 Lior Greenbaum1,2,3*
Lior Greenbaum1,2,3* Ben Pode-Shakked1,3Shlomit Eisenberg-Barzilai1Michal Dicastro-Keidar1Anat Bar-Ziv1Nurit Goldstein1Haike Reznik-Wolf1Hana Poran1Amihai Rigbi4Ortal Barel5Aida M. Bertoli-Avella6Peter Bauer6Miriam Regev1,3Annick Raas-Rothschild1,3Elon Pras1,3Michal Berkenstadt1,3*
Ben Pode-Shakked1,3Shlomit Eisenberg-Barzilai1Michal Dicastro-Keidar1Anat Bar-Ziv1Nurit Goldstein1Haike Reznik-Wolf1Hana Poran1Amihai Rigbi4Ortal Barel5Aida M. Bertoli-Avella6Peter Bauer6Miriam Regev1,3Annick Raas-Rothschild1,3Elon Pras1,3Michal Berkenstadt1,3*- 1The Danek Gertner Institute of Human Genetics, Sheba Medical Center, Tel Hashomer, Israel
- 2The Joseph Sagol Neuroscience Center, Sheba Medical Center, Tel Hashomer, Israel
- 3Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- 4Faculty of Education, Beit Berl College, Kfar Saba, Israel
- 5Sheba Cancer Research Center, Sheba Medical Center, Tel Hashomer, Israel
- 6Centogene AG, Rostock, Germany
Prenatal ultrasound (US) abnormalities often pose a clinical dilemma and necessitate facilitated investigations in the search of diagnosis. The strategy of pursuing fetal whole-exome sequencing (WES) for pregnancies complicated by abnormal US findings is gaining attention, but the reported diagnostic yield is variable. In this study, we describe a tertiary center’s experience with fetal WES from both terminated and ongoing pregnancies, and examine the clinical factors affecting the diagnostic rate. A total of 45 consecutive families of Jewish descent were included in the analysis, for which clinical fetal WES was performed under either single (fetus only), trio (fetus and parents) or quatro (two fetuses and parents) design. Except one, all families were non-consanguineous. In 41 of the 45 families, WES was sought following abnormal fetal US findings, and 18 of them had positive relevant family history (two or more fetuses with US abnormalities, or single fetus with US abnormalities and an affected parent). The overall diagnostic yield was 28.9% (13/45 families), and 31.7% among families with fetal US abnormalities (13/41). It was significantly higher in families with prenatal US abnormalities and relevant family history (10/18, 55.6%), compared to families with prenatal US abnormal findings and lack of such history (3/23, 13%) (p = 0.004). WES yield was relatively high (42.9–60%) among families with involvement of brain, renal or musculoskeletal US findings. Taken together, our results in a real-world setting of genetic counseling demonstrates that fetal WES is especially indicated in families with positive family history, as well as in fetuses with specific types of congenital malformation.
Introduction
Recent years have seen the growing implementation of next-generation sequencing (NGS) techniques into widespread clinical use, revolutionizing the diagnostic odyssey for many families with monogenic disorders (Yang et al., 2013; Stranneheim and Wedell, 2016). While whole-exome sequencing (WES) and whole-genome sequencing (WGS) are more commonly utilized as a tool for molecular diagnosis of affected pediatric and adult patients (Lee et al., 2014; Taylor et al., 2015; Sawyer et al., 2016), data regarding their utility in routine use at the prenatal setting is still relatively limited (Jelin and Vora, 2018).
Initially, several preliminary reports described the successful use of fetal WES/WGS in single families (Talkowski et al., 2012; Filges et al., 2014). Since then, several groups have reported their experience with exome sequencing in the context of fetuses detected by ultrasound (US) to have structural anomalies (for example, Carss et al., 2014; Drury et al., 2015; Fu et al., 2018; Normand et al., 2018). The widely variable diagnostic rates in literature (ranging from around 6.2% to as high as 80%) may be attributable to differences in inclusion criteria, single (fetus only) versus trio sequencing and sample size (Best et al., 2018). Diagnostic yield was higher in cases of, for instance, parental consanguinity, previous normal karyotype and chromosomal microarray analysis (CMA), fetuses with multiple congenital anomalies or fetal features suggestive of a specific syndrome, related to variants in multiple known causative genes (Vora et al., 2017; Best et al., 2018; Daum et al., 2019). Recently, two large scales studies of fetal WES were published. In the Prenatal Assessment of Genomes and Exomes (PAGE) study, which included exome sequencing in 610 fetuses (and 1202 parental samples) with structural anomalies detected by US, a diagnostic genetic variant was found in 8.5% of the fetuses, and additional 3.9% of fetuses had variant of unknown significance (VUS) with potential clinical usefulness (Lord et al., 2019). In another WES study of fetuses with structural anomalies, diagnostic rate of 10.3% among 234 fetus-parents trios was reported (Petrovski et al., 2019).
We describe herein our experience with clinical fetal WES in 45 consecutive families from either terminated or ongoing pregnancies, and discuss the added value and challenges associated with this diagnostic strategy. The study represents a real-world setting in genetic counseling service, in which mixture of various indications for WES is encountered, and WES is performed for clinical purposes and paid by families.
Materials and Methods
Sample Recruitment
In this consecutive series study, we included all families who were consulted at the Danek Gertner Institute of Human Genetics at Sheba Medical Center (SMC) between January 2015 and September 2018, and for whom at least one fetal WES was performed, from a terminated or an ongoing pregnancy. A total of 45 families were retrospectively included in the current study.
All families were of Jewish ancestry, 44 were non-consanguineous and in one family the parents were first degree cousins. Prior to pregnancy or at its first stages, all families were instructed to undertake prenatal genetic carrier screening for prevalent diseases, according to their ethnic origin. All couples received genetic counseling at the SMC genetic institute and a detailed clinical evaluation, including at least one prenatal US examination, and family medical history were obtained. Fetal magnetic resonance imaging (MRI) was carried out in several pregnancies, in addition to US. We included in this study both fetuses with or without abnormal US findings.
WES was performed as a clinical service, paid for directly by the families (for two families, internal funding of the genetic institute was used), and final WES report was issued by the performing lab. Both parents provided written informed consent for the clinical WES, after they had received an explanation regarding the benefits and limitations of the test.
Fetal DNA was extracted following amniocentesis, chorionic villus sampling (CVS) or from fetal material taken during termination of pregnancy (TOP) procedure, and was stored for genetic testing. All TOPs were approved by the institutional committee. Parental DNA was extracted from peripheral blood samples. In addition, prior to WES, CMA was completed in 42 of the families (in at least single fetus) at the SMC cytogenetic laboratory, as a routine clinical service. In all these cases, no cytogenetic findings that explained the phenotype were detected. In the three families with missing CMA, molecular diagnosis was found by WES.
Whole-Exome Sequencing
Thirty three families performed trio (fetus-mother-father) WES, 7 families quatro (two fetuses-mother-father) WES and in 5 families a singleton WES was sent, depending on availability of fetal DNA from previously terminated pregnancies, as well as financial considerations.
The majority of WES (32 out of 45) were performed at Centogene laboratories1. Briefly, commercial capture kits for exome sequencing were used, and sequencing was performed on Ilumina platform, to obtain an average coverage depth of approximately 100X. Bioinformatics analysis included alignments of reads to reference genome, filtering out low quality reads and artifacts, and annotation of variants as described previously (Trujillano et al., 2017). Disease causing variants within the Human Gene Mutation Database (HGMD)2, ClinVar3 or in CentoMD4 as well as variants with a minor allele frequency (MAF) of less than 1% in the Exome Aggregation Consortium (ExAC)5 were considered, focusing on exons and flanking intronic bases. The family history and clinical description provided were used to evaluate the identified variants.
Eight of WES were performed at the bioinformatics unit of SMC Cancer Research Center, using similar WES and bioinformatics methodology, and five in CeGaT laboratories6. In five cases, the WES raw data was further transferred to an independent bioinformatitian for re-analysis, paid for directly by the family.
Exome data were interpreted according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015), and variants were classified as either pathogenic, likely pathogenic, uncertain significance, likely benign or benign.
Molecular Diagnosis
The final molecular diagnosis to each family was given by the SMC genetic institute team, based on the WES report variant classification and consistency between suggested genes and phenotype. A family was considered as molecularly diagnosed when pathogenic or likely pathogenic variants were found in genes that were pertinent to the clinical phenotype and in line with the suspected inheritance pattern. When variants were classified as VUS in the original WES report, re-evaluation was performed based on clinical considerations, bioinformatics and segregation analysis in additional family members. Team decision was made accordingly.
Data Analysis
Families were divided into three groups:
(1) Abnormal prenatal US findings and positive relevant family history: defined as families with two or more fetuses with abnormal prenatal US findings (in current and previous pregnancies), or families with a single fetus with US abnormalities and an affected parent, mother or father (with a resembling phenotype, or potentially manifesting carrier mother). This relates to options of autosomal recessive, autosomal dominant and X-linked models of inheritance, and even of gonadal mosaicism.
(2) Abnormal prenatal US findings without relevant family history: referring to families with a single fetus affected by abnormal US findings, but without any previous fetuses or family members (including adult siblings) with abnormal prenatal US or a medical history relevant to the findings.
(3) Fetuses without any US abnormalities (with or without first degree relative affected by a severe genetic disorder that was excluded in the fetus prior to WES, by Sanger sequencing).
For the diagnostic rate calculations, each family was counted as a single case. To evaluate the effect of fetal US abnormalities type on WES referral and diagnostic yield, we classified the families into five phenotypic groups (based on findings in at least one fetus): brain, renal, cardiac or musculoskeletal system abnormalities and increased nuchal translucency (NT)/edema/hydrops signs. Part of the families matched to more than one group, while others to none.
For statistical analysis, we used T-test, Pearson chi-square or Fisher’s exact test (two sided), as appropriate. P-value < 0.05 was considered statistically significant.
Results
Characteristics of 45 fetal WES families included in the presented study and type of WES (single/trio/qautro) are presented in Table 1. In 41 of the 45 families, WES was performed due to a wide range of prenatal US abnormalities. Average maternal age at the pregnancy of the index fetus was 33.5 ± 4.2 years (range, 23–43 years). The pregnant women were at an average gestational age of 23 ± 7.5 weeks (range, 12.5–36 weeks) at first genetic counseling in our clinic (for the index fetus). For 23 families, WES was sent after TOP (51.1%), and in 22 families during an ongoing pregnancy (48.9%). Turnaround time from WES submission to results was 15 to 20 working days for ongoing pregnancies, and up to 2 months following TOP.
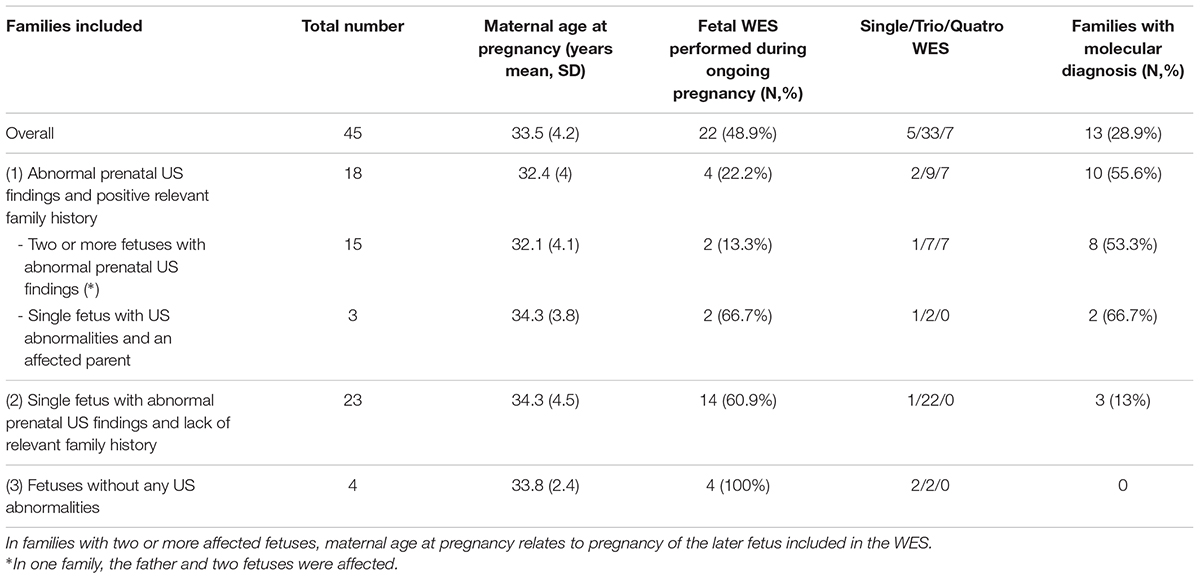
Table 1. Characteristics of 45 fetal WES families included in the presented study, type of WES and rate of molecular diagnosis.
Overall, 13 of the 45 families met criteria for a molecular diagnosis, reaching diagnostic yield of 28.9% (Table 1). Among families with US abnormalities, the yield was 13/41 (31.7%). We detected three de novo heterozygous causative variants, one autosomal dominant causative variant inherited from the mother, eight autosomal recessive cases (three compound heterozygous and five with homozygous variants) and one X-linked causative variant. Osteogenesis imperfecta due to de novo causative variant in the COL1A2 gene was found in two families, while all other conditions occurred once in a single family only. All diagnosed families are described in Table 2, which includes two cases that were previously reported by our group: diaphanospondylodysostosis (BMPER gene) (Greenbaum et al., 2019) and LMOD3-associated Nemaline Myopathy (Berkenstadt et al., 2018). Description of US findings in families without molecular diagnosis is presented in Supplementary Table 1.
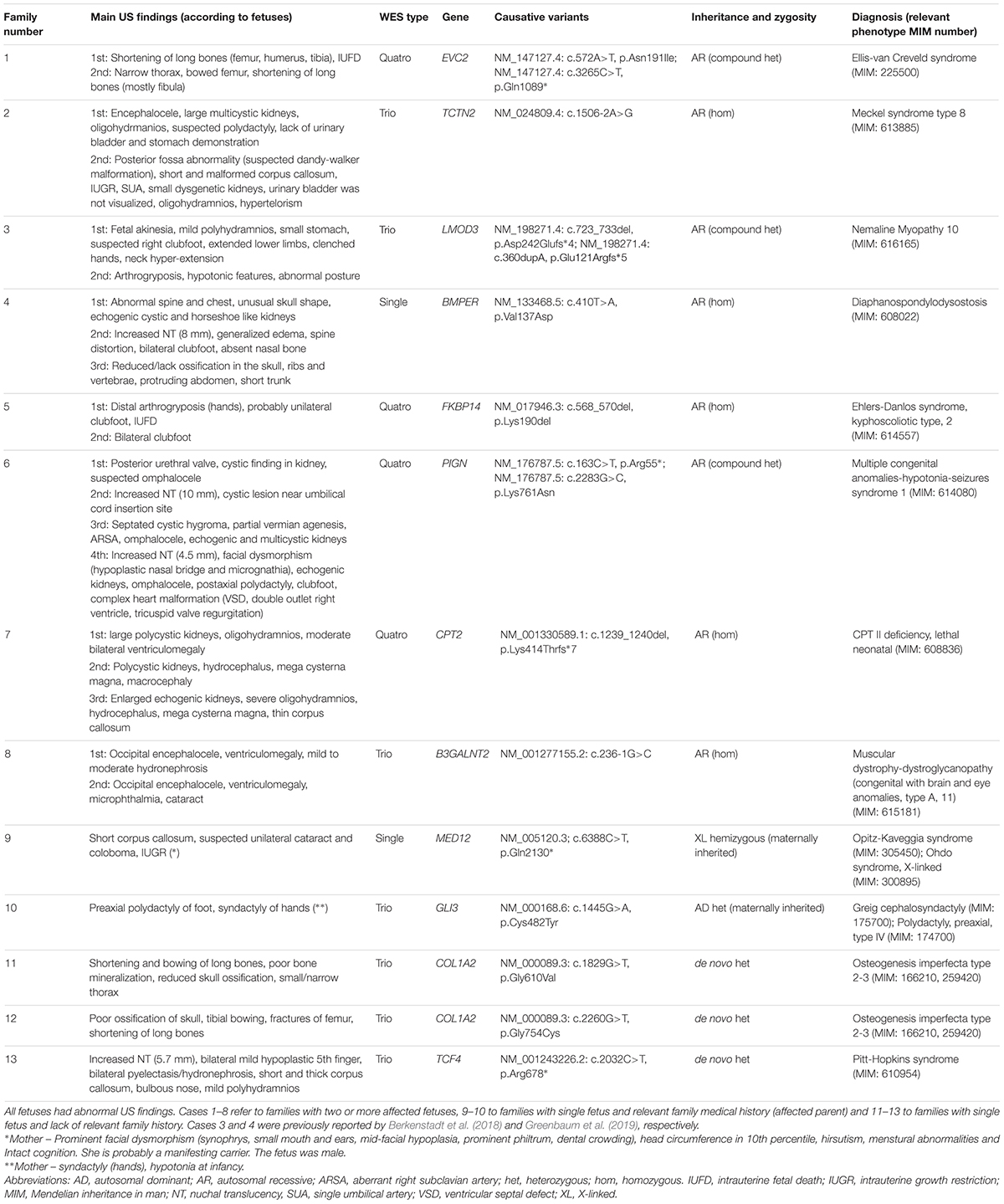
Table 2. Resolved 13 families- summary of main US findings, WES results and final molecular diagnosis.
In none of the four families without prenatal abnormal US findings was a molecular diagnosis reached. Among the 18 families with prenatal US abnormalities and positive relevant family history, a molecular diagnosis was found in 10 (55.6%). This was significantly higher compared to the 23 families with abnormal prenatal US findings and lack of relevant family history, in which the molecular cause was detected in only 3 (13%) (χ2(1) = 8.43, p = 0.004). There was no significant difference in maternal age between the two groups.
In a sub-analysis, the diagnostic rate in families with two or more fetuses with abnormal prenatal US findings was 8 out of 15 (53.3%), significantly higher than in families with prenatal US abnormalities in a mere single fetus, regardless of a relevant family history (5 out of 26, 19.2%; p = 0.038, Fisher’s exact test).
Looking at specific organ/body system involvement, higher diagnostic rates (more than the overall rate) were observed among families with fetal US abnormalities in the brain (60%), renal (42.9%) and musculoskeletal system (55.6%), compared to cardiac US abnormalities (16.7%) or increased NT/edema/hydrops signs (27.3%) (Table 3).

Table 3. Referral indications and WES yield, according to US findings in specific organ or body systems (among 41 families with US abnormalities).
Among ongoing pregnancies with US abnormalities, molecular diagnosis was found in 2 of 18 families (11.1%), both with positive relevant family history (the pregnancies were carried on). Interestingly, among 41 families with US abnormalities, significantly more families without relevant history performed WES during ongoing pregnancy (14/23, 60.9%), compared to families with positive history (4/18, 22.2%) (χ2(1) = 6.12, p = 0.013).
For all families with molecular diagnosis, options for future family planning were discussed, including prenatal diagnosis by amniocentesis or pregestational diagnosis (PGD) before next pregnancy (in autosomal dominant, recessive or X-linked inheritance) and amniocentesis to rule out recurrence due to gonadal mosaicisim (for de novo mutations).
Representative Clinical Cases
Illustrative cases of two families with molecular diagnosis are presented below (numbered according to Table 2), emphasizing the challenges in the genetic counseling and the necessity of accurate US phenotyping. In the first family, with two affected fetuses (suspected recessive inheritance model), the list of potential disease causing genes in the differential diagnosis was long, and WES shortened the time of investigation. In the second family, without relevant family history, no specific diagnosis was suspected by the clinicians, and WES was probably the most cost-effective method for genetic work-up.
Family 2
A healthy couple of Jewish Ethiopian origin was referred for genetic counseling at 33 weeks and 2 days of gestation in their second pregnancy. A previous pregnancy was terminated at week 25, due to abnormal US findings, including encephalocele, large multicystic kidneys, oligohydramnios, suspected polydactyly and lack of urinary bladder and stomach demonstration. In the second pregnancy, NT was normal. US at early stage of pregnancy showed brain and renal malformations, but the couple did not continue follow up. At 33 week of gestation, US demonstrated posterior fossa abnormality (suspected dandy-walker malformation), a short and malformed corpus callosum, intrauterine growth retardation (IUGR), single umbilical artery (SUA), small dysgenetic kidneys, hypertelorism, and oligohydramnios. Urinary bladder was visualized. In follow up fetal brain MRI, a large cystic finding in posterior fossa was noticed, in addition to ventriculomegaly, heterotopic foci at the ventricular wall and short corpus callosum. From this deceased fetus, DNA was extracted, CMA was normal, and the couple sent a trio WES few months later. The combination of brain malformations, polydactcyly and kidneys abnormalities suggested diagnosis of Meckle–Gruber syndrome. However, since many genes are related to this disorder, and to exclude other potential disorders which were on the differential diagnosis, WES was regarded as optimal diagnostic approach. A homozygous NM_024809.4: c.1506-2A>G variant in the TCTN2 gene (intron 13) was found in the fetus. This variant is predicted to disrupt a highly conserved acceptor splice site, and was previously described as disease causing for Meckel–Gruber type 8 syndrome in consanguineous Arab family (Shaheen et al., 2011). Other mutations in TCTN2 were found to cause Joubert syndrome (Huppke et al., 2015). Both parents were heterozygous for this variant. The couple was advised to consider PGD in following pregnancies.
Family 13
A Healthy Jewish couple, both 31 years old (father from Iraq origin and mother of Ashkenazi origin) was consulted at 31 weeks (w)+5 days (d) of gestation of their first pregnancy. NT was 2.9 mm. On 16w+3d of gestation, US demonstrated elevated nuchal fold (5.7 mm), bilateral mild hypoplastic 5th finger and bilateral pyelectasis. Fetal echocardiogram was normal. Amniocentesis performed and CMA was normal. At 22w+3d, relative short and thick corpus callosum was noticed, as well as nuchal fold of 5.1–5.5 mm and mild pyelectasis. Further multiple US were carried by several experts, indicating short corpus callosum, hydronephrosis (10–13 mm), bulbous nose and mild polyhydramnios. Fetal brain MRI (at 30 weeks of gestation) demonstrated dysmorphic and short corpus callosum (<3rd percentile). The couple decided to terminate the pregnancy. In post mortem observation, facial dysmorphism, including coarseness and a bulbous nose, was noticed. Trio WES revealed a de novo heterozygous variant in TCF4 gene (NM_001243226.2: c.2032C>T, p.Arg678∗) in the fetus, consistent with the diagnosis of Pitt-Hopkins syndrome. The fetus facial dysmorphism and the short corpus callosum supported the diagnosis. This variant creates a premature stop codon, and was previously reported in this disorder (Maduro et al., 2016). In a following pregnancy, amniocentesis was performed to exclude mutation in the fetus, due to option of germline mosaicism.
Discussion
In this retrospective study, representing a clinical experience and practice within a tertiary referral hospital in Israel, we investigated the yield of fetal WES, from terminated and ongoing pregnancies.
The overall yield of molecular diagnosis was 28.9% (13 out of 45 families), and in families with US abnormalities, the yield was 31.7%. We noticed a significantly higher diagnostic rate in families with abnormal prenatal US findings and positive relevant family history (mainly an affected fetus from previous pregnancies), compared to families without a relevant history. This shows the importance of fetal WES indication for pregnancies with US abnormalities and positive family history, suggesting an underlining genetic disease.
Our diagnostic rate is within the range of 21–32%, seen in several studies of diagnostic yield for exome sequencing performed on fetal samples (Drury et al., 2015; Fu et al., 2018; Normand et al., 2018; Daum et al., 2019), and close to a diagnostic rate of 36.7% from exome sequencing of 278 infants in an intensive care unit (Meng et al., 2017). However, it is substantially higher than 8.5% and 10.3% found in two recent large scale fetal WES studies (Lord et al., 2019; Petrovski et al., 2019). This gap may be explained by enrichment of cases with positive family history in our cohort, and indeed our diagnostic rate in families without such history was only 13%. Interestingly, a study of 146 fetal exomes (Normand et al., 2018) has not found difference in diagnostic rate between sporadic cases and cases with significant family history. Therefore, the effect of medical family history on fetal WES yield should be further studied.
In addition, WES yield was in the range of 42.9–60% among families with involvement of brain, renal or musculoskeletal US abnormalities (higher than the overall diagnostic rate), and lower among families with cardiac malformations or increased NT/edema/hydrops signs (16.7 and 27.3%, respectively). However, since our sample size is relatively small, any interpretation of this sub-analysis should be cautious.
The resolved cases resulted in identification of causative variants in 12 genes. Some of the diagnoses are relatively common in the setting of prenatal diagnosis, such as osteogenesis imperfecta, while others are rare and include novel variants neither previously reported nor found in the databases. The latter are challenging to interpretate, due to the uncertain impact of the variants. As expected in the clinical setting, due to time and budget restrictions, no functional analysis at the molecular level was available for our cases. Thus, segregation of the variant in other family members was crucial to support diagnosis. In light of these limitations, we cannot definitely exclude that some of the molecular diagnoses are false positive. Two illustrative cases, families with molecular diagnosis of Meckel–Gruber type 8 and Pitt-Hopkins syndromes, demonstrate the clinical and genetic work-up during pregnancy and after termination, the utility of fetal WES, and its contribution for future family planning.
Well-described in the postnatal setting, prenatal WES also harbors special challenges, including numerous ethical issues which should be contemplated and addressed (Hillman et al., 2015; Jelin and Vora, 2018). The potential identification of secondary findings (also designated as incidental) and/or VUS, may have profound effect when found in ongoing pregnancies, either on parental decision making with regard to termination of pregnancy, or on the parental psychological burden following the birth of an affected child (Hillman et al., 2015; van den Veyver and Eng, 2015). One suggested strategy to circumvent some of these concerns in the prenatal setting is targeted exome testing, in which a premeditated and limited list of genes is analyzed (Pangalos et al., 2016; Chandler et al., 2018). On the other hand, according to the ACMG guidelines, no incidental findings in its selected list of actionable genes should be reported in prenatal samples.
Other concerns include issues related to turnaround time, lack of full coverage of potentially relevant genes, and inaccurate and limited phenotyping capabilities for fetuses, resulting in insufficient data for the WES interpretation (Aarabi et al., 2018; Best et al., 2018). Postnatal clinical evaluations or autopsies may be of great value, improving the diagnostic accuracy and yield, with new data not observed in US examination (Aarabi et al., 2018).
Our study is based on a cohort of consecutive (non-selected) series of families for which WES was performed in certified clinical laboratories, on a clinical base (not for research purpose as the main goal), and fully paid for, usually by families. In this regard, the study represents a real-world situation in prenatal counseling practicality, where the diagnosis is pursued due to a clinical need, and not within a research setup. Of note, most families with US abnormalities and lack of relevant family history asked to perform WES during ongoing pregnancy (14/23, 60.9%), while most families with positive history sent it after TOP (14/18, 77.8%). Four families asked to perform fetal WES during ongoing pregnancy even when no US abnormalities were found, mainly for detection of de novo pathogenic variants or recessive disorders. Three of the four families had offsprings affected by a severe single-gene condition. In these cases, WES was sent even when the specific pathogenic variant in the family was excluded in the fetus prior to WES submission (by Sanger sequencing), since families asked to search for other potential genetic disorders in the fetus.
The current study population is relatively homogenous in terms of demographic characteristics, composed of 45 families from Jewish ancestry, without consanguinity (except one family), all living in Israel and followed by a single prenatal genetic clinic. For most families in our cohort, genetic evaluation in addition to WES included CMA as well as parental screening for common genetic diseases in the Jewish population. In this aspect, the reported cohort might not be representative of other patient populations. For example, it is plausible that diagnostic yield may be even higher in WES pursued for fetuses of consanguineous couples.
The majority of families with fetal abnormal US findings seen in our clinic (several hundred each year), decided not to perform fetal WES, due to multiple considerations, including financial issues. Therefore, our study is biased toward families with high motivation to reach molecular diagnosis and available financial resources. An additional limitation of the present study is that WES was sent to three different clinical services. Therefore, sequencing was performed using different protocols and kits, and diverse bioinformatics pipelines were used for data analysis, potentially affecting the diagnostic yield due to inter-laboratory differences. Uniform sequencing and analysis in one laboratory, applied to all families, may have been an advantage. In addition, revision of WES data was not done for most unresolved cases. Together with our relative small sample size, the results should be considered as preliminary, and warrant further study.
To conclude, our study demonstrates that fetal WES is especially indicated when positive relevant family history is present, or in fetuses with specific types of congenital malformation. Following careful patient selection, clinical fetal WES is an important tool for evaluating fetuses with abnormal US findings, with important implications for both ongoing and future pregnancies.
Ethics Statement
The study was carried out in accordance with the recommendations of the Institutional Review Board (IRB) committee of SMC, and was approved by it.
Author Contributions
LG and MB designed the study, provided genetic counseling to families, researched the data, and wrote the manuscript. BP-S and EP contributed to study design and interpretations of data. SE-B, MD-K, AB-Z, NG, HP, MR, and AR-R provided genetic counseling to families, collected the data, and revised the manuscript. AR assisted in statistical analysis. HR-W, AB-A, PB, and OB participated in bioinformatics analysis and revised the manuscript.
Conflict of Interest Statement
AB-A and PB are employees of Centogene AG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00425/full#supplementary-material
Footnotes
- ^ https://www.centogene.com/
- ^ http://www.hgmd.cf.ac.uk/
- ^ https://www.ncbi.nlm.nih.gov/clinvar/
- ^ https://www.centomd.com/
- ^ http://exac.broadinstitute.org/
- ^ https://www.cegat.de/
References
Aarabi, M., Sniezek, O., Jiang, H., Saller, D. N., Bellissimo, D., Yatsenko, S. A., et al. (2018). Importance of complete phenotyping in prenatal whole exome sequencing. Hum. Genet. 137, 175–181. doi: 10.1007/s00439-017-1860-1
Berkenstadt, M., Pode-Shakked, B., Barel, O., Barash, H., Achiron, R., Gilboa, Y., et al. (2018). LMOD3-associated nemaline myopathy: prenatal ultrasonographic, pathologic, and molecular findings. J. Ultrasound Med. 37, 1827–1833. doi: 10.1002/jum.14520
Best, S., Wou, K., Vora, N., Van Der Veyver, I. B., Wapner, R., and Chitty, L. S. (2018). Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat. Diagn. 38, 10–19. doi: 10.1002/pd.5102
Carss, K. J., Hillman, S. C., Parthiban, V., Mcmullan, D. J., Maher, E. R., Kilby, M. D., et al. (2014). Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum. Mol. Genet. 23, 3269–3277. doi: 10.1093/hmg/ddu038
Chandler, N., Best, S., Hayward, J., Faravelli, F., Mansour, S., Kivuva, E., et al. (2018). Rapid prenatal diagnosis using targeted exome sequencing: a cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet. Med. 20, 1430–1437. doi: 10.1038/gim.2018.30
Daum, H., Meiner, V., Elpeleg, O., and Harel, T. (2019). Fetal exome sequencing: yield and limitations in a single tertiary center. Ultrasound Obstet Gynecol. 53, 80–86. doi: 10.1002/uog.19168
Drury, S., Williams, H., Trump, N., Boustred, C., Lench, N., Scott, R. H., et al. (2015). Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat. Diagn. 35, 1010–1017. doi: 10.1002/pd.4675
Filges, I., Nosova, E., Bruder, E., Tercanli, S., Townsend, K., Gibson, W. T., et al. (2014). Exome sequencing identifies mutations in KIF14 as a novel cause of an autosomal recessive lethal fetal ciliopathy phenotype. Clin. Genet. 86, 220–228. doi: 10.1111/cge.12301
Fu, F., Li, R., Li, Y., Nie, Z. Q., Lei, T., Wang, D., et al. (2018). Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obstet Gynecol. 51, 493–502. doi: 10.1002/uog.18915
Greenbaum, L., Gilboa, Y., Raas-Rothschild, A., Barel, O., Kol, N., Wolf, H. R., et al. (2019). Diaphanospondylodysostosis: refining the prenatal diagnosis of a rare skeletal disorder. Eur. J. Med. Genet. 62, 167–171. doi: 10.1016/j.ejmg.2018.07.004
Hillman, S. C., Willams, D., Carss, K. J., Mcmullan, D. J., Hurles, M. E., and Kilby, M. D. (2015). Prenatal exome sequencing for fetuses with structural abnormalities: the next step. Ultrasound Obstet. Gynecol. 45, 4–9. doi: 10.1002/uog.14653
Huppke, P., Wegener, E., Bohrer-Rabel, H., Bolz, H. J., Zoll, B., Gartner, J., et al. (2015). Tectonic gene mutations in patients with joubert syndrome. Eur. J. Hum. Genet. 23, 616–620. doi: 10.1038/ejhg.2014.160
Jelin, A. C., and Vora, N. (2018). Whole exome sequencing: applications in prenatal genetics. Obstet. Gynecol. Clin. North Am. 45, 69–81. doi: 10.1016/j.ogc.2017.10.003
Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., et al. (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887. doi: 10.1001/jama.2014.14604
Lord, J., Mcmullan, D. J., Eberhardt, R. Y., Rinck, G., Hamilton, S. J., Quinlan-Jones, E., et al. (2019). Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 393, 747–757. doi: 10.1016/S0140-6736(18)31940-8
Maduro, V., Pusey, B. N., Cherukuri, P. F., Atkins, P., Du Souich, C., Rupps, R., et al. (2016). Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet. J. Rare Dis. 11:62. doi: 10.1186/s13023-016-0439-6
Meng, L., Pammi, M., Saronwala, A., Magoulas, P., Ghazi, A. R., Vetrini, F., et al. (2017). Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171:e173438. doi: 10.1001/jamapediatrics.2017.3438
Normand, E. A., Braxton, A., Nassef, S., Ward, P. A., Vetrini, F., He, W., et al. (2018). Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected mendelian disorder. Genome Med. 10:74. doi: 10.1186/s13073-018-0582-x
Pangalos, C., Hagnefelt, B., Lilakos, K., and Konialis, C. (2016). First applications of a targeted exome sequencing approach in fetuses with ultrasound abnormalities reveals an important fraction of cases with associated gene defects. PeerJ 4:e1955. doi: 10.7717/peerj.1955
Petrovski, S., Aggarwal, V., Giordano, J. L., Stosic, M., Wou, K., Bier, L., et al. (2019). Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 393, 758–767. doi: 10.1016/S0140-6736(18)32042-7
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sawyer, S. L., Hartley, T., Dyment, D. A., Beaulieu, C. L., Schwartzentruber, J., Smith, A., et al. (2016). Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 89, 275–284. doi: 10.1111/cge.12654
Shaheen, R., Faqeih, E., Seidahmed, M. Z., Sunker, A., Alali, F. E., Alqahtani, K., et al. (2011). A TCTN2 mutation defines a novel meckel gruber syndrome locus. Hum. Mutat. 32, 573–578. doi: 10.1002/humu.21507
Stranneheim, H., and Wedell, A. (2016). Exome and genome sequencing: a revolution for the discovery and diagnosis of monogenic disorders. J. Intern. Med. 279, 3–15. doi: 10.1111/joim.12399
Talkowski, M. E., Ordulu, Z., Pillalamarri, V., Benson, C. B., Blumenthal, I., Connolly, S., et al. (2012). Clinical diagnosis by whole-genome sequencing of a prenatal sample. N. Engl. J. Med. 367, 2226–2232. doi: 10.1056/NEJMoa1208594
Taylor, J. C., Martin, H. C., Lise, S., Broxholme, J., Cazier, J. B., Rimmer, A., et al. (2015). Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 47, 717–726. doi: 10.1038/ng.3304
Trujillano, D., Bertoli-Avella, A. M., Kumar Kandaswamy, K., Weiss, M. E., Koster, J., Marais, A., et al. (2017). Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182. doi: 10.1038/ejhg.2016.146
van den Veyver, I. B., and Eng, C. M. (2015). Genome-wide sequencing for prenatal detection of fetal single-gene disorders. Cold Spring Harb. Perspect Med. 5:a023077. doi: 10.1101/cshperspect.a023077
Vora, N. L., Powell, B., Brandt, A., Strande, N., Hardisty, E., Gilmore, K., et al. (2017). Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet. Med. 19, 1207–1216. doi: 10.1038/gim.2017.33
Keywords: whole-exome sequencing (WES), prenatal diagnosis, ultrasound abnormalities, clinical genetics, congenital anomalies
Citation: Greenbaum L, Pode-Shakked B, Eisenberg-Barzilai S, Dicastro-Keidar M, Bar-Ziv A, Goldstein N, Reznik-Wolf H, Poran H, Rigbi A, Barel O, Bertoli-Avella AM, Bauer P, Regev M, Raas-Rothschild A, Pras E and Berkenstadt M (2019) Evaluation of Diagnostic Yield in Fetal Whole-Exome Sequencing: A Report on 45 Consecutive Families. Front. Genet. 10:425. doi: 10.3389/fgene.2019.00425
Received: 17 November 2018; Accepted: 17 April 2019;
Published: 25 June 2019.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Nelson L. S. Tang, The Chinese University of Hong Kong, ChinaQing Zhou, Zhejiang University, China
Copyright © 2019 Greenbaum, Pode-Shakked, Eisenberg-Barzilai, Dicastro-Keidar, Bar-Ziv, Goldstein, Reznik-Wolf, Poran, Rigbi, Barel, Bertoli-Avella, Bauer, Regev, Raas-Rothschild, Pras and Berkenstadt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michal Berkenstadt, bS5iZXJrZW5Ac2hlYmEuaGVhbHRoLmdvdi5pbA==; Lior Greenbaum, bGlvci5ncmVlbmJhdW1Ac2hlYmEuaGVhbHRoLmdvdi5pbA==