 Tao Liu1,2*†
Tao Liu1,2*† Xumin Wang2†
Xumin Wang2† Guoliang Wang3,4†
Guoliang Wang3,4† Shangang Jia5*
Shangang Jia5* Guiming Liu6Guangle Shan4Shan Chi1,7Jing Zhang8
Guiming Liu6Guangle Shan4Shan Chi1,7Jing Zhang8 Yahui Yu1Ting Xue9*
Yahui Yu1Ting Xue9* Jun Yu3,4*
Jun Yu3,4*- 1College of Marine Life Science, Ocean University of China, Qingdao, China
- 2College of Life Sciences, Yantai University, Yantai, China
- 3CAS Key Laboratory of Genome Sciences and Information, Beijing Key Laboratory of Genome and Precision Medicine Technologies, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China
- 4University of Chinese Academy of Sciences, Beijing, China
- 5College of Grassland Science and Technology, China Agricultural University, Beijing, China
- 6Beijing Agro-Biotechnology Research Center, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
- 7Qingdao Haida Blue Tek Biotechnology Co., Ltd, Qingdao, China
- 8College of Biological Engineering, Qilu University of Technology, Shandong Academy of Sciences, Jinan, China
- 9The Public Service Platform for Industrialization Development Technology of Marine Biological Medicine and Product of State Oceanic Administration, College of Life Sciences, Fujian Normal University, Fuzhou, China
Saccharina, as one of the most important brown algae (Phaeophyceae) with multicellular thallus, has a very remarkable evolutionary history, and globally accounts for most of the economic marine aquaculture production worldwide. Here, we present the 580.5 million base pairs of genome sequence of Saccharina japonica, whose current assembly contains 35,725 protein-coding genes. In a comparative analysis with Ectocarpus siliculosus, the integrated virus sequence suggested the genome evolutionary footprints, which derived from their co-ancestry and experienced genomic arrangements. Furthermore, the gene expansion was found to be an important strategy for functional evolution, especially with regard to extracelluar components, stress-related genes, and vanadium-dependent haloperoxidases, and we proposed a hypothesis that gene duplication events were the main driving force for the evolution history from multicellular filamentous algae to thallus algae. The sequenced Saccharina genome paves the way for further molecular studies and is useful for genome-assisted breeding of S. japonica and other related algae species.
Introduction
Brown algae are a large group of multicellular algae, which displays a huge biomass dominating cool temperate intertidal and subtidal zone water, due to its macro soma and large biomes. Brown algae are fundamentally different from green and red algae, as green and red algae acquired plastids from cyanobacteria during primary endosymbiosis, while brown algae descend from secondary endosymbiosis (Valentin and Zetsche, 1990). Therefore, brown algae may acquire both cyanobacterial genes via EGT (Endosymbiotic Gene Transfer) and eukaryotic sequences from the nucleus of the red algal endosymbiont (Lane and Archibald, 2008), which makes the genome more complicated to interpret. Therefore, knowledge about the brown algal genome is crucial for understanding its evolution path. There are currently approximately 1500–2000 species of brown algae worldwide. The morphology of brown algae ranges from slender filaments (e.g., Ectocarpus siliculosus) to giant thallus (e.g., Saccharina japonica). Meanwhile, brown algae exhibit a diverse range of life cycles, for example the haploid–diploid cycle of the genus Saccharina is quite different from its close relative the genus Ectocarpus which lacks the parenchyma stage. This may have indicated key adaptive events in their evolution. One important question is which genes or evolutionary events underlie the structural evolution from filamentous brown algae (Ectocarpus) to heteromorphic haploid-diploid algae (Saccharina).
In addition, genus Saccharina, one of the most important genera of brown algae, has been recently separated from genus Laminaria based on molecular evidence from nuclear, plastid, and mitochondrial genome sequences (Draisma et al., 2001; Yoon et al., 2001; Erting et al., 2004; Lane et al., 2006). Many species from Saccharina are known to constitute the marine forests in the Asian coastal areas, which are the primary producers in marine ecosystem and traditionally indispensable in a diet with high industrial value (Zemke-White and Ohno, 1999). They are a potential source of renewable energy (Demirbas, 2010), as well as of polysaccharides, e.g., laminarans, alginic acids, and fucoidans (Vishchuk et al., 2011). S. japonica is not only a common seafood in China and many other countries, but has also been documented as a drug in traditional Chinese medicine for over a thousand years, being rich in polysaccharides, e.g., alginate, fucoidin, fucoidan galactosan sulfate and alginic acids, mannitol and trace elements. With the development of modern science and technology, the medical applications of S. japonica have been gradually revealed, such as its capacity to regulate blood lipids, blood sugar, and blood pressure, and its activities such as anticoagulation, antioxidant, anti-tumor and anti-radiation, etc. (Xue et al., 2001; Zhao et al., 2004, 2012; Kim et al., 2006; Wang et al., 2008, 2012; Huang et al., 2010; Mizuta and Yasui, 2010; Vishchuk et al., 2012).
A previous study on the transcriptome of S. japonica has facilitated the understanding of the genome background of S. japonica along the detailing of vanadium-dependent haloperoxidase family (Liang et al., 2014). Previous studies have shown the biosynthesis pathways of important cellular components (alginate and fucoidan) (Chi et al., 2018a) and complex halogen metabolism of Saccharina (Ye et al., 2015). The previous genome sequencing in S. japonica assembled 13,327 scaffolds (537-Mb) which covered 98.5% of the estimated genome and predicted 18,733 protein-coding genes, consisting of 13,327 scaffolds (Ye et al., 2015). It focused on the evolutionary adaptation and the functional diversification of the polysaccharide biosynthesis and iodine concentration mechanisms of S. japonica. However, a better genome assembly is necessary to provide a complete understanding of S. japonica, and further reveal the evolution significance of multicellular parenchymatous thallus. In particular, the Hi-C (High-through chromosome conformation capture) data can be used for clustering the scaffolds into chromosomes, by exploiting chromatin interaction data (Korbel and Lee, 2013). A complete genome assembly of S. japonica will further facilitate our research with the evolution pattern of tissue differentiation and heteromorphic generational alternation. Comparative genomics analysis will give us a deeper insight into the gene family duplication, differential expansion/contraction and evolution of conserved non-coding sequences. Meanwhile, a detailed investigation of S. japonica genome will accelerate the metabolic expatiation of mannitol, laminarin, alginates and trehalose pathways.
Here, we surveyed the genome of S. japonica with the help of high throughput sequencing. We intend to include a detailed analysis of the updated genome data of S. japonica, which will pave the way of its genetic research and breeding. We reported a preliminary analysis of its genome organization and gene content, including gene annotations, GC content, repeat elements and SSRs. Subsequently, phylogenetic analysis, motif analysis and exon-intron organization of some important genes were also investigated. We believe the S. japonica genome generated in this study will enhance both fundamental and applied research in related areas.
Materials and Methods
Sample Collection and DNA Preparation
A thermotolerant and high-yielding Saccharina cultivar “Rongfu” was selected as the genomic DNA source for whole genome sequencing. Samples were collected from Rongcheng, Shandong Province, P. R. China, and were provided by the Culture Collection of Seaweed at the Ocean University of China. Genomic DNA was extracted from fresh sporophyte with improved CTAB method (Guillemaut and Maréchal-Drouard, 1992).
Genome Sequencing and Assembly
We constructed three paired-end libraries and three mate-paired libraries according to Illumina standard operating procedure. Sequencing of each library was performed on an Illumina HiSeq 2000 instrument to produce the raw data. We then filtered out low-quality and short reads to obtain a set of usable reads.
We then assembled the reads into contigs using SOAPdenovo with varying parameters, and mate-paired relationships between the reads were used to construct scaffolds. The genome assembly was improved by exploiting chromatin interaction data from Hi-C data, and grouping contigs into pseudomolecules/chromosomes. Chromatins were cut by the restriction enzyme Mbol, and ligated together in situ after biotinylation. DNA was extracted, and sheared before end repairing. DNA fragments were enriched by using interaction of biotin and streptavidin, and subject to Hiseq sequencing with a paired end length of 150 bp. After trimming and quality control, paired-end reads were aligned to the genome assembly, and the reads with more than one hit were discarded. The reads without the restriction site of Mbol were filtered out. Then, paired-end reads were analyzed for the valid ones, which were aligned to two different enzyme fragments, for the estimated insert size to meet the expectations, by using HiC-Pro v2.7.8 (Servant et al., 2015). Based on the relationships among valid reads, the order and directions of scaffolds/contigs were clustered into the 31 pseudomolecules/chromosomes by LACHESIS (Burton et al., 2013).
Repeat Analysis and Genome Annotation
Both homology-based and de novo prediction analyses were used to identify the repeat content in the Saccharina genome. For the homology-based analysis, we used Repbase (version 20140131) to perform a TE search with RepeatMasker (open-4.0) and the ncbi RMblast search engine. For the de novo prediction analysis, we used RepeatModeler to construct a TE library, and classified elements within the library using a homologous search with Repbase and TEClass.
Approaches including homology detection, expression-evidence-based predictions and ab initio gene predictions were used for gene model construction. To identify homology patterns in S. japonica, the BLASTX search was performed against the NCBI non-redundant protein database with E-value < 10-5. For expression evidences, published ESTs, transcripts and RNAseq datasets from the OneKP database1 were aligned to the genome. After measuring and comparing a variety of programs, AUGUSTUS was used for ab initio gene prediction. Gene model parameters for the programs were trained from long transcripts and known Saccharina genes processed by PASA. And then all these predictions were combined into consensus gene structures using EVM and optimized by manual corrections.
Functional classification of Gene Ontology of the genes was performed with InterProScan (Zdobnov and Apweiler, 2001). The EuKaryotic Orthologous Groups (KOG) classification was performed against KOG database (Tatusov et al., 2003). Pathway analyses were performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation service KAAS (Kanehisa et al., 2004).
Gene Family Analysis
Related protein sequences were downloaded from NCBI and local BLAST was used to get full-length sequences in S. japonica, with tBLASTn module of WU-BLAST 2.0 and an E-value cut-off of 1 × 10-5. Nucleotide sequences were transferred to amino acid sequences using MEGA with option of standard genetic code and then aligned (Tamura et al., 2011). NCBI BLAST was run on proteins sequences sets of S. japonica, E. siliculosus, Aureococcus anophagefferens, Nannochloropsis gaditana, Phaeodactylum tricornutum, and Thalassiosira pseudonana, which were downloaded from the NCBI GenBank, and then were clustered to orthogroups using Orthomcl (Li et al., 2003). The S. japonica assembly was compared to the available E. siliculosus genome2 using lastz (Frith et al., 2010) with – chain and – gapped parameter, and then the alignments were plotted for synteny analysis.
Phylogeny Analysis
In phylogenetic analysis, full amino acid sequences of housekeeping genes from archaea, proteobacteria, cyanobacteria, tracheophytes, and algae, were aligned using MEGA6 (Tamura et al., 2013) software and edited manually. MrBayes v3.1.2 (Ronquist and Huelsenbeck, 2003) software was used to investigate evolutionary relationships based on amino acid sequences. Bayesian analysis was performed by two separate sequence analyses for four Markov chains (using default heating values), which were run for 500,000 generations until the average standard deviation of split frequencies was below 0.01 (Posada and Crandall, 1998). In addition, trees were sampled every 100 generations with the first 25% of trees discarded as the burn-in. Remaining trees were used to build a 50% majority rule consensus tree, accompanied with posterior probability values. FigTree v1.3.13 was used for displaying phylogenetic trees.
Results and Discussion
Genome Assembly
The genome sequence was assembled using 14.86 and 15.06 Gb mate-paired sequences from libraries with 3 and 5 kb inserts, respectively, plus 46.54 Gb paired-end reads from small-insert libraries (Supplementary Table S1). The assembled nuclear genome of S. japonica contains 418,683 contigs and 236,802 scaffolds, and the total length was 580.5 Mb (Table 1 and Supplementary Table S2), with an N50 of 13,636,083 bp and 48.72% GC content. There are 1,602 scaffolds longer than 100 kb, a total of 5,257 longer than 20 kb, and a total of 11,156 that are longer than 10 kb. We further improved the assembly by using the Hi-C data. After trimming, a total of 237,795,214 pair-end reads, and 105,783,030 pair-end reads were determined as valid, which were aligned to two different enzyme fragments, and the estimated insert size met the expectation. Based on the relationships among valid reads, the order and directions of 46,865 scaffolds/contigs were clustered into the 31 pseudomolecules/chromosomes (Supplementary Table S3), which account for 517,689,860 bp, about 89.19% of the whole genome. This was an updated genome assembly and provided a better resource than the previous assembly (Ye et al., 2015).
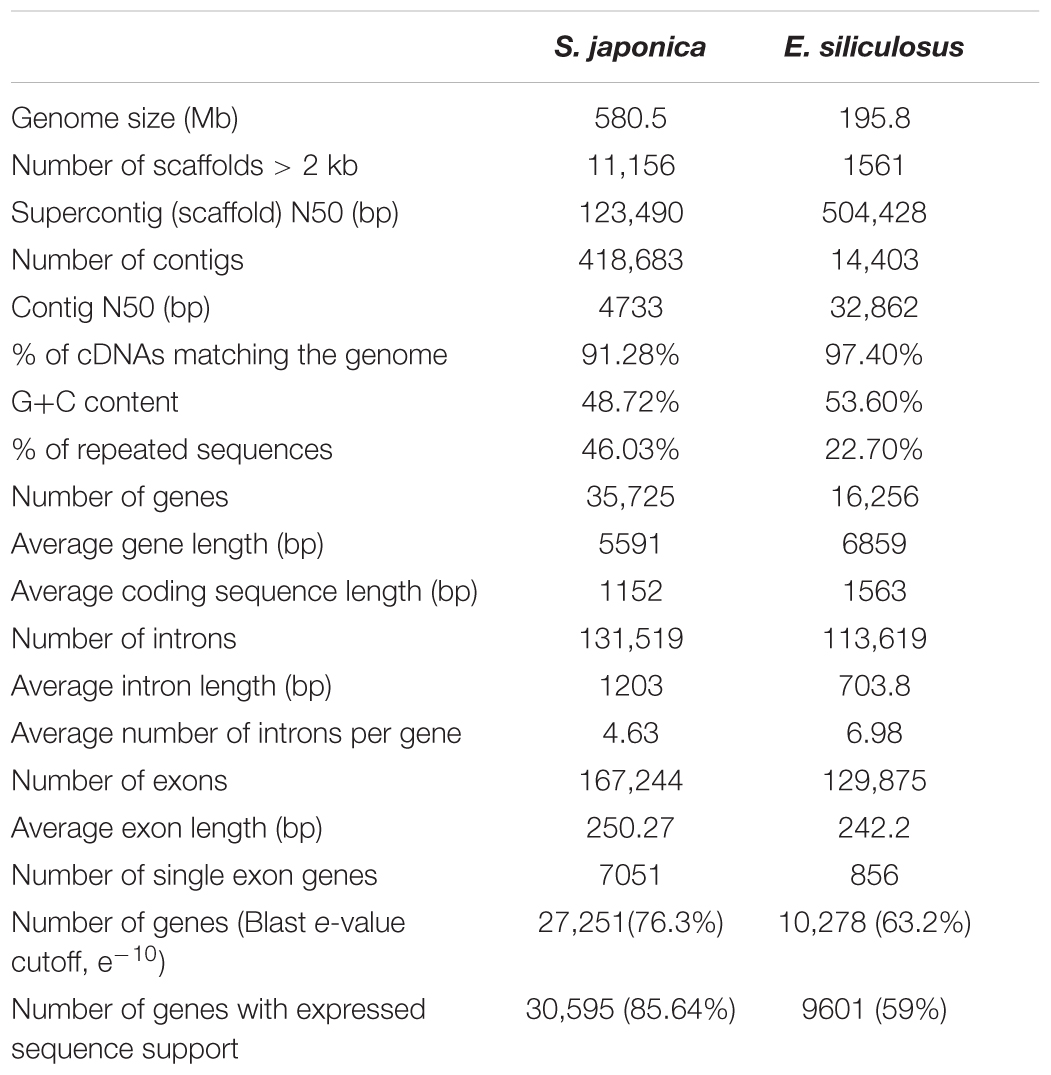
Table 1. Genome statistics of S. japonica and E. siliculosus.
A combination of expert and automatic annotation predicts 35,725 gene models, with an overlap of 23,930 ones with E. siliculosus (Supplementary Figure S1). Genes are rich in introns (4.63 per gene on average). However, the analysis of GO annotation, KOG and KEGG discovered a similar distribution between S. japonica and E. siliculosus (Supplementary Figures S2–S4). A gene family clustering analysis across the whole genome was conducted by using protein sequences of S. japonica and E. siliculosus. Totally, there were 16,954 genes in 9658 orthogroups in S. japonica, 13,063 genes in 9615 orthogroups in E. siliculosus, with 18,771 and 3471 genes unassigned, respectively (Table 2). We extended our ortholog search to more species, i.e., A. anophagefferens, N. gaditana, P. tricornutum, and T. pseudonana, and found that the gene number in orthogroups of S. japonica is the highest, suggesting that the gene duplication strategy was preferred during the genome evolution (Table 2).

Table 2. Orthologous gene number in six algal species.
Integration of Virus Genome in Co-ancestry Period of Laminariales and Ectocarpales
Laminariales and Ectocarpales algae show a close phylogenetic relationship with each other. Analysis of the E. siliculosus genome sequence identified the presence of E. siliculosus virus-1 (EsV-1) (Cock et al., 2010). EsV-1 only infects free-swimming gametes or spores. The viral DNA integrates into the cellular genome after infection and then, through mitosis, it spreads to all cells of a developing host (Bräutigam et al., 1995; Lee et al., 1998). Viral DNA has been detected in some species’ genomes in Ectocarpus and Feldmannia (Meints et al., 2008; Cock et al., 2010). In our analysis, we found some genomic fragments of phaeovirus EsV-1 integrated in S. japonica genome, which were also found in E. siliculosus genome (Figure 1). This result shows that this virus integration may happen before the divergency of Laminariales and Ectocarpales. After BLAST against EsV-1 sequences, we also found that the integrated virus sequences are quite different in these two genomes, as E. siliculosus genome contains large-fragment clusters (310,438 bp of 313,838 bp EsV-1 genome, 98.92%, and contains 173 orthologs genes of 231 EsV-1 ones), and in S. japonica the orthologous regions consist of more fragments and fewer large segments (8,256 bp). So the integration would have occurred before the differentiation of Laminariales and Ectocarpales, and experienced the isolated evolution events after the differentiation. This indicates the diverse algal genome evolutionary mechanisms between Laminariales and Ectocarpales, which may represent the potential driving force for advanced brown algae evolutionary. There may be large-scale rearrangement in Laminariales genomes during evolution from filaments to the thallus, accompanied by loss of some genes.
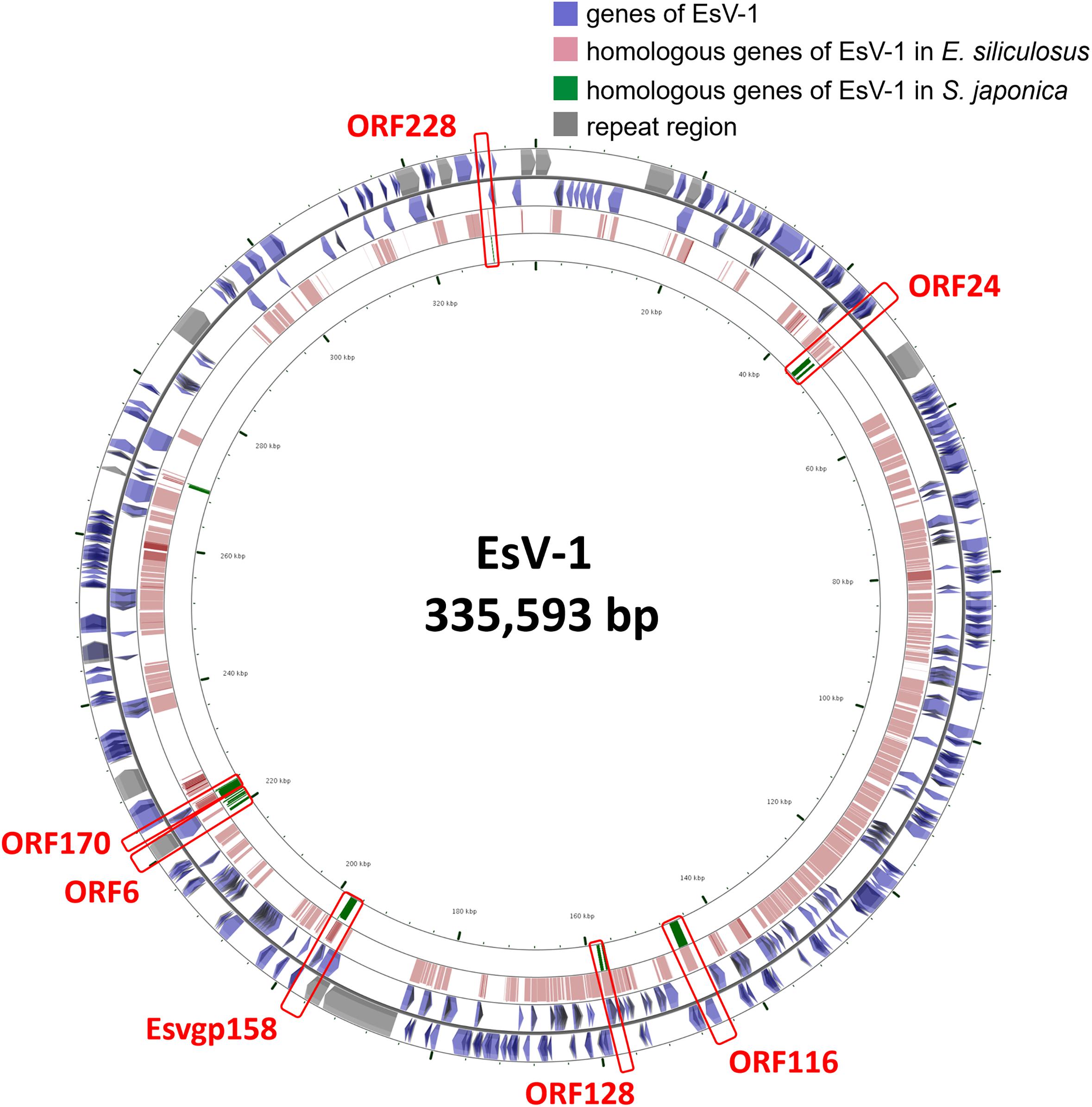
Figure 1. The viral EsV-1 genome and its fragments integrated in the S. japonica and E. siliculosus genomes. The genes highlighted in red bar are found in both S. japonica and E. siliculosus genomes.
Repeats, Introns and Non-coding Regions
We downloaded the genome sequences of brown algae species E. siliculosus (Michel et al., 2010), and conducted a synteny analysis. In total, the conserved regions covered 23,193,396 bases from S. japonica and 15,108,600 bases from E. siliculosus, and no significant replication of large fragments were shown (Supplementary Figure S5). We found that 43.12% of the S. japonica genome assembly could be attributed to repeat sequences, while 22.7% only was found for the reported 214 Mb genome of E. siliculosus, a multicellular filamentous brown alga. LTR, LINE, SINE and simple repeat are the major components of repeat sequences (Supplementary Figure S6).
In total, 98,386 SSRs conforming to the definitions (i.e., unit/minimum number of repeats 1/20, 2/10, 3/7, 4/5, 5/4, and 6/4) were recovered from 15,173 sequences (6.28% of the total sequences) in the S. japonica genome sequences. Provided that the size of the S. japonica genome is 580.5 Mb, the frequency of occurrence of the above SSRs was estimated to be one SSR per 59 kb. Compound SSRs accounted for 17.75% and the average SSR density was 169 SSR/Mb. Di-, tri-, and tetra-nucleotide SSRs accounted for 4.51, 46.67, and 17.21% of the identified SSRs, respectively.
When it comes to the di-nucleotide SSRs, AG/CT pattern was the most frequent type representing 41.06% of di-nucleotide repeat units, followed by AT/AT (35.74%), and AC/GT (23.09%) (Supplementary Figure S7). AGC/CTG was the most abundant trimer motif (34.11%), and ACT/AGT was second at 23.77%. Among the tetra-nucleotide SSRs, ACAG/ATGT (18.20%) was the most common and ACCCG/CGGGT (13.57%) was the most common hexamer (5.00%).
Genome analysis of S. japonica predicted 35,725 genes with average intron length of 1,203 bp, 4.63 introns per gene. Introns constitute 29% of the whole genome, which is lower than that of E. siliculosus (37.3%). A total of 15,820 simple sequence repeats (SSRs) were identified. Among the SSRs, the trinucleotide and dinucleotide repeat types are the most abundant, which is similar to other published algae genome data.
Housekeeping Genes
Just like the diverse archaea, bacteria, tracheophytes and algal species, a wealth of candidate housekeeping gene sequences, such as actin, α-Tubulin (TUA), ß-Tubulin (TUB), Elongation factor 1-alpha (EF1-α), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were also detected in the genome of S. japonica. Therefore, we built phylogenetic trees that display relationships of full amino acid sequences of housekeeping genes from archaea, proteobacteria, cyanobacteria, tracheophytes, and algae (only representative candidates are included to save space) based on Bayesian method. In the consensus tree of four housekeeping genes (EF1α, GAPDH, TUA, and TUB), almost all the Phaeophyceae algae (including S. japonica) formed a well-supported clade with oomycetes or protists, which indicates their origin from endosymbiosis host (Figure 2). The remaining actin genes from brown algae have a complex evolutionary history. Our phylogenetic analysis shows they may have arisen from multiple origins. Three copies of S. japonica actin cluster into separate clades: one is related to oomycetes (e.g., S. japonica 1), and the other groups with cyanobacteria and bacteria (e.g., S. japonica 2, 3). Therefore, S. japonica may acquire actin from different ancestors, while S. japonica 1 is with an endosymbiotic host origin, and S. japonica 2 and 3 have a cyanobacterial origin through endosymbiosis gene transfer (EGT) or acquired from non-cyanobacterial proteobacteria via horizontal gene transfer (HGT).

Figure 2. Phylogenetic tree topologies of housekeeping genes using MrBayes software. (A) EF1α, (B) GAPDH, (C) Actin, (D) TUA, and (E) TUB. ChlP, Chlorophyta; Rho, Rhodophyta; Fun, Fungi; Pha, Phaeophyceae; Dia, Diatom; Oom, Oomycetes; Cya, Cyanobacteria; Bac, Bacteria; Arc, Archaea; Hap, Haptophyta; Cry, Cryptophyta; Gla, Glaucophyta.
Gene Expansion
During the evolution of single cells to multicellular, housekeeping genes, signal transduction and cell junction pathway related genes play an important role (Supplementary Figures S2, S3), while genes and pathways of extracellular component may be more important from multicellular filamentous to multicellular thallus, due to gene expansion (Table 3).
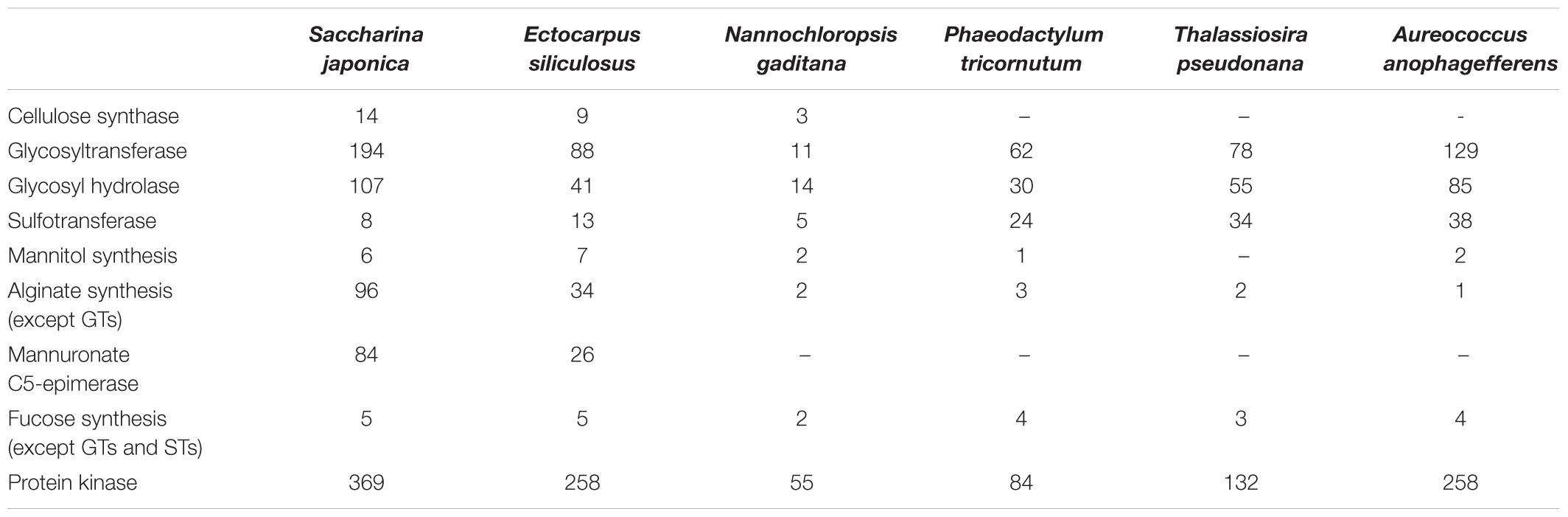
Table 3. Gene number of different metabolisms and gene families in sequenced Heterokontophyta algae.
In phylum Heterokontophyta, there is a very close relationship between orders Laminariales and Ectocarpales, which is consistent with phylogenetic analysis of chloroplast genome. In the present study, in S. japonica the genome size is almost 3 times that of E. siliculosus, and the total gene number is nearly 2.1 times that of E. siliculosus (Tables 2, 3). S. japonica and E. siliculosus genes were subjected to the KEGG database, and the annotated gene comparison revealed a similar distribution of gene types among most categories, which indicated the increases in organismal complexity are not associated with pathway changes. The similarity of gene orthologous were also compared, which suggests that the gigantic increasing of S. japonica genes may depend on gene replication leading to multiple copies. Interestingly, analysis of gene duplication indicated a significant gain of the ones associated with cell wall component metabolisms such as alginate and cellulose synthesis pathways. For instance, Saccharina genome contains a number of 14 candidate cellulose synthase and cellulose synthase-like genes, which is nearly 1.6 times that of Ecutocarpus (Table 3). The modifier gene MC5E in alginate synthesis pathway reached 84 copies in Saccharina genome, about 3.2 times that of Ecutocarpus. The gene number of glycosyltransferases (GTs) and glycosyl hydrolases (GHs), which are likely involved in cell wall polysaccharide metabolisms, also endures an expansion of more than two times in Saccharina genome. The same situation occurred in protein kinases. These genes include membrane-spanning receptor kinases, which may play key roles in developmental processes such as differentiation (De Smet et al., 2009). This indicated that the strategy of gene expansion may contribute a lot to the evolution from the multicellular filamentous brown alga to complex thallus alga.
Saccharina japonica is the most efficient iodine accumulator among all living organisms, owing to the activities of vanadium-dependent haloperoxidases (vHPOs) (Leblanc et al., 2006). The halogen accumulation level of foliaceous S. japonica is much higher than that of filamentous E. siliculosus and the halogen metabolism ability is significantly enhanced. In the genome of S. japonica, we identified 89 vHPOs, including 21 vanadium-dependent bromoperoxidases (vBPOs) and 68 vanadium-dependent iodoperoxidases (vIPOs). In contrast, the previous genomic study on E. siliculosus and S. japonica has, respectively, predicted one (Table 4) and 76 vHPO genes (Ye et al., 2015) involved in halogen metabolism. In addition, 32 dehalogenation related genes were predicted. Brown algae usually have active halogen metabolism, and many halogen related genes were found in the E. siliculosus and Chondrus crispus genome (Saenko et al., 1978; Leblanc et al., 2006). Hypohalous acids and organo-halogenated compounds produced by halogenations are considered substances which can participate in algae defense reactions. The large size of these halogen-related gene families is a result of specific evolutionary adaptation to the marine environment, and it may be evolved in various defense mechanisms and produce more rich secondary metabolites in evolution from small algae to large algae.
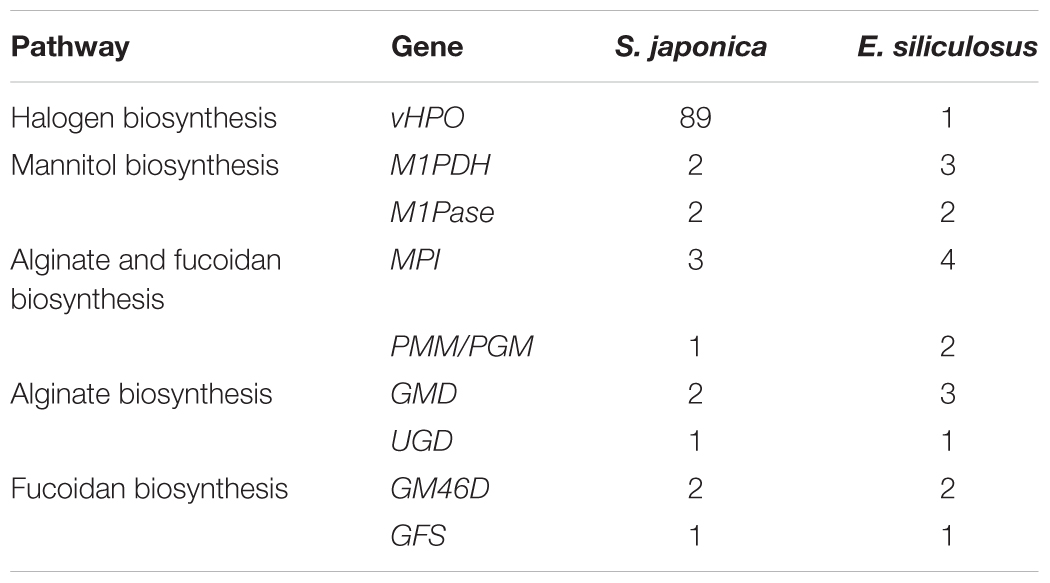
Table 4. Gene copies in the important gene pathways of S. japonica and E. siliculosus.
Plants respond to various biotic or abiotic stimuli with different mechanisms. Heat shock proteins (Hsps) are essential components in plant tolerance mechanism under various abiotic stresses. The function of calcium-based intracellular signaling system is to combine extracellular stimuli with their specific intracellular responses (Edel et al., 2017). Three major elements play a role in the generation and translation of a stimulus-induced Ca2+ signal: influx, efflux and decoding (Edel et al., 2017). In a comparison with E. siliculosus, S. japonica experienced a gene expansion in Hsp family. The number of hsp genes including Hsp20, Hsp33, Hsp40, Hsp70, and Hsp90 in S. japonica is nearly 1.5 times of that of E. siliculosus (Table 5). In the genome of S. japonica, we identified 3 protein families of Ca2+ influx, 2 protein families of Ca2+ efflux and 2 protein families of calcium decoding (Table 6). The number of TPC, MCA, and CaM genes were higher than in E. siliculosus (Table 6). These gene expansion events may contribute to the differentiation and evolution from filamentous Ectocarpus to complex organized Saccharina. In addition, we also found more gene copies of Hsp40, Hsp70, TPC, MCA, and CPK in our S. japonica “Rongfu” genome than in the previous S. japonica genome (Ye et al., 2015). It indicated that the Saccharina high-temperature-resistant variety “Rongfu” possessed more genes of Hsp and calcium-based signaling system which might be caused by artificial selection.

Table 5. Gene number of Hsp gene families in sequenced Saccharina variety “Rongfu,” S. japonica and E. siliculosus.

Table 6. Gene number of the calcium-based signaling system in sequenced Saccharina variety “Rongfu,” S. japonica and E. siliculosus.
The above results indicated that the expansion of Saccharina genome was mainly due to gene family expansion, especially when it contributed to cell wall and halogen biosynthesis (Tables 3–6). The differentiation and evolution from filamentous Ectocarpus to complex organized Saccharina may be related to these gene expansion events.
However, we did not discover a huge replication in housekeeping, signal-transduction-related and cell-communication-related genes. For example, mannitol represents up to 15–26% of dry weight, as one of the primary photosynthetic products and storage compounds in Laminariales, and its biosynthesis involves two major enzymes, mannitol-1-P dehydrogenase (M1PDH) and mannitol-1-phosphatase (M1Pase) (Chi et al., 2018b). Two unigenes of M1PDH1 and M1PDH2 and two M1Pase homologs of M1Pase1 and M1Pase2 were found in the S. japonica genome, while there are three M1PDH unigenes and two M1Pase copies in E. siliculosus (Table 4). Meanwhile, in a comparison of E. siliculosus, S. japonica did not experience a significant family expansion in alginate and fucoidan biosynthesis, which involves 6–8 genes/families (Chi et al., 2018a). Mannose-6-phosphate isomerase (MPI), phosphomannomutase (PMM), and mannose-1-phosphate guanylyltransferase (MPG) are involved in converting fructose-6-phosphate into GDP-mannose. GDP-mannose may be used in the two biosynthesis ways: the alginate biosynthesis which involves GDP-mannose/UDP-glucose 6-dehydrogenase (GMD/UGD), mannuronan synthase (MS), and MC5E; the fucoidan biosynthesis which involves GDP-mannose 4,6-dehydratase (GM46D), GDP-fucose synthetase (GFS), and et al. It showed that S. japonica contained less gene copies of MPI, PMM/PGM, and GMD, compared to E. siliculosus (Table 4).
In addition, we also found more gene copies in our S. japonica than in the previous S. japonica genomes (Ye et al., 2015), for example, the vHPO gene family. It indicated that the Saccharina varieties possess the different gene clusters, which might be caused by artificial selection, and provide important resources for algal breeding on high alginate and iodine.
Data Availability
The raw data is deposited in NCBI, with Bioproject accession of PRJNA280905, and the accession in Short Read Archive of SRP057092, including four running datasets, SRR1972526, SRR1972528, SRR1972529, and SRR1972530.
Conclusion
Large alga S. japonica has a large complex thallus tissue, and possesses a huge genome and gene expansion. In spite of high similarity in gene composition and classification, the structures of the two genomes of E. siliculosus and S. japonica are different in non-coding regions, repeat sequences, introns length and gene number. In particular, the number of genes related to extracellular components and halogen biosynthesis in S. japonica is significantly higher than that of E. siliculosus, which may be the main motive force for evolution of filament to thallus. In addition, the integration of viral genome in the S. japonica and E. siliculosus genomes during their co-ancestry period further demonstrated their close genetic relationship, genome rearrangements and gene duplication events after their differentiation.
Author Contributions
TL, TX, XW, and JY designed the study. SC, JZ, and YY maintained and prepared the plant materials. GL and GS prepared the sequencing libraries and conducted the sequencing. TX conducted the Hi-C sequencing. GW, SJ, and XW assembled the draft sequence, and conducted the analysis. GW and SC drafted the manuscript. SJ modified the manuscript. All authors reviewed and approved the final manuscript.
Funding
This study was funded by the National Natural Science Foundation of China (41376143); Science and Technology Major Project of Fujian Province (2019NZ08003); Leading Talents Program in Taishan Industry of Shandong Province; the seed industry innovation and industrialization project of Fujian Province (2017FJSCZY01); the 13th Five-Year Plan for the Marine Innovation and Economic Development Demonstration Projects (FZHJ1); and China Agriculture Research System-50.
Conflict of Interest Statement
SC was employed by Qingdao Haida Blue Tek Biotechnology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00378/full#supplementary-material
Footnotes
- ^ https://db.cngb.org/onekp/
- ^ http://bioinformatics.psb.ugent.be/genomes/view/Ectocarpus-siliculosus
- ^ http://tree.bio.ed.ac.uk/software/figtree/
References
Bräutigam, M., Klein, M., Knippers, R., and Müller, D. G. (1995). Inheritance and meiotic elimination of a virus genome in the host Ectocarpus szlzculosus (Phaeophyceae). J. Phycol. 31, 823–827. doi: 10.1111/j.0022-3646.1995.00823.x
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31:1119. doi: 10.1038/nbt.2727
Chi, S., Liu, T., Wang, X., Wang, R., Wang, S., Wang, G., et al. (2018a). Functional genomics analysis reveals the biosynthesis pathways of important cellular components (alginate and fucoidan) of Saccharina. Curr. Genet. 64, 259–273. doi: 10.1007/s00294-017-0733-4
Chi, G., Liu, T., Wang, X., Liu, C., Jin, Y., et al. (2018b). Transcriptomic and proteomic analysis of genes associated with mannitol metabolism in Saccharina. Genom. Proteom. Bioinform. (in press).
Cock, J. M., Sterck, L., Rouzé, P., Scornet, D., Allen, A. E., Amoutzias, G., et al. (2010). The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465, 617–621. doi: 10.1038/nature09016
De Smet, I., Voß, U., Jürgens, G., and Beeckman, T. (2009). Receptor-like kinases shape the plant. Nat. Cell Biol. 11:1166. doi: 10.1038/ncb1009-1166
Demirbas, A. (2010). Use of algae as biofuel sources. Energy Convers. Manag. 51, 2738–2749. doi: 10.1016/j.enconman.2010.06.010
Draisma, S. G., Prud’homme Van Reine, W. F., Stam, W. T., and Olsen, J. L. (2001). A reassessment of phylogenetic relationships within the Phaeophyceae based on RUBISCO large subunit and ribosomal DNA sequences. J. Phycol. 37, 586–603. doi: 10.1046/j.1529-8817.2001.037004586.x
Edel, K. H., Marchadier, E., Brownlee, C., Kudla, J., and Hetherington, A. M. (2017). The evolution of calcium-based signalling in plants. Curr. Biol. 27, R667–R679. doi: 10.1016/j.cub.2017.05.020
Erting, L., Daugbjerg, N., and Møller Pedersen, P. (2004). Nucleotide diversity within and between four species of Laminaria (Phaeophyceae) analysed using partial LSU and ITS rDNA sequences and AFLP. Eur. J. Phycol. 39, 243–256. doi: 10.1080/09670260410001712563
Frith, M. C., Wan, R., and Horton, P. (2010). Incorporating sequence quality data into alignment improves DNA read mapping. Nucleic Acids Res. 38:e100. doi: 10.1093/nar/gkq010
Guillemaut, P., and Maréchal-Drouard, L. (1992). Isolation of plant DNA: a fast, inexpensive, and reliable method. Plant Mol. Biol. Rep. 10, 60–65. doi: 10.1007/BF02669265
Huang, L., Wen, K., Gao, X., and Liu, Y. (2010). Hypolipidemic effect of fucoidan from Laminaria japonica in hyperlipidemic rats. Pharm. Biol. 48, 422–426. doi: 10.3109/13880200903150435
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Kim, K.-H., Kim, Y.-W., Kim, H. B., Lee, B. J., and Lee, D. S. (2006). Anti-apoptotic activity of laminarin polysaccharides and their enzymatically hydrolyzed oligosaccharides from Laminaria japonica. Biotechnol. Lett. 28, 439–446. doi: 10.1007/s10529-005-6177-9
Korbel, J. O., and Lee, C. (2013). Genome assembly and haplotyping with Hi-C. Nat. Biotechnol. 31:1099. doi: 10.1038/nbt.2764
Lane, C. E., and Archibald, J. M. (2008). The eukaryotic tree of life: endosymbiosis takes its TOL. Trends Ecol. Evol. 23, 268–275. doi: 10.1016/j.tree.2008.02.004
Lane, C. E., Mayes, C., Druehl, L. D., and Saunders, G. W. (2006). A multi-gene molecular investigation of the kelp (Laminariales, Phaeophyceae) supports substantial taxonomic re-organization. J. Phycol. 42, 493–512. doi: 10.1111/j.1529-8817.2006.00204.x
Leblanc, C., Colin, C., Cosse, A., Delage, L., La Barre, S., Morin, P., et al. (2006). Iodine transfers in the coastal marine environment: the key role of brown algae and of their vanadium-dependent haloperoxidases. Biochimie 88, 1773–1785. doi: 10.1016/j.biochi.2006.09.001
Lee, A. M., Ivey, R. G., and Meints, R. H. (1998). Repetitive DNA insertion in a protein kinase ORF of a latent FSV (Feldmannia sp. Virus) genome. Virology 248, 35–45. doi: 10.1006/viro.1998.9245
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liang, X., Wang, X., Chi, S., Wu, S., Sun, J., Liu, C., et al. (2014). Analysis of Saccharina japonica transcriptome using the high-throughput DNA sequencing technique and its vanadium-dependent haloperoxidase gene. Acta Oceanol. Sin. 33, 27–36. doi: 10.1007/s13131-014-0438-1
Meints, R. H., Ivey, R. G., Lee, A. M., and Choi, T.-J. (2008). Identification of two virus integration sites in the brown alga Feldmannia chromosome. J. Virol. 82, 1407–1413. doi: 10.1128/jvi.01983-07
Michel, G., Tonon, T., Scornet, D., Cock, J. M., and Kloareg, B. (2010). The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus. Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytol. 188, 82–97. doi: 10.1111/j.1469-8137.2010.03374.x
Mizuta, H., and Yasui, H. (2010). Significance of radical oxygen production in sorus development and zoospore germination in Saccharina japonica (Phaeophyceae). Bot. Mar. 53, 409–416. doi: 10.1515/bot.2010.047
Posada, D., and Crandall, K. A. (1998). Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Saenko, G. N., Kravtsova, Y. Y., Ivanenko, V. V., and Sheludko, S. I. (1978). Concentration of iodine and bromine by plants in the seas of Japan and Okhotsk. Mar. Biol. 47, 243–250. doi: 10.1007/bf00541002
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C.-J., Vert, J.-P., et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16:259. doi: 10.1186/s13059-015-0831-x
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Valentin, K., and Zetsche, K. (1990). Rubisco genes indicate a close phylogenetic relation between the plastids of Chromophyta and Rhodophyta. Plant Mol. Biol. 15, 575–584. doi: 10.1007/BF00017832
Vishchuk, O. S., Ermakova, S. P., and Zvyagintseva, T. N. (2011). Sulfated polysaccharides from brown seaweeds Saccharina japonica and Undaria pinnatifida: isolation, structural characteristics, and antitumor activity. Carbohyd. Res. 346, 2769–2776. doi: 10.1016/j.carres.2011.09.034
Vishchuk, O. S., Tarbeeva, D. V., Ermakova, S. P., and Zvyagintseva, T. N. (2012). Structural characteristics and biological activity of fucoidans from the brown algae Alaria sp. and Saccharina japonica of different reproductive status. Chem. Biodivers. 9, 817–828. doi: 10.1002/cbdv.201100266
Wang, J., Wang, F., Yun, H., Zhang, H., and Zhang, Q. (2012). Effect and mechanism of fucoidan derivatives from Laminaria japonica in experimental adenine-induced chronic kidney disease. J. Ethnopharmacol. 139, 807–813. doi: 10.1016/j.jep.2011.12.022
Wang, J., Zhang, Q., Zhang, Z., and Li, Z. (2008). Antioxidant activity of sulfated polysaccharide fractions extracted from Laminaria japonica. Int. J. Biol. Macromol. 42, 127–132. doi: 10.1016/j.ijbiomac.2007.10.003
Xue, C.-H., Fang, Y., Lin, H., Chen, L., Li, Z.-J., Deng, D., et al. (2001). Chemical characters and antioxidative properties of sulfated polysaccharides from Laminaria japonica. J. Appl. Phycol. 13, 67–70. doi: 10.1023/A:1008103611522
Ye, N., Zhang, X., Miao, M., Fan, X., Zheng, Y., Xu, D., et al. (2015). Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 6:6986. doi: 10.1038/ncomms7986
Yoon, H. S., Lee, J. Y., Boo, S. M., and Bhattacharya, D. (2001). Phylogeny of Alariaceae, Laminariaceae, and Lessoniaceae (Phaeophyceae) based on plastid-encoded RuBisCo spacer and nuclear-encoded ITS sequence comparisons. Mol. Phylogenet. Evol. 21, 231–243. doi: 10.1006/mpev.2001.1009
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zemke-White, W. L., and Ohno, M. (1999). World seaweed utilisation: an end-of-century summary. J. Appl. Phycol. 11, 369–376. doi: 10.1023/A:1008197610793
Zhao, X., Dong, S., Wang, J., Li, F., Chen, A., and Li, B. (2012). A comparative study of antithrombotic and antiplatelet activities of different fucoidans from Laminaria japonica. Thromb. Res. 129, 771–778. doi: 10.1016/j.thromres.2011.07.041
Keywords: Saccharina japonica, genome sequencing, virus genome, phylogenetic analysis, extracellular components, halogen biosynthesis
Citation: Liu T, Wang X, Wang G, Jia S, Liu G, Shan G, Chi S, Zhang J, Yu Y, Xue T and Yu J (2019) Evolution of Complex Thallus Alga: Genome Sequencing of Saccharina japonica. Front. Genet. 10:378. doi: 10.3389/fgene.2019.00378
Received: 12 December 2018; Accepted: 09 April 2019;
Published: 02 May 2019.
Edited by:
Gen Hua Yue, Temasek Life Sciences Laboratory, SingaporeReviewed by:
Chaotian Xie, Jimei University, ChinaJian Xu, Chinese Academy of Fishery Sciences (CAFS), China
Copyright © 2019 Liu, Wang, Wang, Jia, Liu, Shan, Chi, Zhang, Yu, Xue and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Liu, bGl1dGFvQG91Yy5lZHUuY24= Shangang Jia, anNnMjAwODMwQDE2My5jb20= Ting Xue, eHVldGluZ0Bmam51LmVkdS5jbg== Jun Yu, anVueXVAYmlnLmFjLmNu
†These authors have contributed equally to this work