998). SOD activities in P. pruinosa were significantly higher than those in P. euphratica when the seeds were exposed to concentrations of NaCl above 0.4%, while CAT activities in P. pruinosa were also higher than those in P. pruinosa when seeds were treated with 1.0% NaCl. Both antioxidases could play a crucial role in scavenging redundant ROS (H2O2) induced by salt stress. Altogether, a significant proportion of the antioxidants induced by salt stress were secondary metabolites, such as a vast amount of compounds primarily derived by the phenylpropanoid pathway (Dixon and Paiva, 1995).
Brassinosteroids are involved in a wide range of growth and development aspects in plants (Kagale et al., 2007). One of the most interesting influences of brassinosteroids is their ability to confer resistance to various abiotic stresses. Several brassinosteroid biosynthesis genes have been identified by molecular genetic analysis and reverse genetic analysis (Takahashi et al., 2005). Among the gene families enriched in the brassinosteroid pathway, DET2 and BAS1 were highly expressed in P. euphratica and exhibited relatively low expression in P. pruinosa, while ROT3 was highly expressed in P. pruinosa but was not detected in P. euphratica (Figures 6, 8). Moreover, DWF4 and BR6OX2 each contain two copies, and each copy exhibited a different expression pattern between the two poplars. The results suggest that the fine regulation of the synthesis of brassinosteroids in desert poplars contributes to their environmental adaptation.
Conclusion
In this study, a multidimensional transcriptome dataset allowed us to discern highly dynamic and coordinated gene expression, as well as functional and regulatory shifts exhibited by the germinating seeds of two species in response to continuous salinity stress. Based on these results, we conclude that the fine regulation of the synthesis of flavonoids and brassinosteroids in desert poplars contributes to their environmental adaptation.
Data Availability
The Illumina sequencing data sets are available at the NCBI Sequence Read Archive (SRA) database with the project accession number: PRJNA484685.
Author Contributions
DW conceived and designed the experiments. CZ, WL, and YL conducted the bioinformatic work and wrote the manuscript. XuZ, XB, and ZN contributed to conducting experiments for physiology and transcript analysis. XiZ and ZL provided assistance in sample collection. All authors read, revised and approved the final manuscript.
Funding
This research was supported by the National Science Foundation of China (Nos. 31470620 and 31870580).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00231/full#supplementary-material
FIGURES S1, S2 | Reproducibility of each trio of biological replicates. The samples were collected at different time points, and total RNA isolation was used to construct RNA-seq libraries for them independently. FPKM values of all the genes expressed in at least one of the 36 sequenced samples are shown in scatter plots and were used as input for the Pearson product-moment correlation coefficient analysis. The correlations between the biological replicates were high in both two species [average r = 0.945 in P. euphratica (Supplementary Figure S1) and r = 0.939 in P. pruinosa (Supplementary Figure S2)].
FIGURE S3 | Number of genes expressed at each time point (A) and hierarchical clustering of six time points for P. euphratica and P. pruinosa (B).
FIGURE S4 | Hierarchical clustering of six time points for P. euphratica and P. pruinosa. A to B, the expression patterns of co-expression modules of P. euphratica (A) and P. pruinosa (B), ordered according to the sample time points of their peak expression. (C) The gene numbers and the expression fitted curves of all the modules in A and B. For each gene, the FPKM value normalized by the maximum value of all FPKM values of the gene over all time points is shown.
FIGURE S5 | GO function enrichment of the DEGs for seed germination processes.
FIGURE S6 | KEGG function enrichment of the DEGs for seed germination processes.
FIGURE S7 | The expression pattern of the hormone-related genes in the two poplars. Expression patterns of hormone-related genes. Normalized expression levels of genes related to ethylene, GA and ABA are shown.
FIGURE S8 | GO function enrichment of the DEGs for salt tolerance variety of the two species.
FIGURE S9 | KEGG function enrichment of the DEGs for salt tolerance variety of the two species.
TABLE S1 | Overview of the data size of all the samples of P. euphratica and P. pruinosa.
TABLE S2 | Summary of the illumine sequencing reads and the matches in the P. euphratica and P. pruinosa.
TABLE S3 | The details of abbreviations.
TABLE S4 | Primer used for real-time quantitative PCR in this study.
Footnotes
References
Allen, D. K., Ohlrogge, J. B., and Shachar-Hill, Y. (2009). The role of light in soybean seed filling metabolism. Plant J. 58, 220–234. doi: 10.1111/j.1365-313X.2008.03771.x
Almansouri, M., Kinet, J.-M., and Lutts, S. (2001). Effect of salt and osmotic stresses on germination in durum wheat (Triticum durum Desf.). Plant Soil 231, 243–254. doi: 10.1023/A:1010378409663
Alscher, R. G. (1989). Biosynthesis and antioxidant function of glutathione in plants. Physiol. Plant. 77, 457–464. doi: 10.1111/j.1399-3054.1989.tb05667.x
Angelovici, R., Fait, A., Fernie, A. R., and Galili, G. (2011). A seed high-lysine trait is negatively associated with the TCA cycle and slows down Arabidopsis seed germination. New Phytol. 189, 148–159. doi: 10.1111/j.1469-8137.2010.03478.x
Annunziata, M. G., Ciarmiello, L. F., Woodrow, P., Maximova, E., Fuggi, A., and Carillo, P. (2017). Durum wheat roots adapt to salinity remodeling the cellular content of nitrogen metabolites and sucrose. Front. Plant Sci. 7:2035. doi: 10.3389/fpls.2016.02035
Arbona, V., Manzi, M., Ollas, C. D., and Gómez-Cadenas, A. (2013). Metabolomics as a tool to investigate abiotic stress tolerance in plants. Int. J. Mol. Sci. 14, 4885–4911. doi: 10.3390/ijms14034885
Bartels, D., and Sunkar, R. (2005). Drought and salt tolerance in plants. Crit. Rev. Plant Sci. 24, 23–58. doi: 10.1080/07352680590910410
Bewley, J. (1982). “Protein and nucleic acid synthesis during seed germination and early seedling growth,” in Nucleic Acids and Proteins in Plants 1. Encyclopedia of Plant Physiology, eds D. Boulter and B. Parthier (New York, NY: Springer), 559–591.
Bewley, J., and Black, M. (1984). Physiology and biochemistry of seeds in relation to germination. Plant Ecol. 57, 71–74.
Bewley, J. D. (1997). Seed germination and dormancy. Plant Cell 96, 1055–1066. doi: 10.1105/tpc.9.7.1055
Bewley, J. D., Bradford, K., and Hilhorst, H. (2012). Seeds: Physiology of Development, Germination and Dormancy. New York, NY: Springer.
Biligetu, B., Schellenberg, M., Mcleod, J., Borges, E., Borges, R., Buckeridge, M., et al. (2011). Seeds: physiology of development and germination. Alterações Fisiológicas E Bioquímicas Durante A 39:26.
Bradford, K. J. (1990). A water relations analysis of seed germination rates. Plant Physiol. 94, 840–849. doi: 10.1104/pp.94.2.840
Brinker, M., Brosché, M., Vinocur, B., Abo-Ogiala, A., Fayyaz, P., Janz, D., et al. (2010). Linking the salt transcriptome with physiological responses of a salt-resistant Populus species as a strategy to identify genes important for stress acclimation. Plant Physiol. 154, 1697–1709. doi: 10.1104/pp.110.164152
Burritt, D. J., and Mackenzie, S. (2003). Antioxidant metabolism during acclimation of Begonia× erythrophylla to high light levels. Ann. Bot. 91, 783–794. doi: 10.1093/aob/mcg076
Carillo, P., Cirillo, C., De Micco, V., Arena, C., De Pascale, S., and Rouphael, Y. (2019). Morpho-anatomical, physiological and biochemical adaptive responses to saline water of Bougainvillea spectabilis Willd. trained to different canopy shapes. Agric. Water Manage. 212, 12–22. doi: 10.1016/j.agwat.2018.08.037
Chang, S., Puryear, J., and Cairney, J. (1993). A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116. doi: 10.1007/BF02670468
Chen, S., Jiang, J., Li, H., and Liu, G. (2012). The salt-responsive transcriptome of Populus simonii× Populus nigra via DGE. Gene 504, 203–212. doi: 10.1016/j.gene.2012.05.023
Chen, S., Li, J., Fritz, E., Wang, S., and Hüttermann, A. (2002). Sodium and chloride distribution in roots and transport in three poplar genotypes under increasing NaCl stress. For. Ecol. Manage. 168, 217–230. doi: 10.1016/S0378-1127(01)00743-5
Cohn, M. A., Butera, D. L., and Hughes, J. A. (1983). Seed dormancy in red rice III. Response to nitrite, nitrate, and ammonium ions. Plant Physiol. 73, 381–384. doi: 10.1104/pp.73.2.381
D’Auria, J. C., and Gershenzon, J. (2005). The secondary metabolism of Arabidopsis thaliana: growing like a weed. Curr. Opin. Plant Biol. 8, 308–316. doi: 10.1016/j.pbi.2005.03.012
Dixon, R. A., and Paiva, N. L. (1995). Stress-induced phenylpropanoid metabolism. Plant Cell 7, 1085–1097. doi: 10.1105/tpc.7.7.1085
Flowers, T. (2004). Improving crop salt tolerance. J. Exp. Bot. 55, 307–319. doi: 10.1093/jxb/erh003
Frazee, A. C., Pertea, G., Jaffe, A. E., Langmead, B., Salzberg, S. L., and Leek, J. T. (2015). Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 33, 243–246. doi: 10.1038/nbt.3172
Fujii, H., and Zhu, J.-K. (2009). Arabidopsis mutant deficient in 3 abscisic acid-activated protein kinases reveals critical roles in growth, reproduction, and stress. Proc. Natl. Acad. Sci. U.S.A. 106, 8380–8385. doi: 10.1073/pnas.0903144106
Goffman, F. D., Alonso, A. P., Schwender, J., Shachar-Hill, Y., and Ohlrogge, J. B. (2005). Light enables a very high efficiency of carbon storage in developing embryos of rapeseed. Plant Physiol. 138, 2269–2279. doi: 10.1104/pp.105.063628
Hasegawa, P. M., Bressan, R. A., Zhu, J. K., and Bohnert, H. J. (2000). Plant cellular and molecular responses to high salinity. Annu. Rev. Plant Biol. 51, 463–499. doi: 10.1146/annurev.arplant.51.1.463
Hendricks, S., and Taylorson, R. (1974). Promotion of seed germination by nitrate, nitrite, hydroxylamine, and ammonium salts. Plant Physiol. 54, 304–309. doi: 10.1104/pp.54.3.304
Hilhorst, H. W., and Karssen, C. M. (1989). Nitrate reductase independent stimulation of seed germination in Sisymbrium officinale L. (hedge mustard) by light and nitrate. Ann. Bot. 63, 131–137. doi: 10.1093/oxfordjournals.aob.a087715
Holdsworth, M. J., Bentsink, L., and Soppe, W. J. (2008). Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 179, 33–54. doi: 10.1111/j.1469-8137.2008.02437.x
Hukin, D., Cochard, H., Dreyer, E., Le Thiec, D., and Bogeat, M. B. (2005). Cavitation vulnerability in roots and shoots: does Populus euphratica Oliv., a poplar from arid areas of Central Asia, differ from other poplar species? J. Exp. Bot. 56, 2003–2010. doi: 10.1093/jxb/eri198
Imit, Y.,Taxi, Z., and Cyffka, B. (2015). Seed germination characteristics of Populus euphratica from different provenances under NaCl stess. J. Northwest For. Univ. 30, 88–94. doi: 10.3969/j.issn.1001-7461.2015.06.15
Janz, D., Behnke, K., Schnitzler, J.-P., Kanawati, B., Schmitt, P., and Polle, A. (2010). Pathway analysis of the transcriptome and metabolome of salt sensitive and tolerant poplar species reveals evolutionary adaption of stress tolerance mechanisms. BMC Plant Biol. 10:150. doi: 10.1186/1471-2229-10-150
Joosen, R. V. L., Arends, D., Li, Y., Willems, L. A., Keurentjes, J. J., Ligterink, W., et al. (2013). Identifying genotype-by-environment interactions in the metabolism of germinating Arabidopsis seeds using generalized genetical genomics. Plant Physiol. 162, 553–566. doi: 10.1104/pp.113.216176
Kagale, S., Divi, U. K., Krochko, J. E., Keller, W. A., and Krishna, P. (2007). Brassinosteroid confers tolerance in Arabidopsis thaliana and Brassica napus to a range of abiotic stresses. Planta 225, 353–364. doi: 10.1007/s00425-006-0361-6
Kaya, M. D., Ipek, A., and Öztürk, A. (2003). Effects of different soil salinity levels on germination and seedling growth of safflower (Carthamus tinctorius L.). Turk. J. Agric. For. 27, 221–227.
Khajeh-Hosseini, M., Powell, A., and Bingham, I. (2003). The interaction between salinity stress and seed vigour during germination of soyabean seeds. Seed Sci. Technol. 31, 715–725. doi: 10.15258/sst.2003.31.3.20
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kucera, B., Cohn, M. A., and Leubner, G. (2005). Plant hormone interactions during seed dormancy release and germination. Seed Sci. Res. 15, 281–307. doi: 10.1079/SSR2005218
Lee, J. H., and Kim, W. T. (2003). Molecular and biochemical characterization of VR-EILs encoding mung bean ETHYLENE INSENSITIVE3-LIKE proteins. Plant Physiol. 132, 1475–1488. doi: 10.1104/pp.103.022574
Lepiniec, L., Debeaujon, I., Routaboul, J. M., Baudry, A., Pourcel, L., Nesi, N., et al. (2006). Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 57, 405–430. doi: 10.1146/annurev.arplant.57.032905.105252
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ma, J., He, X., Bai, X., Niu, Z., Duan, B., Chen, N., et al. (2016). Genome-wide survey reveals transcriptional differences underlying the contrasting trichome phenotypes of two sister desert poplars. Genes 7:111. doi: 10.3390/genes7120111
Ma, T., Wang, J., Zhou, G., Yue, Z., Hu, Q., Chen, Y., et al. (2013). Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 4:2797. doi: 10.1038/ncomms3797
Ma, T., Wang, K., Hu, Q., Xi, Z., Wan, D., Wang, Q., et al. (2018). Ancient polymorphisms and divergence hitchhiking contribute to genomic islands of divergence within a poplar species complex. Proc. Natl. Acad. Sci. U.S.A. 115, E236–E243. doi: 10.1073/pnas.1713288114
Meyer, E. H., Tomaz, T., Carroll, A. J., Estavillo, G., Delannoy, E., Tanz, S. K., et al. (2009). Remodeled respiration in ndufs4 with low phosphorylation efficiency suppresses Arabidopsis germination and growth and alters control of metabolism at night. Plant Physiol. 151, 603–619. doi: 10.1104/pp.109.141770
Müller, K., Carstens, A. C., Linkies, A., Torres, M. A., and Leubner, G. (2009). The NADPH-oxidase AtrbohB plays a role in Arabidopsis seed after-ripening. New Phytol. 184, 885–897. doi: 10.1111/j.1469-8137.2009.03005.x
Munns, R. (2002). Comparative physiology of salt and water stress. Plant Cell Environ. 25, 239–250. doi: 10.1046/j.0016-8025.2001.00808.x
Murillo-Amador, B., López-Aguilar, R., Kaya, C., Larrinaga-Mayoral, J., and Flores-Hernández, A. (2002). Comparative effects of NaCl and polyethylene glycol on germination, emergence and seedling growth of cowpea. J. Agron. Crop Sci. 188, 235–247. doi: 10.1046/j.1439-037X.2002.00563.x
Nambara, E., and Marion-Poll, A. (2005). Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 56, 165–185. doi: 10.1146/annurev.arplant.56.032604.144046
Nonogaki, H., Bassel, G. W., and Bewley, J. D. (2010). Germination-still a mystery. Plant Sci. 179, 574–581. doi: 10.1016/j.plantsci.2010.02.010
North, H., Baud, S., Debeaujon, I., Dubos, C., Dubreucq, B., Grappin, P., et al. (2010). Arabidopsis seed secrets unravelled after a decade of genetic and omics-driven research. Plant J. 61, 971–981. doi: 10.1111/j.1365-313X.2009.04095.x
Ottow, E. A., Brinker, M., Teichmann, T., Fritz, E., Kaiser, W., Brosché, M., et al. (2005). Populus euphratica displays apoplastic sodium accumulation, osmotic adjustment by decreases in calcium and soluble carbohydrates, and develops leaf succulence under salt stress. Plant Physiol. 139, 1762–1772. doi: 10.1104/pp.105.069971
Ouaked, F., Rozhon, W., Lecourieux, D., and Hirt, H. (2003). A MAPK pathway mediates ethylene signaling in plants. EMBO J. 22, 1282–1288. doi: 10.1093/emboj/cdg131
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Preston, J., Tatematsu, K., Kanno, Y., Hobo, T., Kimura, M., Jikumaru, Y., et al. (2009). Temporal expression patterns of hormone metabolism genes during imbibition of Arabidopsis thaliana seeds: a comparative study on dormant and non-dormant accessions. Plant Cell Physiol. 50, 1786–1800. doi: 10.1093/pcp/pcp121
Pritchard, S. L., Charlton, W. L., Baker, A., and Graham, I. A. (2002). Germination and storage reserve mobilization are regulated independently in Arabidopsis. Plant J. 31, 639–647. doi: 10.1046/j.1365-313X.2002.01376.x
Qiu, Q., Ma, T., Hu, Q., Liu, B., Wu, Y., Zhou, H., et al. (2011). Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 31, 452–461. doi: 10.1093/treephys/tpr015
Ren, Z. H., Gao, J. P., Li, L. G., Cai, X. L., Huang, W., Chao, D. Y., et al. (2005). A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat. Genet. 37, 1141–1146. doi: 10.1038/ng1643
Roberts, E. (1964). The distribution of Oxidation-reduction enzymes and the effects of respiratory inhibitors and oxidising agents on dormancy in rice seed. Physiol. Plant. 17, 14–29. doi: 10.1111/j.1399-3054.1964.tb09013.x
Roberts, L. M., and Lord, J. M. (1979). Developmental changes in the activity of messenger RNA isolated from germinating castor bean endosperm. Plant Physiol. 64, 630–634. doi: 10.1104/pp.64.4.630
Rosental, L., Nonogaki, H., and Fait, A. (2014). Activation and regulation of primary metabolism during seed germination. Seed Sci. Res. 24, 1–15. doi: 10.1017/S0960258513000391
Ruuska, S. A., Schwender, J., and Ohlrogge, J. B. (2004). The capacity of green oilseeds to utilize photosynthesis to drive biosynthetic processes. Plant Physiol. 136, 2700–2709. doi: 10.1104/pp.104.047977
Shalata, A., and Tal, M. (1998). The effect of salt stress on lipid peroxidation and antioxidants in the leaf of the cultivated tomato and its wild salt-tolerant relative Lycopersicon pennellii. Physiol. Plant. 104, 169–174. doi: 10.1034/j.1399-3054.1998.1040204.x
Sun, J., Chen, S., Dai, S., Wang, R., Li, N., Shen, X., et al. (2009). NaCl-induced alternations of cellular and tissue ion fluxes in roots of salt-resistant and salt-sensitive poplar species. Plant Physiol. 149, 1141–1153. doi: 10.1104/pp.108.129494
Sun, T. P., and Gubler, F. (2004). Molecular mechanism of gibberellin signaling in plants. Annu. Rev. Plant Biol. 55, 197–223. doi: 10.1146/annurev.arplant.55.031903.141753
Taji, T., Ohsumi, C., Iuchi, S., Seki, M., Kasuga, M., Kobayashi, M., et al. (2002). Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 29, 417–426. doi: 10.1046/j.0960-7412.2001.01227.x
Takahashi, N., Nakazawa, M., Shibata, K., Yokota, T., Ishikawa, A., Suzuki, K., et al. (2005). shk1-D, a dwarf Arabidopsis mutant caused by activation of the CYP72C1 gene, has altered brassinosteroid levels. Plant J. 42, 13–22. doi: 10.1111/j.1365-313X.2005.02357.x
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Ueguchi-Tanaka, M., Nakajima, M., Motoyuki, A., and Matsuoka, M. (2007). Gibberellin receptor and its role in gibberellin signaling in plants. Annu. Rev. Plant Biol. 58, 183–198. doi: 10.1146/annurev.arplant.58.032806.103830
Umezawa, T., Sugiyama, N., Mizoguchi, M., Hayashi, S., Myouga, F., Yamaguchi, K., et al. (2009). Type 2C protein phosphatases directly regulate abscisic acid-activated protein kinases in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 106, 17588–17593. doi: 10.1073/pnas.0907095106
Van Dongen, J. T., Gupta, K. J., Ramírez, S. J., Araújo, W. L., Nunes, A., and Fernie, A. R. (2011). Regulation of respiration in plants: a role for alternative metabolic pathways. J. Plant Physiol. 168, 1434–1443. doi: 10.1016/j.jplph.2010.11.004
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Wang, H., Han, L., and Jia, W. (2013). Response of seeds germination of Populus euphratica and Populus pruinosa to slat stress. J. Desert Res. 33, 743–750.
Wang, J., Wu, Y., Ren, G., Guo, Q., Liu, J., and Lascoux, M. (2011). Genetic differentiation and delimitation between ecologically diverged Populus euphratica and P. pruinosa. PLoS One 6:e26530. doi: 10.1371/journal.pone.0026530
Wang, Y., Li, L., Ye, T., Zhao, S., Liu, Z., Feng, Y. Q., et al. (2011). Cytokinin antagonizes ABA suppression to seed germination of Arabidopsis by downregulating ABI5 expression. Plant J. 68, 249–261. doi: 10.1111/j.1365-313X.2011.04683.x
Wang, W. Q., Møller, I. M., and Song, S. Q. (2012). Proteomic analysis of embryonic axis of Pisum sativum seeds during germination and identification of proteins associated with loss of desiccation tolerance. J. Proteomics 77, 68–86. doi: 10.1016/j.jprot.2012.07.005
Weitbrecht, K., Müller, K., and Leubner, G. (2011). First off the mark: early seed germination. J. Exp. Bot. 62, 3289–3309. doi: 10.1093/jxb/err030
Winkel-Shirley, B. (2002). Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Biol. 5, 218–223. doi: 10.1016/S1369-5266(02)00256-X
Wullschleger, S. D., Weston, D., DiFazio, S. P., and Tuskan, G. A. (2013). Revisiting the sequencing of the first tree genome: Populus trichocarpa. Tree Physiol. 33, 357–364. doi: 10.1093/treephys/tps081
Yamaguchi-Shinozaki, K., and Shinozaki, K. (2005). Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 10, 88–94. doi: 10.1016/j.tplants.2004.12.012
Yang, M. F., Liu, Y. J., Liu, Y., Chen, H., Chen, F., and Shen, S. H. (2009). Proteomic analysis of oil mobilization in seed germination and postgermination development of Jatropha curcas. J. Proteome Res. 8, 1441–1451. doi: 10.1021/pr800799s
Yang, P., Li, X., Wang, X., Chen, H., Chen, F., and Shen, S. (2007). Proteomic analysis of rice (Oryza sativa) seeds during germination. Proteomics 7, 3358–3368. doi: 10.1002/pmic.200700207
Ye, C. Y., Zhang, H. C., Chen, J. H., Xia, X. L., and Yin, W. L. (2009). Molecular characterization of putative vacuolar NHX-type Na+/H+ exchanger genes from the salt-resistant tree Populus euphratica. Physiol. Plant. 137, 166–174. doi: 10.1111/j.1399-3054.2009.01269.x
Zhang, J., Feng, J., Lu, J., Yang, Y., Zhang, X., Wan, D., et al. (2014). Transcriptome differences between two sister desert poplar species under salt stress. BMC Genomics 15:337. doi: 10.1186/1471-2164-15-337
Zhang, J., Xie, P., Lascoux, M., Meagher, T. R., and Liu, J. (2013). Rapidly evolving genes and stress adaptation of two desert poplars, Populus euphratica and P. pruinosa. PLoS One 8:e66370. doi: 10.1371/journal.pone.0066370
Zhu, J. K. (2001). Plant salt tolerance. Trends Plant Sci. 6, 66–71. doi: 10.1016/S1360-1385(00)01838-0
Keywords: transcriptome, salt stress, seed germination, differentially expressed gene, desert poplar species
Citation: Zhang C, Luo W, Li Y, Zhang X, Bai X, Niu Z, Zhang X, Li Z and Wan D (2019) Transcriptomic Analysis of Seed Germination Under Salt Stress in Two Desert Sister Species (Populus euphratica and P. pruinosa). Front. Genet. 10:231. doi: 10.3389/fgene.2019.00231
Received: 16 October 2018; Accepted: 04 March 2019;
Published: 25 March 2019.
Edited by:
Ancha Baranova, George Mason University, United StatesReviewed by:
Petronia Carillo, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyAndrés A. Borges, Spanish National Research Council (CSIC), Spain
Copyright © 2019 Zhang, Luo, Li, Zhang, Bai, Niu, Zhang, Li and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongshi Wan, d2FuZHNoQGx6dS5lZHUuY24=
†These authors have contributed equally to this work