 Hui Wang1Yuan Lv1Cao Wang1Dongjing Leng1Yan Yan1Moyondafoluwa Blessing Fasae1
Hui Wang1Yuan Lv1Cao Wang1Dongjing Leng1Yan Yan1Moyondafoluwa Blessing Fasae1 Syeda Madiha Zahra1Yanan Jiang1,2Zhiguo Wang1
Syeda Madiha Zahra1Yanan Jiang1,2Zhiguo Wang1 Baofeng Yang1,2*
Baofeng Yang1,2* Yunlong Bai1,2*
Yunlong Bai1,2*- 1Department of Pharmacology (State-Province Key Laboratories of Biomedicine–Pharmaceutics of China, Key Laboratory of Cardiovascular Medicine Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin, China
- 2Chronic Disease Research Institute, Translational Medicine Research and Cooperation Center of Northern China, Heilongjiang Academy of Medical Sciences, Harbin, China
Hepatocellular carcinoma (HCC) is an extremely fatal malignancy. Intestinal microRNAs, which can be detected in fecal samples in humans may be involved in the pathological process of HCC. Therefore, screening for functional intestinal microRNAs in fecal samples and investigating their potential roles in the molecular progression of HCC are necessary. Quantitative real-time PCR (qRT-PCR) has been widely used in microRNA expression studies. However, few genes have been reported as reference genes for intestinal microRNAs in fecal samples. In order to obtain a more accurately analyzed intestinal microRNAs expression, we first searched for reliable reference genes for intestinal microRNAs expression normalization during qRT-PCR, using three software packages (GeNorm, NormFinder, and Bestkeeper). Next we screened and predicted the target genes of the differentially intestinal microRNAs of control and HCC mice through quantitative RT-PCR or miRtarBase. Finally, we also analyzed the mRNA targets for enrichment of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using the DAVID Bioinformatic Resources database. This study has successfully screened relatively suitable reference genes and we have discovered that the differential intestinal microRNAs play significant roles in the development of HCC. The top reference genes identified in this study could provide a theoretical foundation for the reasonable selection of a suitable reference gene. Furthermore, the detection of intestinal microRNAs expression may serve as a promising therapeutic target for the diagnosis and treatment of HCC.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers in the world, and the overall 5-year survival rate for HCC is less than 12% (Torre et al., 2015). It is usually diagnosed at the late stage and is characterized by a low resection rate, high postoperative recurrence, and poor drug treatment results (Galun et al., 2015). At present, the only drug approved for the treatment of liver cancer is sorafenib. Oxaliplatin has been shown to be effective in the treatment of advanced liver cancer, but the drug resistance potentially limits its efficacy (Petrelli et al., 2014). Finding sensitive and specific early diagnostic biomarkers, and understanding the mechanisms involved in the development and progression of HCC are a major issue in need of urgent resolution.
MicroRNAs are small non-coding RNAs, with transcripts that are just 18-25 nucleotides in length, and they bind mainly to the 3′ untranslated regions (3′ UTRs) of target RNAs through the microRNAs’ seed sequences (microRNA nucleotides 2–7) (Parodi et al., 2016). They are highly conservative and tissue-specific and exists widely in nematodes, drosophila, and plants, as well as in humans (Gillan et al., 2017; You et al., 2018). MicroRNAs regulate the expression of protein-coding gene expression in a sequence-specific manner through cleavage or translational repression in the genomes (Kagiya, 2016; Xiao et al., 2018). The abnormal expression of microRNAs is related to many diseases, including cancer, and miRNAs can also be used as tumor-suppressor genes or oncogenes. During the development and progression of human cancers, microRNAs have been observed to regulate cell proliferation, survival, differentiation, invasion and metastasis (Davis-Dusenbery and Hata, 2010). In patients with HCC caused by a hepatitis B virus infection, mir-29c could control cell proliferation and leads to apoptosis by targeting TNFAIP3 (, Wang et al., 2011); mir-101 produces a pro-apoptotic effect by targeting Mcl-1 (Su et al., 2009). Since the discovery of their essential roles in the occurrence and development of diseases, microRNAs have been intensely studied as prognostic and diagnostic biomarkers and predictors of drug responses (Li et al., 2014).
Past research has focused mainly on post-transcriptional and translational controls regulated by non-coding RNAs (Ranjha and Paul, 2013). MicroRNAs could efficiently interrupt the synthesis of proteins excluding further transcriptional activation and any subsequent mRNA processing steps. Therefore, it provides cells with a more accurate, direct and energy-efficient way to manipulate protein expression (Xi et al., 2007; Ge et al., 2015; Connerty et al., 2016). Liu et al. (2016) have illustrated that microRNAs in feces, which originate from intestinal cells, can enter into bacteria and regulate their gene expression and growth. These special microRNAs are called intestinal microRNAs. They specifically show that mir-515-5p and mir-1226-5p can promote the growth of Fn and E. coli, respectively. Thus, knowing the differential expressions of intestinal microRNAs in fecal samples could be critical to understand physiological and pathological conditions. They could even be used as a potential marker for a new therapeutic strategy. However, the expression profiles and functions of the intestinal microRNAs present in the fecal samples of patients with HCC remains unclear.
Due to the instability of microRNAs, their low expression level and minor differences in sequence, highly sophisticated analytical methods are required. Quantitative real-time PCR (qRT-PCR) plays an important role in studying the biological functions of microRNA with strong specificity and high sensitivity (Daud and Scott, 2008).With the development of qRT-PCR, the selection of reference genes is crucial for the accuracy of the relative quantification of gene expressions (Liu et al., 2018; Zhou et al., 2018). However, only a few studies have been published that systematically evaluated the normalization targets in intestinal microRNAs qRT-PCR assays. Successfully screening for suitable reference genes first will provide reliable evidence for the quantitative analysis of intestinal microRNAs in fecal samples. Once the expression profile of intestinal microRNAs in HCC is confirmed by qRT-PCR, the functions of differentiated intestinal microRNAs could be investigated during the biological process.
This study aimed to find relatively stable reference genes and to clarify whether intestinal microRNAs from fecal samples play an important role in indicating HCC development. RNA was extracted from mouse fecal samples and microRNA expressions were detected by qRT-PCR. Four conventional housekeeping genes and 18 microRNAs were selected as candidate genes. Their expression stability was evaluated using GeNorm, NormFinder, and Bestkeeper software. These statistical analysis software programs were primarily used to identify appropriate reference genes in qRT-PCR experiments. We identified three genes (mir-23a, GAPDH and let-7i) which may be used as suitable reference genes for intestinal microRNAs. Next, we detected the expression levels of some intestinal microRNAs, which had already been observed in humans and mice, between the control and HCC group. Finally, we analyzed the mRNA targets of the differentiated intestinal microRNAs. Then GO terms and KEGG pathways were built using these databases. In this study, relatively suitable reference genes of intestinal microRNAs in fecal samples are first screened for, with a potential diagnosis and treatment for patients with HCC is then proposed.
Materials and Methods
Samples Preparation and the Ethics Approval
Professor Feng Hai from Harbin Medical University supplied fecal samples from five control and five HCC-suffering mice. The C57BL/6 mice (14th postnatal days) were injected DEN at a single intraperitoneal dose of 25 mg/kg and the animals were observed until 10 months. At 10 months of age, mice injected with DEN developed striking liver HCCs with tumors that could be clearly observed. The mice were allowed to defecate normally, and the first three fecal pellets of each animal were collected into an empty 1.5 ml tube with a sterile toothpick. A new toothpick was used for each mouse. The tubes were closed immediately and placed into liquid nitrogen, finally transferred into a −80°C refrigerator for storage. The methods were performed in accordance with the National Guidelines for Experimental Animal Welfare (the Ministry of Science and Technology, People’s Republic of China, 2006). All experimental protocols were pre-approved by the Experimental Animal Ethics Committee of Harbin Medical University, China.
Total RNA Extraction
One ml of Trizol isolation reagent (Invitrogen, Carlsbad, CA) was added per 40–60 mg of frozen fecal samples and total RNAs was extracted according to the manufacturer’s protocol. The only difference from the usual RNA extraction protocol used in fecal samples was the need for repeated extraction using chloroform. The concentrations and purity of the total RNA extracted were measured by ultraviolet spectrophotometer NanoDro 2000 (Thermo Scientific, Waltham, USA). Purity requirements were considered to be met if the extracted RNA had an A260/A280 value between 1.80 and 2.00.
Quantitative Real-Time PCR Assays
For reverse transcription, 1 μg of total RNA was reverse transcribed using a ReverTra Ace® qPCR RT Kit (Toyobo, Osaka, Japan). The reverse transcription used a 20 μl reaction with random primers or gene-specific stem-loop primers, designed as described previously (Chen et al., 2005). All microRNA RT primer sequences are shown in Table S1. At the beginning of the experiment, the amplification efficiencies of the primers were first tested. According to the relative standard curve, the amplification efficiency of primers was about 93–116%. The cDNA was amplified by real-time PCR using SYBR® Green Realtime PCR Master Mix (Toyobo, Osaka, Japan) on the ABI 7500 fast Real Time PCR system (Applied Biosystems, Carlsbad, CA, USA). The primer sequences of the primers (Invitrogen, Shanghai, China) are listed in Table S1. The quantitative RT-PCR reaction mixture was run in a 20 μl volume reaction. The mixture consisted of 10 μl SYBR Green PCR Master Mix, 1.0 μl of each specific primer, 2.0 μl cDNA template, and 6.0 μl RNase-free water. The reactions performed were as follows: 1 min at 95°C for pre-denaturation, 40 cycles of 15 s at 95°C for denaturation, 15 s at 60°C for annealing, 45 s at 72°C for extension.
Data Preprocessing
Bestkeeper uses the cycle threshold (Ct) value directly from the qRT-PCR to analyze the stability of the gene, whereas the Ct value needs to be converted to a Q value before analysis by GeNormer and NormFinder. The formula for calculating the Q value is: Q = 2^-(Ct/sample-Ct/min). The calculated results were then imported into Excel according to the rules of GeNormer and NormFinder as they only identify a specific format (Vandesompele et al., 2002, Andersen et al., 2004).
MicroRNA Target Prediction and Functional Classification
The miRTarBase database (http://miRTarBase.mbc.nctu.edu.tw/) is a commonly used database for microRNA-target interactions. Using miRTarBase, all the validated mRNA targets of the differentially expressed intestinal microRNAs of control and HCC mice were listed. To create more accurate and reliable predicted results, only target genes validated by at least one experimental method were chosen. The DAVID Bioinformatic Resources database (https://david.ncifcrf.gov/) was then used to analyze the mRNA targets for enrichment of GO terms and KEGG pathways. A P value of <0.05 was used as the significance cut-off.
Statistical Analysis
All measurement data were presented as mean ± SEM and analyzed by GraphPad Prism 5. Differences between groups were analyzed by Student’s t-test, where P< 0.05 was considered as indicative of a statistically significant difference.
Results
A Schematic Diagram for the Experimental Process
In order to identify the suitable reference genes for intestinal microRNAs in fecal samples and to systematically analyze intestinal microRNAs expression in HCC, a schematic flow diagram of sample processing and microRNA analysis was first designed (Figure 1). Due to the novelty and scarcity of data in this area of study, 17 intestinal microRNAs, commonly expressed both in human and mouse fecal samples, were selected as candidate genes (Liu et al., 2016). Generally, GAPDH, U6 snRNA, 16S rRNA and 5S rRNA were also selected as they are usually used as reference genes.
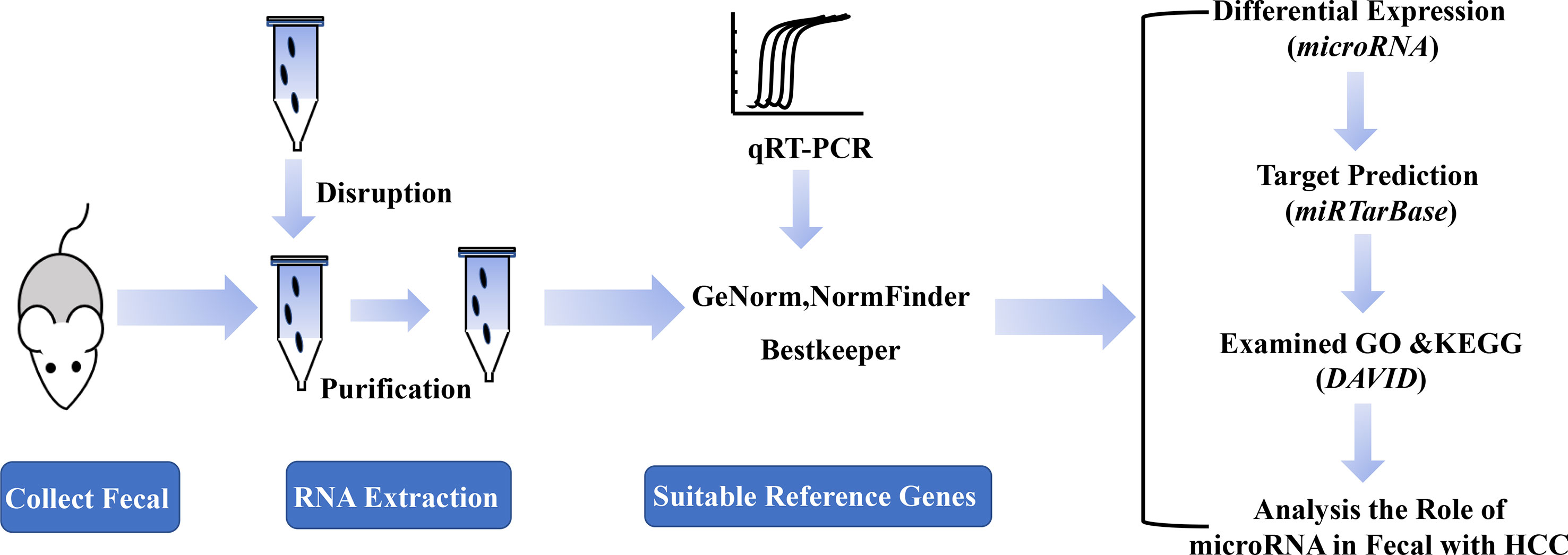
Figure 1 A schematic diagram of the experimental process.
Comparison of Ct Values Between Candidate Reference Genes
Even in different tissues or different growth stages of the same organ, there may be differences in the expression of the same gene. The Ct value of the gene is inversely proportional to the amount of target nucleic acid present. The lower the Ct value, the higher the amount of the gene present. According to its Ct value from real-time PCR, we analyzed the expression of each candidate reference gene (Figure 2A). The Ct values of 22 candidate reference genes ranged between 11 and 37. Large differences and a wide distribution range were found between the expressions of the candidate genes. The traditional reference genes 5S rRNA had the highest expression level, where the Ct value was between 11.67-14.94; the expressions of U6 snRNA, mir-192, mir-574, mir-194, let-7i, and mir-1224 were relatively high; Mir-15a and mir-155 showed a larger Ct value with low expressions. Additionally, the box plots illustrated that the expression range of each candidate reference gene varied significantly (Figures 2B, C). The Ct value of mir-155, mir-23a, mir-378, GAPDH, mir-574, let-7i and mir-1224 had fluctuating ranges that were comparatively small with a more concentrated distribution. Preliminary Ct values showed that the expression of mir-23a, GAPDH, mir-574, let-7i, and mir-1224 was higher and had few differences between the samples.
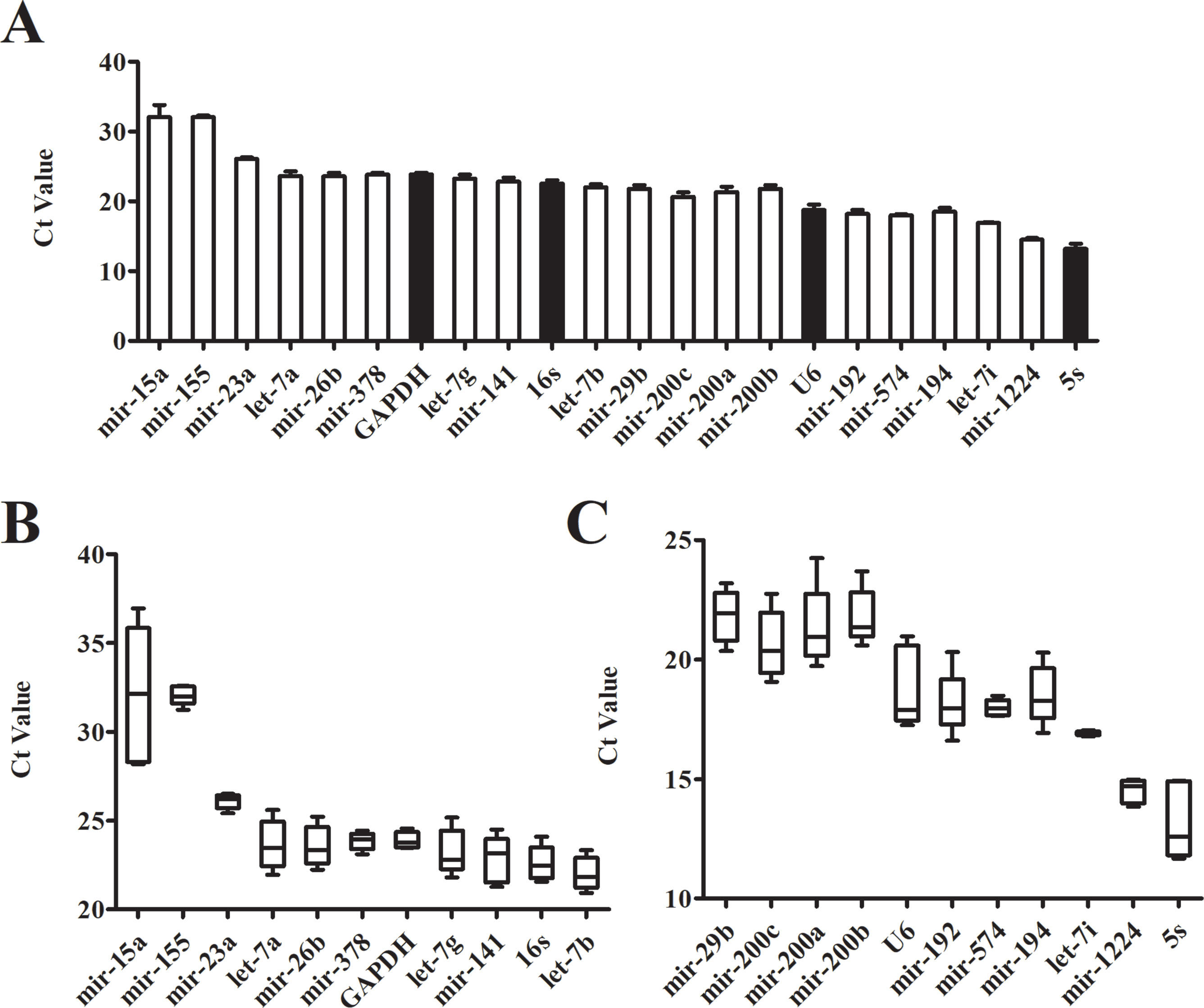
Figure 2 Expression level of candidate genes by real-time PCR. (A) Cycle threshold(Ct) values for the 22 candidate reference genes, n = 5. (B, C) Distribution of Ct values of candidate reference genes. Box representative the range of the Ct value distribution.
Expression Stability of Candidate Reference Genes
Three software packages (GeNorm, NormFinder, and Bestkeeper) were used for analyzing the average expression stabilities of the candidate reference genes. GeNorm and NormFinder software was specifically used for selecting reference genes. Before the analysis, it was necessary to convert the qRT-PCR result (Ct value) to a Q value. GeNorm software was written by Vandesompele in 2002 for screening reference genes where two or more reference genes can be chosen (Vandesompele et al., 2002). For the screening and selection of reference genes, an M value is usually displayed according to the output file of the GeNorm program. The default critical M value of the software is 1.5, where an M value of less than 1.5 indicates that the gene expression is relatively stable. The smaller the M value, the more stable the gene is. Through GeNorm software, we analyzed the M values of 22 candidate reference genes (Figure 3A). The data showed that the M value of mir-200b was the largest, whereas GAPDH and mir-23a were the smallest, indicating that GAPDH and mir-23a were the most stable genes. However, the traditional reference genes U6 snRNA and 5S rRNA remained rather unstable. Another program, NormFinder, was written in 2004 by Claus, and was used to screen the stability of qualitative reference genes (Andersen et al., 2004). The calculation principle of this program is similar to that of GeNorm and the stability value used is also the M value. The results showed that mir-23a, mir-378, and GAPDH were relatively more constant (Figure 3B). Mir-200b and mir-15a were discarded in the results since they had M values that were too large. These results were similar to those obtained from the GeNorm software. The Bestkeeper software was also used to evaluate the stability of the candidate genes (Pfaffl et al., 2004). The Ct values of 22 candidate reference genes were calculated, along with the standard deviation (SD) and coefficient of variation (CV). Our results showed that higher gene stability is related to a smaller SD and CV. The data showed that let-7i had the lowest SD and CV values of 0.06 and 0.36, respectively, followed by mir-23a, mir-574, and GAPDH (Figure 3C, Table S2). The results from Bestkeeper were somewhat similar to those from GeNorm and NormFinder.
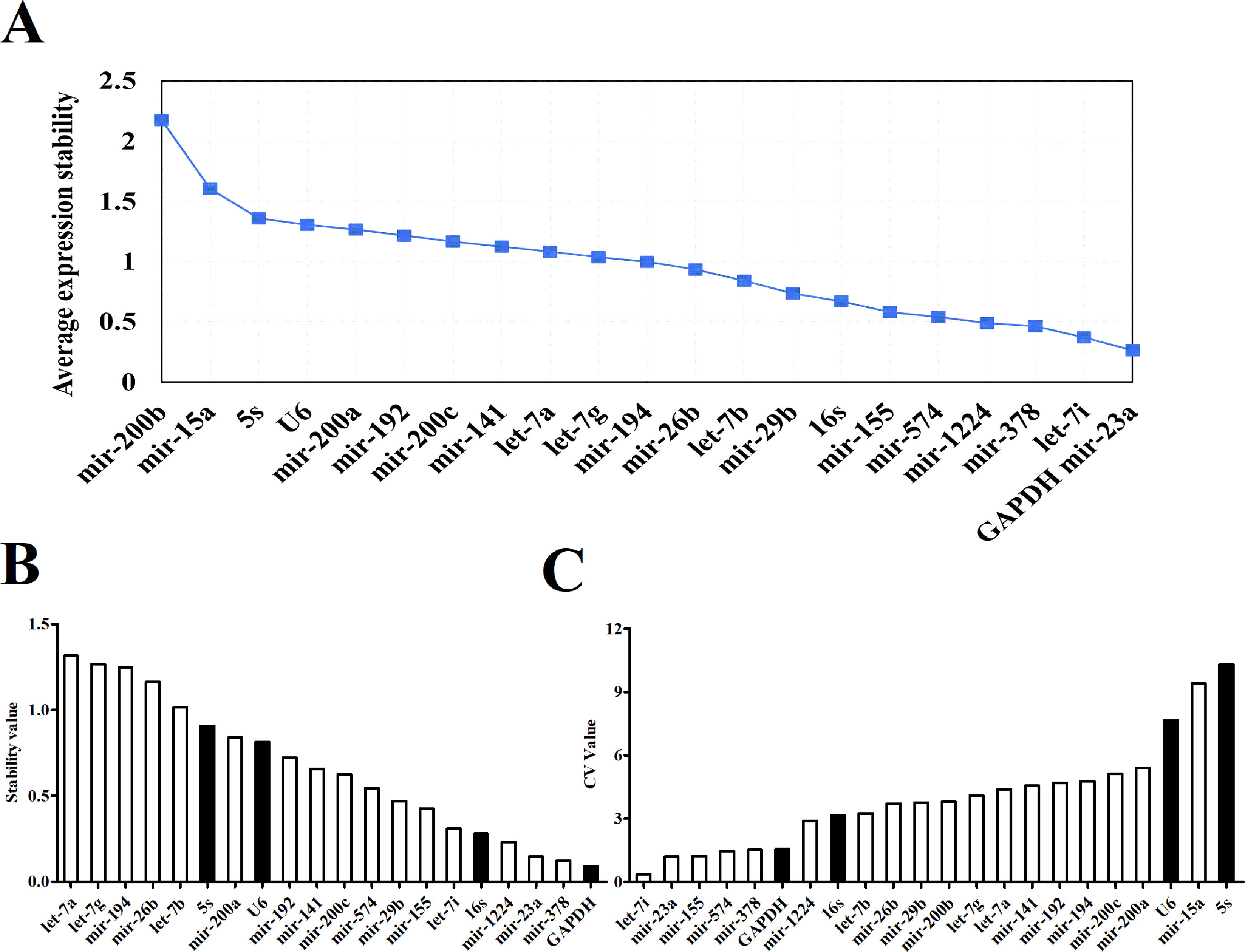
Figure 3 The expression stabilities of candidate reference genes analyzed by software. (A) Average expression M values analysis of candidate reference genes by geNorm software. (B) Average expression stability values analysis of candidate reference genes by NormFinder software. (C) Average expression CV values analysis of candidate reference genes by Bestkeeper software.
Comprehensive Evaluation of Candidate Reference Genes in the Three Software Programs
When comparing the results of the three software programs, we found that despite the similar results obtained, there were still differences among them. A geometric mean method was thus used to evaluate the stability of each candidate reference gene. The candidate genes’ stabilities were ranked in each of the software programs and then the rankings were averaged across the three software programs (Table 1). The top three genes were mir-23a, GAPDH and let-7i. These three genes were found to be the most suitable reference genes for intestinal microRNAs. Considering that the expression and distribution of let-7i was the most concentrated and the highest, it was selected as a reference gene for the subsequent analysis. To further identify the stability of let-7i as a reference gene, a validated experiment was carried out in different animal models. We collected the fecal samples from some disease models whose incidence is higher, such as HCC, Type 2 diabetes (T2DM) and Myocardial Infarction (MI). The Ct value dispersion of the let-7i was detected by qRT-PCR. The result showed that there was no significant difference between different disease animal models (Figure S1A). Additionally, the boxplot also indicated that the distribution of let-7i was concentrated within groups (Figure S1B). These assays demonstrated that let-7i displayed higher stability which could be used as a reference gene for microRNA qRT-PCR in fecal samples.
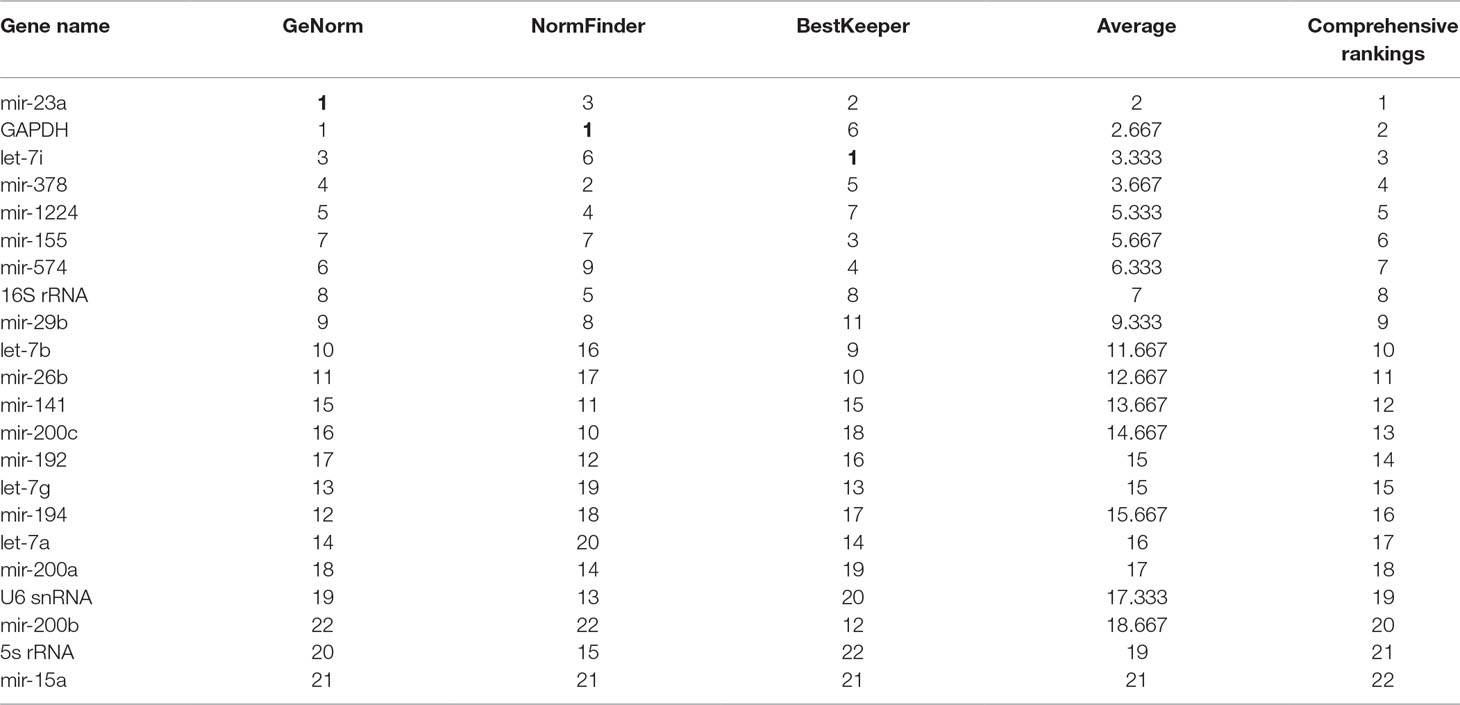
Table 1 The overall ranking of expression stability for candidate reference genes.
Altered Intestinal MicroRNAs Expression Profiles in HCC Mice
In order to explore the functional intestinal microRNAs in HCC, we detected the expression levels of microRNAs which had been observed both in humans and mice. According to the comprehensive evaluation of candidate reference genes in the three software programs and their expression levels (Ct value), we chose let-7i as an internal reference gene. We demonstrated the presence of 11 miRNAs that displayed a decrease (miR-155, miR-378, mir-23a, mir-26b, mir-29b, mir-194, mir-192, mir-15a, let-7a, mir-200a, and mir-200b), in the HCC group compared with the control group, while mir-200c, mir-1224, mir-574, mir-141, let-7b, and let-7g showed no significant differences between the groups (Figure 4).
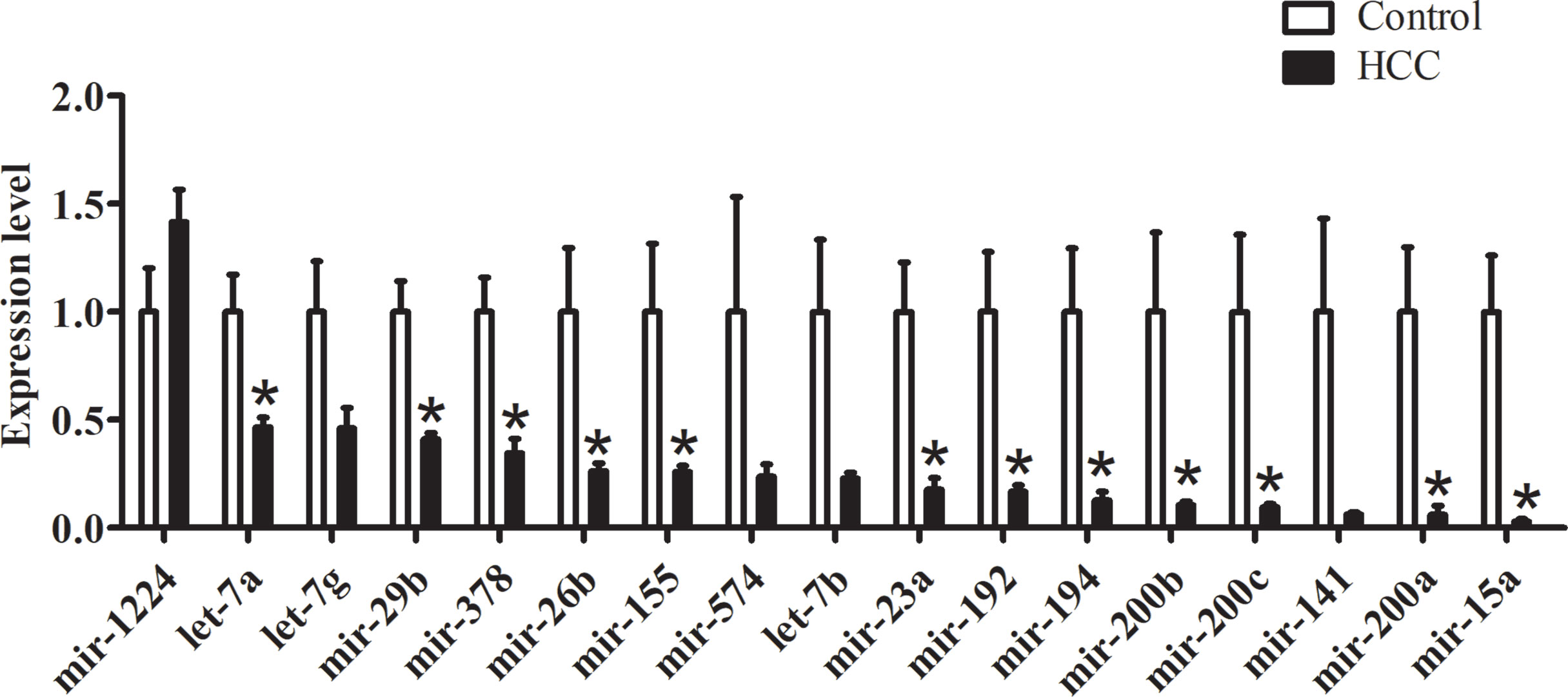
Figure 4 qRT-PCR validations of intestinal microRNA expression within HCC mice, n = 5, *P < 0.05.
Enrichment of Predicted mRNA Targets for Differentiated Intestinal MicroRNAs
To clarify downstream target gene networks for the intestinal microRNAs with HCC, we first listed all the potential mRNA targets of the differentiated intestinal microRNAs. These were predicted using miRTarBase. In order to improve the accuracy of the predicted mRNA targets, we only chose targets with strong evidence which had been supported by the reporter assay, western blotting or qPCR. The GO terms and KEGG pathways were then examined using the DAVID database. The results obtained from GO terms showed that the major processes in the pathology of HCC includes apoptosis, cell proliferation and the Wnt signaling pathway (Figure 5A). The KEGG pathway analysis explained that mRNA targets were mainly concentrated in the Jak-STAT pathway, apoptosis, and the p53 signaling pathway (Figure 5B). These results suggested that the differential intestinal microRNAs are likely to indicate the occurrence of HCC through predicted targets and pathways.
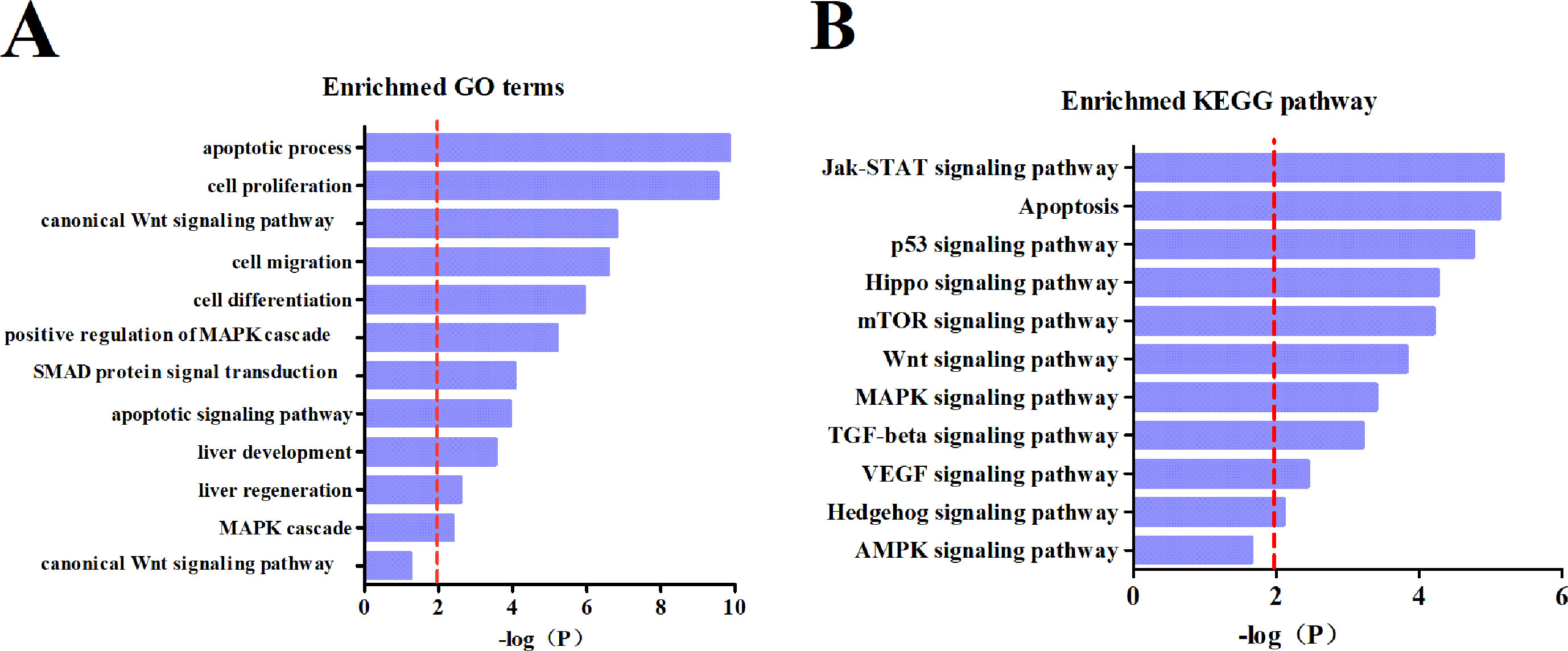
Figure 5 Subpathway enrichment analysis of microRNA target. (A) Enrichment of GO terms. (B) Enrichment of KEGG pathways.
Discussion
MicroRNAs are expected to become potential biomarkers for cancer diagnosis and new targets for disease treatments as aberrant expressions of microRNAs may be related to specific diseases. In recent years, quantitative RT-PCR assays have become the main method of studying the relative expression of microRNA. However, the quality of the results may be affected by some non-specific variables, such as the quality of RNA, efficiency of PCR amplification and the selection of reference genes (Yang et al., 2017). Appropriate normalization is a key part of quantitative gene expression analysis which is often ignored. The reference gene is also known as a housekeeping gene and is usually used to correct experimental errors when testing the gene expression levels. The ideal internal reference gene needs to satisfy the following conditions: the gene’s expression should be stable in different tissues, experimental conditions and growth stages (Lv et al., 2017, Molina et al., 2018). By using stable reference genes, the data in quantitative RT-PCR assays can be standardized. However, no reference genes have been found to be stable under all experimental conditions. Therefore, when studying specific gene expressions, it is necessary to select the most stable gene as the reference gene to correct the data and to ensure the accuracy of the results.
Interestingly, a new study suggests that fecal microRNAs could be identified as potential markers for intestinal malignancy (Ahmed et al., 2009). These fecal microRNAs exist in the gut lumen and feces and are called intestinal microRNAs. Some specific microRNAs that have been found in HCC cells and tissues may participate in tumor development, or could be a notable biological feature (Nelson and Weiss, 2008). The expression of microRNA in feces is therefore likely to become a new diagnostic marker in HCC. To achieve this potential, relatively accurate expression profiles of intestinal microRNA in HCC is a critical requirement. Because the reference gene of intestinal microRNA has not been identified yet, our first process step was to seek out a stable normalizer for the subsequent microRNA qRT-PCR experiments. Beginning with screening potentially stable reference genes, four traditional reference genes (5S rRNA, U6 snRNA, GAPDH and 16S rRNA) and 22 intestinal microRNAs co-expressed in humans and mice were used as candidate genes. Considering that normal samples are necessary in all experiments and conditions, we initiated our studies using control mice. First of all, the dispersion of the candidate genes’ Ct values was detected by qRT- PCR. There was a particularly large range of Ct values from 11 to 37 (Figure 2A). The boxplot indicated that Ct value distribution of let-7i was the most concentrated whereas mir-23a, mir-378 and GAPDH had slightly dispersed Ct values (Figures 2B, C). Preliminarily assays demonstrated that these four genes with little dispersion also displayed higher stability.
However, the Ct value distribution map could only roughly assess the stability of the candidate reference genes. In order to determine the stability of a reference gene more accurately, it is necessary to further evaluate it using reference gene analysis software. There are a number of software programs available for analyzing reference genes, including GeNorm, NormFinder and Bestkeeper. They are three different statistical models that are widely used to select the most appropriate reference gene (Shen et al., 2011). Both GeNorm and NormFinder evaluates the stability of the gene by measuring the M value. When the maximum expression of the gene’s stability is achieved, it will be accompanied by a low M value. Bestkeeper is a comprehensive evaluation software program which depends on the SD and CV. The principle of determination is that the smaller the SD and CV is, the better the stability of the reference gene is. When SD is greater than one, it directly indicates that the expression of the reference gene is unstable. In our study, the stability of reference genes was analyzed by all three of these software programs. According to the qRT-PCR results, NormFinder showed that GAPDH was the most stable reference gene with the smallest M value (Figure 3B). This result was consistent with GeNorm software, with only one difference, GeNorm had the two best candidate reference genes and the other one was mir-23a (Figure 3A). This is in accordance with previous studies where mir-23a was considered to be a novel microRNA normalizer in profiling studies of cervical tissues (Shen et al., 2011). Nevertheless, the results of Bestkeeper were slightly different from those of GeNorm and NormFinder. The results showed that let-7i was one of the best reference genes with the smallest SD and CV (Figure 3C, Table S1), and was thus considered the most stable gene by the Bestkeeper software. Since different software programs have different algorithms, some differences are expected between the results obtained from each program. Therefore, to obtain relatively stable reference genes, we comprehensively analyzed the results from the three software programs. As presented in Table 1, it can be seen that the stabilities of the expression levels of the 22 candidate reference genes from the highest to the lowest were mir-23a > GAPDH > let-7i > mir-378 > mir-1224 > mir-155 > mir-574 > 16S Rrna > mir-29b > let-7b > mir-26b > mir-141mir-200c > mir-192 > let-7g > mir-194 > let-7a > mir-200a > U6 snRNA > mir-200b > 5s Rrna > mir-15a. Commonly used conventional reference genes in our microRNA qRT-PCR assays, like 5S rRNA and U6 snRNA were particularly unstable. This phenomenon indicates that traditional reference genes are not applicable to all samples. Therefore, gene stability should always be evaluated and verified before use. Conclusively, mir-23a, GAPDH and let-7i are the optimal reference genes for intestinal microRNA in normal mouse feces. This information could provide recommendations for intestinal microRNA qRT-PCR studies with diseases statuses or other conditions.
MicroRNAs could modulate multiple target mRNA degradations or translations in the same pathway. Therefore, it has been proposed to identify miRNA regulatory profiles through predicted target genes and established miRNA-mRNA expression modules (Fu et al., 2012). In order to explore the possible mechanism of intestinal microRNAs regulating HCC, we constructed the intestinal miRNA-mRNA expression modules. Intestinal microRNA expression levels with HCC were first detected by quantitative RT-PCR. Let-7i was chosen as the reference gene since it showed a higher expression (Ct < 15) than mir-23a and GAPDH (Figure 2A). Results showed that there were differential intestinal microRNAs in the HCC group compared to the control group (Figure 4). Unexpectedly, mir-23a displayed a significant change in the HCC group, even though it has never been reported that mir-23a is a novel microRNA normalizer for relative quantification in human uterine cervical tissues. Indeed, this also clarified that the expression of reference genes is not always stable, and an appropriate reference gene under one experimental condition may not be suitable for all experimental conditions.
To better understand the biological functions of dysregulation intestinal microRNAs in HCC, a functional analysis of miRNA-mRNA was performed. Thus, an analysis of the differential microRNAs for GO terms and KEGG pathways were conducted. The targets of the intestinal microRNAs were significantly enriched in cell proliferation, migration and differentiation, which is closely related to the TGF-beta, Wnt and VEGF signaling pathway that is involved in tumorigenesis, metastasis and therapy (Figure 5A). miR-29b plays a protective role in cardiac remodeling by targeting the TGF-beta/Smad3 pathway (Zhang et al., 2014). mTOR is the key point of various important signaling pathways in cells, as well as being activated in a subset of HCCs (Fikret et al., 2004). The enriched KEGG pathway indicated that differential intestinal microRNA targets were likely to be involved in hepatocellular carcinogenesis through the mTOR pathway (Figure 5B). Previous research has clarified that mir-155 enhances mTOR activity in the progression of breast carcinomas (Martin et al., 2014). It is evident that mRNA targets of differential intestinal microRNAs were significantly enriched in the apoptotic process and apoptosis pathway (Figure 5), where apoptosis was indicated to be involved in liver cancer development. Low mir-15a expression has shown negative regulation by targeting Bcl-2 in human leukemia that is directly or indirectly involved in the biological process of apoptosis (Calin et al., 2008). The above evidence supports that intestinal miRNA could be a potential oncogene and antioncogene that is involved in HCC development.
In conclusion, the top three genes that were comprehensively ranked (mir-23a, GAPDH and let-7i), provide recommendations for intestinal microRNA qRT-PCR studies with disease statuses or other conditions. Additionally, differential intestinal microRNAs in fecal samples were predicted to be a potential oncogene or antioncogene involved in HCC development and may provide a novel strategy for diagnosing HCC and other related diseases.
Author Contributions
HW and YL conceptualized and designed the experiments in this study. CW, DL, and YY performed experimental procedures related to the study. MB, SM, and YJ proofread and discussed articles relating to this research. ZW provides great help for revise article. YB and BY provided fnancial support and reviewed experimental directions. All authors contributed to the generation of experimental data for this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to extend our gratitude to Howard L. Weiner and Shirong Liu from Brigham and Women’s Hospital, Harvard Medical School for providing help in intestinal microRNA isolation. Also, to Professor Feng Hai from Harbin Medical University for supplying fecal samples from HCC mice. We also thank Chiam Kai Wen Rachel for modifying the language used in this article. This project was supported by the National Natural Science Foundation of China (Grant No. 81674326 and Grant No. 81730012).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00687/full#supplementary-material
Figure S1 | The expression level of let-7i by real-time PCR. (A) Cycle threshold (Ct) values for let-7i, n=5. (B) Distribution of Ct values of let-7i. Box representative the range of the Ct value distribution.
References
Ahmed, F. E., Jeffries, C. D., Vos, P. W., Flake, G., Nuovo, G. J., Sinar, D. R., et al. (2009). Diagnostic MicroRNA markers for screening sporadic human colon cancer and active ulcerative colitis in stool and tissue. Cancer Genomics Proteomics 6 (5), 281.
Andersen, C. L., Jensen, J. L., Orntoft, T. F. (2004). Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64 (15), 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
Calin, G. A., Amelia, C., Muller, F., Manuela, F., Wojcik, S. E., Masayoshi, S., et al. (2008). MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. U. S. A. 105 (13), 5166–5171. doi: 10.1073/pnas.0800121105
Chen, C., Ridzon, D. A., Broomer, A. J., Zhou, Z., Lee, D. H., Nguyen, J. T., et al. (2005). Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33 (20), e179. doi: 10.1093/nar/gni178
Connerty, P., Bajan, S., Remenyi, J., Fuller-Pace, F. V., Hutvagner, G. (2016). The miRNA biogenesis factors, p72/DDX17 and KHSRP regulate the protein level of Ago2 in human cells. Biochim. Biophys. Acta 1859 (10), 1299–1305. doi: 10.1016/j.bbagrm.2016.07.013
Daud, I. I., Scott, M. E. (2008). Validation of reference genes in cervical cell samples from human papillomavirus-infected and -uninfected women for quantitative reverse transcription-PCR assays. Clin. Vaccine Immunol. 15 (9), 1369–1373. doi: 10.1128/CVI.00074-08
Davis-Dusenbery, B. N., Hata, A. (2010). MicroRNA in Cancer: the involvement of aberrant MicroRNA biogenesis regulatory pathways. Genes Cancer 1 (11), 1100–1114. doi: 10.1177/1947601910396213
Fikret, S., Rajesh, K., Onikepe, A., Jianzhou, W., Gloria, S., Michael, T. (2004). mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 10 (24), 8421–8425. doi: 10.1158/1078-0432.CCR-04-0941
Fu, J., Tang, W., Du, P., Wang, G., Chen, W., Li, J., et al. (2012). Identifying microRNA-mRNA regulatory network in colorectal cancer by a combination of expression profile and bioinformatics analysis. BMC Syst. Biol. 6, 68. doi: 10.1186/1752-0509-6-68
Galun, D., Basaric, D., Zuvela, M., Bulajic, P., Bogdanovic, A., Bidzic, N., et al. (2015). Hepatocellular carcinoma: from clinical practice to evidence-based treatment protocols. World J. Hepatol. 7 (20), 2274–2291. doi: 10.4254/wjh.v7.i20.2274
Ge, Y., Yan, X., Jin, Y., Yang, X., Yu, X., Zhou, L., et al. (2015). MiRNA-192 [corrected] and miRNA-204 Directly Suppress lncRNA HOTTIP and Interrupt GLS1-Mediated Glutaminolysis in Hepatocellular Carcinoma. PLoS Genet. 11 (12), e1005726. doi: 10.1371/journal.pgen.1005726
Gillan, V., Maitland, K., Laing, R., Gu, H., Marks, N. D., Winter, A. D., et al. (2017). Increased expression of a MicroRNA correlates with anthelmintic resistance in parasitic nematodes. Front. Cell. Infect. Microbiol. 7, 452. doi: 10.3389/fcimb.2017.00452
Kagiya, T. (2016). MicroRNAs: potential biomarkers and therapeutic targets for alveolar bone loss in periodontal disease. Int. J. Mol. Sci. 17 (8), 1317. doi: 10.3390/ijms17081317
Li, X., Yang, W., Ye, W., Jin, L., He, J., Lou, L. (2014). microRNAs: novel players in hepatitis C virus infection. Clin. Res. Hepatol. Gastroenterol. 38 (6), 664–675. doi: 10.1016/j.clinre.2014.04.008
Liu, S., da Cunha, A. P., Rezende, R. M., Cialic, R., Wei, Z., Bry, L., et al. (2016). The host shapes the gut microbiota via fecal MicroRNA. Cell Host Microbe 19 (1), 32–43. doi: 10.1016/j.chom.2015.12.005
Liu, S., Meng, C., Xu, G., Jian, H., Wang, F. (2018). Validation of reference genes for reverse transcription real-time quantitative PCR analysis in the deep-sea bacterium Shewanella psychrophila WP2. FEMS Microbiol. Lett. 365 (7), fny048. doi: 10.1093/femsle/fny048
Lv, Y., Zhao, S. G., Lu, G., Leung, C. K., Xiong, Z. Q., Su, X. W., et al. (2017). Identification of reference genes for qRT-PCR in granulosa cells of healthy women and polycystic ovarian syndrome patients. Sci. Rep. 7 (1), 6961. doi: 10.1038/s41598-017-07346-x
Martin, E. C., Rhodes, L. V., Elliott, S., Krebs, A. E., Nephew, K. P., Flemington, E. K., et al. (2014). microRNA regulation of mammalian target of rapamycin expression and activity controls estrogen receptor function and RAD001 sensitivity. Mol. Cancer 13 (1), 229. doi: 10.1186/1476-4598-13-229
Molina, C. E., Jacquet, E., Ponien, P., Munoz-Guijosa, C., Baczko, I., Maier, L. S., et al. (2018). Identification of optimal reference genes for transcriptomic analyses in normal and diseased human heart. Cardiovasc. Res. 114 (2), 247–258. doi: 10.1093/cvr/cvx182
Nelson, K. M., Weiss, G. J. (2008). MicroRNAs and cancer: past, present, and potential future. Mol. Cancer Ther. 7 (12), 3655–3660. doi: 10.1158/1535-7163.MCT-08-0586
Parodi, F., Carosio, R., Ragusa, M., Di Pietro, C., Maugeri, M., Barbagallo, D., et al. (2016). Epigenetic dysregulation in neuroblastoma: a tale of miRNAs and DNA methylation. Biochim. Biophys. Acta 1859 (12), 1502–1514. doi: 10.1016/j.bbagrm.2016.10.006
Petrelli, F., Coinu, A., Borgonovo, K., Cabiddu, M., Ghilardi, M., Lonati, V., et al. (2014). Oxaliplatin-based chemotherapy: a new option in advanced hepatocellular carcinoma. A systematic review and pooled analysis. Clin. Oncol. 26 (8), 488–496. doi: 10.1016/j.clon.2014.04.031
Pfaffl, M. W., Tichopad, A., Prgomet, C., Neuvians, T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: bestKeeper – excel-based tool using pair-wise correlations. Biotechnol. Lett. 26 (6), 509–515. doi: 10.1023/B:BILE.0000019559.84305.47
Ranjha, R., Paul, J. (2013). Micro-RNAs in inflammatory diseases and as a link between inflammation and cancer. Inflamm. Res. 62 (4), 343–355. doi: 10.1007/s00011-013-0600-9
Shen, Y., Li, Y., Ye, F., Wang, F., Wan, X., Lu, W., et al. (2011). Identification of miR-23a as a novel microRNA normalizer for relative quantification in human uterine cervical tissues. Exp. Mol. Med. 43 (6), 358–366. doi: 10.3858/emm.2011.43.6.039
Su, H., Yang, J. R., Xu, T., Huang, J., Xu, L., Yuan, Y., et al. (2009). MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 69 (3), 1135–1142. doi: 10.1158/0008-5472.CAN-08-2886
Torre, L. A., Bray, F., Siegel, R. L., Ferlay, J., Lortet-Tieulent, J., Jemal, A. (2015). Global cancer statistic, 2012. CA Cancer J. Clin. 65 (2), 87–108. doi: 10.3322/caac.21262
Vandesompele, J., Preter, K. D., Pattyn, F., Poppe, B., Roy, N. V., Paepe, A. D., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3 (7), research0034.1–research0034.11. doi: 10.1186/gb-2002-3-7-research0034
Wang, C. M., Wang, Y., Fan, C. G., Xu, F. F., Sun, W. S., Liu, Y. G., et al. (2011). miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 411 (3), 586–592. doi: 10.1016/j.bbrc.2011.06.191
Xi, Y., Nakajima, G., Gavin, E., Morris, C. G., Kudo, K., Hayashi, K., et al. (2007). Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 13 (10), 1668–1674. doi: 10.1261/rna.642907
Xiao, S., Wang, R., Wu, X., Liu, W., Ma, S. (2018). The Long Noncoding RNA TP73-AS1 Interacted with miR-124 to modulate glioma growth by targeting inhibitor of apoptosis-stimulating protein of p53. DNA Cell Biol. 37 (2), 117–125. doi: 10.1089/dna.2017.3941
Yang, Y., Li, Z., Cao, J., Li, Y., Li, H., Yang, Q., et al. (2017). Identification and evaluation of suitable reference genes for normalization of MicroRNA expression in helicoverpa armigera (Lepidoptera: Noctuidae) using quantitative real-time PCR. J. Insect Sci. 17 (2), 1–10. doi: 10.1093/jisesa/iex007
You, S., Fulga, T. A., Van Vactor, D., Jackson, F. R. (2018). Regulation of circadian behavior by astroglial MicroRNAs in drosophila. Genetics 208 (3), 1195–1207. doi: 10.1534/genetics.117.300342
Zhang, Y., Huang, X. R., Wei, L. H., Chung, A. C., Yu, C. M., Lan, H. Y. (2014). miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 22 (5), 974–985. doi: 10.1038/mt.2014.25
Keywords: intestinal microRNAs, quantitative real-time PCR assays, reference genes, hepatocellular carcinoma, feces
Citation: Wang H, Lv Y, Wang C, Leng D, Yan Y, Blessing Fasae M, Madiha Zahra S, Jiang Y, Wang Z, Yang B and Bai Y (2019) Systematic Analysis of Intestinal MicroRNAs Expression in HCC: Identification of Suitable Reference Genes in Fecal Samples. Front. Genet. 10:687. doi: 10.3389/fgene.2019.00687
Received: 03 September 2018; Accepted: 01 July 2019;
Published: 13 August 2019.
Edited by:
Thomas S. Wingo, Emory University, United StatesReviewed by:
Chenghua Li, Ningbo University, ChinaVenugopal Thayanithy, Medical School, University of Minnesota, United States
Copyright © 2019 Wang, Lv, Wang, Leng, Yan, Blessing Fasae, Madiha Zahra, Jiang, Wang, Yang and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baofeng Yang, eWFuZ2JmQGVtcy5ocmJtdS5lZHUuY24=; Yunlong Bai, YmFpeXVubG9uZ0BlbXMuaHJibXUuZWR1LmNu