 Silvio D. Brugger
Silvio D. Brugger Julia G. Kraemer1,4†
Julia G. Kraemer1,4† Lindsey Bomar
Lindsey Bomar Markus Hilty
Markus Hilty- 1Institute for Infectious Diseases, Faculty of Medicine, University of Bern, Bern, Switzerland
- 2Department of Oral Medicine, Infection and Immunity, Harvard School of Dental Medicine, Boston, MA, United States
- 3Department of Infectious Diseases and Hospital Epidemiology, University Hospital Zurich – University of Zurich, Zurich, Switzerland
- 4Institute for Work and Health, University of Lausanne, University of Geneva, Épalinges, Switzerland
- 5Functional Genomics Center Zurich, Swiss Federal Institute of Technology Zurich, University of Zurich, Zurich, Switzerland
- 6Department of Microbiology, The Forsyth Institute, Cambridge, MA, United States
Acute bacterial otitis media is usually caused by otopathogens ascending to the middle ear from the nasopharynx (NP). However, it is unknown if the nasopharyngeal microbiota of children with acute otitis media (AOM) can serve as an age-dependent or independent proxy for the microbial communities of the middle ear fluid (MEF) as there is a lack of 16S rRNA amplicon sequencing studies simultaneously analyzing the microbial communities of the two sites. Within this study, we performed 16S rRNA next generation sequencing on a total of 286 nasopharyngeal swabs (NPSs) collected between 2004 and 2013 within a Swiss national AOM surveillance program from children (0–6 years) with AOM. In addition, 42/286 children had spontaneous tympanic membrane perforation and, therefore, those MEF could also be analyzed. We found that alpha [Richness, Shannon diversity index (SDI) and Evenness] and beta diversity measurements of the nasopharyngeal bacterial microbiota showed a clear dependency of the increasing age of the children. In more detail, bacterial richness and personalized profiles (measured by beta dispersion) were higher and more frequent in older children, respectively. Dissimilarity values based on the binary distance matrix of the microbiota patterns of the NP and the MEF also correlated with increasing age. In general, positive (PPV) and negative predictive values (NPV) of the most abundant operational taxonomic units (OTUs) in the NP were moderately and well predictive for their presence in the MEF, respectively. This data is crucial to better understand polymicrobial infections and therefore AOM pathogenesis.
Introduction
Acute otitis media (AOM) is one of the most frequent pediatric diseases with a peak incidence at 6–12 months of age and is still responsible for high rates of antibiotic prescription (Lieberthal et al., 2013). Conventionally, bacterial or viral culturing of the MEF has been used as the gold standard for establishing microbial etiology of AOM (Yatsyshina et al., 2016). Traditionally, Streptococcus pneumoniae, non-typeable Haemophilus influenzae and Moraxella catarrhalis have been reported as the most common bacterial causes of AOM (Post et al., 1995; Kilpi et al., 2001; van Dongen et al., 2013; Kaur et al., 2014, 2017). However, the incidences of the pathogens causing AOM seem to be age-dependent, e.g., it has been reported that the incidence of pneumococcal AOM peaks around 12 months of age, whereas the incidence of M. catarrhalis AOM first peaks at 6 months and H. influenzae AOM at 20 months (Kilpi et al., 2001; Kaur et al., 2014).
A clinically important question to address, is whether sampling the NP provides an accurate estimate of the presence of the various microorganisms involved in otitis media as access to the NP is much less invasive than that to the middle ear (tympanocentesis) (van Dongen et al., 2013). Furthermore, it is hypothesized that bacterial AOM is caused by otopathogens residing in the nasal passages by ascension through the Eustachian tube under disturbance of the normal homeostasis (Coticchia et al., 2013). It has been described that nasopharyngeal colonization with the bacterial species most known to cause AOM is associated with middle ear disease with odds ratios (ORs) of approximately 2 (Faden et al., 1997; Revai et al., 2008). However, all of these studies are culture based or used specific PCR protocols targeting distinct species (Yatsyshina et al., 2016). There is a lack of 16S rRNA next generation sequencing studies characterizing the whole bacterial microbiota, rather than single species, with paired samples of NPSs and MEF of patients with AOM as well as of studies investigating the nasopharyngeal microbiota in toddlers and prepubertal children. Some very recent studies by us and others analyzing either the middle ear (Sillanpaa et al., 2017) and/or the NP (Hilty et al., 2012; Chonmaitree et al., 2017; Lappan et al., 2018) have already shown that a broader, sequencing-based survey of the bacterial microbiota provides additional important information which cannot be derived by bacterial cultures. Therefore, the overarching aim of this study was to characterize the bacterial, nasopharyngeal microbiota in Swiss children with AOM and to simultaneously characterize the bacterial microbiota of MEF and NPS in patients with AOM who also had a perforated tympanic membrane, at different ages. In more detail we aimed at (a) characterizing the nasopharyngeal microbiota of children with AOM aged <7 years and at (b) simultaneously characterizing the microbiota of NP and MEF of 42 children with spontaneous tympanic membrane perforation in different age groups in order to predict if and to what extend the microbiota of NPS may serve as proxy for the microbiota of MEF.
Materials and Methods
Study Design
This study included outpatients with AOM between 2004 and 2013. All children aged less than 7 years were included. Children were observed within a Swiss nationwide prospective surveillance system for various diseases, which is ongoing since 1998 and has been explained previously (Muhlemann et al., 2003; Allemann et al., 2017). The diagnosis of AOM episodes was done according to Centers for Disease Control and Prevention definitions which included two or more of the following symptoms: fever (>38°C), inflamed or painful or retracted tympanic membrane, reduced mobility of the tympanic membrane or fluid behind the tympanic membrane on otoscopy. Patients were eligible to be signed up repeatedly for each new episode of AOM (Muhlemann et al., 2003; Allemann et al., 2017). Participating physicians seeing AOM patients sampled the NP and provided a sample of the MEF in case of spontaneous rupture of the tympanic membrane. In total, 42/286 of participants provided both, MEF and a NPS. Tympanocentesis for routine surveillance was not intended in the surveillance system and was fully up to the decision of the treating physician. Participating physicians were only asked to swab MEF of spontaneously ruptured tympanic membranes. Nasopharyngeal samples analysis included the data from a previous study for children age 0–1 years (Hilty et al., 2012). NPS samples from the other age groups (>1 year but <7 years) and MEF samples are presented for the first time in this study. The overall AOM surveillance program is part of the governmental public health surveillance and is therefore exempted from approval by Institutional Review Boards. Informed consent was received from the parents.
Sample Collection
Nasopharyngeal swabs were obtained through the nose reaching the NP using twisted wire rayon tipped applicators (Copan Ventury Transystem; Copan, Italy), and MEF were gained using the identical or cotton tip swabs (Allemann et al., 2017). Concerning the latter, the amount of time between spontaneous perforation and collection of samples was not recorded. With each sample, practitioners submitted a standardized questionnaire comprising the following patient data: age, gender, vaccination status, preceding antibiotic therapy, day care attendance, AOM history (number of otitis media episodes within the last 12 months) and place of residence. Culture and subsequent serotyping for S. pneumoniae was done for all NPS and MEF as described (Allemann et al., 2017).
PCR Amplification and Amplicon Sequencing of 16S rRNA Genes
Amplification by PCR and amplicon sequencing has been described elsewhere (Hilty et al., 2012). In brief, DNA was extracted using 200 μl of transport medium, followed by amplification of bacterial 16S rRNA variable regions V3-V5 by MID tagged primer pair 341F/907R (Escherichia coli numbering). The final primer sequences including MID and adaptor sequences were A-341F, 5′-cgtatcgcctccctcgcgccatcag-[NNNNNNNNNN]-ACTCCTACGGGAGGCAGCAG-3′ and B-926R, 5′-ctatgcgccttgccagcccgctcag-[NNNNNNNNNN]-CCGTCAATTCMTTTGAGTTT-3′. PCR reactions were eluted by 40 μl double distilled water and the eluate was measured by Agilent 2100 Bioanalyzer (Agilent Technologies, Basel, Switzerland). PCR products with a concentration <1.0 ng/μl were excluded from the study. This corresponds to <1 pg/μl bacterial DNA as evaluated by quantitative PCR of pneumococcal DNA at different dilutions (Mika et al., 2015). A minimum of 1 pg/μl of bacterial DNA has been suggested as the cut-off when dealing with low density sample material (Biesbroek et al., 2012). Resulting PCR products were evenly pooled (40 ng), whereas every MID was used once, resulting in eight amplicon pools (Hilty et al., 2012). The amplicon pools were sequenced using the 454 sequencing platform. The resulting reads were submitted to the National Center for Biotechnology Information Sequence Read Archive (accession numbers SRR077426, SRR086165, SRR086166, and PRJEB23228).
Analysis of Sequence Reads
Several quality control steps were performed for the “raw” 454 sequence reads to circumvent artificial inflation of diversity estimates (Kunin et al., 2010). First, forward reads were trimmed to 230 base pairs (bp) with shorter reads being rejected. The length of 230 bp was selected because the quality scores generally dropped for bases after this cut off as we have already previously shown (Hilty et al., 2012). After the additional trimming of MID and primer sequences, the actual sequences subjected to microbial diversity analysis were 200 bp long. It has been previously hypothesized that the 200 bp long forward reads are appropriate for accurate microbial community analysis (Liu et al., 2007). Second, reads having any unresolved bases (“N”s) or homopolymers longer than 8 bp were excluded, as well as singleton reads (reads, which appear only once in all samples and are therefore likely to be the product of polymerase errors). Finally, trimmed reads were filtered with a modified version of the quality filtering script in Pyrotagger (Kunin and Hugenholtz, 2010). The script removes first reads containing sequence error(s) in its MID and/or primer, and then discards reads in which over 15% of bases show a quality score lower than 20.
Taxonomic Assignments and Calculation of Relative Bacterial Abundances of Operational Taxonomic Units (OTUs) and Bacterial Families
Pyrotagger was also used for the definition of OTUs, estimation of chimeras, and taxonomy assignments as previously described (Hilty et al., 2012). In brief, reads were clustered at a 97% sequence identity threshold using the Pyroclust algorithm. Putative chimera clusters flagged by Pyrotagger were discarded. The most abundant unique sequence was then chosen as a cluster representative and classified by comparison to Phylodb, which contains reference sequences exported from Greengene and SILVA databases (Kunin and Hugenholtz, 2010). An in house perl script was additionally used to collate family level taxonomic abundance for each sample by summing the number of reads belonging to a given family.
Within this study, the five most abundant families were analyzed separately, whereas all the remaining families were grouped as “others.” These analyses included the calculations of mean values and SEM (standard error of the mean) of the relative abundances per age group (0–1, 1–2, 2–3, 3–4, 4–5, 5–6, and 6–7 years).
Alpha Diversity Calculations of NPS
Alpha diversity [Richness (S), SDI, and Evenness], referred to as within-community diversity (Lemon et al., 2012), was assessed for OTUs based on 97% sequencing identity.
Richness is defined by the total number of OTUs while evenness refers to the similarity in OTU relative abundance in a bacterial community. Shannon’s index accounts for both abundance and evenness of the species present. Values were calculated in R1, using the respective function of the vegan package. SDI and Richness values were log-transformed to achieve normally distributed residuals. Values of log-transformed Richness (S), log-transformed SDI, and Evenness were grouped according to the age of the study participant and regression analysis was performed. Shannon or Shannon–Weaver (or Shannon–Wiener) index is defined as H = -sum p_i log(b) p_i, where p_i is the proportional abundance of species i and b is the base of the logarithm.
Evenness index is defined as J′ = H′/H′max, where H′ is the number derived from the SDI and H′max is the maximum possible value of H′. To correct for the dependency nature of the study, a linear mixed model including fixed effects [different age groups, the year of sampling, previous antibiotic treatment of the patient (yes or no), region of origin (West or East of Switzerland) and day care attendance (yes or no)] was done using the package lme4 in R (lm command). SDI and Richness values were log-transformed to achieve normally distributed residuals and outcomes were visualized according to the different age groups [e.g., for SDI: model.sdilog < -lm(df.SDI$SDIlog∼df.SDI$age + df.SDI$year + df.SDI$antibiotic + df.SDI$Region + df.SDI$day_care, na.action = na.exclude)].
Beta Diversity Analyses of NPS
Beta-diversity (between-sample diversity) was measured by the weighted Ružička index (abundance-based) and the unweighted Jaccard index (presence/absence-based) of dissimilarity. Jaccard index is computed as 2B/(1+B), where B is Bray–Curtis (d[jk] = (sum abs(x[ij]-x[ik])/(sum (x[ij]+x[ik]))) dissimilarity using the binary = TRUE flag. Ružička index is calculated identical to the Jaccard index but binary = FALSE. Pairwise distances between samples were received by the vegdist function and the resulting matrices were included to create NMDS plots (metaMDS function). Significant groupings between samples were assessed by a permutational multivariate analysis of variance using 1000 Monte Carlo permutation tests (PERMANOVA; adonis function). The multivariate dispersion of each sample group was determined by calculating the average distance (based on Jaccard and Ružička indices) to the sample type’s centroid using the betadisper function, and significant differences were assessed with Tukey’s Honest Significant Difference Test (TukeyHSD function). Boxplots and NMDS plots were generated in R utilizing the ggplot2 package.
Analyses of Paired Samples (NPS and MEF)
Heat maps of the most abundant OTUs were plotted using the ComplexHeatmap function in R. Dissimilarity values between paired NP and MEF samples were derived by the weighted Ružička (abundance-based) and the unweighted Jaccard (presence/absence-based) distance matrices. Sensitivity/specificity and PPV/NPV including 95% CIs for the most abundant OTUs and pneumococcal serotypes were calculated. Linear correlation of weighted (Ružička) or unweighted (Jaccard) paired dissimilarity values and age (in months) of the patients with AOM were calculated using Graph pad prism.
Results
Study Population and Sample Processing
A total of 286 NPS from children (<7 years) with AOM were collected from 2004–2013. Samples were roughly equally distributed for sex and region of origin (i.e., west as compared to east). Very young children (0–3 years of age) were more likely to attend a day care and were more likely to be vaccinated with either PCV7 or PCV13. Detailed information on the study population, including previous antibiotic exposure and previous otitis media episodes, is listed in Table 1. In addition, 42/286 children were reported having tympanic membrane perforation and, therefore, provided both, MEF and a NPS. The microbiota of all samples was analyzed. After exclusion of low-quality reads, this resulted in 299’966 (mean, 1048.8; SD), ±940) and 39’726 (mean, 945.9; SD, ±580.9) high quality sequence reads for the NPS and MEF samples, respectively. Comparing the values for extrapolated true richness and extrapolated estimated richness indices revealed a coverage of 66.8 and 65.4% for the NPS (Supplementary Figure 1) and MEF (Supplementary Figure 2), respectively.
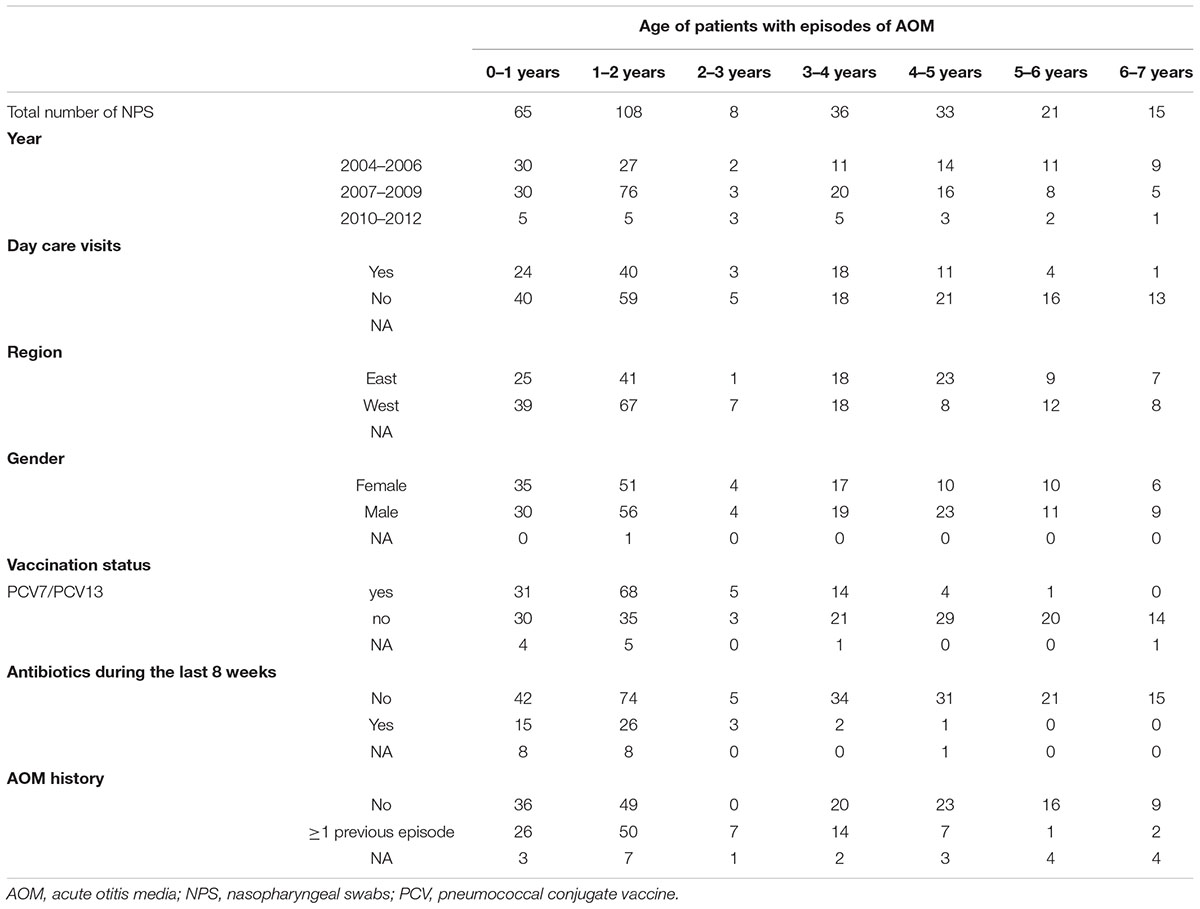
Table 1. Overview of study population/samples.
Dynamics of Bacterial Families Within NPS
We analyzed the dynamics of the five most abundant bacterial families (Moraxellaceae, Streptococcaceae, Corynebacteriaceae, Pasteurellaceae, and Staphylococcaceae) and all remaining families (“others”). Moraxellaceae was the most abundant bacterial family with a maximum relative abundance within the first year of life (mean 54.7%) (Figure 1A). However, a clear decrease of Moraxellaceae from the 3rd to the 6th year of life was noted. In contrast, we observed an increase of Staphylococcaceae and Corynebacteriaceae with increasing age (Figures 1B,C). No clear monotonic changes were noted for Streptococcaceae, Pasteurellaceae and others (Figures 1D–F) though the relative abundance of Staphylococcaceae and Corynebacteriacecae decreased at age 2–3 and increased between 3–4 years, and, the relative abundance of Streptococcaceae slightly decreased between 3–4 years. Due to the low number of NP samples at age 2–3 (n = 8), the observed non-monotonic changes might be coincidental.
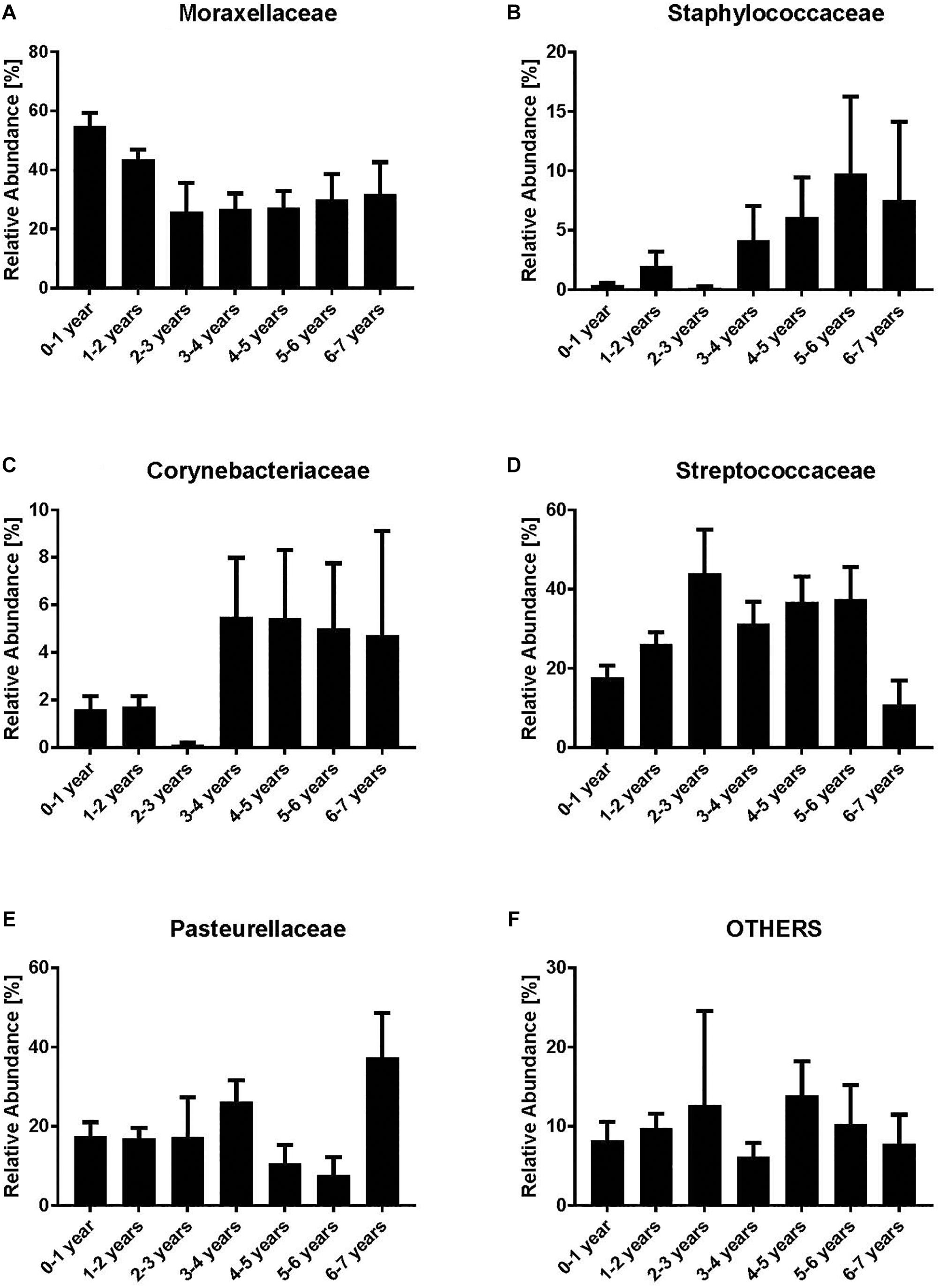
Figure 1. Relative abundances of bacterial families [Moraxellaceae (A), Staphylococcaceae (B), Corynebacteriaceae (C), Streptococcaceae (D), Pasteurellaceae (E) and Others (F)]. Indicated are the most abundant bacterial families while all the remaining are grouped as “others.” Bars represent mean values according to the age of the children with acute otitis media on the x-axis (in years). Moraxellaceae is the most abundant family within the younger age groups, whereas in older age groups Corynebacteriaceae and Staphylococcaceae are most abundant.
Bacterial Richness of the Nasopharyngeal Microbiota Increases With Age in Children With AOM
We next determined the values for the alpha diversity indices of richness (S), SDI, and evenness based on 97% OTUs (Figures 2A–F). The residuals of the models for S and SDI were not normally distributed and, therefore, were log transformed (Figures 2A–D). To correct for the dependency nature of the study, linear mixed models including fixed effects were done (Figures 2B,D,F). Results showed that the bacterial richness (S) based on OTUs with 97% sequence identity, increased with age and was highest in children with 6 years of age (Figures 2A,B). Increase was significant using a linear mixed effect (LM) model with fixed effects age, year of sample collection, previous antibiotic usage, day care attendance and geographical origin (Figure 2B) (P < 0.001). In contrast, there was no such trend for SDI (Figures 2C,D), whereas for evenness, we observed a significant decrease between younger and older age groups (P < 0.001) and values were lowest at 6 years of age (Figures 2E,F).
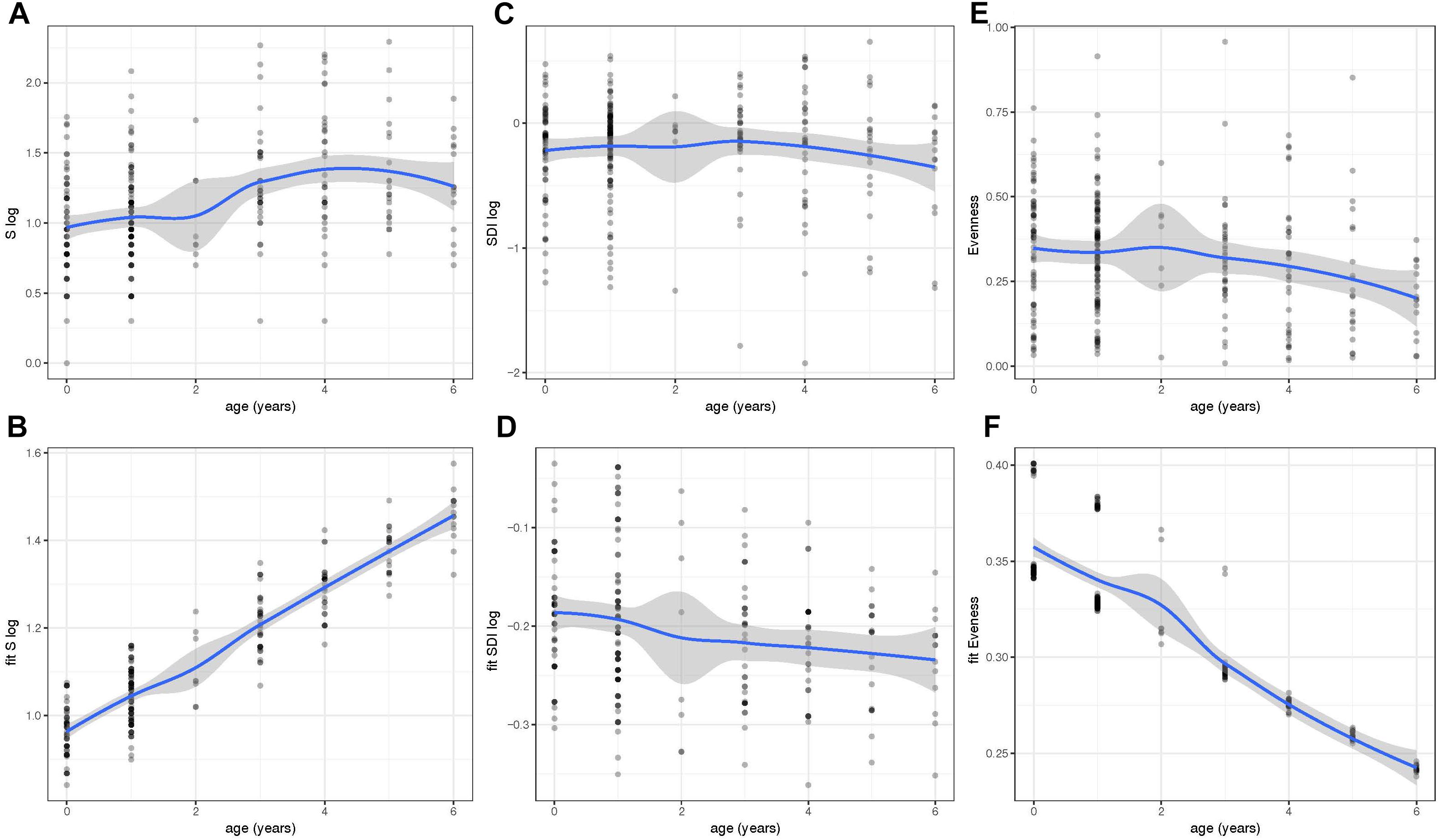
Figure 2. Within-community diversity [Richness (S), Shannon Diversity Index (SDI) and Evenness] analysis in different age groups. Values of S (A), SDI (C), and Evenness (E) were obtained at different ages (0–6 years of age). However, the residuals of the models for S and SDI were not normally distributed and, therefore, were log transformed (A–D). To correct for the dependency nature of the study, linear mixed models including fixed effects were also done and fitted values of S (B), SDI (D), and evenness (F) were plotted throughout time. Richness and Evenness values reveal an increase and decrease in older children, respectively. Fitted values were obtained using the lme4 R package and plots were generated using ggplot2.
Beta Diversity Analyses Revealed Clustering of NPS According to Age
We then used weighted (Figures 3A,B) and unweighted distance matrices (Figures 3C,D) to create ordination method based NMDS (Figures 3A,C) and multivariate dispersion plots (Figures 3B,D) for the analysis of Beta diversity. The ordination method based NMDS plots (Figures 3A,C) showed a distinct clustering of the microbiota of the NP of children with AOM according to age (PERMANOVA, unweighted: F-value: 6.6, P < 0.01, weighted: F-value: 6.3, P < 0.01). This indicates a very strong effect of age on the human bacterial microbiota of the NP. Interestingly, children at a very young age seemed to display a significantly lower dispersion as compared to older children (unweighted distances from the centroid; Tukey’s HSD test; P < 0.001; Figure 3D), indicating that an increase in age leads to a more heterogeneous microbial community structure. In contrast, all comparisons of weighted distances from the centroid were non-significant (Figure 3B), suggesting a stronger effect of community structure compared to community composition on multivariate dispersion across groups.
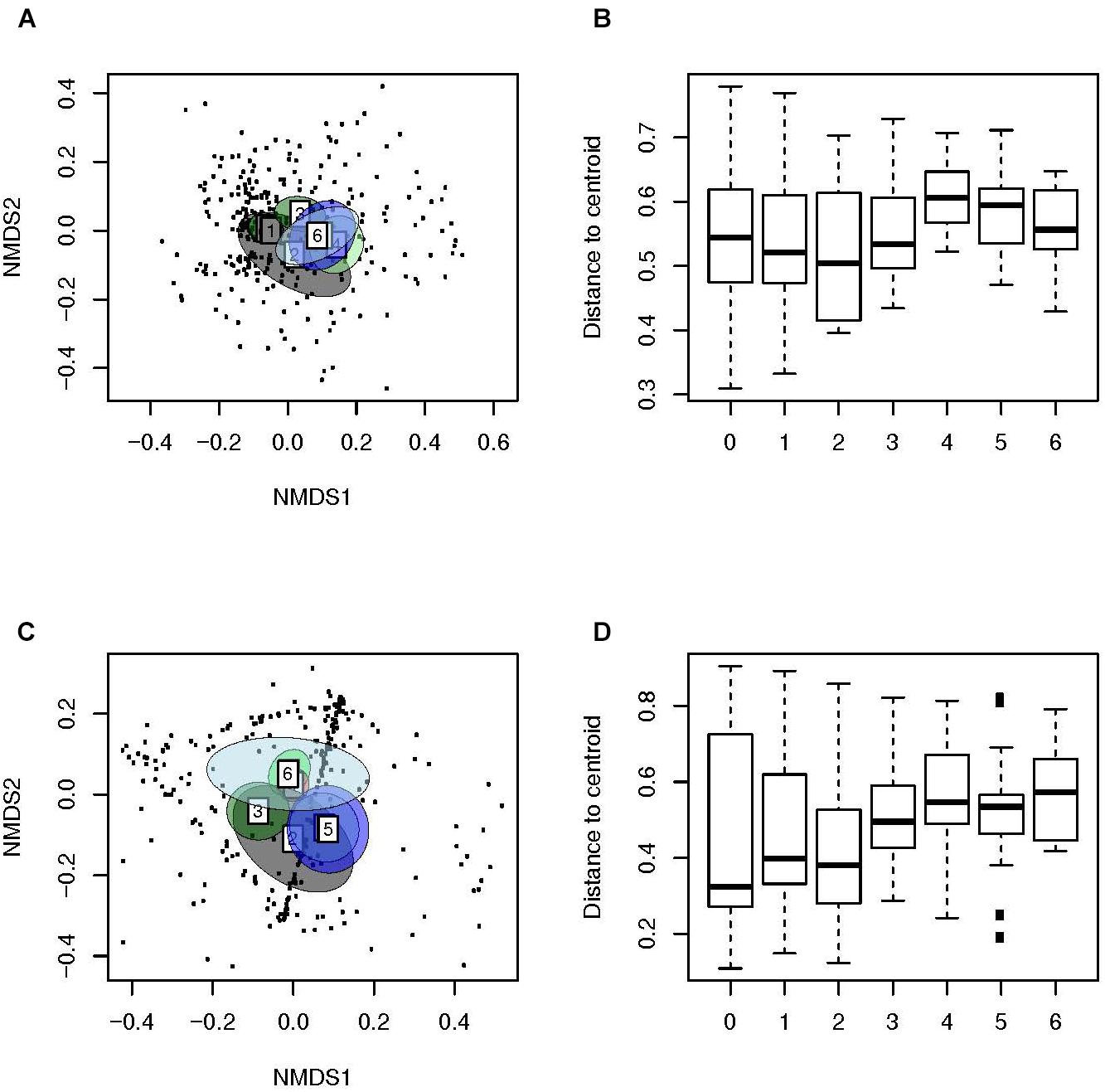
Figure 3. Beta-diversity analyses of samples of children with AOM suggest a more personalized bacterial microbiota with age. Illustrated are (A) Weighted (Ružička) and (C) unweighted (Jaccard) distances in microbiota composition, reduced in a 2D-space by using NMDS, 95% confidence ellipse for the group centroid shown. In addition, shown are the beta-dispersion based on (B) Ružička and (D) Jaccard dissimilarity indices in each sample type. The boxplots represent median (midline), interquartile ranges (shaded boxes), and ranges (whiskers). Data is shown according to the age (years) of the children with AOM.
Microbiota of NP and MEF Show Highest Similarity When S. pyogenes, H. influenzae or S. pneumoniae Are the Predominant OTUs
We then compared the NPS microbiota profiles with the corresponding MEF profiles from 42 children. The 11 most abundant OTUs are illustrated (Figure 4) while all others were grouped (“others”). High dissimilarity values between the microbiota profiles of the NPS and the MEF were received if Staphylococcus spp. and “others” were the predominate OTUs within the MEF. Increased similarity was seen if the predominate OTUs within the MEF were S. pyogenes, Streptococcus mitis group (includes S. pneumoniae) and H. influenzae.
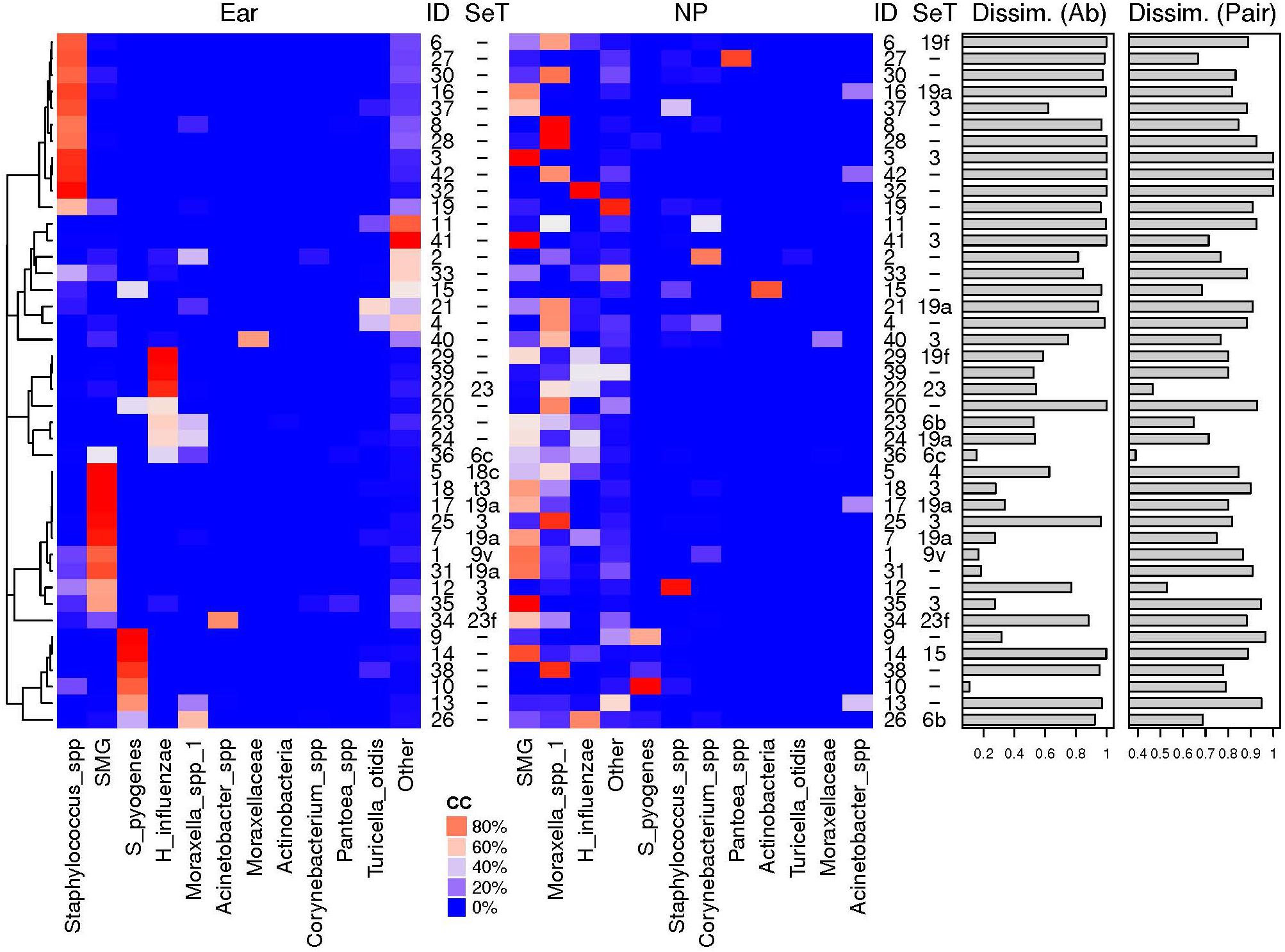
Figure 4. Relative abundances of most abundant OTUs in MEF from ear and NP. Shown are the heat maps of the relative abundances of the most abundant OTUs in % [illustrated by a distinct color code (CC)]. The pneumococcal serotpyes (SeT) are indicated. Dissimilarly values based on abundance based Ružička (Dissim. Ab) and presence-absence based Jaccard (Dissim. Pair) distance values between the two different sample types of the same individual (ID) are shown as bar plots. High similarity of the bacterial microbiota of NPS and MEF when S. pyogenes, H. influenzae or S. pneumoniae were the predominant OTUs and overall high negative predictive values for NPS based prediction of MEF (see Table 2 and text for more details). SMG, Streptococcus mitis group members (including Streptococcus pneumoniae).
Calculation of the correlation between dissimilarity values and age of the children with AOM (Figures 5A,B) revealed a significant positive correlation for the presence/absence-based dissimilarity values (Figure 5B), which was not the case for the abundance-based values (Figure 5A).
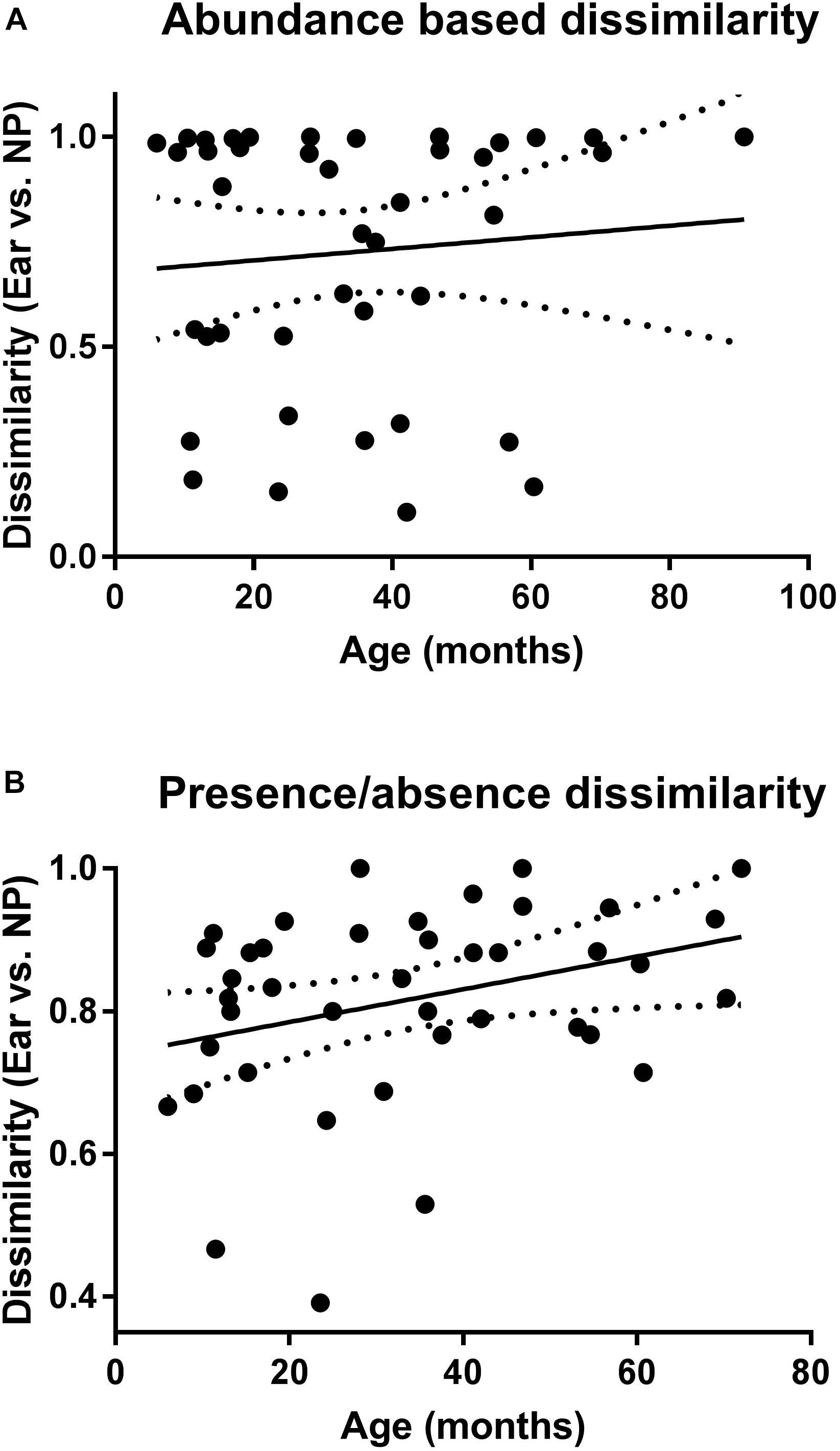
Figure 5. Correlation of the microbiota of NP and MEF according to age. Shown are the abundance (A) and presence/absence (B) based dissimilarity values (for NP and MEF) for the children with AOM at different ages. Linear regression lines are indicated. P-value is significant for the presence/absence based dissimilarities (P = 0.04).
Finally, we determined the sensitivity/specificity of individual OTUs and the pneumococcal serotypes for the prediction of corresponding MEF results (Table 2). Highest sensitivity/specificity values were achieved for S. pyogenes and the pneumococcal serotypes. For one patient, the serotyping results were discordant which might be due to multiple colonization of different S. pneumoniae (Brugger et al., 2009, 2010). Concerning the other OTUs, some were more often found in the MEF (especially Turicella spp.) while the opposite was true for e.g., Corynebacterium spp., Moraxella spp. and H. influenzae. In general, positive predictive values (PPV) revealed a considerable, pathogen-dependent variability while the negative predictive values (NPV) were found to be consistently high for most of the OTUs (Table 2).
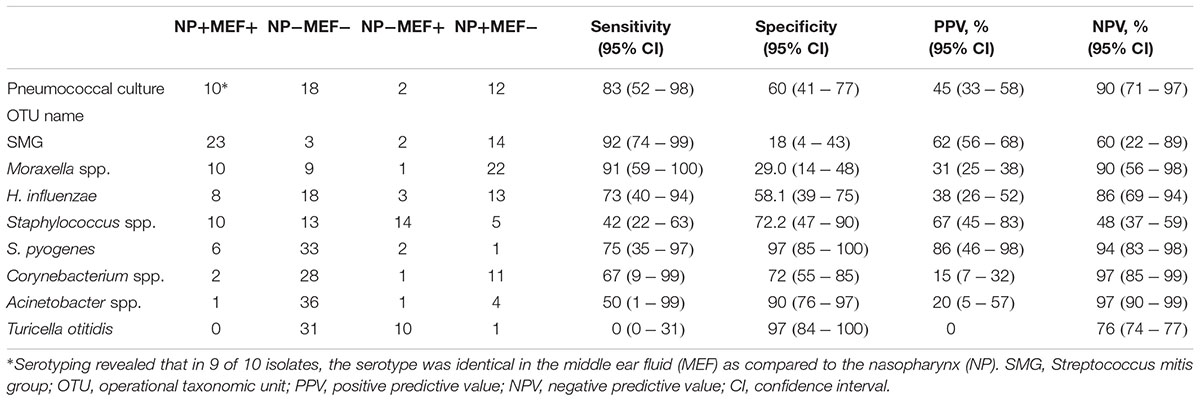
Table 2. Sensitivity and specificity MEF/NP based on pneumococcal culture and presence and absence of the eight most frequent OTUs in the NPS and ears, respectively.
Discussion
This study analyzed the bacterial microbiota profiles from 286 children with AOM (286 NPS and 42 MEF). In the NP, we noted an increase in bacterial richness and beta dispersion with age and a decrease in evenness (Figures 2, 3). This indicates the development of a more distinct bacterial microbiota profile toward the end of the sixth year of life. In addition, we found high similarity of the bacterial microbiota of NPS with MEF if the predominant OTU within the MEF was S. pyogenes, H. influenzae, or S. pneumoniae. The opposite was true for Staphylococcus spp. and others (Figure 4 and Table 2).
We performed 454 sequencing in our study, a technique which has been replaced by more high-throughput platforms, e.g., Illumina. However, the nasal microbiota has a rather simple, low-diversity profile (Escapa et al., 2018) and in order to achieve an approximate OTU coverage of 70% (which is mainly recommended) there is no need for a large amount of sequencing reads (Hilty et al., 2012). In addition, 454 sequencing produced longer reads (as compared to Illumina) which increased the resolution for OTUs. Most importantly, our protocols have been systematically investigated as we have previously done validation work on mock communities using a similar design (Brugger et al., 2012). This included using different primers [i.e., 8F (for V1–V5) versus 341 F (for V3–V5)] and investigating the detection limit of bacteria (i.e., mock communities) which are very well known to be present in the NP of children. Using the primer 8F, the detection limit was about 30 genome copies μl-1 with the exception of H. influenzae and M. catarrhalis (100 copies μl-1). As for the observed difference of the detection limit, this is probably due to a SNP present in the conserved region of the forward primer 8F and therefore suboptimal PCR efficiency. There are no SNPs in the primer regions of 341F and 907R for the bacteria mentioned above and we therefore concluded this primer pair being appropriate for the study of the bacterial microbiota of the NP. Using Illumina MiSeq, the same primer design cannot be used as the reads are not long enough.
There is a lack of studies like ours describing the nasopharyngeal bacterial microbiota in children with AOM up to 7 years of age. However, data exists for infants at a younger age describing a distinct profile for the first 3 months of life with increased relative abundances of Staphylococcaceae and Corynebacteriaceae (Laufer et al., 2011; Biesbroek et al., 2014a,b; Mika et al., 2015; Teo et al., 2015; Bomar et al., 2017; Salter et al., 2017). In our study we found an increase of Staphylococcaceae and Corynebacteriaceae in children older than 3 years. This may indicate the transition toward a more “adult”-like composition of the microbiota as these two families are very often identified within the adult microbiome (Frank et al., 2010; Camarinha-Silva et al., 2014; Bomar et al., 2017). This transition may take place due to a more mature immune response resulting in less episodes of AOM in older children. In addition, common to our and other studies using molecular or conventional culture methods are the high (relative) abundances of Moraxellaceae and Streptococcaceae within the first 2 years of life (Faden et al., 1994, 1997; Bogaert et al., 2004, 2011; Hilty et al., 2012). However, in our study, we show that the abundance of Moraxellaceae drastically decreases in children aged more than 2 years.
A further major finding of our study was the discovery of a significant increasing correlation of age and dissimilarly of the microbiota of NP as compared to the MEF. Based on bacterial cultures, a recent study has observed that S. pneumoniae and non-typeable Haemophilus influenzae nasopharyngeal colonization increases significantly between 6 and 30–36 months of age, but as children get older the relationship between potential otopathogen in the NP with detection in MEF is weakened (Kaur et al., 2014). Similarly, a high prevalence of nasopharyngeal colonization but low frequency of S. pneumoniae as an etiology of AOM with age, especially after children became >18 months of age has been reported (Syrjanen et al., 2005). Our bacterial microbiota-based analyses showed similar findings as compared to culture-based studies. However, NP microbiota patterns from children below 2 years of age are still not very good surrogates of MEF cultures in children as dissimilarity values were quite high.
A recent systematic review of the literature reported the concordance between culture test results of the NP and MEF samples for the most prevalent microorganisms in children with otitis media (van Dongen et al., 2013). Culture-based AOM studies revealed higher PPVs (for H. influenzae and S. aureus) and lower PPVs (for M. catarrhalis) as compared to our study, respectively (van Dongen et al., 2013). As for S. aureus, this might be in part due to the fact that all MEF in this study were derived from cases with spontaneous tympanic membrane perforation which have been reported to contain a higher proportion of S. aureus (Brook and Gober, 2009).
However, with the exception of S. aureus, we received much higher NPVs as compared to culture-based studies. Our NPVs were also higher if compared to a study which was using a series of polymerase chain reactions in order to detect bacteria (Yatsyshina et al., 2016). This means that high-throughput sequencing of the bacterial microbiota of the NP results in decent NPVs for finding the pathogens within the MEF while this is not the case for the PPVs (with the exception of S. pyogenes).
Recently, Man et al. (2018) compared the microbiota of NP and MEF in almost 100 Dutch children under 5 years of age with AOM in children with tympanostomy tubes and also reported a high overall NPV (on OTU level) suggesting that the NP might be the main source of MEF bacteria in otitis media. The overall PPV was 0.4 (95%CI 0.39 – 0.42) which is in line with our results. However, significant quantitative correlations between NP and tympanostomy tube otorrhea (TTO) samples for Haemophilus influenzae, S. aureus, and P. aeruginosa abundance were reported (Man et al., 2018). In agreement with our results, T. otitis was associated with MEF supporting its role as an otopathogen (Man et al., 2018).
This study has some major strengths. First, we have included a large number of NPS (n = 286) from children with AOM at different ages (up to <7 years of age). Patients were recruited from different outpatient settings from a well-defined sentinel network and medical diagnosis was done based on CDC case definition. In addition, we have included 42 paired samples of NPS and MEF allowing the investigation of the appropriateness of the NP as a proxy for the microbiota of the MEF. Apart from 16S rRNA sequencing, we also performed culturing for S. pneumoniae and subsequent serotyping for all positive samples.
This study has also some major limitations. Within our study, we did not investigate homogeneity of nasopharyngeal microbiota at the different locations of the nose of children with AOM. However, results from a recent study suggest that the microbiota at the different locations of the nose of children with AOM is almost homogeneous, irrespective of the clinical signs (Kaieda et al., 2005). We also only analyzed the bacterial microbiota and did not include virus detection in this study (e.g., by shotgun metagenomics sequencing). In addition, MEF collection was biased toward aggressive forms of AOM as only material of spontaneous perforations was collected which resulted in only 48 NP/MEF paired analyses. Also, based on the design of the surveillance program the information about the exact duration of the AOM episode as well as the time of tympanic membrane perforation was not recorded. Furthermore, sample acquisition was not standardized (i.e., no tympanocentesis samples) which could have resulted in contamination with skin flora. Thus, we cannot rule out that otitis externa might have been diagnosed as AOM in some cases (this could be reflected by the higher relative abundance of Staphylococcaceae in MEF compared to NPS, however, the pneumococcal isolation from NPS highly matched with pneumococcal isolation in MEF).
Further studies investigating the development of the bacterial microbiome including AOM should follow a prospective design (using a power calculation based on our results) to reduce the risk of unmeasured confounders.
Conclusion
In conclusion, this study revealed varying nasopharyngeal bacterial microbiota patterns according to age and moderate to high NPVs for the prediction of MEF OTUs based on analyses from the NP in a subset of children with spontaneous tympanic membrane rupture where we simultaneously characterized the bacterial microbiota of NPS and MEF. The NP of children with AOM serves as a moderate and insufficient proxy for the bacterial communities of the MEF at a very young and more advanced age, respectively. Given these results as well as previous culture (or species-specific PCR) based studies, the nasopharyngeal bacterial microbiota does not necessarily reflect the one of the middle ear in AOM and should only be used with caution for clinical decision decision-making. This might be in part due to the complex disease pathology in AOM, which also involves viruses and biofilms.
Data Availability
The datasets generated for this study can be found in National Center for Biotechnology Information Sequence Read Archive, SRR077426, SRR086165, SRR086166, and PRJEB23228.
Ethics Statement
The overall AOM surveillance program has been part of the governmental public health surveillance and has therefore been exempted from approval by Institutional Review Boards and the need for written consent from parents. Informed oral consent was received from the parents.
Author Contributions
SB and MH were investigators in this study and contributed to the study design. SB, JK, WQ, LB, AO, and MH contributed to data collection and analysis. SB, JK, and MH contributed to data interpretation. All authors contributed to the writing, review of the report, and approved the final version of the manuscript.
Funding
The prospective surveillance study on the surveillance of acute otitis media within Sentinella including the investigations of MEF and NPS has been supported by the Federal Office of Public Health (FOPH). SB was supported by a grant (P3SMP3_155315) from the Swiss National Science Foundation (SNSF) and the Swiss Foundation for Grants in Biology and Medicine (SFGBM). This work was also partly supported by Swiss National Science Foundation (SNF) grant 320030_159791 to MH.
Conflict of Interest Statement
MH received an educational grant from Pfizer AG for partial support of this project. However, Pfizer AG had no role in the data analysis and content of the manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Marianne Küffer and Chantal Studer for their extraordinary assistance. We are also very thankful to all the physicians who actively take and took part in the Sentinella surveillance system and who provided non-invasive MEF and NPS.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00555/full#supplementary-material
Abbreviations
CI, confidence interval; MEF, middle ear fluid; MID, multiplex identifier; nMDS, non-metric multidimensional scaling; NP, nasopharynx; NPS, nasopharyngeal swab; OTU, operational taxonomic unit; PCV, pneumococcal conjugate vaccine; rRNA, ribosomal RNA; SD, standard deviation; SDI, shannon diversity index.
Footnotes
References
Allemann, A., Frey, P. M., Brugger, S. D., and Hilty, M. (2017). Pneumococcal carriage and serotype variation before and after introduction of pneumococcal conjugate vaccines in patients with acute otitis media in Switzerland. Vaccine 35, 1946–1953. doi: 10.1016/j.vaccine.2017.02.010
Biesbroek, G., Bosch, A. A., Wang, X., Keijser, B. J., Veenhoven, R. H., Sanders, E. A., et al. (2014a). The impact of breastfeeding on nasopharyngeal microbial communities in infants. Am. J. Respir. Crit. Care Med. 190, 298–308. doi: 10.1164/rccm.201401-0073OC
Biesbroek, G., Tsivtsivadze, E., Sanders, E. A., Montijn, R., Veenhoven, R. H., Keijser, B. J., et al. (2014b). Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am. J. Respir. Crit. Care Med. 190, 1283–1292. doi: 10.1164/rccm.201407-1240oc
Biesbroek, G., Sanders, E. A., Roeselers, G., Wang, X., Caspers, M. P., Trzcinski, K., et al. (2012). Deep sequencing analyses of low density microbial communities: working at the boundary of accurate microbiota detection. PLoS One 7:e32942. doi: 10.1371/journal.pone.0032942
Bogaert, D., Keijser, B., Huse, S., Rossen, J., Veenhoven, R., van Gils, E., et al. (2011). Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One 6:e17035. doi: 10.1371/journal.pone.0017035
Bogaert, D., van Belkum, A., Sluijter, M., Luijendijk, A., de Groot, R., Rumke, H. C., et al. (2004). Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363, 1871–1872. doi: 10.1016/s0140-6736(04)16357-5
Bomar, L., Brugger, S. D., and Lemon, K. P. (2017). Bacterial microbiota of the nasal passages across the span of human life. Curr. Opin. Microbiol. 41, 8–14. doi: 10.1016/j.mib.2017.10.023
Brook, I., and Gober, A. E. (2009). Bacteriology of spontaneously draining acute otitis media in children before and after the introduction of pneumococcal vaccination. Pediatr. Infect. Dis. J. 28, 640–642. doi: 10.1097/INF.0b013e3181975221
Brugger, S. D., Frei, L., Frey, P. M., Aebi, S., Muhlemann, K., and Hilty, M. (2012). 16S rRNA terminal restriction fragment length polymorphism for the characterization of the nasopharyngeal microbiota. PLoS One 7:e52241. doi: 10.1371/journal.pone.0052241
Brugger, S. D., Frey, P., Aebi, S., Hinds, J., and Muhlemann, K. (2010). Multiple colonization with S. pneumoniae before and after introduction of the seven-valent conjugated pneumococcal polysaccharide vaccine. PLoS One 5:e11638. doi: 10.1371/journal.pone.0011638
Brugger, S. D., Hathaway, L. J., and Muhlemann, K. (2009). Detection of Streptococcus pneumoniae strain cocolonization in the nasopharynx. J. Clin. Microbiol. 47, 1750–1756. doi: 10.1128/JCM.01877-08
Camarinha-Silva, A., Jauregui, R., Chaves-Moreno, D., Oxley, A. P., Schaumburg, F., Becker, K., et al. (2014). Comparing the anterior nare bacterial community of two discrete human populations using illumina amplicon sequencing. Environ. Microbiol. 16, 2939–2952. doi: 10.1111/1462-2920.12362
Chonmaitree, T., Jennings, K., Golovko, G., Khanipov, K., Pimenova, M., Patel, J. A., et al. (2017). Nasopharyngeal microbiota in infants and changes during viral upper respiratory tract infection and acute otitis media. PLoS One 12:e0180630. doi: 10.1371/journal.pone.0180630
Coticchia, J. M., Chen, M., Sachdeva, L., and Mutchnick, S. (2013). New paradigms in the pathogenesis of otitis media in children. Front. Pediatr. 1:52. doi: 10.3389/fped.2013.00052
Escapa, I. F., Chen, T., Huang, Y., Gajare, P., Dewhirst, F. E., and Lemon, K. P. (2018). New insights into human nostril microbiome from the expanded human oral microbiome database (eHOMD): a resource for the microbiome of the human aerodigestive tract. mSystems 3:e187-18. doi: 10.1128/mSystems.00187-18
Faden, H., Duffy, L., Wasielewski, R., Wolf, J., Krystofik, D., and Tung, Y. (1997). Relationship between nasopharyngeal colonization and the development of otitis media in children. J. Infect. Dis. 175, 1440–1445. doi: 10.1086/516477
Faden, H., Harabuchi, Y., and Hong, J. J. (1994). Epidemiology of Moraxella catarrhalis in children during the first 2 years of life: relationship to otitis media. J. Infect. Dis. 169, 1312–1317. doi: 10.1093/infdis/169.6.1312
Frank, D. N., Feazel, L. M., Bessesen, M. T., Price, C. S., Janoff, E. N., and Pace, N. R. (2010). The human nasal microbiota and Staphylococcus aureus carriage. PLoS One 5:e10598. doi: 10.1371/journal.pone.0010598
Hilty, M., Qi, W., Brugger, S. D., Frei, L., Agyeman, P., Frey, P. M., et al. (2012). Nasopharyngeal microbiota in infants with acute otitis media. J. Infect. Dis. 205, 1048–1055. doi: 10.1093/infdis/jis024
Kaieda, S., Yano, H., Okitsu, N., Hosaka, Y., Okamoto, R., and Inoue, M. (2005). Investigation about the homogeneity of nasopharyngeal microflora at the different location of nasopharynx of children with acute otitis media. Int. J. Pediatr. Otorhinolaryngol. 69, 959–963. doi: 10.1016/j.ijporl.2005.01.036
Kaur, R., Czup, K., Casey, J. R., and Pichichero, M. E. (2014). Correlation of nasopharyngeal cultures prior to and at onset of acute otitis media with middle ear fluid cultures. BMC Infect. Dis. 14:640. doi: 10.1186/s12879-014-0640-y
Kaur, R., Morris, M., and Pichichero, M. E. (2017). Epidemiology of acute otitis media in the postpneumococcal conjugate vaccine era. Pediatrics 140:e20170181. doi: 10.1542/peds.2017-0181
Kilpi, T., Herva, E., Kaijalainen, T., Syrjanen, R., and Takala, A. K. (2001). Bacteriology of acute otitis media in a cohort of finnish children followed for the first two years of life. Pediatr. Infect. Dis. J. 20, 654–662. doi: 10.1097/00006454-200107000-00004
Kunin, V., Engelbrektson, A., Ochman, H., and Hugenholtz, P. (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123. doi: 10.1111/j.1462-2920.2009.02051.x
Kunin, V., and Hugenholtz, P. (2010). PyroTagger : a fast, accurate pipeline for analysis of rRNA amplicon pyrosequence data. Open J. 1, 1–8.
Lappan, R., Imbrogno, K., Sikazwe, C., Anderson, D., Mok, D., Coates, H., et al. (2018). A microbiome case-control study of recurrent acute otitis media identified potentially protective bacterial genera. BMC Microbiol. 18:13. doi: 10.1186/s12866-018-1154-3
Laufer, A. S., Metlay, J. P., Gent, J. F., Fennie, K. P., Kong, Y., Pettigrew, M. M., et al. (2011). Microbial communities of the upper respiratory tract and otitis media in children. mBio 2:e245-210. doi: 10.1128/mBio.00245-10
Lemon, K. P., Armitage, G. C., Relman, D. A., and Fischbach, M. A. (2012). Microbiota-targeted therapies: an ecological perspective. Sci. Transl. Med. 4:137rv5. doi: 10.1126/scitranslmed.3004183
Lieberthal, A. S., Carroll, A. E., Chonmaitree, T., Ganiats, T. G., Hoberman, A., Jackson, M. A., et al. (2013). The diagnosis and management of acute otitis media. Pediatrics 131, e964–e999. doi: 10.1542/peds.2012-3488
Liu, Z., Lozupone, C., Hamady, M., Bushman, F. D., and Knight, R. (2007). Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 35:e120. doi: 10.1093/nar/gkm541
Man, W. H., van Dongen, T. M. A., Venekamp, R. P., Pluimakers, V. G., Chu, M., van, Houten MA, et al. (2018). Respiratory microbiota predicts clinical disease course of acute otorrhea in children with tympanostomy tubes. Pediatr. Infect. Dis. J. 38, e116–e125. doi: 10.1097/INF.0000000000002215
Mika, M., Mack, I., Korten, I., Qi, W., Aebi, S., Frey, U., et al. (2015). Dynamics of the nasal microbiota in infancy: a prospective cohort study. J. Allergy Clin. Immunol. 135, 905.e11–912.e11. doi: 10.1016/j.jaci.2014.12.1909
Muhlemann, K., Matter, H. C., Tauber, M. G., and Bodmer, T. (2003). Nationwide surveillance of nasopharyngeal Streptococcus pneumoniae isolates from children with respiratory infection, Switzerland, 1998-1999. J. Infect. Dis. 187, 589–596. doi: 10.1086/367994
Post, J. C., Preston, R. A., Aul, J. J., Larkins-Pettigrew, M., Rydquist-White, J., Anderson, K. W., et al. (1995). Molecular analysis of bacterial pathogens in otitis media with effusion. JAMA 273, 1598–1604. doi: 10.1001/jama.273.20.1598
Revai, K., Mamidi, D., and Chonmaitree, T. (2008). Association of nasopharyngeal bacterial colonization during upper respiratory tract infection and the development of acute otitis media. Clin. Infect. Dis. 46, e34–e37. doi: 10.1086/525856
Salter, S. J., Turner, C., Watthanaworawit, W., de, Goffau MC, Wagner, J., Parkhill, J., et al. (2017). A longitudinal study of the infant nasopharyngeal microbiota: the effects of age, illness and antibiotic use in a cohort of South East Asian children. PLoS Negl. Trop. Dis. 11:e0005975. doi: 10.1371/journal.pntd.0005975
Sillanpaa, S., Kramna, L., Oikarinen, S., Sipila, M., Rautiainen, M., Aittoniemi, J., et al. (2017). Next-generation sequencing combined with specific PCR assays to determine the bacterial 16S rRNA gene profiles of middle ear fluid collected from children with acute otitis media. mSphere 2:e00006-17. doi: 10.1128/mSphere.00006-17
Syrjanen, R. K., Auranen, K. J., Leino, T. M., Kilpi, T. M., and Makela, P. H. (2005). Pneumococcal acute otitis media in relation to pneumococcal nasopharyngeal carriage. Pediatr. Infect. Dis. J. 24, 801–806. doi: 10.1097/01.inf.0000178072.83531.4f
Teo, S. M., Mok, D., Pham, K., Kusel, M., Serralha, M., Troy, N., et al. (2015). The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 17, 704–715. doi: 10.1016/j.chom.2015.03.008
van Dongen, T. M., van der Heijden, G. J., van Zon, A., Bogaert, D., Sanders, E. A., Schilder, A. G., et al. (2013). Evaluation of concordance between the microorganisms detected in the nasopharynx and middle ear of children with otitis media. Pediatr. Infect. Dis. J. 32, 549–552. doi: 10.1097/INF.0b013e318280ab45
Yatsyshina, S., Mayanskiy, N., Shipulina, O., Kulichenko, T., Alyabieva, N., Katosova, L., et al. (2016). Detection of respiratory pathogens in pediatric acute otitis media by PCR and comparison of findings in the middle ear and nasopharynx. Diagn. Microbiol. Infect. Dis. 85, 125–130. doi: 10.1016/j.diagmicrobio.2016.02.010
Keywords: nasopharyngeal microbiota, bacterial families, toddlers, acute otitis media, middle ear fluid, age
Citation: Brugger SD, Kraemer JG, Qi W, Bomar L, Oppliger A and Hilty M (2019) Age-Dependent Dissimilarity of the Nasopharyngeal and Middle Ear Microbiota in Children With Acute Otitis Media. Front. Genet. 10:555. doi: 10.3389/fgene.2019.00555
Received: 21 March 2019; Accepted: 24 May 2019;
Published: 19 June 2019.
Edited by:
Regie Santos-Cortez, University of Colorado Denver, United StatesReviewed by:
Paola Marchisio, University of Milan, ItalyTheodora Katsila, National Hellenic Research Foundation, Greece
Copyright © 2019 Brugger, Kraemer, Qi, Bomar, Oppliger and Hilty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Hilty, TWFya3VzLkhpbHR5QGlmaWsudW5pYmUuY2g=
†These authors have contributed equally to this work