 Xiaokai Fang*
Xiaokai Fang* Yonghu Sun
Yonghu Sun- Shandong Provincial Hospital of Dermatology, Shandong University, Jinan, China
Background: Xeroderma pigmentosum (XP) is a rare autosomal, recessive, inherited disease. XP patients exhibit high sensitivity to sunlight and increased incidence of skin cancer. The different XP subtypes, which are caused by mutations of eight distinct genes, show some specific clinical manifestations. XP variant (XPV) is caused by mutations in the gene encoding DNA polymerase eta (POLH).
Case Presentation: We report a family that included two XP patients whose parents were first cousins. The proband is a 36-year-old male who developed a large number of pigmented freckle-like lesions starting at 4 years of age; later, he displayed typical psoriasis manifestation, abnormal renal function and hyperglycaemia. He was suspected as suffering from dyschromatosis symmetrica hereditaria (DSH), but negative results were obtained in candidate gene analyses. Whole-exome sequencing was performed in four subjects, including the two patients and two controls, and a new pathogenic homozygous nonsense mutation (c.353dupA, p. Y118_V119delinsX) of the POLH gene, which was identified in all nine family members by Sanger sequencing, was detected in the patients.
Conclusion: A novel XPV pathogenic homozygous nonsense mutation in the POLH gene was identified. Our case proves that next-generation sequencing is an effective method for the rapid diagnosis and determination of XP genetic etiology.
Background
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder resulting from deficiency in base excision repair caused by single-nucleotide mutations, especially in skin exposed to sunlight (Okamura et al., 2015). XP is classified into eight subtypes. Patients with XP show light sensitivity and skin pigmental changes in sun-exposed areas and have a higher incidence of neurological abnormalities, skin cancer, and other tumors (Ben Rekaya et al., 2018). Although the different XP subtypes present some specific clinical manifestations, the conditions are usually difficult to diagnose, and subtypes are defined only based on clinical manifestations if patients display mild phenotype or early-stage uncharacteristic manifestations.
The gene responsible for each type of XP has been identified. Most of the eight distinct genes encode proteins associated with the nucleotide excision repair (NER) function (Lehmann et al., 2011). The XP variant (XPV) type is the only variant that does not involve mutation in NER pathway components and instead results from mutations in the XPV gene. XPV is also named the DNA polymerase eta (POLH) gene and encodes the Y-DNA polymerase that participates in the repair of damaged DNA (Ohmori et al., 2001). POLH mutations weaken DNA of replication under exposure to ultraviolet light (Cordonnier et al., 1999).
In this study, we performed whole-exome sequencing of an XP family, which had been suspected as having dyschromatosis symmetrica hereditaria (DSH), and a novel XP pathogenic homozygous nonsense mutation (c.353dupA, p. Y118_V119delinsX) was identified in the POLH gene (NM_006502). Our case proves that next-generation sequencing (NGS) is an effective method for the rapid diagnosis and determination of XP genetic etiology.
Case Presentation
We report a family that included two patients whose parents are first cousins. The proband (Figure 1: IV:5) is a 36-year-old male who developed many pigmented freckle-like patches in the skin starting at 4 years of age. These changes were particularly located in UV-exposed areas, such as the face (Figure 2a), upper thorax, upper limbs, dorsal hands (Figure 2b), feet and legs, and gradually worsened before 18 years of age, after which the condition stabilized. Over the entire time period, sunlight aggravated the disease. The palms, planta pedis, scalp, mucous membranes and nervous system were not affected. Histopathological examination of the skin lesions was performed and revealed melanin pigmentation in the basal layer and extension of the furcella into the dermis with a bud shape (Figures 2c,d). The patient presented a typical psoriasis manifestation at 35 years of age. Irregular geographic erythema appeared mainly in the extremities and gradually progressed. The skin lesions were covered with thick scales that had characteristics of the wax droplet phenomenon, the membrane phenomenon and dotty hemorrhage (Figure 3a). Upon histopathological examination of the skin lesions, the epidermal layer showed continuous parakeratosis and Munro’s microabscesses. The dermis layer exhibited dilated superficial vessels and infiltration of a few lymphocytes (Figure 3b). Abnormal renal function was also found, with elevated levels of creatinine 152.2 μmol/L (normal 44–132.6 μmol/L) and uric acid 552 μmol/L (normal 44–132.6 μmol/L), at the age of 35. At 36 years of age, the patient was found to have hyperglycaemia, with elevated fasting blood glucose in multiple tests. No skin tumors were found. The proband’s older sister (IV:1) is a 41-year-old female without any concomitant disease. The sister presented pigmented lesions that were similar to those observed in the proband. Other members in this pedigree were healthy.
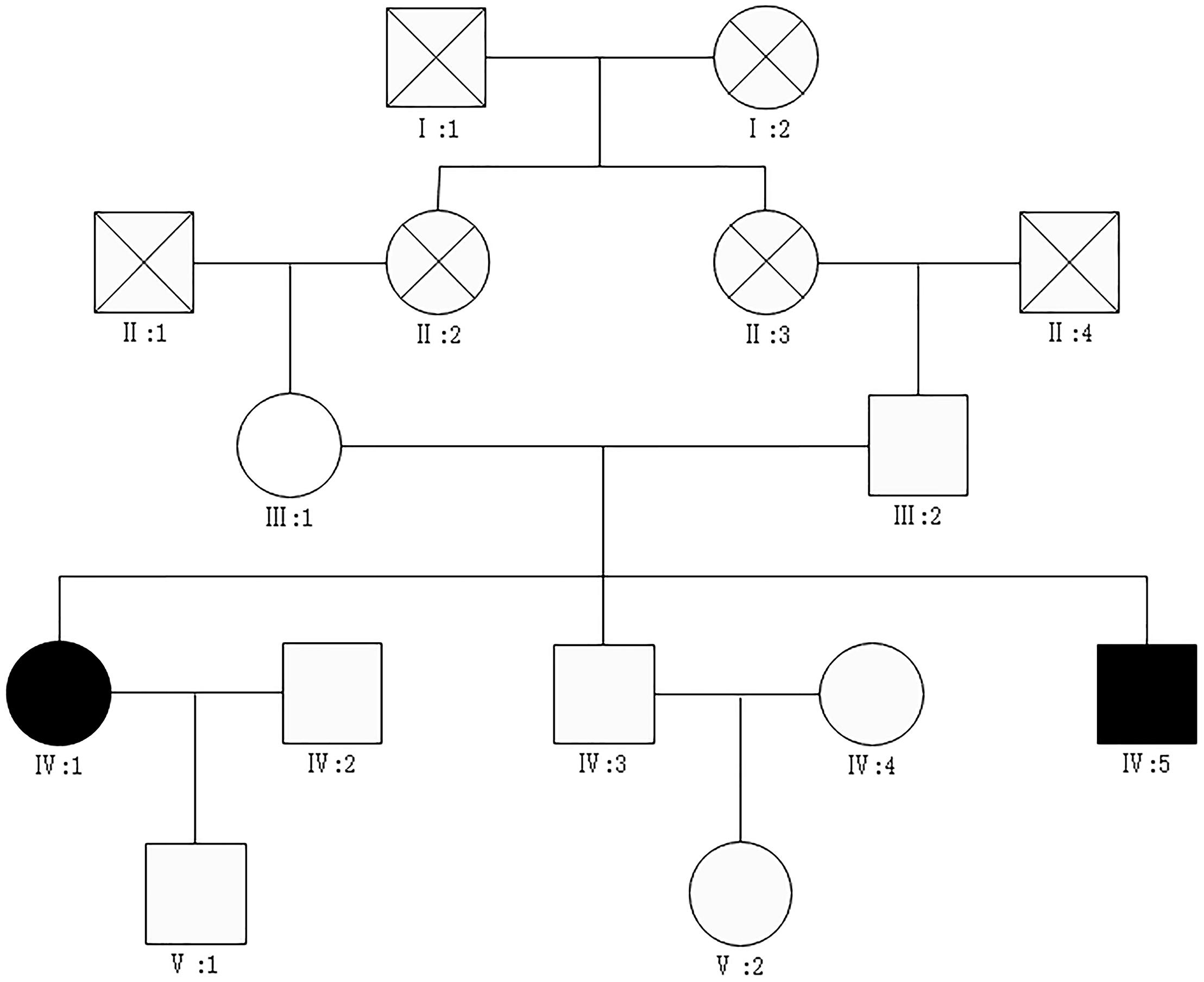
Figure 1. The pedigrees of the patients.  affected male,
affected male,  affected female,
affected female,  unaffected male,
unaffected male,  unaffected female, X mark: deceased.
unaffected female, X mark: deceased.
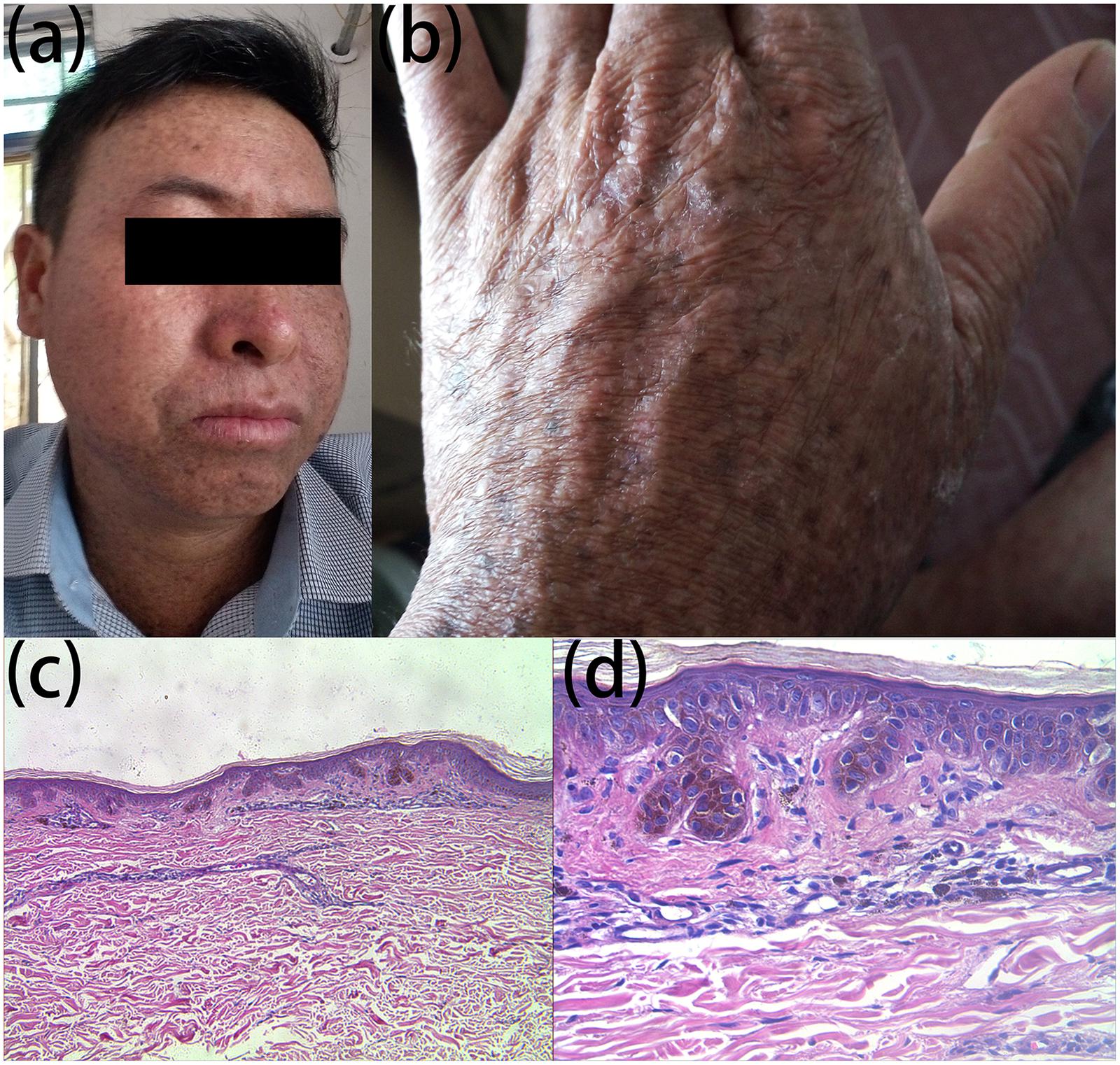
Figure 2. Clinical and histopathological appearance of hyperpigmentation. Hyperpigmented lesions on the face (a) and dorsal hand (b); melanin pigmentation in the basal layer and extension of the furcella into the dermis in a bud shape (c) HE stain, × 100; (d) HE stain, × 400.
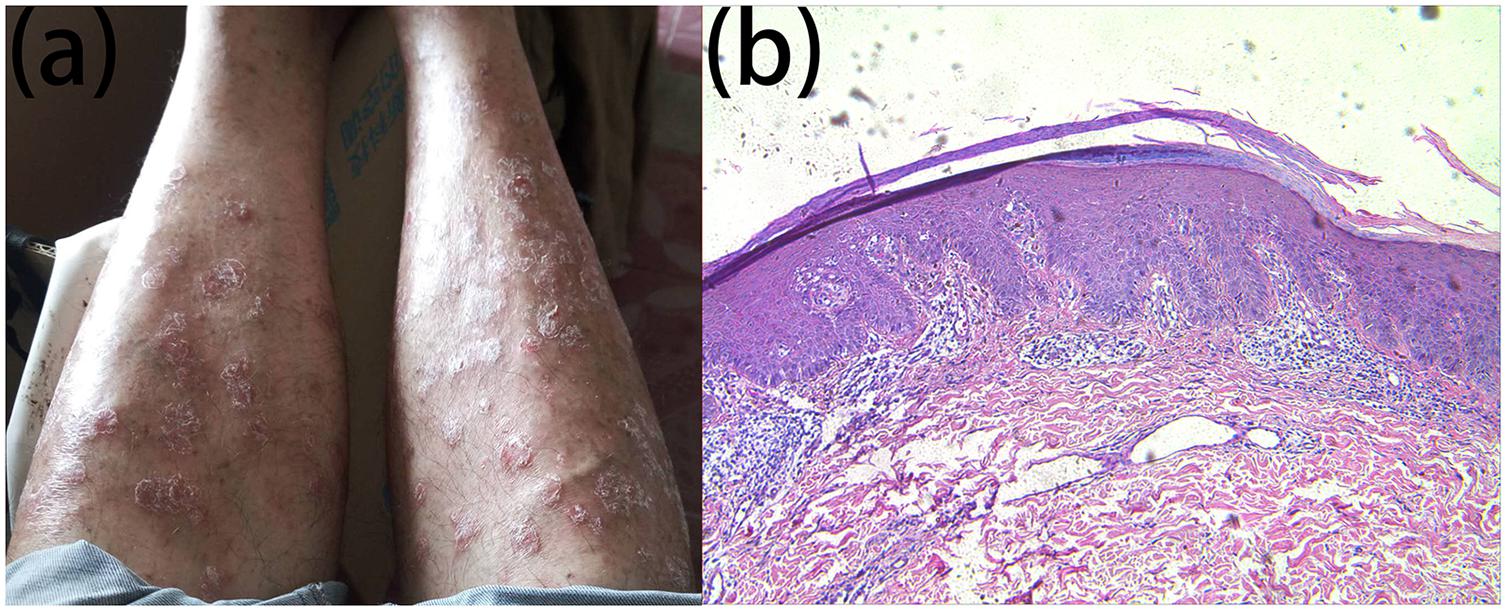
Figure 3. Clinical and histopathological appearance of psoriasis. Lesions on the lower limbs (a) continuous parakeratosis and Munro microabscesses in the epidermal layer, dilated superficial vessels, and infiltration of a few lymphocytes in the dermis layer (b) HE stain, × 100.
This study was approved by the Ethical Committee and was carried out according to the Declaration of Helsinki Principles. Nine people, including the two patients in the family, provided written consent to join the study, including authorization to extract peripheral anticoagulation blood and to publish these case details.
Description of Laboratory Investigations and Diagnostic Tests
Whole-Exome Sequencing and Variant Analysis
Genomic DNA was isolated from whole blood samples using a Flexigene® DNA kit based on the manufacturer’s protocol. DNA was available for all nine people in the family, including the two patients.
Whole-exome sequencing was conducted for the patients (IV:1, IV:5) and normal controls (IV:4, V:2). At least 0.6 μg of DNA from each of the subjects was fragmented into 180–280 bp segments using a Covaris S220 sonicator. The Agilent SureSelect Human All Exon V6 kit was employed to enrich, hybridize and capture these fragments following the manufacturer’s specifications. Qubit 2.0 and Agilent 2100 were used for preliminary quantification and detection of the library insert size. qPCR was used for accurate quantification of the effective concentration to ensure the library quality, and an Illumina HiSeq 2000 was utilized for library sequencing.
The Burrows-Wheeler Alignment tool (BWA) was used to match the clean reads without adapters or debased reads to the human reference genome (UCSC hg19)1, (Li and Durbin, 2009). Duplicate reads were marked by Picard after the deletion or insertion of nucleotide fragments, and single-nucleotide polymorphisms (SNPs) were identified by Sequence Alignment/Map tools (SAMtools) (Li et al., 2009). The variants were filtered with SNP database 147 (dbSNP147) (Sherry et al., 2001), 1000 Genomes Project (version 2015 August) (Genomes Project et al., 2012), and NHLBI Exome Sequencing Project (ESP) 6500. Sorting Intolerant from Tolerant (SIFT) (Ng and Henikoff, 2003) and Polymorphism Phenotyping version2 (PolyPhen-2) were employed to predict protein function (Adzhubei et al., 2013). Highly suspicious mutations were noted by Annotate Variation (ANNOVAR) software (Wang et al., 2010).
We generated over 40 GB of data by exome sequencing. Among the four samples, there were 77,073,558, 74,946,888, 67,801,236, and 76,068,580 clean data reads obtained, and 99.87, 99.90, 99.84, and 99.93% of the reads were mapped to the human reference genome; the average sequencing depths were 126.61×, 112.91×, 104.50×, and 117.42×, respectively. At least 99.6, 99.3, 97.9, and 99.3% of the targeted exomes were covered and exceeded 10 × depths, which was considered a meaningful standard for identifying SNPs and insertion or deletion mutations. The bases with quality above 99.9% accuracy represented over 90% of the total data. Because XP is an extremely rare genetic disorder, we excluded variants, including those in the 1000 Genomes Project database (Genomes Project et al., 2012), with minor allele frequencies over 0.5%, dbSNP147 (Sherry et al., 2001) with a frequency higher than 0.1%, and NHLBI ESP6500. PolyPhen-2 (Adzhubei et al., 2013) and SIFT (Ng and Henikoff, 2003) were used to predict functional changes due to the candidate mutations as a reference. Ten possible mutation sites, including a novel homozygous nonsense mutation (c.353dupA, p. Y118_V119delinsX) in the POLH gene and a homozygous missense mutation (c. T214C, p. S72P) in the t-complex-associated-testis-expressed 1 (TCTE1) gene (NM_182539) were identified in the two patients. Exome sequencing processing showed that these mutation sites were not present in the normal controls (IV:4, V:2).
Sanger Sequencing and Functional Prediction
To verify pathogenic mutations, the entire pedigree was subjected to direct Sanger sequencing using an ABI3500 sequencer (Applied Biosystems, Foster City, CA, United States). Primer sequences for candidate pathogenic variants were designed, and segmented primer sequences for the ADAR gene were also designed to exclude DSH. The change in a potential pathogenic variant’s function was predicted by MutationTaster (Schwarz et al., 2014). Protein models of DNA polymerase eta were constructed using Swiss-Model (Waterhouse et al., 2018).
Based on Sanger sequencing for the 10 possible mutation sites in all 9 family members, eight sites not fitting the phenotype were excluded (Supplementary Table 1). The POLH variant (c.353dupA, p. Y118_V119delinsX) and the TECE1 variant (c. T214C, p. S72P) were confirmed by Sanger sequencing and identified as being heterozygous in the patients’ normal parents and another family member (III:1, III:2, V1; Figures 4c,f). They were both homozygous variants in the patients (IV:1, IV:5; Figures 4b,e). However, the variants were not found in the rest of the family (IV:2, IV:3, IV:4, V2; Figures 4a,d) and was not reported in the databases searched or previous genome-wide association studies. The homozygous POLH variant co-segregating with the disease phenotype in the family was predicted to lead to a change in amino acid sequence and a premature termination codon and to affect protein features and splice site changes, as assessed by MutationTaster (Schwarz et al., 2014); this may be a morbigenous variant in this pedigree. According to Swiss-Model (Waterhouse et al., 2018), the protein model of DNA polymerase eta exhibits different lengths and configurations in the normal controls and patients with mutations in the POLH gene, suggesting significant functional deficiency (Supplementary Figure 1). TCTE1 and POLH are both on 6p21.1, and the mutation in TCTE1 also co-segregated with the phenotype in the entire family.
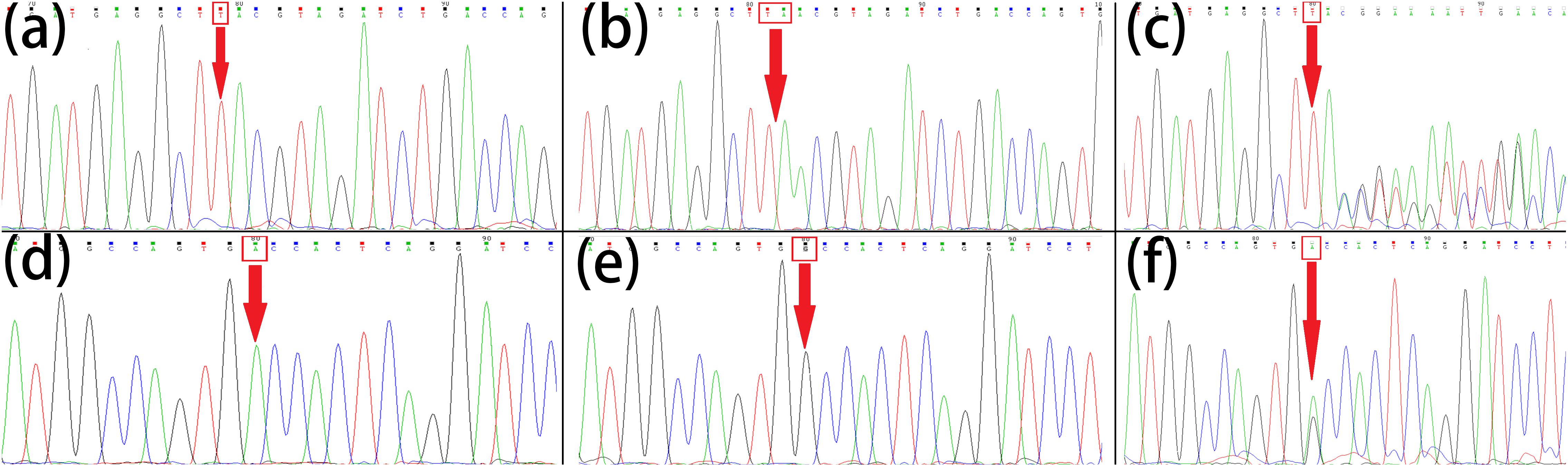
Figure 4. Sanger sequencing of POLH and TCTE1. Normal control (IV:2, IV:3, IV:4, V2) (a), homozygous mutations (IV:1, IV:5) (b) and heterozygous mutations (III:1, III:2, V1) (c) of POLH (c.353dupA, p. Y118_V119delinsX); normal control (IV:2, IV:3, IV:4, V2) (d), homozygous mutations (IV:1, IV:5) (e) and heterozygous mutations (III:1, III:2, V1 (f) of TCTE1 (c. T214C, p. S72P). The mutational bases were arrowed.
Discussion of the Underlying Pathophysiology and the Novelty of the Case
The proband (IV:5) is a 36-year-old male who presented with hyperpigmented macules that appeared in early childhood. His parents are of a consanguineous marriage, and his sister has features similar to his own. We investigated some genetic pigmentation diseases, such as Bloom syndrome, Rothmund-Thomson syndrome, Peutz-Jeghers syndrome, and Cockayne syndrome. Combined with the medical history, DSH was suspected. Indeed, because histopathological examination revealed melanin pigmentation in the basal layer, the diagnosis of DSH seemed reasonable. DSH is caused by a heterozygous mutation in the adenosine deaminase RNA-specific gene (ADAR) on chromosome 1q21 and shows a high-penetrance, autosomal dominant inheritance pattern (Xing et al., 2003). Although the genetic model of this family appears to fit recessive inheritance and the patients did not have hypopigmented macules, Sanger sequencing of the ADAR gene for five family members, including the two patients (III:1, III:2, IV:1, IV:3 and IV:5), was performed to eliminate possible errors in penetrance and information collection. However, no ADAR mutation was detected in this family. Considering that the proband had concomitant symptoms, including psoriasis and multiple organ damage, diagnosis of the disease remained challenging.
Whole-exome sequencing analyses genetic information in a rapid and effective manner, allowing hereditary speculation of complex and monogenic genetic diseases, including skin pathology (Choi et al., 2009). For rare disorders, the application of whole-exome sequencing can minimize mistakes in detecting mutations in hot-spot regions. Specifically, for Mendelian disorders, NGS can be helpful for inferring pathogenesis and exploring new mutations of genes associated with the disease (Yang et al., 2013). Furthermore, this method is very effective for the study of genetic diseases with recessive genetic patterns because it can effectively identify homozygous pathogenic mutations through contrast analysis of the coding sequence between affected and unaffected individuals (Bamshad et al., 2011).
Because the sequencing results for the ADAR gene were negative, we utilized high-throughput sequencing to identify the genetic nature of the disease. Through whole-exome and Sanger sequencing, the POLH mutation was found to be the pathogenic factor. Combined with the medical history, clinical manifestations, laboratory findings and previous reports (Inui et al., 2008), the diagnosis of XPV caused by a POLH mutation was confirmed. This gene encodes certain DNA polymerases that repair DNA damage and inhibit the mutagenicity of UV-induced DNA changes. In addition, POLH is proposed to be related to hypermutation in the process of immunoglobulin class switch recombination. To date, approximately one hundred POLH mutations have been shown to be associated with XPV pathogenesis (Yuasa et al., 2000; Inui et al., 2008; Opletalova et al., 2014; Karass et al., 2015). In most situations, these mutations result in premature termination codons in mRNA that lead to degradation by the nonsense-mediated decay (NMD) system (Inui et al., 2008). Opletalova et al. (Opletalova et al., 2014) summarized the relationship of phenotype/genotype in 23 XPV patients, showing that the type of missense mutation was clearly related to the clinical severity. For patients with truncating mutations, life-cumulated UV exposure is probably the best predictor of cancer incidence, and it is very necessary to avoid sun exposure. In our case, the variant was a nonsense mutation. The proband showed a stable general condition without any tumor, consistent with another report (Ben Rekaya et al., 2011).
TCTE1 has not been previously reported as a pathogenic gene. Kwiatkowski et al. (1991) identified highly informative dinucleotide repeat polymorphisms in the TCTE1 locus that were previously called D6S46. As our proband had some concomitant diseases, such as psoriasis, abnormal renal function and hyperglycaemia, the missense mutation in TCTE1 may be due to multiple unknown factors or may be meaningless. It is worth mentioning that the homozygous missense TCTE1 variant completely co-segregated with the disease phenotype in the pedigree and was expected to lead to changes in amino acid sequences and protein features, as well as splice site changes and disease, as assessed by MutationTaster. Both of the mutated genes are located at 6p21.1. Although pure linkage inheritance is highly possible, we cannot exclude the possibility that it is a pathogenic factor related to XPV, a possibility that requires further exploration.
Xeroderma pigmentosum variant patients constitute approximately 20% of all patients with XP. Compared with XP groups A-G, XPV is more benign, presenting with mild skin lesions and low tumor incidence. In addition, symptoms occur late, and patients have a longer life expectancy, with few developing neurological abnormalities (Gratchev et al., 2003). The individuals affected by analogous pigmented diseases and sun sensitivity, such as in XPV, are usually underdiagnosed because patients do not display attentional symptoms, such as apparent damage or carcinomatous degeneration of the light-exposed skin, until a late age. In some cases, skin lesions in XPV patients can vary greatly in the degree of severity (Inui et al., 2008). Overall, molecular diagnosis remains very challenging for these patients (Ben Rekaya et al., 2018).
Xeroderma pigmentosum variant is a cancer-prone syndrome that results in photosensitivity, dermatic and ocular injury, and skin precancerous lesions after sun exposure (Broughton et al., 2002). Tumors of various pathological forms and neurological symptoms are concomitant symptoms. The proband in the current study presented with psoriasis, renal dysfunction and hyperglycaemia. However, the relationship between the primary disease and associated symptoms is unknown. Ezzedine et al. (2008) reported XP accompanied by psoriasis. The pathogenesis of both diseases is completely different, and psoriasis may be another primary disease or may be influenced by XPV. Further investigations are required to determine how these likely irrelevant diseases can coexist. Some previous XP cases exhibited manifestations of end-stage renal disease (Radwan et al., 2015), renal cell carcinoma (Loghin et al., 2016) and diabetes (Das et al., 2018). There are many connections between these conditions, but it is difficult to determine the specificity because renal dysfunction and hyperglycaemia appear to be more similar to the primary disease.
The use of NGS technology is a double-edged sword. Because we cannot accurately diagnose diseases and because known genes have many exons, first-generation sequencing of all exons is difficult and time-consuming (Ben Rekaya et al., 2018). Diseases with unknown mutations have yet to be discovered, and NGS is more efficient than first-generation sequencing approaches, with similar costs (Kleijer et al., 2008). Nonetheless, Ortega-Recalde et al. (2014) reported a bottleneck of downstream bioinformatics analysis of data, and transformation of next-generation technologies into clinical practice remains challenging (Smith et al., 2014). Although some distinct bioinformatics tools have been implemented, much time and high costs have been invested in establishing a reliable NGS platform.
Regardless, our experience proves that whole-exome sequencing is an efficient method that can be used for XP diagnosis and etiology classification in the future. Through various technologies, our understanding of XP will gradually deepen, thus promoting continuous improvement in treating this disease.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: http://genome.ucsc.edu/.
Ethics Statement
This study has been approved by the Ethical Committee and was conducted according to the Declaration of Helsinki Principles. Nine people, including two patients in the family, provided a written consent form to participate in the study, which included an authorization to extract peripheral anticoagulation blood and to publish these case details.
Author Contributions
XF made substantial contributions to the conception design, wrote the manuscript, and collected the materials and blood samples of the patients. YS performed data analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the patients for their participation in the study and their consent to publication of clinical data and photos.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00495/full#supplementary-material
Footnotes
References
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7:Unit7.20. doi: 10.1002/0471142905.hg0720s76
Bamshad, M. J., Ng, S. B., Bigham, A. W., Tabor, H. K., Emond, M. J., Nickerson, D. A., et al. (2011). Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755. doi: 10.1038/nrg3031
Ben Rekaya, M., Messaoud, O., Mebazaa, A., Riahi, O., Azaiez, H., Kefi, R., et al. (2011). A novel POLH gene mutation in a xeroderma pigmentosum-V Tunisian patient: phenotype-genotype correlation. J. Genet. 90, 483–487. doi: 10.1007/s12041-011-0101-y
Ben Rekaya, M., Naouali, C., Messaoud, O., Jones, M., Bouyacoub, Y., Nagara, M., et al. (2018). Whole Exome Sequencing allows the identification of two novel groups of Xeroderma pigmentosum in Tunisia, XP-D and XP-E: impact on molecular diagnosis. J. Dermatol. Sci. 89, 172–180. doi: 10.1016/j.jdermsci.2017.10.015
Broughton, B. C., Cordonnier, A., Kleijer, W. J., Jaspers, N. G., Fawcett, H., Raams, A., et al. (2002). Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc. Natl. Acad. Sci. U.S.A. 99, 815–820. doi: 10.1073/pnas.022473899
Choi, M., Scholl, U. I., Ji, W., Liu, T., Tikhonova, I. R., Zumbo, P., et al. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 106, 19096–19101. doi: 10.1073/pnas.0910672106
Cordonnier, A. M., Lehmann, A. R., and Fuchs, R. P. (1999). Impaired translesion synthesis in xeroderma pigmentosum variant extracts. Mol. Cell Biol. 19, 2206–2211. doi: 10.1128/mcb.19.3.2206
Das, S., Purkayastha, S., Roy, H., Sinha, A., and Choudhury, Y. (2018). Polymorphisms in DNA repair genes increase the risk for type 2 diabetes mellitus and hypertension. Biomol. Concep. 9, 80–93. doi: 10.1515/bmc-2018-0008
Ezzedine, K., Simonart, T., Candaele, M., Malvy, D., and Heenen, M. (2008). Concomitant Xeroderma pigmentosum and disseminated small plaque psoriasis: first case of an antinomic association. Cases J. 1:74. doi: 10.1186/1757-1626-1-74
Genomes Project, C., Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
Gratchev, A., Strein, P., Utikal, J., and Sergij, G. (2003). Molecular genetics of Xeroderma pigmentosum variant. Exp. Dermatol. 12, 529–536. doi: 10.1034/j.1600-0625.2003.00124.x
Inui, H., Oh, K. S., Nadem, C., Ueda, T., Khan, S. G., Metin, A., et al. (2008). Xeroderma pigmentosum-variant patients from America, Europe, and Asia. J. Invest. Dermatol. 128, 2055–2068. doi: 10.1038/jid.2008.48
Karass, M., Naguib, M. M., Elawabdeh, N., Cundiff, C. A., Thomason, J., Steelman, C. K., et al. (2015). Xeroderma pigmentosa: three new cases with an in depth review of the genetic and clinical characteristics of the disease. Fetal Pediatr. Pathol. 34, 120–127. doi: 10.3109/15513815.2014.982336
Kleijer, W. J., Laugel, V., Berneburg, M., Nardo, T., Fawcett, H., Gratchev, A., et al. (2008). Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair 7, 744–750. doi: 10.1016/j.dnarep.2008.01.014
Kwiatkowski, T. J. Jr., Beaudet, A. L., Trask, B. J., and Zoghbi, H. Y. (1991). Linkage mapping and fluorescence in situ hybridization of TCTE1 on human chromosome 6p: analysis of dinucleotide polymorphisms on native gels. Genomics 10, 921–926. doi: 10.1016/0888-7543(91)90180-m
Lehmann, A. R., McGibbon, D., and Stefanini, M. (2011). Xeroderma pigmentosum. Orphanet J. Rare Dis. 6:70. doi: 10.1186/1750-1172-6-70
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Loghin, A., Banescu, C., Nechifor-Boila, A., Chibelean, C., Orsolya, M., Nechifor-Boila, A., et al. (2016). XRCC3 Thr241Met and XPD Lys751Gln gene polymorphisms and risk of clear cell renal cell carcinoma. Cancer Biomark 16, 211–217. doi: 10.3233/CBM-150558
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Ohmori, H., Friedberg, E. C., Fuchs, R. P., Goodman, M. F., Hanaoka, F., Hinkle, D., et al. (2001). The Y-family of DNA polymerases. Mol. Cell. 8, 7–8.
Okamura, K., Toyoda, M., Hata, K., Nakabayashi, K., and Umezawa, A. (2015). Whole-exome sequencing of fibroblast and its iPS cell lines derived from a patient diagnosed with xeroderma pigmentosum. Genom. Data 6, 4–6. doi: 10.1016/j.gdata.2015.07.008
Opletalova, K., Bourillon, A., Yang, W., Pouvelle, C., Armier, J., Despras, E., et al. (2014). Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations. Hum. Mutat. 35, 117–128. doi: 10.1002/humu.22462
Ortega-Recalde, O., Vergara, J. I., Fonseca, D. J., Rios, X., Mosquera, H., Bermudez, O. M., et al. (2014). Whole-exome sequencing enables rapid determination of xeroderma pigmentosum molecular etiology. PLoS One 8:e64692. doi: 10.1371/journal.pone.0064692
Radwan, W. M., Elbarbary, H. S., and Alsheikh, N. M. (2015). DNA repair genes XPD and XRCC1 polymorphisms and risk of end-stage renal disease in Egyptian population. Ren. Fail. 37, 122–128. doi: 10.3109/0886022X.2014.967646
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi: 10.1093/nar/29.1.308
Smith, A., Boycott, K. M., and Jarinova, O. (2014). Lake Louise mutation detection meeting 2013: clinical translation of next-generation sequencing requires optimization of workflows and interpretation of variants. Hum. Mutat. 35, 265–269. doi: 10.1002/humu.22480
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi: 10.1093/nar/gky427
Xing, Q. H., Wang, M. T., Chen, X. D., Feng, G. Y., Ji, H. Y., Yang, J. D., et al. (2003). A gene locus responsible for dyschromatosis symmetrica hereditaria (DSH) maps to chromosome 6q24.2-q25.2. Am. J. Hum. Genet. 73, 377–382. doi: 10.1086/377007
Yang, Y., Muzny, D. M., Reid, J. G., Bainbridge, M. N., Willis, A., Ward, P. A., et al. (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511. doi: 10.1056/NEJMoa1306555
Keywords: whole-exome sequencing, xeroderma pigmentosum (XP), DNA polymerase eta (POLH) gene, novel mutation, psoriasis
Citation: Fang X and Sun Y (2019) Whole-Exome Sequencing Enables the Diagnosis of Variant-Type Xeroderma Pigmentosum. Front. Genet. 10:495. doi: 10.3389/fgene.2019.00495
Received: 15 February 2019; Accepted: 06 May 2019;
Published: 24 May 2019.
Edited by:
Andrew Landstrom, Duke University, United StatesReviewed by:
Muhammad Ansar, Quaid-i-Azam University, PakistanSaadullah Khan, Kohat University of Science and Technology, Pakistan
Copyright © 2019 Fang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaokai Fang, ZmFuZ3hpYW9rYWkxOTk0QHFxLmNvbQ==