 Xiaofeng Xu
Xiaofeng Xu Tao Liu
Tao Liu Yijin Wang
Yijin Wang Jian Fu4
Jian Fu4 Qian Yang
Qian Yang Jun Wu
Jun Wu Huaijun Zhou
Huaijun Zhou- 1Department of Gynecology, The Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing, China
- 2Medical College, Nanjing University, Nanjing, China
- 3Medical College, Southeast University, Nanjing, China
- 4Department of Gynecology, Suqian People’s Hospital of Nanjing Drum Tower Hospital Group, Suqian, China
- 5Department of Gynecology and Obstetrics, The Pukou Hospital of Nanjing, The Fourth Affiliated Hospital of Nanjing Medical University, Nanjing, China
Although various factors may contribute to its initiation and progression, the etiology and prognostic factors of endometrial carcinoma (EC) remains not fully understood. We sought to understand the role of changes in transcriptome during the progress of EC by exploring public datasets. The aberrant expression characteristics of EC based on RNA-Seq and miRNA-seq data from The Cancer Genome Atlas (TCGA) were analyzed. Kaplan–Meier analysis was performed to assess the relationship between differently expressed genes (DEGs) and patient survival. As a result, 320 out of 4,613 differently expressed mRNAs (DE mRNAs) and 68 out of 531 differently expressed miRNAs (DE miRNAs) with a significantly poorer survival were determined. We predicted eight paired DE miRNAs and DE mRNAs through TargetScan. Patients with three out of the eight paired low rate of miRNA/mRNA (miR-497/EMX1, miR-23c/DMBX1, and miR-670/KCNS1) expression had a significantly poorer survival. Furthermore, the simultaneous presence of these selected low miRNA/mRNA pairs occurred in most patients and resulted in a significantly poorer survival rate. Luciferase reporter assay identified that EMX1 was a direct target of miR-497. Both low expression of miR-497 and overexpression of EMX1 were significantly associated with more advanced clinicopathologic characteristics (stage, tumor status, grade, and histology) besides survival (all P values < 0.05). Multivariate analysis also demonstrated that miR-497 remained an independent prognostic variable for overall survival. In summary, we identified that a series of DE mRNAs and miRNAs, including eight paired DE miRNAs and mRNAs, were associated with survival in EC. Clinical evaluation of downregulated miR-497 and paired upregulated EMX1 confirmed the value of our prediction analysis. The simultaneous presence of low rate of these selected low miRNA/mRNA pairs (miR-497/EMX1, miR-23c/DMBX1, and miR-670/KCNS1) might have a better prediction value. Therefore, further studies are required to validate these findings.
Introduction
Endometrial cancer (EC) is one of the most prevalent gynecological malignancies in the word (Siegel et al., 2018). The incidence has increased by 1.2% per year from 2005 to 2014, along with the mortality rates, which similarly increased during this period (Smith et al., 2018). Importantly, EC is one of only two common cancers that defy the general trend of improvement in morbidity and mortality, with a worse survival rate today than in the 1970s (Siegel et al., 2018). The American Cancer Society (ACS) estimates that 63,230 women will be diagnosed with EC and that more than 11,000 women will die from this disease in 2018 (Siegel et al., 2018). Clearly, the burden of EC is increasing in the USA and worldwide, hence increasing the need to investigate its causes and improve prevention, early diagnosis, and treatment (Binder and Mutch, 2014). Although various endocrine, genetic, and external factors may contribute to its initiation and progression, the etiology and prognostic factors of endometrial carcinoma remain not fully understood.
Previous analysis of the human genome revealed that while ∼85% is transcribed, only ∼1% is protein-coding mRNA (Dunham et al., 2012; Djebali et al., 2012; Dykes and Emanueli, 2017). With time, more and more attention was being paid to the research of non-coding RNAs, including microRNAs (miRNAs), which have important regulatory roles in cancer cellular biology. Studies demonstrated that mature miRNAs can regulate the expression of their target genes by imprecise complementation to the 3′-untranslated regions (UTRs), 5′-UTRs, and even coding sequences of the mRNAs to repress their translation (Ambros, 2004; Duursma et al., 2008; Sacco and Masotti, 2012). Srivastava et al. reviewed the amount of miRNAs differentially expressed in EC versus normal endometrial tissue, including the increased expression of miR-9, miR-92a, miR-141, miR-182, miR-183, miR-186, miR-200a, miR-205a, miR-222, miR-223, miR-410, miR-429, miR-449, and miR-1228 and downregulation of miR-99b,miR-143, miR-145, miR-193b, and miR-204 (Srivastava et al., 2017). Numerous miRNAs could regulate EC cells by silencing their target genes (Zhou et al., 2012; Chen et al., 2016). Though expression patterns of several miRNAs were found to be associated with the International Federation of Gynaecology and Obstetrics (FIGO) stage, grade, relapse, and nodal metastases in EC (Torres et al., 2013; Canlorbe et al., 2016; Srivastava et al., 2017), few studies focused on the relationship between both miRNAs and their target genes with patient survival.
The Cancer Genome Atlas (TCGA) studies have defined the molecular genetic landscape of EC and highlighted the molecular genetic diversity of both endometrioid and non-endometrioid cancers (Burk et al., 2017). Recently, accompanied by the advent era of sharing information, more and more cancer researches including EC were carried out based on TCGA database (Dellinger et al., 2016; Reyes et al., 2017; Wang et al., 2018; Wu and Zhang, 2018). However, the previous studies mainly focused on the single factor, such as differently expressed mRNAs (DE mRNAs), differently expressed miRNAs (DE miRNAs), or differently expressed long non-coding RNAs (DE lncRNAs) to find the potential etiology and prognostic factor of tumor.
In this study, considering the silence effect to their target genes of miRNAs, we performed survival analysis on both DE mRNAs and DE miRNAs comparing EC and normal samples from TCGA database. As a result, we revealed a group of DE mRNAs and DE miRNAs associated with survival. Furthermore, we identified that low expression of miR-497 and overexpression of its potential target gene Empty Spiracles Homeobox 1 (EMX1) were both related to more advanced clinicopathologic characteristics.
Methods
Data Collection
Expression profiles of RNA-Seq and miRNA-seq for TCGA-UCEC project were downloaded from TCGA official website (https://cancergenome.nih.gov) in July 2018. RNA expression data of 543 EC cases and 35 normal cases were downloaded from the database, while miRNA expression data of 539 EC and 33 normal samples were included. The corresponding clinical information was downloaded from http://www.cbioportal.org. Data were collated and extracted for analysis.
Identification of Differently Expressed Genes (DEGs)
We applied the expression profiles to the edgeR package in R language and calculated for differential expression between tumor and normal group samples after normalization and filtration. The adjusted P values (adj. P) were applied to correct the false-positive results. Then the significant DEGs (adj. P < 0.05 and fold-change value larger than 2) were selected out for the next step analysis.
Survival Analysis
In this study, survival analysis refers to the overall survival (OS) Kaplan–Meier estimate. We performed Kaplan–Meier analysis (R package “survival”) to assess survival and relapse difference across cases with DEGs. For each gene, patients were divided into high-expression group and low-expression group according to the median expression level. Based on the survival curves of each group, these upregulated and downregulated DEGs with a poorer survival rate were within our consideration. P value < 0.05 was set as the cutoff point.
Functional and Pathway Enrichment Analysis of DEGs
The Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) is a biological database regularly used to facilitate functional annotation and pathway analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis aims to identify and visualize significantly enriched pathways of molecular interactions, reactions, and relations. Gene Ontology (GO) analysis uses hypergeometric tests to perform enrichment analysis on gene sets (Falcon and Gentleman, 2007). We uploaded selected DEGs associated with survival to DAVID to perform KEGG pathway and GO enrichment analysis. The human genome was selected as the background list parameter, and P value < 0.05 was set as the cutoff point.
Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether a predefined set of genes shows significant differences between two biological phenotypes under study (Subramanian et al., 2005). Gene set permutations were performed 1,000 times for each analysis. The nominal P value (NOM P value) and normalized enrichment score (NES) were used to sort the pathways enriched in each phenotype.
Prediction of DE miRNA-Targeted DE mRNAs Associated With OS
In order to improve the validity of our search results, we further excavated the relationship between the selected DE mRNAs and DE miRNAs through TargetScan (http://www.targetscan.org/vert_72/). TargetScan predicts biological targets of miRNAs by searching for the presence of conserved sites that match the seed region of each miRNA (Fromm et al., 2015). In brief, we not only searched for the presence of conserved sites among these DE mRNA that matched the seed region of DE miRNA but also searched for DE miRNA containing matched seed region for DE mRNA. These poorly conserved sites were excluded. Only these enrolled miRNA–mRNA pairs with opposite expression levels in EC samples as compared with normal tissues both presented in our dataset were adapted for further study.
Cell Culture and Luciferase Reporter Assay
Cell culture and Luciferase reporter assay were very similar with those in our previous study (Zhou et al., 2012). Briefly, 293T cell and human endometrial cancer Ishikawa cell were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and penicillin (100 U/ml). The 300-nt-long 3′-UTR (wild type or mutant) of EMX1 containing the predicted conserved binding sites for miR-497 was cloned into H306 pMIR-LUC vectors purchased from Obio Technology (Shanghai, China). Cells were plated at a density of 1 × 105 in 12-well plate. After 24 h, the pMIR-LUC reporters were co-transfected with either miR-497 mimics or control using Lipofectamine 3000. Luciferase activity normalized to the Renilla Luciferase was measured by the Dual-Luciferase assay (Promega, USA) according to the manufacturer’s instructions after 48 h on the Luminometer (Promega, USA). The assay was repeated three times.
RNA Extraction and Quantitative RT-PCR
Total RNA was extracted from cells with isolator reagent (Vazyme, China). After measurement of the RNA concentration, cDNAs were generated from reverse transcription with the HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, China). The expression level of miR-497 was measured according to the instructions of the ChamQTM Universal SYBR qPCR Master Mix kit (Vazyme, China) using the ABI-7300 Real-Time PCR Detection System (Applied Biosystems, USA). The bulge-loopTM miRNA Primer Sets (one RT primer and a pair of qPCR primers) specific for miR-497 were purchased from RiboBio (Guangzhou, China). The level of miR-497 was normalized to U6. Fold changes were calculated using the 2−ΔΔCt method. Each plate was run in triplicate.
Statistical Analysis
Logistic regression and t test were used to evaluate the relationship between gene expression and clinical–pathologic features. For gene expression level, the cutoff value was determined by its absolute median expression level (high- and low-expression groups). For the analysis of survival for patients with paired miRNA/mRNA, the median rate of miRNA/mRNA expression was used to divide groups for low and high rates. In logistic regression, the absolute gene expression level (high- and low-expression group) was used as categorical dependent variable, while clinical–pathologic feature was used as independent variable. Among all the clinical–pathologic features, age and BMI were calculated as continuous variables and the rest as categorical variables. Kaplan–Meier method and Cox regression were used to analyze clinicopathologic characteristics associated with OS. The influence of gene expression on survival along with other clinical characteristics was determined by multivariate Cox analysis. P value < 0.05 was set as the cutoff point. *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Identification of DE mRNAs and DE miRNAs With a Poorer Survival in EC
The RNA-Seq profile data of 543 EC and 35 normal cases were downloaded from TCGA database along with the miRNA-seq data of 539 EC and 33 normal samples. Among them, 533 patients shared two expression profiles (Figure 1). Under the threshold of adj. P < 0.05 and fold-change value larger than 2, a total of 4,613 DE mRNAs (3,221 upregulated and 1,392 downregulated) and 531 DE miRNAs (374 upregulated and 157 downregulated) were identified in EC compared with normal samples. The heatmaps and volcano plots are shown in Supplementary Figure 1.
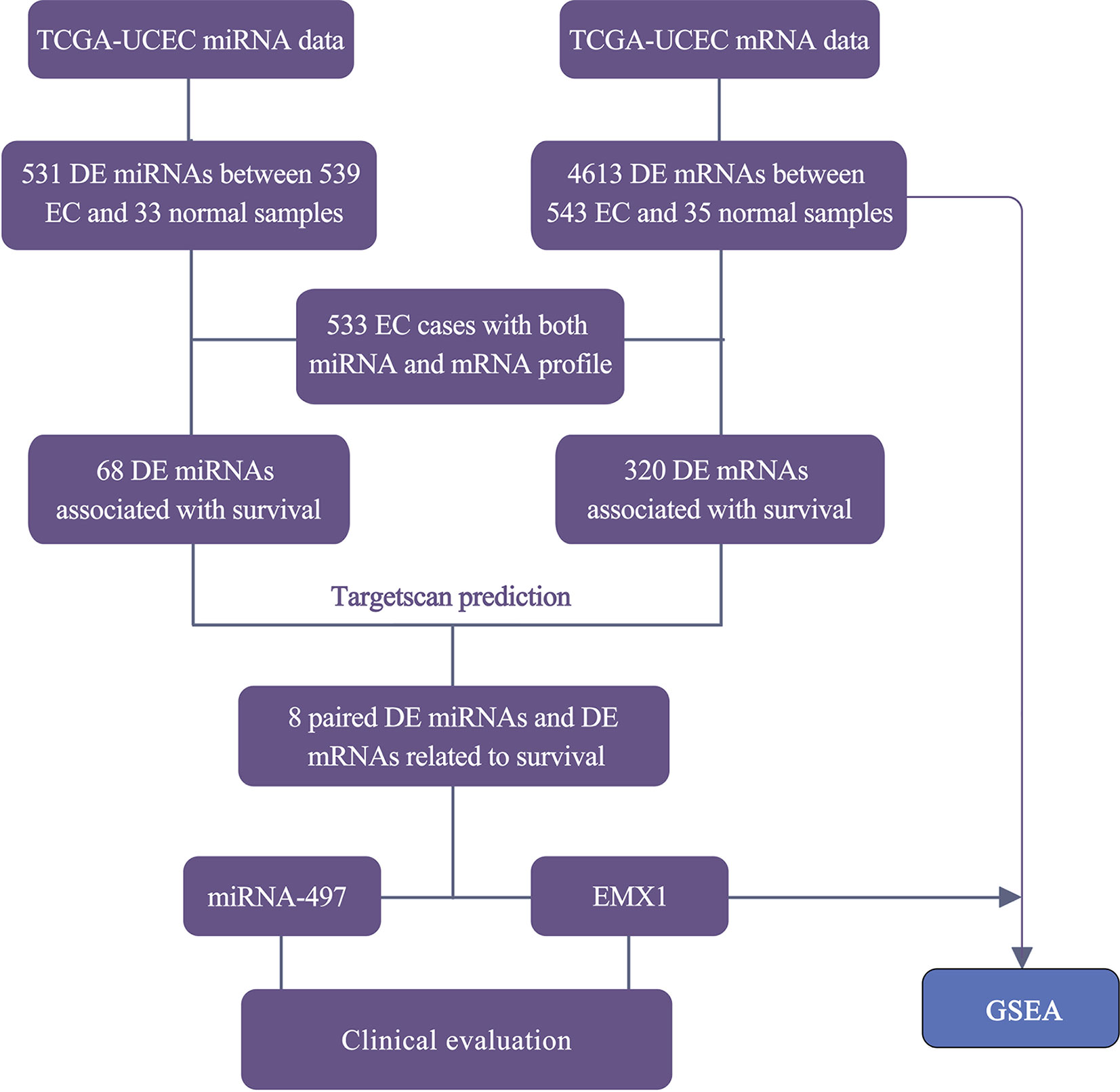
Figure 1 Workflow for the screening and evaluation of DE mRNAs and DE miRNAs between EC and normal samples.
To have a better understanding of the relationship between DEGs and patient survival, we conducted the “survival” package in R language to draw Kaplan–Meier curves according to the median expression level of DEGs among 533 patients. These upregulated and downregulated genes with a significantly poorer survival (P < 0.05), respectively, were determined. A total of 320 (280 upregulated and 40 downregulated) out of 4,613 DE mRNAs and 68 (43 upregulated and 25 downregulated) out of 531 DE miRNAs were chosen for further study (Supplementary Table 1). Kaplan–Meier curves of the partial genes between the high-expression group and low-expression group are shown in Figure 2.
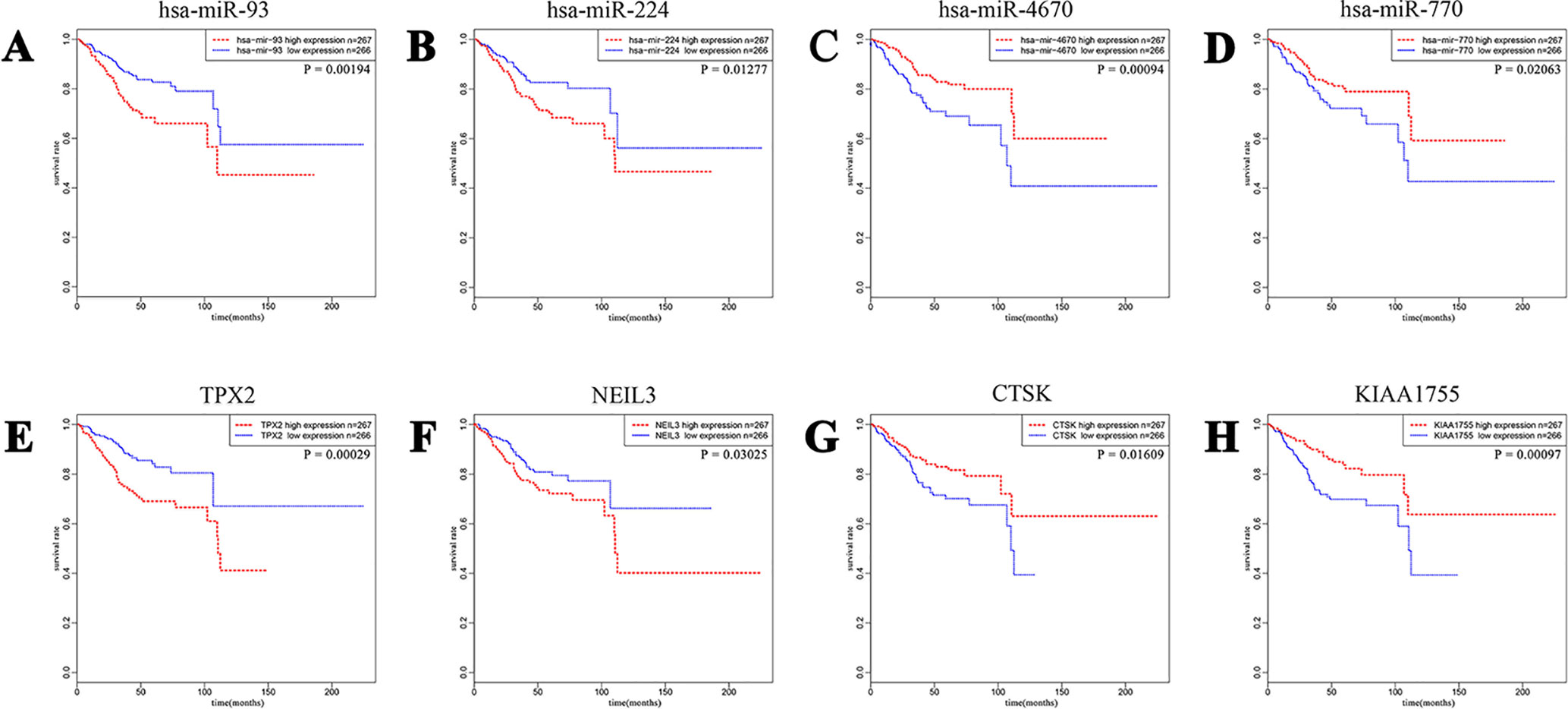
Figure 2 A total of 320 out of 4,613 DE mRNAs and 68 out of 531 DE miRNAs associated with a poorer survival rate were chosen. The eight plots show Kaplan–Meier curves of the partial DE miRNAs and DE mRNAs between cases of high-expression group and low-expression group divided according to the median expression level. (A and B) Upregulated miRNA-93 and miRNA-224. (C and D) Downregulated miRNA-4670 and miRNA-770. (E and F) Upregulated TPX2 and NEIL3. (G and H) Downregulated CTSK and KIAA1755.
Functional and Pathway Enrichment Analysis
The selected 320 DE mRNAs associated with survival were uploaded to DAVID to perform KEGG pathways and GO enrichment analysis. KEGG analysis revealed that these DEGs were mostly enriched in cell cycle, neuroactive ligand–receptor interaction, microRNAs in cancer, oocyte meiosis, and serotonergic synapse signaling pathways (Figure 3A). The top five enriched GO biological process (BP) terms included positive regulation of transcription from RNA polymerase II promoter, cell division, negative regulation of transcription from RNA polymerase II promoter, cell proliferation, and mitotic nuclear division. Nucleus, cytoplasm, nucleoplasm, microtubule, and neuronal cell body were the five most enriched GO terms for cellular component (CC). The top five enriched GO molecular function (MF) terms were protein binding, DNA binding, ATP binding, transcription factor activity, and sequence-specific DNA binding (Figures 3B–D). The most significantly enriched pathways and enrichment terms are also shown in Supplementary Table 2.
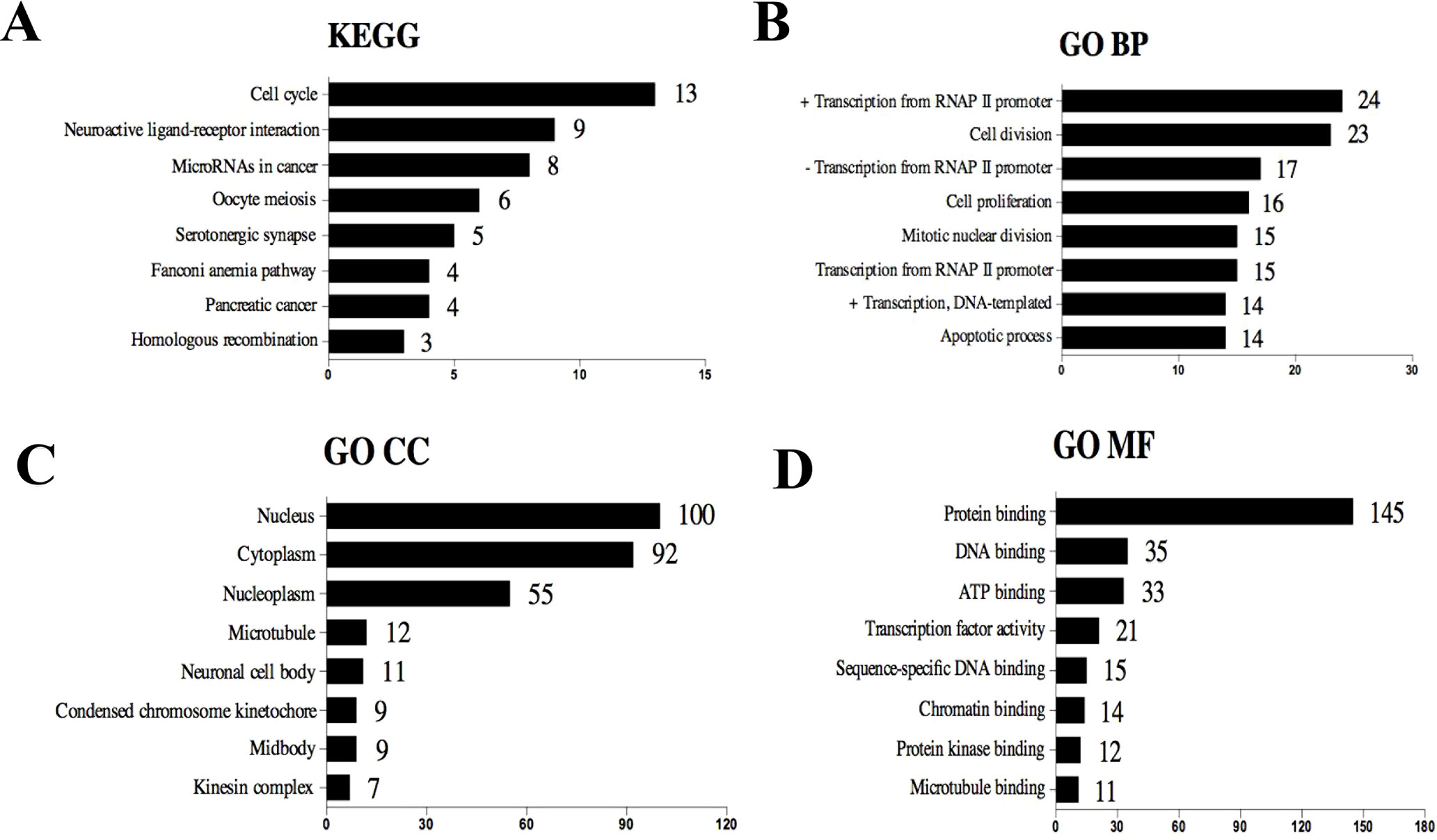
Figure 3 Top eight (sorted by count) KEGG pathways and GO terms identified among DEGs associated with survival using DAVID. (A) KEGG pathways. (B) GO term of Biological Process. (C) GO term of Cellular Component. (D) GO term of Molecular Function. “+” represents positive regulation, and “−” represents negative regulation.
Prediction Roles of Identified DE miRNAs and Their Potential Targeted mRNAs Associated With Survival
In order to better understand the role of DE miRNAs and their potential targeted DE mRNAs related to survival, we made target predictions through TargetScan. These genes containing conserved target sites, which matched the seed regions of miRNAs, were in our consideration. Besides, we only accepted these DE miRNAs and DE mRNAs with opposite expression levels in view of the silence effect of miRNA targeting. As a result, we predicted that 5 out of 68 DE miRNAs could interact with 8 out of 320 DE mRNAs (Figure 4), with one upregulated miRNA paired with two downregulated mRNAs and four downregulated miRNAs paired with six upregulated mRNAs (Table 1). The survival curves are also shown in Figure 5.
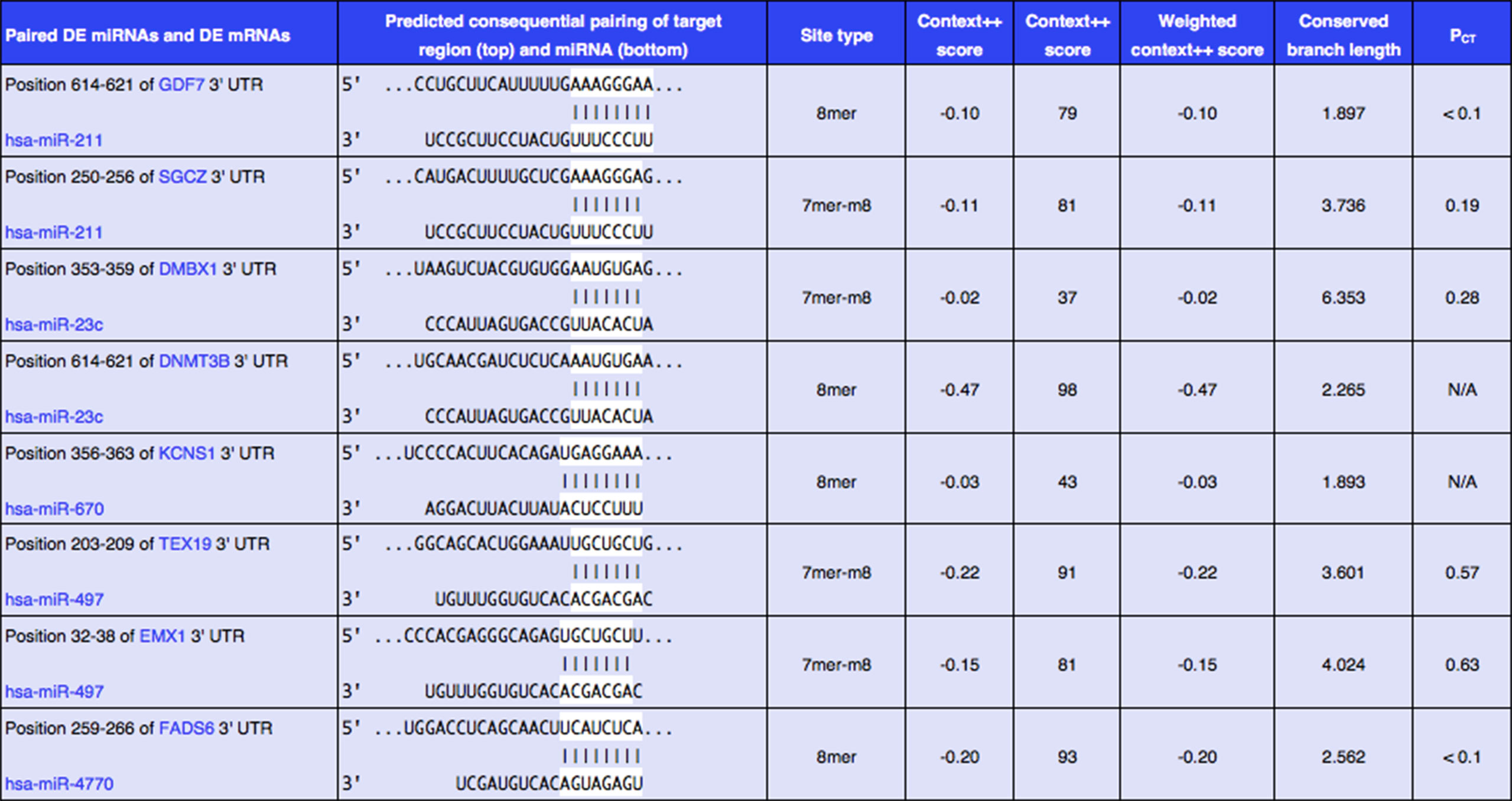
Figure 4 Eight predicted consequential pairings of DE miRNAs and target regions of DE mRNAs associated with survival. These enrolled miRNA–mRNA pairs not only contained conserved binding sites but also had opposite expression levels in EC samples as compared with normal tissues.
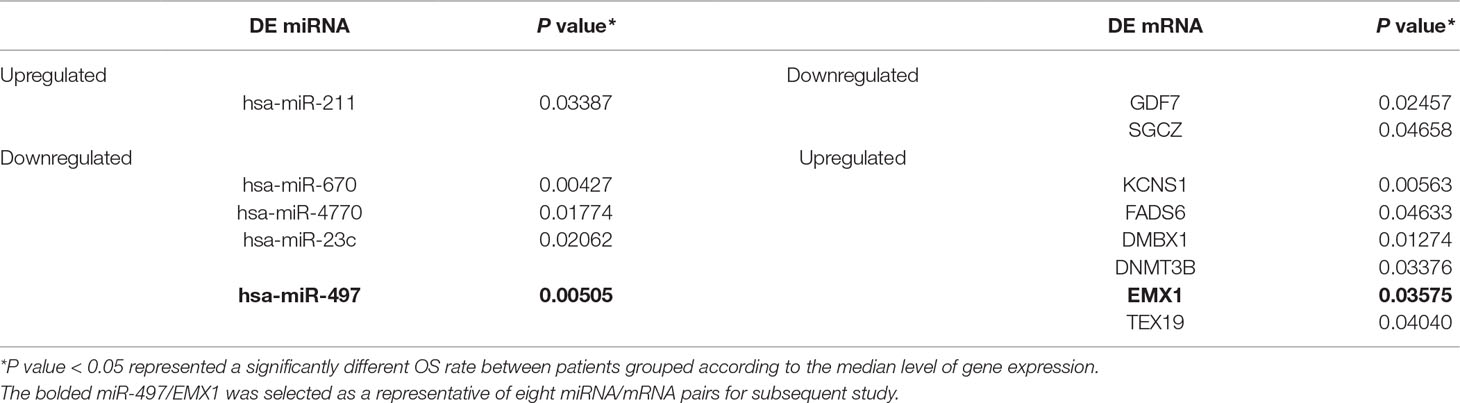
Table 1 DE miRNAs and their potential target DE mRNAs associated with survival.
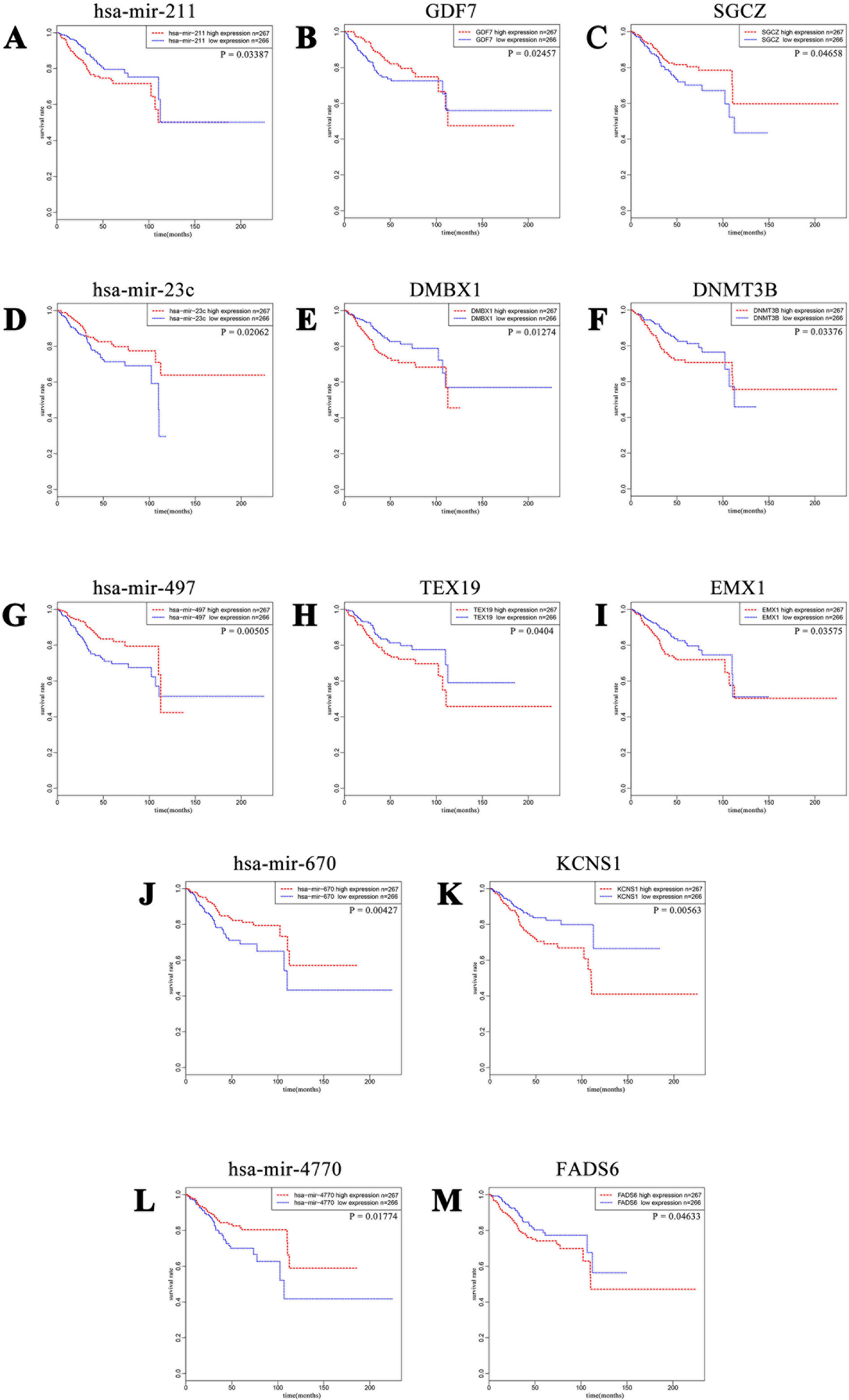
Figure 5 Kaplan–Meier curves of five DE miRNAs and eight potential target DE mRNAs, with one upregulated miRNA (A) miR-211 paired with two downregulated mRNAs (B) GDF7 and (C) SGCZ) and four downregulated miRNAs (D) miR-23c, (G) miR-497, (J) miR-670, and (L) miR-4770) paired with six upregulated mRNAs (E) DMBX1, (F) DNMT3B, (H) TEX19, (I) EMX1, (K) KCNS1, and (M) FADS6). For each gene, patients were divided into high-expression group and low-expression group according to the median expression level.
We further tried to assess the differences between high and low rates of expression levels for the eight miRNA/mRNA pairs under the cutoff of median rate. As shown in Figure 6A, patients with low rate of three paired miRNA/mRNA (miR-497/EMX1, miR-23c/DMBX1, and miR-670/KCNS1) expression had a significantly poorer survival, while the rest seemed no different (data were not shown). Furthermore, under each cutoff of median rate, there were 156 patients carrying low rates of both miR-497/EMX1 and miR-23c/DMBX1 expression, 135 patients carrying low rates of both miR-497/EMX1 and miR-670/KCNS1 expression, 160 patients carrying low rates of both miR-670/KCNS1 and miR-23c/DMBX1 expression, and 92 patients carrying low rates of all the three miRNA/mRNA pairs (Table 2). Surprisingly, these patients with the simultaneous presence of these two or three low miRNA/mRNA pairs had a significantly poorer survival rate (Figure 6B). These results strongly verified the validity and prediction capacity of our analysis.
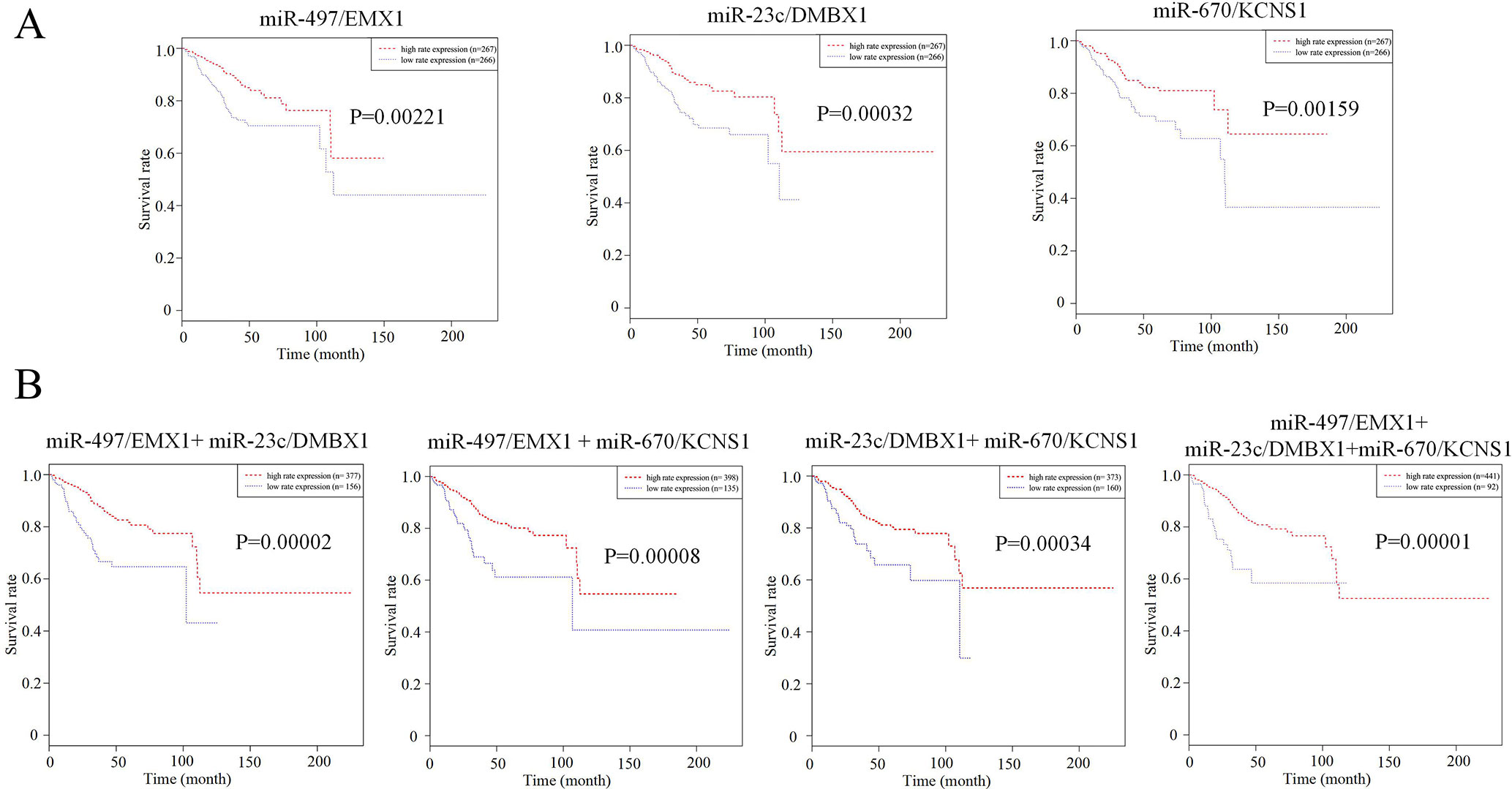
Figure 6 Prediction roles of identified DE miRNAs and their potential targeted mRNAs associated with survival. (A) Kaplan–Meier curves for patients with rate of three single paired miRNA/mRNA (miR-497/EMX1, miR-23c/DMBX1, and miR-670/KCNS1) expression. (B) Kaplan–Meier curves for patients with simultaneous presence of rates for more than one miRNA/mRNA pair among these three selected pairs. The median rate of miRNA/mRNA expression was used to divide groups into low and high rates.
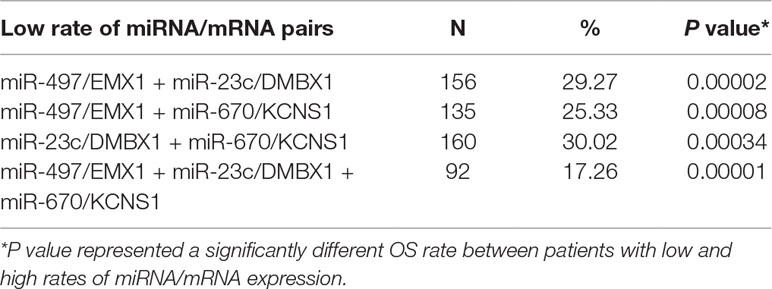
Table 2 Patients with the simultaneous presence of low rates of miRNA/mRNA pairs.
Clinical Evaluation of miR-497 and EMX1 Expression in EC
Patient Characteristics
We downloaded the clinical data of 533 primary EC from http://www.cbioportal.org. Patient characteristics are shown in Table 3. Median age at diagnosis of our study cohort was 64 year old (range 31–90 years old); 72.6% of the study group were white, with the rest scattered among other races. Most of the patients were endometrioid endometrial adenocarcinoma (EA; n = 402, 75.4%), 20.5% (n = 109) were serous, and 22 (4.1%) were mixed serous and endometrioid. The histopathologic distribution of EC was well differentiated (18.2%), moderately differentiated (22.5%), and poorly differentiated (59.3%). The cancer status included 419 cases without tumor (84.5%) and 77 cases with tumor (15.5%). In all patients, 32.7% had deep myometrial invasion, and 14.2% had positive peritoneal washings. About 16.8% of all patients had positive pelvic lymph nodes, while 10.3% had positive para-aortic lymph nodes. Stages I, II, III, and IV comprised 62.9%, 9.0%, 22.8%, and 5.3%, respectively. Median follow-up for subjects alive at last contact was 23.5 months (range 0–190 months).
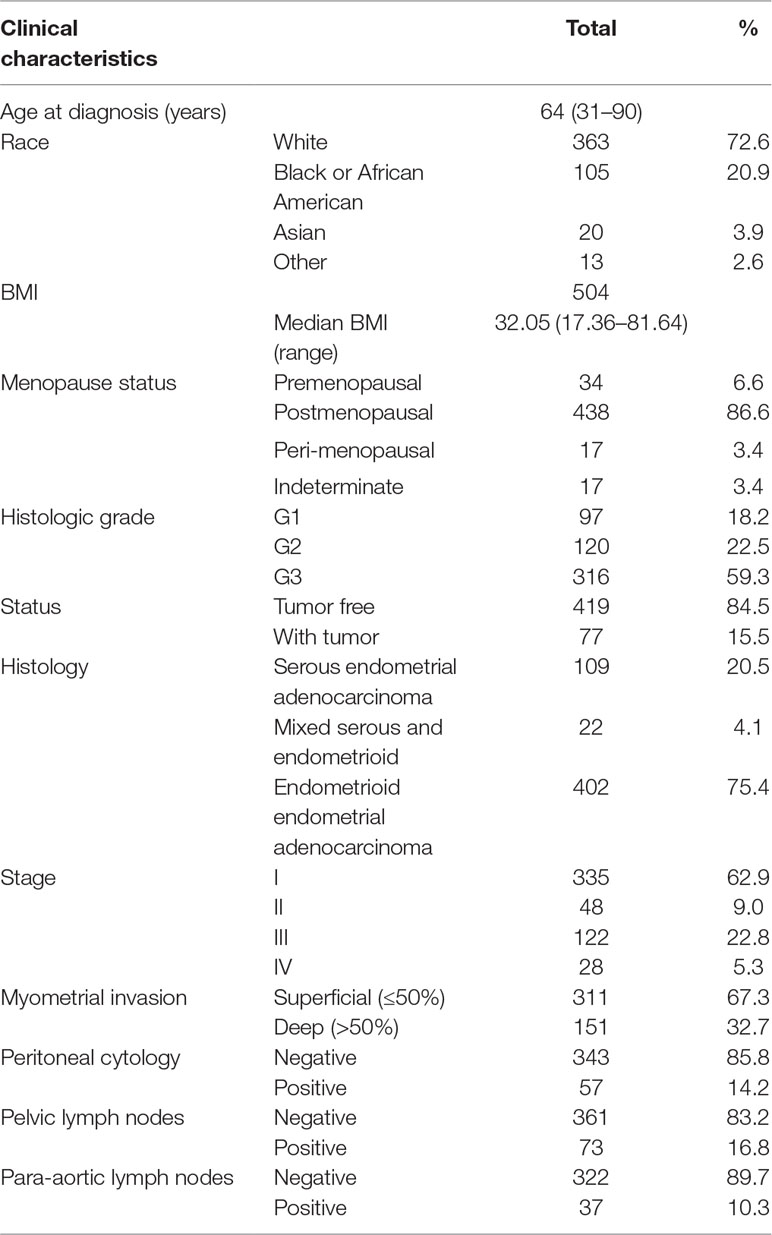
Table 3 Clinical characteristics of TCGA EC patients.
miR-497, EMX1 Expression, and Association With Clinicopathologic Variables
Univariate analysis using logistic regression revealed that miR-497 expression as a categorical dependent variable (based on median expression value) was associated with adverse prognostic clinical pathological features (Table 4). Decreased miR-497 expression in EC was significantly associated with high grade (OR = 0.228 for G1, G2 vs. G3), stage (OR = 0.470 for I vs. II, III, and IV), histology (OR = 0.175 for EA vs. non-EA), tumor status (OR = 0.391 for tumor free vs. with tumor), pelvic lymph node metastasis (OR = 0.294 for positive vs. negative), para-aortic lymph node metastasis (OR = 0.232 for positive vs. negative) and peritoneal washings (OR = 0.510 for positive vs. negative) (all P values < 0.05, Figures 7A–D).
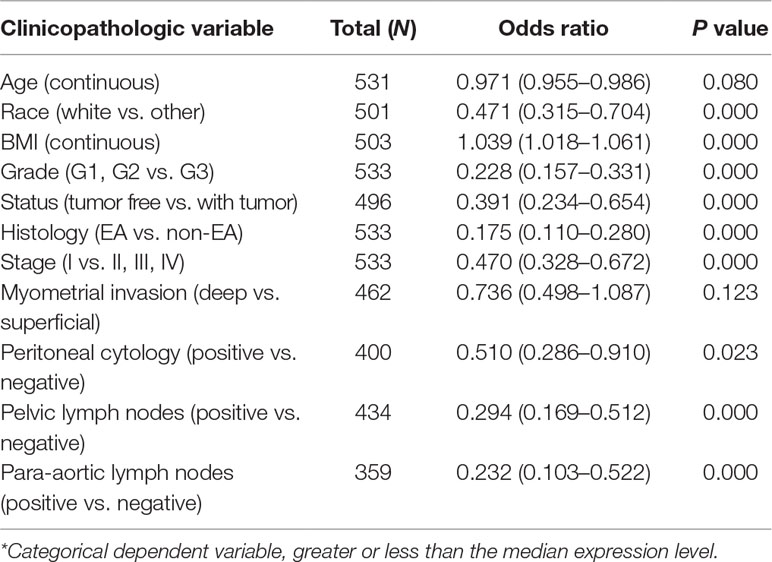
Table 4 Univariate miR-497 expression* association with clinicopathologic characteristics (logistic regression).
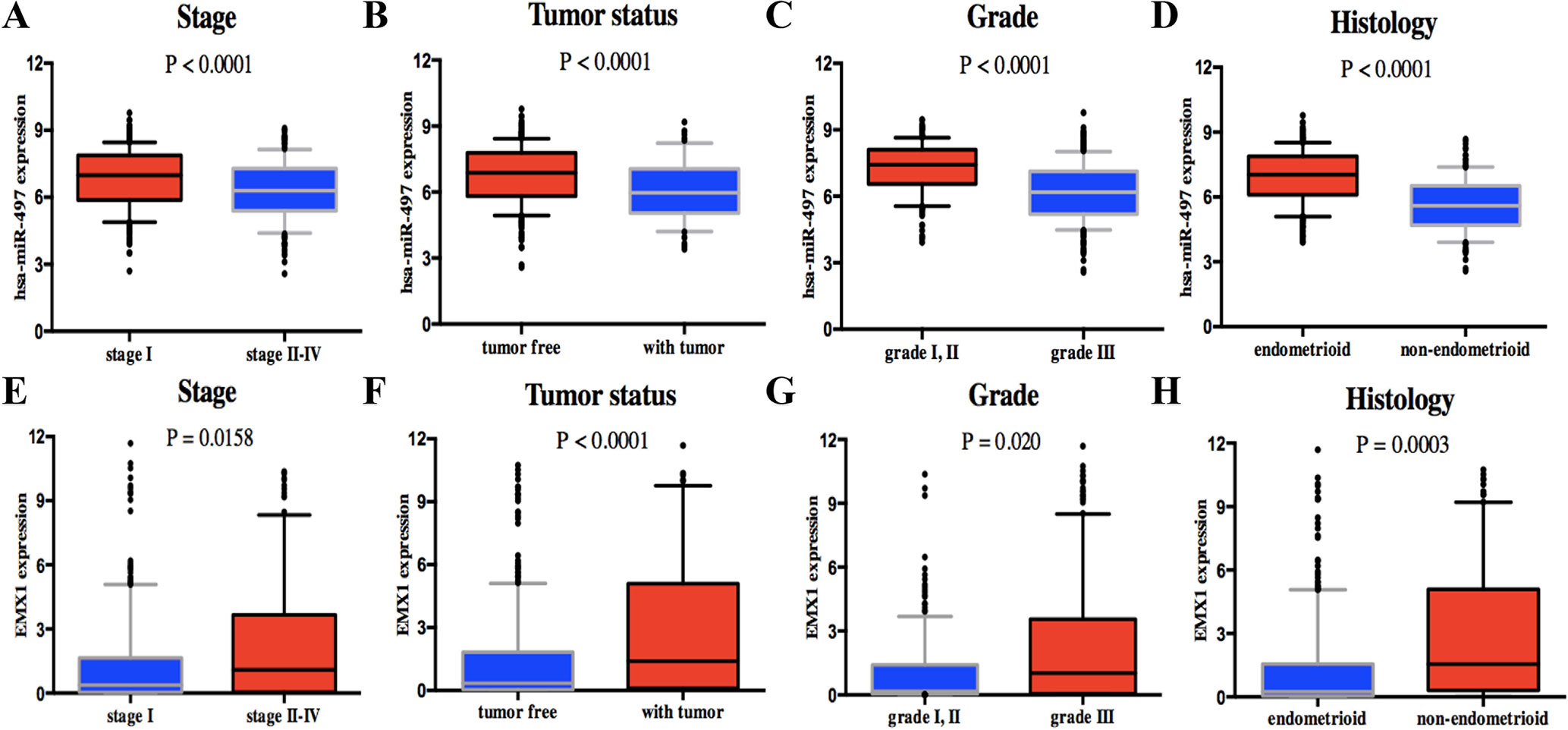
Figure 7 Association with DEG expression and clinicopathologic characteristics, including has-miR-497 expression associated with (A) stage, (B) tumor status, (C) grade, and (D) histology and EMX1 expression associated with (E) stage, (F) tumor status, (G) grade, and (H) histology. P values were calculated using t test.
On the contrary, high EMX1 expression was associated with poor prognostic clinicopathologic characteristics (Table 5). Overexpressed EMX1 in EC was associated with clinically advanced stage (OR = 1.668 for I vs. II, III, and IV), high grade (OR = 2.325 for G1, G2 vs. G3), histology (OR = 3.060 for EA vs. non-EA), tumor status (OR = 2.342 for tumor free vs. with tumor), and para-aortic lymph node metastasis (OR = 2.547 for positive vs. negative) (all P values < 0.05, Figures 7E–H).
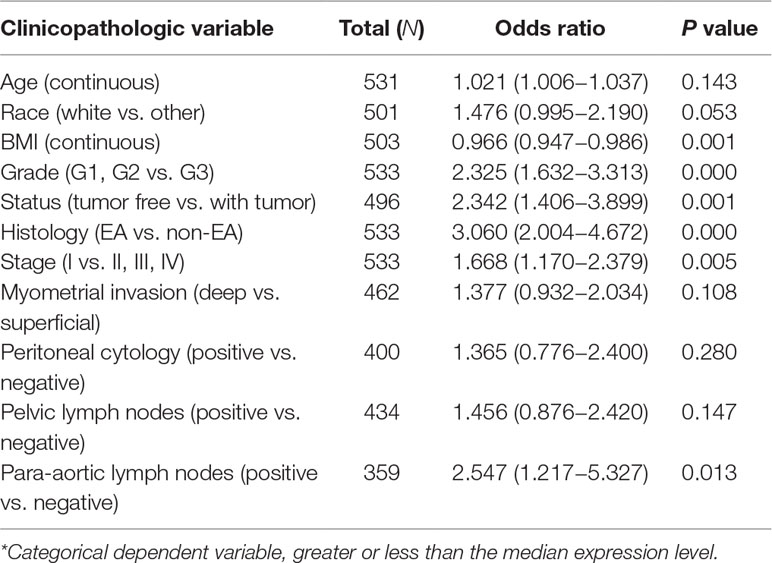
Table 5 Univariate EMX1 expression* association with clinicopathologic characteristics (logistic regression).
These results suggested that EC patients with low expression of miR-497 and high expression of EMX1 were prone to progress to a more advanced stage than those with high expression of miR-497 and low expression of EMX1.
Survival Outcomes and Multivariate Analysis
Kaplan–Meier survival analysis showed that EC with low expression of miR-497 had a worse prognosis than that with high expression (Figure 5G). The univariate analysis revealed that reduced miR-497 correlated significantly with a poor OS (HR: 0.536; 95% CI: 0.345–0.831; P = 0.005).
Meanwhile, Kaplan–Meier survival analysis suggested that patients with high EMX1 expression also had a poorer prognosis than those with low EMX1 expression (Figure 5I). And, univariate analysis revealed that high EMX1 expression correlated significantly with a poorer OS rate (HR: 1.563, 95% CI: 1.019–2.396; P = 0.041).
Other clinicopathologic factors associated with poor survival include high grade, histology, advanced stage, myometrial invasion, peritoneal washings, and lymph node metastasis (Table 6A).
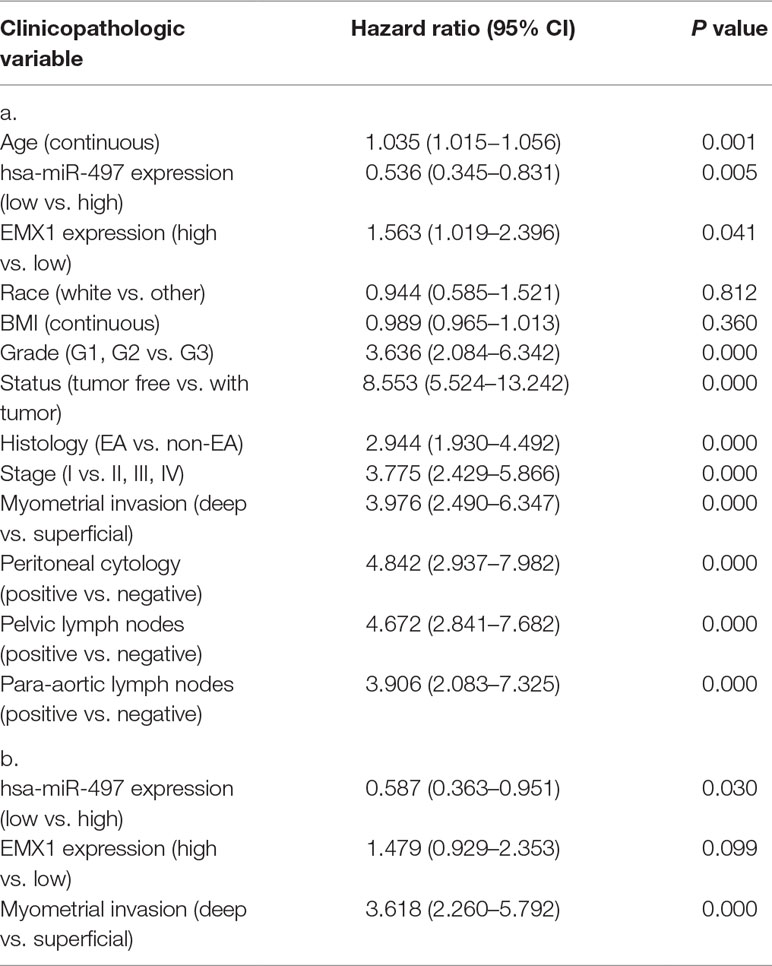
Table 6 a. Overall survival and associations with clinicopathologic characteristics using Cox regression. b. Multivariate survival model after variable selection.
In multivariate analysis (Table 6b), miR-497 remained an independent prognostic variable for OS, with an HR of 0.587 (CI: 0.363–0.951, P = 0.030), along with myometrial invasion.
EMX1 Was a Direct Target of miR-497
To explore whether EMX1 was a direct target of miR-497, we first detected the miR-497 levels in 293T and Ishikawa cells. As shown in Figure 8A, the miR-497 level in Ishikawa cell was very low, while it was much higher in 293T cell. We next performed a Dual-Luciferase reporter assay. The binding sites for miR-497 with EMX1 wild-type 3′-UTR (EMX1 3′-UTR) and EMX1 mutant 3′-UTR (EMX1 mu3′-UTR) are shown in Figure 8B. There was no loss of luciferase activity in Ishikawa cells with co-transfection of miR-497 mimics and mutated 3′-UTR of EMX1 plasmid, while a significant luciferase activity decrease was observed in cells co-transfected with miR-497 mimics and EMX1 3′-UTR plasmid (Figure 8C, *** P = 0.0001). We also performed the luciferase assay in 293T cells, which showed an increased relative luciferase activity of the construct having the mutated binding site compared with that of the wild-type site (Figure 8D, ** P < 0.01). These data indicated that EMX1 was a direct target of miR-497.
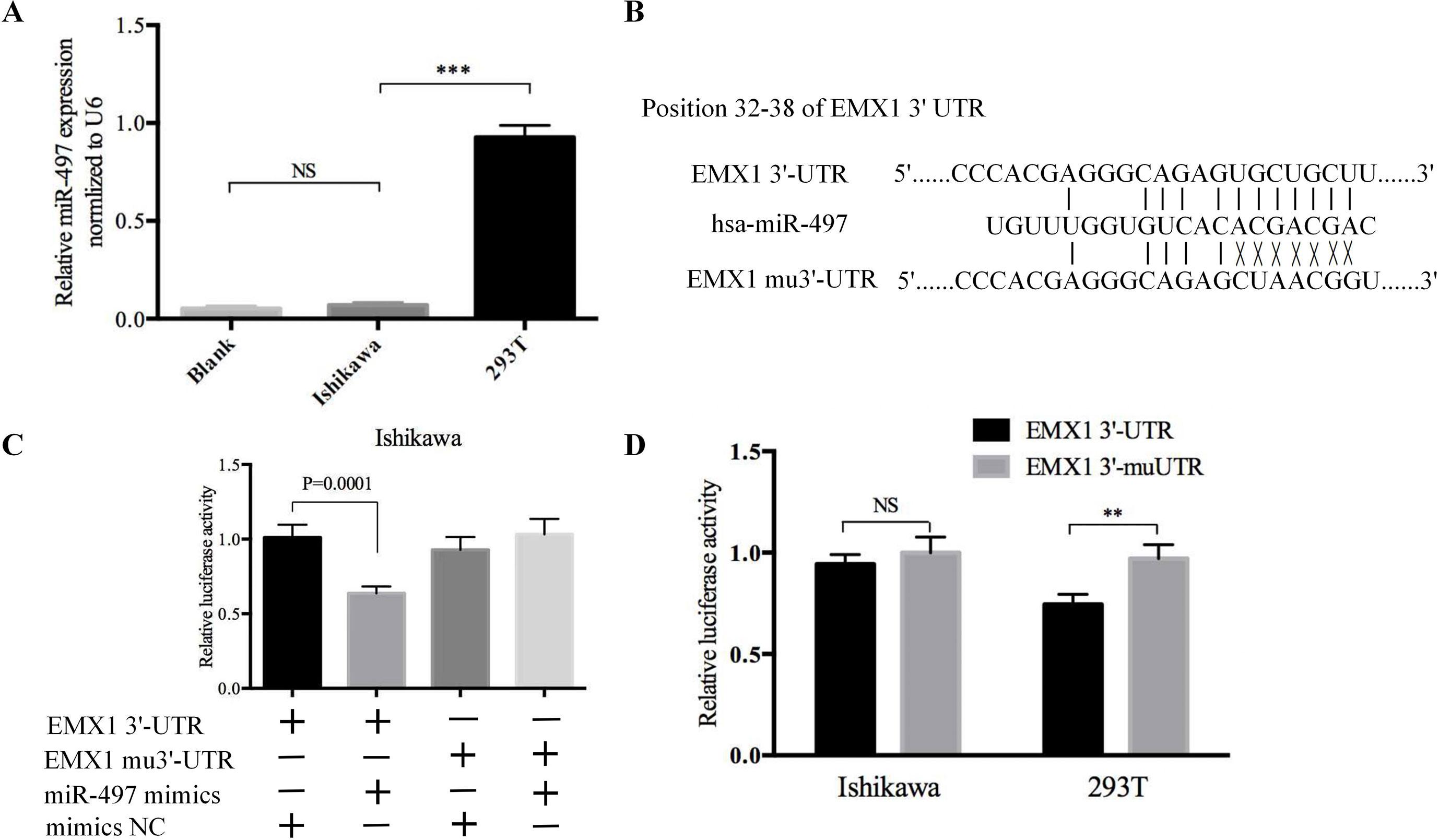
Figure 8 The luciferase reporter assay confirmed that EMX1 was a direct target of miR-497. (A) The expression levels of miR-497 in Ishikawa and 293T cells. (B) The seed regions of miR-497 of EMX1 3′-UTR and mu3′-UTR. (C) The luciferase activity of Ishikawa cells co-transfected with miR-497 mimics or mimics NC, and pMIR-LUC-EMX1-3′-UTR (EMX1 3′-UTR) or pMIR-LUC-EMX1-mu3′-UTR (EMX1 mu3′-UTR). (D) The luciferase activity of Ishikawa and 293T cells transfected with EMX1 3′-UTR or EMX1 mu3′-UTR. **P < 0.01, ***P < 0.001.
GSEA Identified EMX1-Related Oncogenic Signaling Pathways
The GSEA method is used to deal with continuous data and identify gene sets that are enriched at the top (overexpressed vs. control) or bottom (underexpressed) of a ranked gene list (Subramanian et al., 2005). We performed GSEA with the ordered list of preliminary screening DEGs according to their correlation with EMX1 expression. MSigDB Collection (c6.all.v6.2.symbols.gmt), which represents signatures of cellular pathways often dysregulated in cancer, was applied to our GSEA analysis at the phenotype of EMX1 expression level. Twenty-one signaling pathways were significantly enriched based on their NES (NOM P value < 0.05, FDR q-value < 0.25, Table 7). Supplementary Figure 2 shows that part of those oncogenic gene-involved cellular pathways, including PIGF, SRC, JNK KRAS.AMP.LUNG, PTEN, and E2F3, were differentially enriched in EMX1 high-expression phenotype.
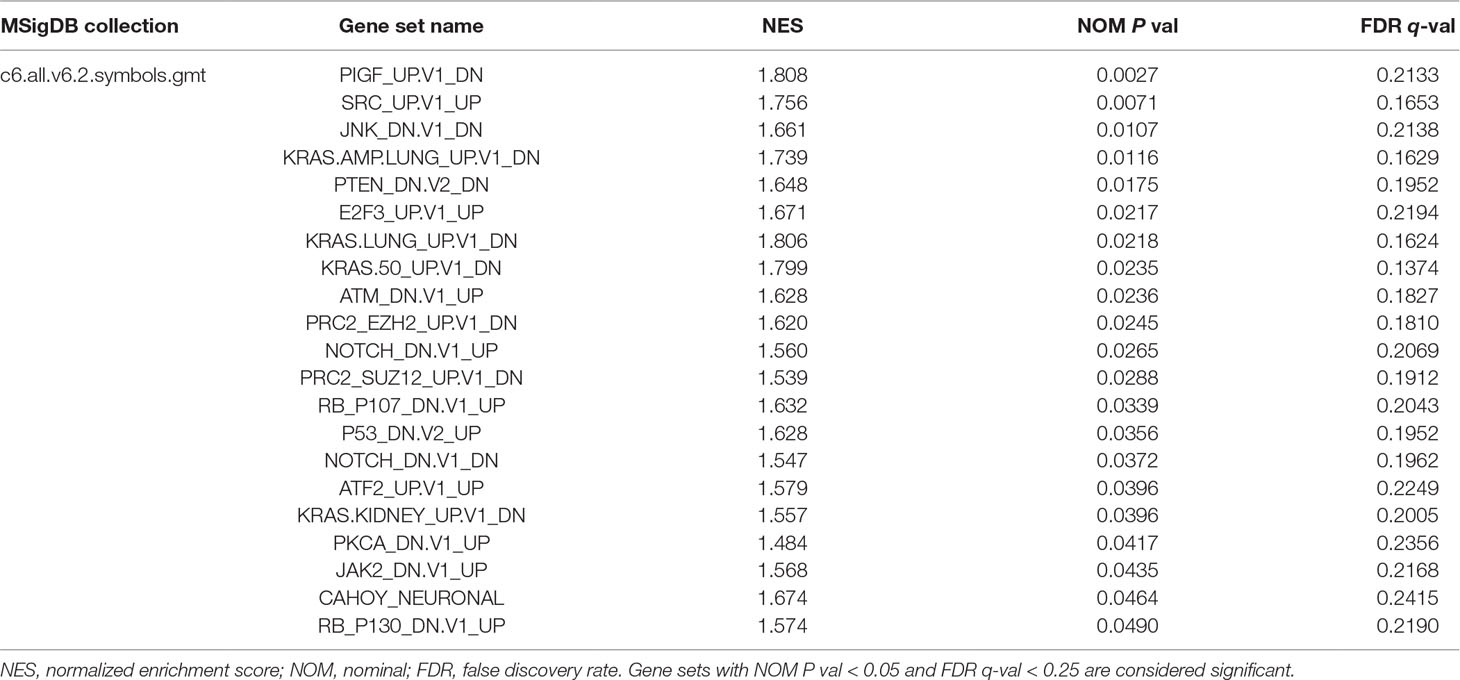
Table 7 Gene Set Enrichment Analysis in EMX1 phenotype.
Discussion
In the recent years, more and more evidence has revealed that miRNAs play important roles in the development and progression of tumors. They may serve as molecular indicators of prognosis and targets for oncotherapy (Jonas and Izaurralde, 2015). In endometrial cancer, a number of studies have looked at the miRNA profiles using tumor tissues or blood samples with a goal to identify disease-specific biomarkers (Srivastava et al., 2017). However, previous studies usually focused on one or two specific miRNAs and their target genes based on their potential to regulate diverse biological processes. Few researchers carried out studies on the relationship between whole miRNA profile and their potential target mRNAs based on their impact on patient survival.
A miRNA-based research data source of which was also a TCGA-UCEC project was carried out not long ago (Wang et al., 2018). It identified miRNAs that were correlated with the occurrence and progression of EC and established a six-miRNA (miR-15a. MIMAT0000068, miR-142. MIMAT0000433, miR-142. MIMAT0000434, miR-3170. MIMAT0015045, miR-1976. MIMAT0009451, and miR-146a. MIMAT0000449) expression signature as a predictor for the OS of patients with EC. In this study, the six-miRNA model was based on these 144 DE miRNAs resulting only from 15 patients with corresponding miRNA expression of the paired adjacent tissues. Another miRNA-based study on TCGA-UCEC project focused on the difference of miRNA profile between metastatic and nonmetastatic ECs (Zhu et al., 2018). It revealed that four miRNAs (miR-1247 miR-3200, miR-150, and miR-301b) were differently expressed between two groups, miR-1247 was associated with metastasis of EC to the lung, and miR-3200 is associated with the clinical stage of EC. The researchers also performed functional enrichment analysis with these predicted potential target genes of the four miRNAs, which showed that they might be involved in multiple pathways of cancer, including the Wnt, NOTCH, and TGF-β signaling pathways and signaling pathways regulating pluripotency of stem cells. However, miRNAs can target multiple genes, and a single gene can be targeted by multiple miRNAs (Ambros, 2004). Unilateral research is not enough to illustrate the problem.
In this study, for the first time, we performed Kaplan–Meier analysis for all the DE mRNAs and DE miRNAs in a larger number of EC cases from TCGA project with the help of bioinformation technology. As a result, 320 out of 4,613 DE mRNAs and 68 out of 531 DE miRNAs with a significantly poorer survival were determined. For the selected DE mRNAs associated with survival, KEGG pathway and GO enrichment analysis revealed that these selected DE mRNAs were significantly enriched in several functional pathways, including microRNAs in cancer. We further made target predictions through TargetScan and found that 5 out of 68 DE miRNAs could interact with eight DE mRNAs from 320 DE mRNAs. However, since the cutoff value for dividing groups (high expression and low expression) was based on the absolute median, most P values for different OS rates of the selected paired genes were of borderline statistical significance. Xie et al. (2018) used median mRNA expression level of ALKBH1 as a separation for high- and low-expression groups based on another TCGA project of glioblastoma (n = 488), which also resulted in a minor difference (P = 0.0386) and provided evidence for ALKBH1 to be a potential therapeutically targetable node. Another way to perform analysis was by using the computing cutoff expression level for the best separation with the smallest P value on survival and different numbers in two groups for each gene, which was also our initial design and resulted in a much larger number of DE mRNAs and miRNAs related to survival. To narrow the results of genes associated with survival, we finally chose the median truncation value, though some genes with potential predictability may be lost in our study.
Since each group of patients with a single miRNA or mRNA was made independently and their composition might be different, it would be interesting to determine differential expression of more than one miRNA or mRNA simultaneously in the patients. We tried to assess the differences between high and low rates of expression levels for the eight miRNA/mRNA pairs. As a result, patients with a low rate of three paired miRNA/mRNA (miR-497/EMX1, miR-23c/DMBX1, and miR-670/KCNS1) had a significantly poorer survival, while the rest seemed no different. As the presence of a single miRNA/mRNA pair is clearly not sufficient to predict the outcome of a patient having EC, we further verified the simultaneous presence of low rate of more than one miRNA/mRNA pair among these three selected pairs in the same group of patients. Surprisingly, the simultaneous presence of these selected low miRNA/mRNA pairs occurred in most EC patients and resulted in a significantly poorer survival rate, which strongly verified the validity and prediction capacity of our analysis. The experimental luciferase reporter assay confirmed that EMX1 was a direct target of miR-497. Clinical evaluation was assessed on miR-497 and EMX1 expression levels. Interestingly, they had an opposite significant relationship with several clinicopathologic characteristics besides survival. Multivariate analysis also demonstrated that miR-497 remained independent prognostic variables for OS. These data also provided evidence for the value of our prediction analysis in EC.
Among 68 DE miRNAs related to a poorer survival, 43 miRNAs were upregulated and 25 were downregulated. In 2013, Torres et al. conducted a study that aimed to reveal the relationship between DE miRNAs and EC (Torres et al., 2013). They found that upregulated miR-1228 was significantly associated with a poorer survival, which was consistent with our research result. Another research revealed that lower expression of miR-101, miR-10b, miR-139-5p, miR-152, miR-29b, and miR-455-5p was significantly correlated with poor OS and that decreased expression of miR-152 was a statistically independent risk factor for OS in EC (Hiroki et al., 2010). Mitamura et al. found that miR-31 was significantly upregulated in the EC patients with a high risk of recurrence compared with that observed in the low-risk patients, and this higher expression correlated with a poorer progression-free survival (Mitamura et al., 2014).
miRNAs mainly function and result in silence effect at posttranscriptional level by base pairing with the 3′-UTR of their target mRNAs completely or incompletely (Moran et al., 2017). In the present study, we predicted eight potential target DE mRNAs for the five DE miRNAs, including miR-211, miR-23c, miR-670, miR-497, and miR-4770. To our knowledge, there is no basic experimental evidence for the eight pairs of miRNAs and mRNAs predicted. One of the mostly studied miRNAs, miR-497, was recognized as a tumor suppressor in many cancers (Zhao et al., 2013; Du et al., 2015; Zhao et al., 2015; Yang et al., 2016), which was also consistent with our result in EC. During our manuscript preparation, a meta-analysis (Feng et al., 2018) on the prognostic role of miR-497 in different cancer patients revealed that high-expression levels of miR-497 are less possible to have lymph node metastasis and have better overall survival, which indicated that miR-497 might be a potential biomarker and could be used to predict the better prognosis of different cancer types. A recent study showed that miR-497 negatively regulated glioma cells by targeting oncogene Wnt3α and that reduced expression of miR-497 was associated with poor disease-free and overall survival rates (Lu et al., 2018). It could also suppress clear cell renal cell carcinoma by targeting PD-L1, which was an immune-related oncogene (Qu et al., 2018). miR-497 could target SERPINE-1 and induce reversion of epithelial-to-mesenchymal transition in cutaneous squamous cell carcinoma (Mizrahi et al., 2018). However, these targets of miR-497 did not appear as a DE mRNA associated with survival in the present study. EMX1, a potential target of miR-497 predicted through TargetScan, also had a very close relationship with EC clinicopathologies and patient survival, which was in contrast to the miR-497 in our study. Asada et al. revealed that a high quartile of EMX1 methylation level had a significant univariate HR and a multivariate-adjusted HR of developing authentic metachronous gastric cancers (Asada et al., 2015). Its methylation level was also differently expressed in hepatocellular carcinoma (HCC), which showed a potential role in the development of HCC (Sun et al., 2018). However, most of the research on EMX1 was focused on brain development (Lim et al., 2015; Kobeissy et al., 2016). The relationship between miR-497/EMX1 and cancer needs further study.
Testis-expressed 19 (TEX19) was another potential target gene of miR-497 in our study. Zhong. et al. revealed that TEX19 exhibited increased expression in high-grade tumors and might represent a novel cancer-testis gene related to the progression of bladder cancer (Zhong et al., 2016). TEX19 was also required to drive cell proliferation in a range of cancer cell types, and its expression was linked to a poor prognosis for breast cancer, kidney cancer, prostate cancer, and glioma cancer (Planells-Palop et al., 2017). However, further study on its role in EC is needed.
Bu Y. et al. identified that miR-211 directly targets to Bmal1 and Clock in Burkitt’s lymphoma, thereby suppressing both circadian oscillation and ongoing protein synthesis to facilitate tumor progression (Bu et al., 2018). miR-23c, as a target of lncRNA MALAT1, could directly repress its target ELAVL1 and inhibit hyperglycemia-induced cell pyroptosis (Li et al., 2017). Shi C. et al. showed evidence that miR-670 could induce cell proliferation in hepatocellular carcinoma by targeting PROX1 (Shi and Xu, 2016), while our study predicted that it might play an important role in EC progression via targeting Potassium Voltage-Gated Channel Modifier Subfamily S Member 1 (KCNS1), which was also a DE mRNA associated with survival in our study. Previous studies on KCNS1 mainly focused on pain (Tsantoulas et al., 2018; Wonkam et al., 2018). Besides the downregulated expression level of KCNS1 in metastatic breast carcinoma (Savci-Heijink et al., 2016), little research has been carried out on the relationship between KCNS1 and cancer.
DNA methyltransferase 3B (DNMT3B), one of the eight paired genes in our study, was a methyltransferase responsible for de novo DNA methylation. A previous study also showed that it was significantly upregulated in both grade I and grade III ECs as compared with normal controls (Jin et al., 2005). DNMT3B-involved divergent DNA methylation pathways and protein synthesis required for posttranscriptional regulation may be implicated in the development of type I and type II ECs (Xiong et al., 2005a; Xiong et al., 2005b). However, little is known about the relationships between EC and the rest of the potential miRNA-targeted DE mRNAs, including growth differentiation factor 7 (GDF7), sarcoglycan zeta (SGCZ), diencephalon/mesencephalon homeobox 1 (DMBX1), and fatty acid desaturase 6 (FADS6). Further research is also needed.
There were emerging researches on EC using TCGA data. Studies revealed that L1CAM (Dellinger et al., 2016) and TFAP2B (Wu and Zhang, 2018) were the two DEGs associated with advanced clinicopathologic characteristics and independent predictors for survival in EC based on TCGA-UCEC data. However, the massive narrowing down process for the DEG screening was not shown. Another study focused on the mutation-expression profile and looked for guidance for EC drug discovery (Wong et al., 2016). Thus, while these studies and ours were all based on TCGA data for EC, the different focus and data analysis approaches led to the discovery of different aspects of the disease.
In this study, we identified a series of DE mRNAs and DE miRNAs associated with survival in EC. Furthermore, we predicted eight pairs of DE miRNAs and their potential target DE mRNAs related to survival. Clinical evaluation of downregulated miR-497 and paired upregulated EMX1 confirmed the value of our prediction analysis in EC. The simultaneous presence of low rate of these selected low miRNA/mRNA pairs (miR-497/EMX1, miR-23c/DMBX1, and miR-670/KCNS1) might have a better prediction value. Therefore, further studies are required to validate these findings.
Author Contributions
XX, JW, and HZ designed the study, checked the data, and prepared the manuscript. TL, YW, JF, and QY performed data collection and statistical analysis. TL searched the literature and took part in the manuscript preparation. JW and HZ conceived and supervised the project.
Funding
Funding was provided by the Natural Science Foundation of Jiangsu Province (BK20151096) and the Key Projects of National Health and Family Planning Commission of Nanjing City (ZKX17015) to HZ and the Fundamental Research Funds for the Central Universities (3332018179) to JW.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Juan Chen from CCN Department of ZTE for her computer technology support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00743/full#supplementary-material
References
Asada, K., Nakajima, T., Shimazu, T., Yamamichi, N., Maekita, T., Yokoi, C., et al. (2015). Demonstration of the usefulness of epigenetic cancer risk prediction by a multicentre prospective cohort study. Gut 64, 388–396. doi: 10.1136/gutjnl-2014-307094
Binder, P. S., Mutch, D. G. (2014). Update on prognostic markers for endometrial cancer. Women’s Heal. 10, 277–288. doi: 10.2217/WHE.14.13
Bu, Y., Yoshida, A., Chitnis, N., Altman, B. J., Tameire, F., Oran, A., et al. (2018). A PERK–miR-211 axis suppresses circadian regulators and protein synthesis to promote cancer cell survival. Nat. Cell Biol. 20, 104–115. doi: 10.1038/s41556-017-0006-y
Burk, R. D., Chen, Z., Saller, C., Tarvin, K., Carvalho, A. L., Scapulatempo-Neto, C., et al. (2017). Integrated genomic and molecular characterization of cervical cancer. Nature 543, 378–384. doi: 10.1038/nature21386
Canlorbe, G., Wang, Z., Laas, E., Bendifallah, S., Castela, M., Lefevre, M., et al. (2016). Identification of microRNA expression profile related to lymph node status in women with early-stage grade 1–2 endometrial cancer. Mod. Pathol. 29, 391–401. doi: 10.1038/modpathol.2016.30
Chen, S., Sun, K., Liu, B., Zong, Z., Zhao, Y. (2016). MicroRNA-505 functions as a tumor suppressor in endometrial cancer by targeting TGF-α. Mol. Cancer 15, 11. doi: 10.1186/s12943-016-0496-4
Dellinger, T. H., Smith, D. D., Ouyang, C., Warden, C. D., Williams, J. C., Han, E. S. (2016). L1CAM is an independent predictor of poor survival in endometrial cancer—an analysis of The Cancer Genome Atlas (TCGA). Gynecol. Oncol. 141, 336–340. doi: 10.1016/j.ygyno.2016.02.003
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Du, M., Shi, D., Yuan, L., Li, P., Chu, H., Qin, C., et al. (2015). Circulating miR-497 and miR-663b in plasma are potential novel biomarkers for bladder cancer. Sci. Rep. 5, 10437. doi: 10.1038/srep10437
Dunham, I., Kundaje, A., Aldred, S. F., Collins, P. J., Davis, C. A., Doyle, F., et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Duursma, A. M., Kedde, M., Schrier, M., le Sage, C., Agami, R. (2008). miR-148 targets human DNMT3b protein coding region. RNA 14, 872–877. doi: 10.1261/rna.972008
Dykes, I. M., Emanueli, C. (2017). Transcriptional and post-transcriptional gene regulation by long non-coding RNA. Genomics Proteomics Bioinformatics 15, 177–186. doi: 10.1016/j.gpb.2016.12.005
Falcon, S., Gentleman, R. (2007). Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258. doi: 10.1093/bioinformatics/btl567
Feng, J., Gu, X., Liu, L., Lu, M., Ma, X., Cao, Y., et al. (2018). Prognostic role of microRNA-497 in cancer patients: a meta-analysis. J. Cancer. 9, 3334–3342. doi: 10.7150/jca.25514
Fromm, B., Billipp, T., Peck, L. E., Johansen, M., Tarver, J. E., King, B. L., et al. (2015). A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 49, 213–242. doi: 10.1146/annurev-genet-120213-092023
Hiroki, E., Akahira, J., Suzuki, F., Nagase, S., Ito, K., Suzuki, T., et al. (2010). Changes in microRNA expression levels correlate with clinicopathological features and prognoses in endometrial serous adenocarcinomas. Cancer Sci. 101, 241–249. doi: 10.1111/j.1349-7006.2009.01385.x
Jin, F., Dowdy, S. C., Xiong, Y., Eberhardt, N. L., Podratz, K. C., Jiang, S. (2005). Up-regulation of DNA methyltransferase 3B expression in endometrial cancers. Gynecol. Oncol. 96, 531–538. doi: 10.1016/j.ygyno.2004.10.039
Jonas, S., Izaurralde, E. (2015). Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 16, 421–433. doi: 10.1038/nrg3965
Kobeissy, F. H., Hansen, K., Neumann, M., Fu, S., Jin, K., Liu, J. (2016). Deciphering the role of Emx1 in neurogenesis: a neuroproteomics approach. Front. Mol. Neurosci. 98, 1–16. doi: 10.3389/fnmol.2016.00098
Li, X., Zeng, L., Cao, C., Lu, C., Lian, W., Han, J., et al. (2017). Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp. Cell Res. 350, 327–335. doi: 10.1016/j.yexcr.2016.12.006
Lim, J. W. C., Donahoo, A.-L. S., Bunt, J., Edwards, T. J., Fenlon, L. R., Liu, Y., et al. (2015). EMX1 regulates NRP1-mediated wiring of the mouse anterior cingulate cortex. Development 142, 3746–3757. doi: 10.1242/dev.119909
Lu, F., Ye, Y., Zhang, H., He, X., Sun, X., Yao, C., et al. (2018). miR-497/Wnt3a/c-jun feedback loop regulates growth and epithelial-to-mesenchymal transition phenotype in glioma cells. Int. J. Biol. Macromol. 120, 985–991. doi: 10.1016/j.ijbiomac.2018.08.176
Mitamura, T., Watari, H., Wang, L., Kanno, H., Kitagawa, M., Hassan, M., et al. (2014). microRNA 31 functions as an endometrial cancer oncogene by suppressing Hippo tumor suppressor pathway. Mol. Cancer 13, 97. doi: 10.1186/1476-4598-13-97
Mizrahi, A., Barzilai, A., Gur-Wahnon, D., Ben-Dov, I. Z., Glassberg, S., Meningher, T., et al. (2018). Alterations of microRNAs throughout the malignant evolution of cutaneous squamous cell carcinoma: the role of miR-497 in epithelial to mesenchymal transition of keratinocytes. Oncogene 37, 218–230. doi: 10.1038/onc.2017.315
Moran, Y., Agron, M., Praher, D., Technau, U. (2017). The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 1, 0027. doi: 10.1038/s41559-016-0027
Planells-Palop, V., Hazazi, A., Feichtinger, J., Jezkova, J., Thallinger, G., Alsiwiehri, N. O., et al. (2017). Human germ/stem cell-specific gene TEX19 influences cancer cell proliferation and cancer prognosis. Mol. Cancer 16,aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa 84. doi: 10.1186/s12943-017-0653-4
Qu, F., Ye, J., Pan, X., Wang, J., Gan, S., Chu, C., et al. (2018). MicroRNA-497-5p down-regulation increases PD-L1 expression in clear cell renal cell carcinoma. J. Drug Target. 0, 1–8. doi: 10.1080/1061186X.2018.1479755
Reyes, H. D., Miecznikowski, J., Gonzalez-Bosquet, J., Devor, E. J., Zhang, Y., Thiel, K. W., et al. (2017). High stathmin expression is a marker for poor clinical outcome in endometrial cancer: An NRG oncology group/gynecologic oncology group study. Gynecol. Oncol. 146, 247–253. doi: 10.1016/j.ygyno.2017.05.017
Sacco, L., Masotti, A. (2012). Recent insights and novel bioinformatics tools to understand the role of micrornas binding to 5' untranslated region. Int. J. Mol. Sci. 14, 480–495. doi: 10.3390/ijms14010480
Savci-Heijink, C. D., Halfwerk, H., Koster, J., van de Vijver, M. J. (2016). A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res. Treat. 156, 249–259. doi: 10.1007/s10549-016-3741-z
Shi, C., Xu, X. (2016). MiR-670-5p induces cell proliferation in hepatocellular carcinoma by targeting PROX1. Biomed. Pharmacother. 77, 20–26. doi: 10.1016/j.biopha.2015.07.030
Siegel, R. L., Miller, K. D., Jemal, A. (2018). Cancer statistics, 2018. CA. Cancer J. Clin. 68, 7–30. doi: 10.3322/caac.21442
Smith, R. A., Andrews, K. S., Brooks, D., Fedewa, S. A., Manassaram-Baptiste, D., Saslow, D., et al. (2018). Cancer screening in the United States, 2018: a review of current American Cancer Society guidelines and current issues in cancer screening. CA. Cancer J. Clin. 68, 297–316. doi: 10.3322/caac.21446
Srivastava, S. K., Ahmad, A., Zubair, H., Miree, O., Singh, S., Rocconi, R. P., et al. (2017). MicroRNAs in gynecological cancers: small molecules with big implications. Cancer Lett. 407, 123–138. doi: 10.1016/j.canlet.2017.05.011
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550. doi: 10.1073/pnas.0506580102
Sun, X.-J., Wang, M.-C., Zhang, F.-H., Kong, X. (2018). An integrated analysis of genome-wide DNA methylation and gene expression data in hepatocellular carcinoma. FEBS Open Bio. 8, 1093–1103. doi: 10.1002/2211-5463.12433
Torres, A., Torres, K., Pesci, A., Ceccaroni, M., Paszkowski, T., Cassandrini, P., et al. (2013). Diagnostic and prognostic significance of miRNA signatures in tissues and plasma of endometrioid endometrial carcinoma patients. Int. J. Cancer 132, 1633–1645. doi: 10.1002/ijc.27840
Tsantoulas, C., Denk, F., Signore, M., Nassar, M. A., Futai, K., McMahon, S. B. (2018). Mice lacking Kcns1 in peripheral neurons show increased basal and neuropathic pain sensitivity. Pain 159, 1641–1651. doi: 10.1097/j.pain.0000000000001255
Wang, Y., Xu, M., Yang, Q. (2018). A six-microRNA signature predicts survival of patients with uterine corpus endometrial carcinoma. Curr. Probl. Cancer. 43, 1–10. doi: 10.1016/j.currproblcancer.2018.02.002
Wong, H. S.-C., Juan, Y.-S., Wu, M.-S., Zhang, Y.-F., Hsu, Y.-W., Chen, H.-H., et al. (2016). Integrative bioinformatic analyses of an oncogenomic profile reveal the biology of endometrial cancer and guide drug discovery. Oncotarget 7, 5909–5923. doi: 10.18632/oncotarget.6716
Wonkam, A., Mnika, K., Ngo Bitoungui, V. J., Chetcha Chemegni, B., Chimusa, E. R., Dandara, C., et al. (2018). Clinical and genetic factors are associated with pain and hospitalisation rates in sickle cell anaemia in Cameroon. Br. J. Haematol. 180, 134–146. doi: 10.1111/bjh.15011
Wu, H., Zhang, J. (2018). Decreased expression of TFAP2B in endometrial cancer predicts poor prognosis: a study based on TCGA data. Gynecol. Oncol. 149, 592–597. doi: 10.1016/j.ygyno.2018.03.057
Xie, Q., Wu, T., Gimple, R., Li, Z., Prager, B., Wu, Q., et al. (2018). N6-methyladenine DNA modification in glioblastoma. Cell. 175, 1–16. doi: 10.1016/j.cell.2018.10.006
Xiong, Y., Dowdy, S. C., Podratz, K. C., Jin, F., Attewell, J. R., Eberhardt, N. L., et al. (2005a). Histone deacetylase inhibitors decrease DNA methyltransferase-3B messenger RNA stability and down-regulate de novo DNA methyltransferase activity in human endometrial cells. Cancer Res. 65, 2684–2689. doi: 10.1158/0008-5472.CAN-04-2843
Xiong, Y., Dowdy, S. C., Xue, A., Shujuan, J., Eberhardt, N. L., Podratz, K. C., et al. (2005b). Opposite alterations of DNA methyltransferase gene expression in endometrioid and serous endometrial cancers. Gynecol. Oncol. 96, 601–609. doi: 10.1016/j.ygyno.2004.11.047
Yang, G., Xiong, G., Cao, Z., Zheng, S., You, L., Zhang, T., et al. (2016). miR-497 expression, function and clinical application in cancer. Oncotarget 7, 21–23. doi: 10.18632/oncotarget.10152
Zhao, W., Wang, Y., An, Z., Shi, C., Zhu, G., Wang, B., et al. (2013). Downregulation of miR-497 promotes tumor growth and angiogenesis by targeting HDGF in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 435, 466–471. doi: 10.1016/j.bbrc.2013.05.010
Zhao, X., Zhao, Z., Xu, W., Hou, J., Du, X. (2015). Down-regulation of miR-497 is associated with poor prognosis in renal cancer. Int. J. Clin. Exp. Pathol. 8, 758–764.
Zhong, J., Chen, Y., Liao, X., Li, J., Wang, H., Wu, C., et al. (2016). Testis expressed 19 is a novel cancer-testis antigen expressed in bladder cancer. Tumor Biol. 37, 7757–7765. doi: 10.1007/s13277-015-4567-8
Zhou, H., Xu, X., Xun, Q., Yu, D., Ling, J., Guo, F., et al. (2012). microRNA-30c negatively regulates endometrial cancer cells by targeting metastasis-associated gene-1. Oncol. Rep. 27, 807–812. doi: 10.3892/or.2011.1574
Keywords: endometrial carcinoma, miR-497, EMX1, survival, TCGA
Citation: Xu X, Liu T, Wang Y, Fu J, Yang Q, Wu J and Zhou H (2019) miRNA–mRNA Associated With Survival in Endometrial Cancer. Front. Genet. 10:743. doi: 10.3389/fgene.2019.00743
Received: 29 September 2018; Accepted: 16 July 2019;
Published: 20 August 2019.
Edited by:
Ge Shan, University of Science and Technology of China, ChinaReviewed by:
Andrés Fernando Muro, International Centre for Genetic Engineering and Biotechnology, ItalyFeng Wang, Emory University School of Medicine, United States
Copyright © 2019 Xu, Liu, Wang, Fu, Yang, Wu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaijun Zhou, emhvdWhqMjAwN0AxMjYuY29t; Jun Wu, aWFtd3VqdW4yMDA4QDE2My5jb20=
†These authors share first authorship.