Abstract
As reliable, efficient genome sequencing becomes ubiquitous, the need for similarly reliable and efficient variant calling becomes increasingly important. The Genome Analysis Toolkit (GATK), maintained by the Broad Institute, is currently the widely accepted standard for variant calling software. However, alternative solutions may provide faster variant calling without sacrificing accuracy. One such alternative is Sentieon DNASeq, a toolkit analogous to GATK but built on a highly optimized backend. We conducted an independent evaluation of the DNASeq single-sample variant calling pipeline in comparison to that of GATK. Our results support the near-identical accuracy of the two software packages, showcase optimal scalability and great speed from Sentieon, and describe computational performance considerations for the deployment of DNASeq.
Introduction
Advancements in sequencing technology (Metzker, 2010; Goodwin et al., 2016) have resulted in an explosion of Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES) (Stephens et al., 2015). As sequencing machines become faster and cheaper (Illumina, 2018b), analysis must speed up as well. It is becoming less and less acceptable for genomic variant calling to take many hours, let alone days, on a single WGS sample. This is important for both medical applications where rapid genomic analysis could mean accurate diagnosis in time to save a patient’s life, as well as in agriculture where massive genotyping efforts are used to refine breeding programs via genomic prediction. Yet the standard software package for variant calling, GATK (Genome Analysis Toolkit; Van der Auwera et al., 2013), still requires many hours to analyze a single WGS sample, even on multiple nodes and after optimization (Heldenbrand et al., 2018).
Since 2014, Sentieon’s DNASeq pipeline (Sentieon, 2018) has been promoted as an ultrafast alternative to GATK (Weber et al., 2016; Freed et al., 2017). Other ultrafast options, such as Genalice (Plüss et al., 2017) and Isaac (Raczy et al., 2013), do not adhere to the original and trusted algorithms of GATK. Additionally, Genalice replaces the commonly used file formats BAM and VCF with the proprietary GAR, which takes hours to convert back to BAM when needed. However, Sentieon claims to follow the GATK Best Practices (DePristo et al., 2011; Van der Auwera et al., 2013) through reimplementation of GATK’s algorithms in C, C++, Python, and ASSEMBLY. It thus constitutes a highly optimized rewrite of the Java-based GATK, and also includes an optimized version of the popular BWA-MEM aligner (Li, 2013) prompting us to choose it for evaluation. Unlike the open-source GATK, however, DNASeq requires a license for use.
Here we present the results of unbiased benchmarking of DNASeq by an independent academic group. Our investigations focused on the broadly applicable and clinically useful case of single-sample variant calling. Speed, accuracy, scalability, and resource utilization were measured. Evaluations of DNASeq’s speed and accuracy are presented in comparison to GATK3.8 and GATK4 (single-threaded), the two most recent versions of GATK at the time of writing. The results suggest that Sentieon offers superior compute speed to GATK without loss of accuracy.
Materials and Methods
Experimental Setup
Software Versions
All DNASeq tests were run using Sentieon version 201711.02, except for one BWA MEM performance benchmark run on version 201711.03. A trial license was provided by Sentieon. GATK3.8 was downloaded from the Broad Institute’s software download page (Broad Institute, 2018), build GATK-3.8-0-ge9d806836. Picard version 2.17.4 and GATK 4.0.1.2 were downloaded from GitHub as pre-compiled jar files. For alignment comparisons, we used BWA-MEM v0.7.16, samtools view from Samtools v1.5, and Novosort from Novocraft v3.04.06.
Tools Benchmarked
We benchmarked “best practices” single-sample germline variant calling pipelines built with GATK3.8, GATK4.0, and Sentieon DNASeq (Table 1; see Tool Comparison Overview: Sentieon vs GATK for exceptions). Both GATK and DNASeq haplotyping steps offer a ploidy option, making them equally applicable to non-human sequencing data. Alignment was performed by BWA-MEM or its optimized alternative from Sentieon (marked with †). A sample benchmarking script can be found at https://github.com/ncsa/Sentieon_DNASeq_Benchmarking.
Table 1
| Pipeline Step | Sentieon | GATK 3.8/4.0 |
|---|---|---|
| Alignment | BWA MEM† | BWA MEM |
| Sorting | Sort utility | NovoSort |
| Deduplication | LocusCollector and Dedup | MarkDuplicates |
| Realignment | Realigner | Not recommended |
| Quality score recalibration | QualCal | BaseRecalibrator |
| Apply new quality scores | Invoked during Haplotyper | PrintReads (3.8)/ApplyBQSR (4.0) |
| Variant callling | Haplotyper | HaplotypeCaller |
Sentieon DNASeq vs. GATK pipelines.
Sentieon’s optimized BWA MEM marked with †.
Data
Three datasets were used in testing: 1) A dataset corresponding to whole genome sequencing (WGS) of NA12878 (Zook et al., 2014; Zook et al., 2018) to ∼20X depth was downloaded from Illumina BaseSpace on Dec 16, 2016. The NA12878 Illumina Platinum variant calls were used as the truth set to assess variant calling accuracy on these reads. 2) A dataset corresponding to WGS of NA24694 (Church, 2005) was downloaded on January 19, 2018 from Genome in a Bottle (JIMB, 2018). The NA24694 data arrived in multiple files, which we combined into several subsets to mimic sequencing depths of 25X, 50X, 75X, and 100X, as recommended in the ftp data download README for the NA24694 dataset. 3) A small synthetic dataset simulating WGS on chromosomes 20-22 was created using NEAT-genReads (Stephens et al., 2016; Stephens, 2018), a synthetic reads generator. The software introduced random mutations into the hg38 reference, and the simulated reads were produced from that mutated reference. The mutations were recorded in a “Golden VCF,” which was used as the truth set when assessing variant calling accuracy on these synthetic data. The command used to generate synthetic reads and the Golden VCF can be found on the github repository mentioned in the Data Availability section. The October 2017 GATK bundle was used for the human reference (hg38), dbSNP (build 138), and the Mills and 1000G gold standard indels.
Hardware
All tests were conducted on Skylake Xeon Gold 6148 processors with 40 cores, 2.40 GHz. Each node had 192 GB and 2,666 MHz RAM. The nodes were stateless, connected to a network-attached IBM GPFS ver. 4.2.1 with custom metadata acceleration. The cluster used EDR InfiniBand with 100 Gb/sec bandwidth, 100 ns latency. Nodes ran Red Hat Enterprise Linux 6.9. Each test was run on a single node. We ran two to three replicates of most tests; the differences across the replicates were negligible, and we are confident that the walltime was not affected by other activity on the cluster.
Tool Comparison Overview: Sentieon vs GATK
Sentieon DNASeq tools largely mirror those of GATK (Table 1), and in both pipelines the steps can be individually skipped or replaced by other tools. Although GATK no longer recommends realignment, we included Sentieon’s Realigner tool in all runs except for those intended to compare directly to GATK because realigning can convey benefits in a Sentieon pipeline. While GATK creates a separate recalibrated BAM by default using PrintReads (GATK3.8) or ApplyBQSR (GATK4.0), Sentieon’s default is to apply BQSR calculations “on the fly” during the Haplotyper step. Although users do have the option to generate a recalibrated BAM with DNASeq’s ReadWriter algorithm, this step is extraneous to Sentieon’s recalibration process. Therefore, there is no equivalent to GATK’s PrintReads/ApplyBQSR tool in our DNASeq pipelines.
Results
Thread-Level Scalability
We tested the single-node scalability of Sentieon DNASeq by running the same pipeline with 4, 8, 16, 24, and 40 (max) threads. Optimal scalability was calculated by projecting the walltime decrease proportionately to the increase in thread count, using the first walltime measurement at 4 threads as the starting point. Each of the constituent tools appeared to scale equally well (data not shown), and the entire DNASeq pipeline scaled near-optimally up to our max of 40 threads/node (Figure 1). The pipeline completed in ∼16 h when running across 4 threads, and ∼2 h when running across 40.
Figure 1
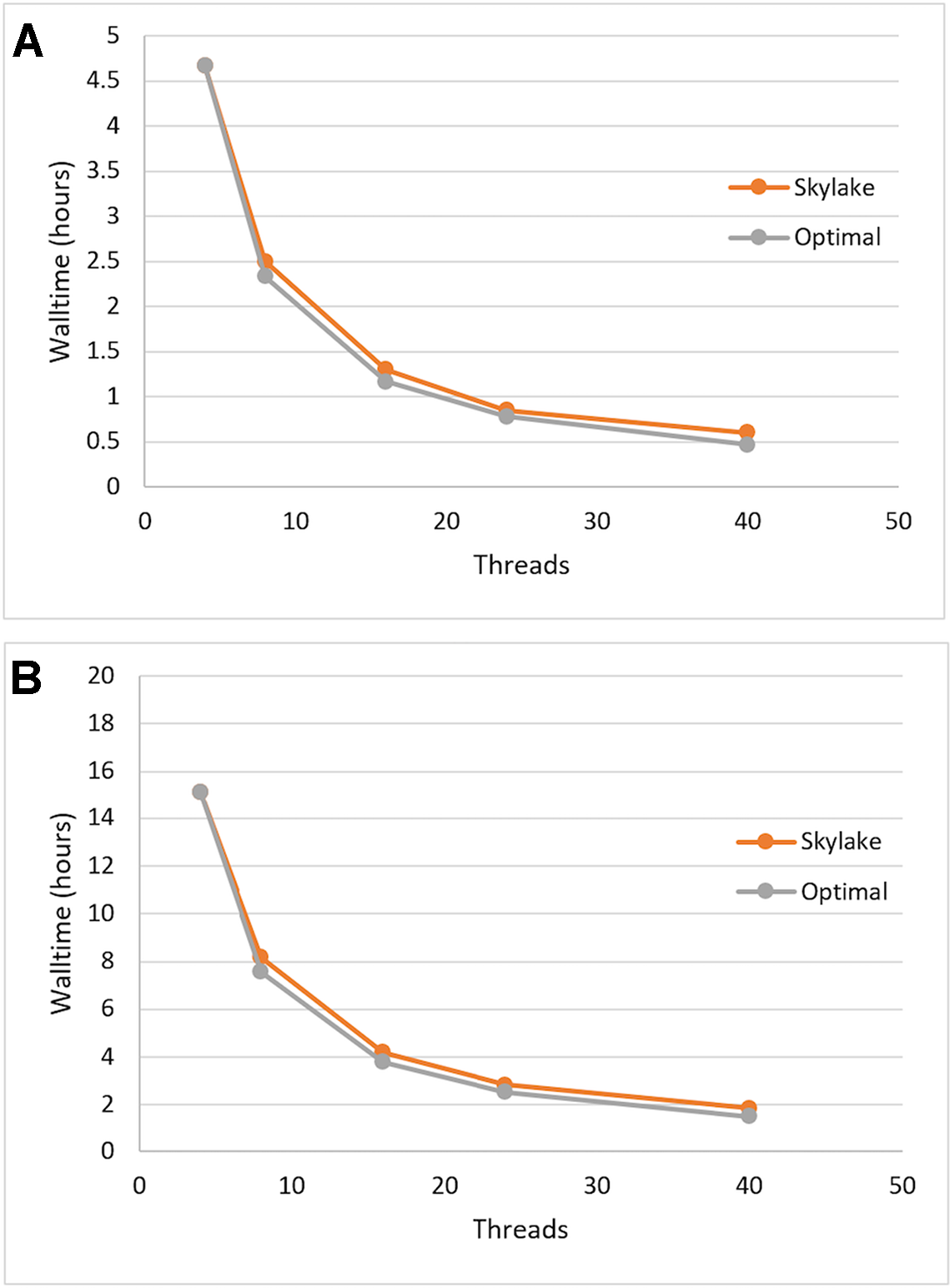
Sentieon DNASeq pipeline: demonstrated scaling across threads on Skylake architecture vs. optimal (linear) scaling. Sample: NA12878, WGS, 20X. Data points reflect averages over two replicates, highlighting (A) post-alignment steps only and (B) the full pipeline including alignment.
Effect of Sequencing Depth on Performance
We investigated the impact of sequencing depth on performance by running our Sentieon DNASeq pipeline on NA24694 WGS data subsets representing 25X, 50X, 75X, and 100X coverage depth. We ran each subset on the maximum available cores/node (40) to minimize runtime. All individual tools in the pipeline, including alignment, appeared to scale equally well. (Figure 2, top). The pipeline as a whole showed a small drop-off in performance between 25X and 50X: walltime increased more than twofold on 50X depth, compared to the 25X dataset, which is half the size (Figure 2, bottom). However, it scaled near-linearly at all depths beyond 25X. Linear scaling was calculated using the median walltime measurement by projecting that an n-fold increase or decrease in depth would lead to an equivalent n-fold increase or decrease in runtime. We do not believe this scaling pattern is due to any specific feature of alignment, as it also holds for the post-alignment portion by itself (data not shown). Considering the I/O-intensive nature of the DNASeq workflow (described below), it is more likely that the 25X data were not large enough to impact performance on our filesystem, resulting in lower run time. Nonetheless, the overall walltime performance remains outstanding: on data sequenced at 100X depth, the pipeline completed in fewer than 13 h. We expect these results to apply, qualitatively if not numerically, to other human data sequenced using Illumina technology. In other species, such as polyploid plants, which have greater content of highly repetitive genomic regions, the scalability pattern might be different due to the extra work required for read realignment, and higher content of multiply mapped reads. An in-depth investigation of these differences was deemed out of scope for this manuscript.
Figure 2
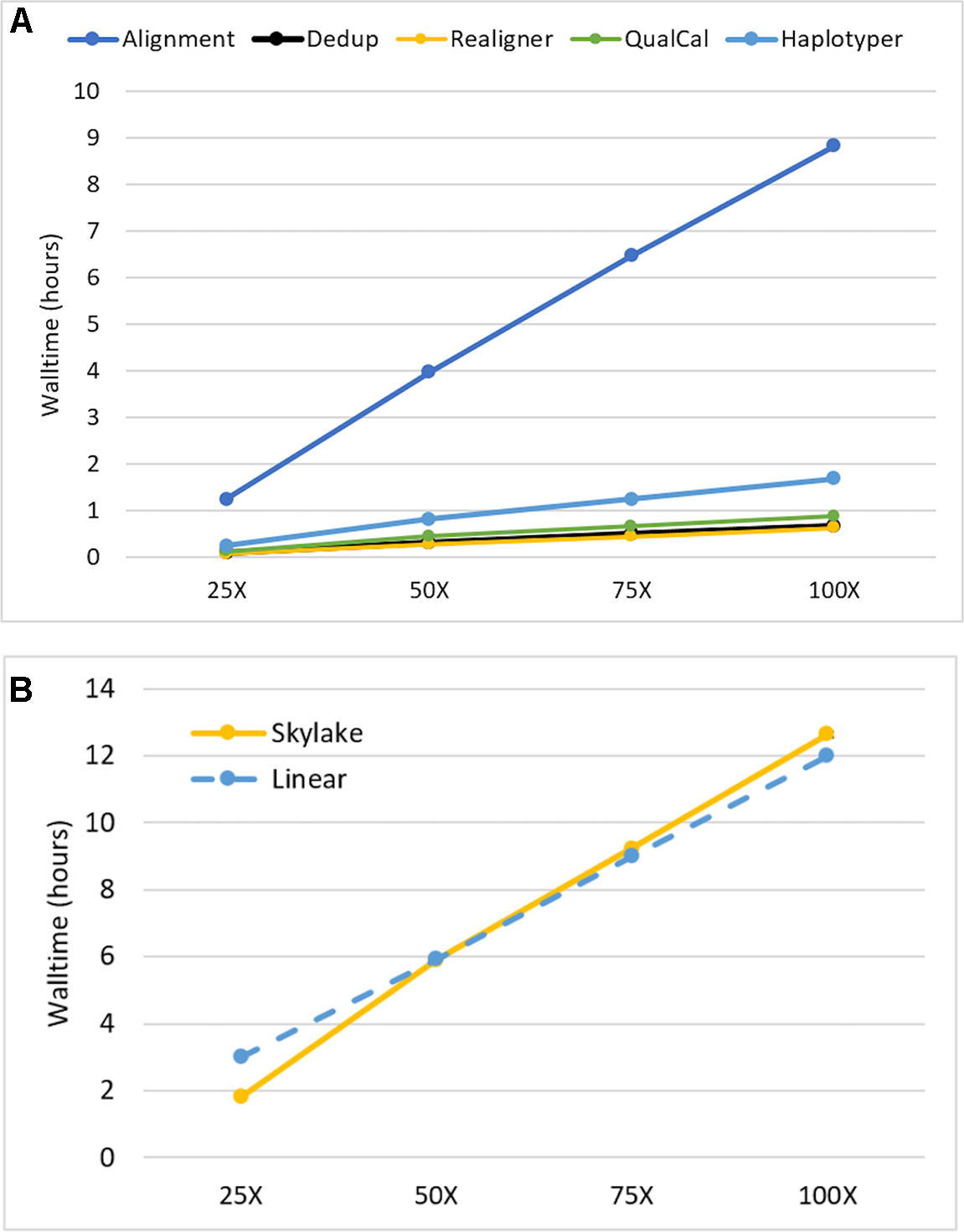
Sentieon DNASeq scalability as a function of sequencing coverage depth (A) by tool and (B) across the entire pipeline. Sample: NA24694, WGS, 25X-100X. Datapoints reflect averages over two replicates. Error bars are included in (B) but are too small to be visible.
Computational Performance Relative to GATK
To understand the extent of the performance improvements introduced by Sentieon, we compared the runtime of GATK vs. DNASeq on NA12878 WGS data. The computational performance of GATK3.8 and GATK4.0 have been reviewed in detail by Heldenbrand et al. (2018). We ran each of the three pipelines with their respective default settings and maximum thread count (40) to establish a “baseline.” Then we ran each pipeline with “optimal” settings: for DNASeq, 40 threads across the pipeline (same as baseline), and for GATK3.8 and GATK4, the optimized parameters recommended in Heldenbrand et al. (2018) (Table 2, reproduced with permission). No data-level parallelization was applied, and each test was performed on a single node. This choice was driven by the fact that both GATK 3.8 and DNASeq do not support cross-node parallelization via MPI, and the GATK4 Spark-enabled cross-node parallelization was not yet mature enough for testing at the time we ran our experiments.
Table 2
| Tool name | GATK3.8 | GATK4.0 | |||
|---|---|---|---|---|---|
| PGC | Tools threads | PGC | Async | AVX threads | |
| MarkDuplicates | 2 threads | 1 | 2 threads | N/A | N/A |
| BaseRecalibrator | 20 threads | −nct 40 | 20 threads | Yes for Samtools, No for Tribble | N/A |
| ApplyBQSR | Off | −nct 3 | Off | N/A | |
| HaplotypeCaller | Off | −nt 1 – nct 39 | Off | 8 | |
Summary of optimized parameter values for GATK3.8 and GATK4.0 in reference to parallel garbage collection (PGC) threads, tool threads, async I/O, and AVX threads.
Reproduced from Heldenbrand et al. (2018) with permission.
Our walltime comparison excludes BWA MEM (not part of GATK; see Comparison of Sentieon BWA to Traditional BWA) and realignment (as the GATK team recommends against it). In this configuration, DNASeq completed in less than half an hour on NA12878 WGS 20X, while the GATK pipelines took between 15 and 25 h on the same dataset (Table 3).
Table 3
| Pipeline | Walltime (h) | Sentieon Speedup |
|---|---|---|
| DNASeq | .49 | – |
| GATK3.8 Baseline | 21.7 | x44 |
| GATK3.8 Optimized | 15.3 | x31 |
| GATK4.0 Baseline | 24.9 | x51 |
| GATK4.0 Optimized | 20.7 | x42 |
Speed comparison: Sentieon DNASeq vs. GATK.
Speedup factor indicates n-fold speedup represented by DNASeq walltime as compared to GATK walltime. Sample: NA12878, WGS, 20X.
Comparison of Sentieon BWA to Traditional BWA
Sentieon DNASeq includes an optimized version of BWA MEM and a proprietary utility for sorting an aligned SAM into a BAM. We juxtaposed Sentieon’s BWA MEM and sort utility with a typical alignment and sorting pipeline consisting of traditional BWA MEM (Li, 2013), samtools view (Li et al., 2009), and NovoSort (Novocraft Technologies, 2014). We compared performance of the two by running both with 40 threads on NA12878 WGS data (no piping). The Sentieon version was ∼28% faster: 1.25 h vs. 1.73 h. This speed-up results from optimization of the klib library, at the cost of almost doubling the memory used by BWA: 22.45 ± 1.58 GB vs 12.13 ± 0.56 GB, measured as the resident set size (no swapping to disk was observed).
Version 201711.03
A new version of Sentieon (201711.03) was released as we completed our testing, featuring a non-trivial speed-up of BWA MEM derived from a complete rewrite of the traditional BWA MEM code. This new version completed in 0.95 h, 25% faster than 201711.02 and 45% faster than “BWA MEM → samtools view → NovoSort.”
Variant Calling Accuracy
The accuracy of Sentieon’s DNASeq pipeline has been demonstrated in several FDA and DREAM Challenges (U.S. Food and Drug Administration, 2016a; U.S. Food and Drug Administration, 2016b; Sage Bionetworks, 2016). As the algorithms underlying Sentieon are nearly identical to those underlying GATK, the two can be expected to produce similar results. Nevertheless, we ran a cursory comparison of Sentieon’s DNASeq accuracy against GATK4. NA12878 and the synthetic chr 20-22 dataset were run through both pipelines. The resultant VCFs were compared to their respective truth sets and to each other, using the vcf-compare tool from the NEAT package (Stephens, 2018). The comparison was limited to the Illumina Platinum confident regions (Illumina, 2018a). In comparing Sentieon and GATK4 directly, we treated the output from GATK4 as the truth set because DNASeq is based on GATK algorithms. The command used to run comparisons is available on github.
The output of vcf-compare provides counts for true positives (TP), false positives (FP), and false negatives (FN). To account for both precision (p) and recall (r), we calculated F-scores based on these values using the formula , where and . Precision and recall values were similar for each comparison.
Sentieon and GATK4 were highly concordant with each other (Table 4) on both datasets, as expected. Using realignment in DNASeq did not meaningfully affect concordance with GATK4; the difference could become more significant on datasets of poorer quality. Both toolkits had high rates of variant detection relative to the truth sets. Significantly, GATK and Sentieon demonstrated effectively identical detection rates on the Illumina Platinum data.
Table 4
| Dataset | Synthetic WGS, chr 20-22 | NA12878 |
|---|---|---|
| Sentieon vs. GATK4 | 0.99 | 0.997 w/ Realigner |
| 0.997 w/o Realigner | ||
| Truth sets | Golden VCF | Illumina Platinum variant calls |
| Sentieon vs. Truth set | 0.96 | 0.96 |
| GATK4 vs. Truth set | 0.95 | 0.96 |
Variant detection accuracy in Sentieon DNASeq and GATK4: F1 scores.
Computer Resource Utilization
Variant calling workflows are notorious for high RAM utilization, high rates of disk I/O, and inefficient data access patterns, all of which can lead to performance issues (Banerjee et al., 2016; Kathiresan et al., 2017). Therefore, we investigated the patterns of compute resource utilization for DNASeq in addition to its speed and accuracy. We recorded CPU load, memory utilization and I/O patterns for each tool while running the DNASeq pipeline on the NA12878 dataset. Our in-house profiling utility memprof (Kindratenko, 2018) accesses/proc/PID for each process it monitors.
CPU Utilization
The resulting profile of DNASeq (Figure 3) shows near-maximum core utilization by all tools except LocusCollector (one of the algorithms used for deduplication). This suggests that the tools are largely CPU-bound, explaining the strong thread-level scalability described above.
Figure 3

CPU utilization, memory usage and I/O of the Sentieon DNASeq tools, excluding BWA MEM. The pipeline steps are labeled in the middle panel, following the –algo options used in the script. CPU utilization in the top panel corresponds to the sum total across the 40 cores on the node. RAM utilization in the middle panel was measured as resident set size (VmRSS) and total RAM reserved for computation (VmSize). I/O rates in the bottom panel were measured in reads and writes per second. Sample: NA12878, WGS, 20X.
RAM Utilization
Haplotyper uses the most memory in the pipeline (up to 6 GB), as expected for the local reassembly subroutine. High RAM utilization toward the end of the process is likely due to processing of the difficult HG38 decoy regions. Nonetheless, DNASeq RAM utilization across all tools is lower than some previous GATK benchmarks (Chapman, 2014) have shown.
I/O Rates
Deduplication and realignment show very active and similar I/O patterns, reaching high rates for both reads from the input BAM and writes to the output BAM. Many intermediary BAM files are created during these steps, resulting in high data-level parallelization at the cost of high I/O (Figure 3, inset in the bottom panel). This could introduce a filesystem bottleneck when analyzing large numbers of samples (hundreds) simultaneously on a cluster, but can be countered by using local disk or SSDs instead of network storage. In contrast, no new BAMs are created during QualCal (BQSR), as it only calculates the required modification of the quality scores and the actual recalibration is applied during the variant calling stage. Thus the rate of writes/sec is very low for QualCal. We also include the near-identical profile for the optional command that applies the recalibration to calculate the post calibration data table (Figure 3, step Recal Data Table). For comparison, GATK’s I/O performance was extensively measured and reviewed by Banerjee et al. (2016).
Discussion
The tests we conducted were intended to a) benchmark Sentieon’s DNASeq speed and scalability and b) compare DNASeq to GATK as an alternative option for germline variant calling. Our results show that DNASeq is fast: for a WGS sample sequenced to approximately 20X depth, the DNASeq pipeline processes from FASTQ to VCF in under 2 h, and from aligned sorted BAM to VCF in less than half an hour. This opens up possibilities for point-of-care patient analysis in the clinic, massive reanalysis of legacy data and high throughput variant calling in livestock and crop breeding programs.
Our scaling tests show that DNASeq scales optimally across threads, which suggests that it will run efficiently on a variety of processors. Our tests were limited to single-node, multi-threaded scaling, although both single-threaded and multi-node tests may be of future interest. DNASeq also scales well as sequencing depth increases, a useful characteristic as deeper sequencing becomes more common. There does appear to be a small slowdown between 25X and 50X, however, which suggests that DNASeq may be most efficient on less deeply sequenced samples.
When compared to GATK, we found Sentieon DNASeq to be equally accurate. Sentieon uses the same algorithms as GATK and reliably puts out new releases in response to GATK version updates. Comparisons to Illumina platinum calls for NA12878 yielded equivalent results, suggesting no meaningful differences in reliability. It is possible that more discrepancies may be revealed when measuring with different metrics.
In terms of runtime, GATK post-alignment processing can take up to a day. Even with parameter optimizations in place, we found GATK3.8 took 15 h, while GATK4 (single-threaded) took 20 h. DNASeq is able to complete the same work over 30x faster, representing a time savings of approximately 97%. This increase in speed doesn’t appear to rely on increased resource consumption: While Sentieon’s BWA MEM implementation did utilize twice as much RAM as traditional BWA MEM, we found that RAM utilization among the other DNASeq tools did not exceed previous results recorded for GATK. Overall, our results suggest that Sentieon’s DNASeq pipeline does present a viable alternative to GATK, offering significantly better speed than GATK3.8 and GATK4 without sacrificing accuracy.
Funding
Major funding was provided by the Mayo Clinic Center for Individualized Medicine and the Todd and Karen Wanek Program for Hypoplastic Left Heart Syndrome.
Statements
Author’s note
The authors of this paper did not receive any benefits or compensation, financial or otherwise, from Sentieon in exchange for testing DNASeq, evaluating its performance, or expressing positive views thereof.
Data availability statement
The NA12878 and NA24694 datasets analyzed in this study can be found in the appropriate Genome in a Bottle repositories:
NA12878: ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/NA12878/NIST_NA12878_HG001_HiSeq_300x
NA24694: ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/ChineseTrio/
The synthetic dataset representing WGS on chromosomes 20–22 will be made available by the authors, without undue reservation, to any qualified researcher.
Benchmarking scripts and commands used to generate synthetic data and accuracy comparisons are available at https://github.com/ncsa/Sentieon_DNASeq_Benchmarking.
Author contributions
LM designed the tests, drawing on previous work with MT. KK conducted the investigation; JH contributed additional research. KK and LM wrote the manuscript and prepared the visuals. The project was administered by DW and LM. Results were discussed with SB, MB, TD, SH, MEH, MH, MK, EK, NM, CR, EW, and MW.
Acknowledgments
This work was a product of the Mayo Clinic and Illinois Alliance for Technology-Based Healthcare. Special thanks for the funding provided by the Mayo Clinic Center for Individualized Medicine and the Todd and Karen Wanek Program for Hypoplastic Left Heart Syndrome. We also thank the Interdisciplinary Health Sciences Institute, the Carl R. Woese Institute for Genomic Biology and the National Center for Supercomputing Applications for their generous support and access to resources. We particularly acknowledge the support of Keith Stewart, M.B., Ch.B., Mayo Clinic/Illinois Grand Challenge Sponsor and Director of the Mayo Clinic Center for Individualized Medicine. Special gratitude to Gay Reed and Amy Weckle for managing the project. Many thanks to the Sentieon team for the evaluation license, as well as consultation and advice on the Sentieon variant calling software.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
BanerjeeS. S.AthreyaA. P.MainzerL. S.JongeneelC. V.HwuW.-M.KalbarczykZ. T.et al. (2016). Efficient and scalable workflows for genomic analyses. In Proceedings of the ACM International Workshop on Data-Intensive Distributed Computing (ACM), 27–36. doi: 10.1145/2912152.2912156
2
Broad Institute (2018). GATK | Archived versions. https://software.broadinstitute.org/gatk/download/archive.
3
ChapmanB. (2014). Benchmarking variation and rna-seq analyses on amazon web services with docker. Blue Collar Bioinformatics.
4
ChurchG. M. (2005). The personal genome project. Mol. Syst. Biol.1. doi: 10.1038/msb4100040
5
DePristoM. A.BanksE.PoplinR.GarimellaK. V.MaguireJ. R.HartlC.et al. (2011). A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat. Genet.43, 491–498. doi: 10.1038/ng.806
6
FreedD.AldanaR.WeberJ. A.EdwardsJ. S. (2017). The sentieon genomics tools — a fast and accurate solution to variant calling from next-generation sequence data. bioRxiv. doi: 10.1101/115717
7
GoodwinS.McPhersonJ. D.McCombieW. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet.17, 333–351. doi: 10.1038/nrg.2016.49
8
HeldenbrandJ. R.BahetiS.BockolM. A.DruckerT. M.HartS. N.HudsonM. E.et al. (2018). Performance benchmarking of GATK3.8 and GATK4. bioRxiv [Preprint]. doi: 10.1101/348565
9
Illumina (2018a). Data from: Illumina platinum confident regions. https://github.com/Illumina/PlatinumGenomes/blob/master/files/2017-1.0.files.
10
Illumina (2018b). Illumina sequencing platforms. https://www.illumina.com/systems/sequencing-platforms.html.
11
JIMB (2018). Data from: Genome in a bottle (giab). http://jimb.stanford.edu/giab/.
12
KathiresanN.TemanniR.AlmabraziH.SyedN.JitheshP. V.Al-AliR. (2017). Accelerating next generation sequencing data analysis with system level optimizations. Sci. Rep.7, 9058. doi: 10.1038/s41598-017-09089-1
13
KindratenkoV. (2018). memprof. GitHub Repository: https://github.com/IGBIllinois/memprof.
14
LiH. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arxiv [Preprint].
15
LiH.HandsakerB.WysokerA.FennellT.RuanJ.HomerN.et al. (2009). The sequence alignment/map format and samtools. Bioinformatics25, 2078–2079. doi: 10.1093/bioinformatics/btp352
16
MetzkerM. L. (2010). Sequencing technologies — the next generation. Nat. Rev. Genet.11, 31–46. doi: 10.1038/nrg2626
17
Novocraft Technologies (2014). Novosort. http://www.novocraft.com/.
18
PlüssM.KoppsA. M.KellerI.MeienbergJ.CasparS. M.DubacherN.et al. (2017). Need for speed in accurate whole-genome data analysis: GENALICE MAP challenges BWA/GATK more than PEMapper/PECaller and Isaac. Proc. Natl. Acad. Sci. U.S.A.114, E8320–E8322. doi: 10.1073/pnas.1713830114
19
RaczyC.PetrovskiR.SaundersC. T.ChornyI.KruglyakS.MarguliesE. H.et al. (2013). Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics29, 2041–2043. doi: 10.1093/bioinformatics/btt314
20
Sage Bionetworks (2016). ICGC-TCGA DREAM Mutation Calling Challenge. https://www.synapse.org/!Synapse:syn312572/wiki/247695.
21
Sentieon (2018). Sentieon DNASeq. https://www.sentieon.com/products/.
22
StephensZ. (2018). Data simulated from: NEAT-genReads. https://github.com/zstephens/neat-genreads.
23
StephensZ. D.HudsonM. E.MainzerL. S.TaschukM.WeberM. R.IyerR. K. (2016). Simulating next-generation sequencing datasets from empirical mutation and sequencing models. PLOS ONE11, 1–18. doi: 10.1371/journal.pone.0167047
24
StephensZ. D.LeeS. Y.FaghriF.CampbellR. H.ZhaiC.EfronM. J.et al. (2015). Big data: astronomical or genomical? PLoS Biol.13, e1002195. doi: 10.1371/journal.pbio.1002195
25
U.S. Food and Drug Administration (2016a). PrecisionFDA Consistency Challenge. https://precision.fda.gov/challenges/consistency/results.
26
U.S. Food and Drug Administration (2016b). PrecisionFDA Truth Challenge. https://precision.fda.gov/challenges/truth/results.
27
Van der AuweraG. A.CarneiroM. O.HartlC.PoplinR.Del AngelG.Levy-MoonshineA.et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics43, 1–33. doi: 10.1002/0471250953.bi1110s43
28
WeberJ.AldanaR.GallagherB.EdwardsJ. (2016). Sentieon DNA pipeline for variant detection — Software-only solution, over 20 faster than GATK 3.3 with identical results. PeerJ PrePrints. doi: 10.7287/peerj.preprints.1672v2
29
ZookJ.McDanielJ.ParikhH.HeatonH.IrvineS. A.TriggL.et al. (2018). Reproducible integration of multiple sequencing datasets to form high-confidence SNP, indel, and reference calls for five human genome reference materials. bioRxiv [Preprint]. doi: 10.1101/281006
30
ZookJ. M.ChapmanB.WangJ.MittelmanD.HofmannO.HideW.et al. (2014). Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol.32, 246–251. doi: 10.1038/nbt.2835
Summary
Keywords
Sentieon, variant calling, benchmarking, GATK, DNASeq
Citation
Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, Hernaez M, Hudson ME, Kalmbach MT, Klee EW, Mattson NR, Ross CA, Taschuk M, Wieben ED, Wiepert M, Wildman DE and Mainzer LS (2019) Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front. Genet. 10:736. doi: 10.3389/fgene.2019.00736
Received
09 May 2019
Accepted
12 July 2019
Published
20 August 2019
Volume
10 - 2019
Edited by
Gustavo Glusman, Institute for Systems Biology (ISB), United States
Reviewed by
Gabriel Rodrigues Alves Margarido, University of São Paulo, Brazil; Sergey Aganezov, Johns Hopkins University, United States
Updates
Copyright
© 2019 Kendig, Baheti, Bockol, Drucker, Hart, Heldenbrand, Hernaez, Hudson, Kalmbach, Klee, Mattson, Ross, Taschuk, Wieben, Wiepert, Wildman and Mainzer.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liudmila S. Mainzer, lmainzer@illinois.edu
This article was submitted to Bioinformatics and Computational Biology, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.