 Yongjian Yue1
Yongjian Yue1 Qijun Huang1
Qijun Huang1 Peng Zhu2Pan Zhao3Xinjuan Tan1
Peng Zhu2Pan Zhao3Xinjuan Tan1 Shengguo Liu1Shulin Li1Xuemei Han1Linling Cheng4
Shengguo Liu1Shulin Li1Xuemei Han1Linling Cheng4 Bo Li5*Yingyun Fu1*
Bo Li5*Yingyun Fu1*- 1Key Laboratory of Shenzhen Respiratory Diseases, Department of Pulmonary and Critical Care Medicine, Shenzhen Institute of Respiratory Disease, The First Affiliated Hospital of Southern University of Science and Technology, The Second Clinical Medical College of Jinan University, Shenzhen People’s Hospital, Shenzhen, China
- 2Central Lab of Shenzhen Pingshan People’s Hospital, Shenzhen, China
- 3Clinical Medical Research Center, The First Affiliated Hospital of Southern University of Science and Technology, The Second Clinical Medical College of Jinan University, Shenzhen People’s Hospital, Shenzhen, China
- 4State Key Laboratory of Respiration Diseases, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 5Department of Pediatric, First Affiliated Hospital of Southern University of Science and Technology, The Second Clinical Medical College of Jinan University, Shenzhen People’s Hospital, Shenzhen, China
Primary ciliary dyskinesia (PCD), a rare genetic disorder, is mostly caused by defects in more than 40 known cilia structure-related genes. However, in approximately 20–35% of patients, it is caused by unknown genetic factors, and the inherited pathogenic factors are difficult to confirm. Kartagener syndrome (KTS) is a subtype of PCD associated with situs inversus, presenting more complex genetic heterogeneity. The aim of this study was to identify pathogenic mutations of candidate genes in Chinese patients with KTS and investigate the activation of the heterotaxy-related NOTCH pathway. Whole-exome sequencing was conducted in five patients with KTS. Pathogenic variants were identified using bioinformatics analysis. Candidate variants were validated by Sanger sequencing. The expression of the NOTCH pathway target genes was detected in patients with KTS. We identified 10 KTS-associated variants in six causative genes, namely, CCDC40, DNAH1, DNAH5, DNAH11, DNAI1, and LRRC6. Only one homozygote mutation was identified in LRRC6 (c.64dupT). Compound heterozygous mutations were found in DNAH1 and DNAH5. Six novel mutations were identified in four genes. Further analyses showed that the NOTCH pathway might be activated in patients with KTS. Overall, our study showed that compound heterozygous mutations widely exist in Chinese KTS patients. Our results demonstrated that the activation of the NOTCH pathway might play a role in the situs inversus pathogenicity of KTS. These findings highlight that Kartagener syndrome might be a complex genetic heterogeneous disorder mediated by heterozygous mutations in multiple PCD- or cilia-related genes.
Introduction
Primary ciliary dyskinesia (PCD) is a sinopulmonary disease caused by dysfunctional immotile or dyskinetic respiratory cilia. It is an autosomal recessive disorder and clinically characterized by respiratory distress, tympanitis, sinusitis, bronchiectasis, and male infertility in approximately 50% of patients. Male infertility usually caused by aberrant sperm flagella function results from immotile or dyskinetic cilia. Further, PCD affects approximately 1 in 15,000 individuals of the general population (Bush et al., 2007). It was reported that patients with heterotaxy and airway ciliary dysfunction were enriched for mutations in the PCD genes, but interestingly, these were all heterozygous (Wilson et al., 2009). Kartagener syndrome (KTS;MIM# 244400), a subtype of PCD presenting with the phenotype situs inversus, might have a more complex genetic heterogeneity.
The nasal nitric oxide measurement and brush biopsy are typical diagnostic methods for PCD. The diagnosis of PCD is challenging due to phenotypic heterogeneity. Moreover, owing to the non-specificity of symptoms and lack of an early diagnostic method, the diagnosis of PCD is usually missed or delayed (Hosie et al., 2014; Lucas et al., 2014). Because of the lack of a reference standard for PCD diagnosis, a combination of tests is required (Lucas et al., 2016). Definitive diagnosis is usually confirmed through transmission electron microscopy (TEM) to visualize the ultrastructure of cilia, but approximately 30% of patients show no defect in TEM. Furthermore, there is a lack of evidence-based medicine in the management of PCD (Lucas et al., 2014). Therefore, the early diagnosis of PCD is important to select an appropriate management to prevent irreversible damage. With the rapid advances in next-generation sequencing, genetic-based studies have increased our understanding of the pathogenesis of even PCD with remarkable genetic heterogeneity. Diagnostic gene panel has been used for several diseases, and a genetic testing panel for PCD diagnosis is being developed (Paff et al., 2018; Takeuchi et al., 2018). As an autosomal recessive disorder, the inherited mutations of ciliary function-related genes are the main genetic risk factors of PCD. Most of the genetic mutations in PCD contribute to defective cilia structure. Some patients with PCD due to mutations also showed normal motility and ultrastructure, but reduced cilia numbers (Wallmeier et al., 2014). Mutations of ciliary function-related genes can cause immotile cilia due to congenital ultrastructural abnormalities. The prevalence of PCD has a relatively higher rate in consanguineous families (Onoufriadis et al., 2014). To date, genetic mutations in over 40 genes, including DNAH5, CCD39, and DYX1C1, account for an estimated 65% of individuals (Olbrich et al., 2002; Merveille et al., 2011; Tarkar et al., 2013). DNAH5 mutations cause approximately 50% of outer dynein arm (ODA) defects, whereas SPAG1 mutations cause approximately 10% of inner dynein arm (IDA) and ODA defects (Hornef et al., 2006; Knowles et al., 2013; Lucas et al., 2014).
The pathogenic genes reported previously have improved genetic-based diagnosis. Based on the pathogenic mutations of known genes involved in PCD, genetic testing presents a possibility to identify high-risk populations among familial siblings with PCD. However, the genetic factors in approximately 20–35% of patients with PCD are unknown, and the pathogenicity of novel mutations in known PCD genesis is also difficult to confirm based on a functional study. The genetic defect has not yet been well elucidated in some patients. Previously, one study demonstrated that GALNT11 is a heterotaxy disease candidate gene (Fakhro et al., 2011). Further, another study showed that GALNT11 O-glycosylation modified NOTCH, which balances motile and immotile cilia in left–right patterning and affects the etiology of heterotaxy (Boskovski et al., 2013). However, whether the NOTCH pathway is associated with KTS remains unclear. In the present study, we aimed to identify pathogenic mutations and investigate the NOTCH pathway activation in Chinese patients with KTS. Our study would expand our knowledge on the spectrum of PCD- or cilia-related genes, thereby contributing to the genetic diagnosis and counseling of KTS patients.
Materials and Methods
Subjects
Five patients with KTS and nine familial control subjects were recruited for the present study conducted in the Second Clinical Medical College of Shenzhen People’s Hospital and Institute of Guangzhou Respiratory Diseases (Guangdong, China) from December 2013 to July 2019. The control subjects included in the study were the parents of KT4 and LEI, father of KT2, mother of KT5, and the father, sister, brother, and uncle of KT1 (Table 1). Patients with KTS were diagnosed based on the presence of chronic sinusitis, situs inversus, bronchiectasis, and ciliary ultrastructural defects. Demographic characteristics and medical history were recorded, including age and clinical test results. Informed consent was obtained from each patient enrolled in the study. Research and ethics approval was obtained from the ethics committee of the Shenzhen People’s Hospital.
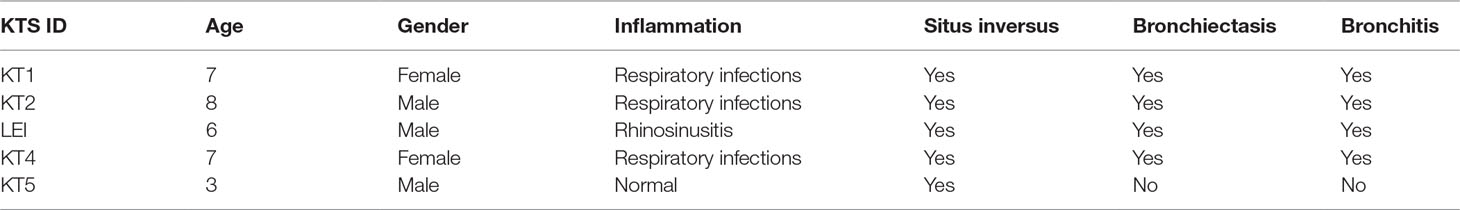
Table 1 Demographics of the KTS participants.
Bronchoscopy and Electron Microscopy
High-resolution computed tomography and magnetic resonance imaging were performed to check for bronchiectasis or situs inversus. Nasal mucosa and bronchial cilia were obtained for electron microscopy. Transmission electron microscopic examination (Kingmed, China) was performed to identify the ultrastructural abnormality of cilia. Bronchoscopic examination was performed for PCD diagnosis.
Next-Generation Sequencing and Bioinformatics Analysis
High-quality genomic DNA was extracted from blood samples of patients, and exome capture was performed using the Agilent SureSelect V6 Capture Kit according to the manufacturer’s protocols (Agilent, CA, USA). An enriched library targeting the exome was subjected to massive parallel sequencing using NovaSeq platform (Illumina). The variants were called using Genome Analysis Toolkit (GATK) and annotated using ANNOVAR (Wang et al., 2010). We only focused on the genes with nonsynonymous, Indel, or deletion variants in the coding region. Later, all variants were filtered by region, frequency (MAF > 5%), and functional prediction. The following databases were used as the reference: 1,000 genome, dbSNP, and gnomAD (http://gnomad.broadinstitute.org/).
Candidate Gene Selection and Pathogenicity Assessment
Previous studies have reported that more than 40 genes are related to PCD. Based on previous panel analyses and genetic studies on PCD (Guo et al., 2017; Takeuchi et al., 2018; Yang et al., 2018), 43 genes were selected as candidate genes, which were highly associated with the pathogenesis of PCD (Supplemental Table S1). A gene-based filtering analysis was conducted among all variants of the five patients with PCD. To assess the pathogenesis of the variants, various protein functions were predicted using bioinformatic programs, including Sift, Polyphen-2, MutationTaster, and CADD (combined annotation dependent depletion). The interpretation of variants followed the American College of Medical Genetics and Genomics (ACMG) guideline (Richards et al., 2015). An evolutionary conservation analysis of the identified variants was conducted using CLUSTALW on orthologs of different mammal species.
Sanger Sequencing Validation
Genomic DNA was extracted from whole blood samples using the QIAamp DNA Blood Kit (QIANGEN, Hilden, Germany) according to the manufacturer’s instruction. All primers were designed using IDT PrimerQuest Tool online software (https://sg.idtdna.com/Primerquest/Home/Index). All gene segments were amplified using the standard PCR. The candidate variants among patients with PCD and control subjects were genotyped using Sanger genotyping platform. The sequences were analyzed using Chromas software (Technelysium Pty Ltd).
Detection of NOTCH Signaling Pathway Activation
Blood samples of KTS and control subjects were collected into Na-citrate-containing Vacutainer tube. Total RNA was extracted from whole blood using animal tissue RNA Purification Kit (TRK-1002, LC Sciences, Houston, USA). To confirm whether the Notch pathway was abnormally activated in patients with PCD, real-time PCR was conducted for the Notch pathway–related and downstream target genes, including ADAM17, Cyclin D3, p21, MYC, and HES1. Primers were obtained from the database of PrimerBank (https://pga.mgh.harvard.edu/primerbank/) or designed using Integrated DNA Technologies online software (Supplemental Table S2).
Statistical Analysis
All statistical analyses were carried out using the software SPSS 19 (IBM, New York, USA). Chi square and Bonferroni (post hoc) tests were used for cross-table and multiple comparisons, respectively. The results with P < 0.05 were considered statistically significant.
Results
Characteristics of Patients and Diagnosis of KTS
All patients with KTS had a history of recurrent cough, expectoration, and rhinorrhea since the age of diagnosis at childhood (Table 1). Chest computed tomography images showed that all the patients had symptoms of KTS, including the classic phenotype of situs inversus (Figure 1). The family members (father) of two subjects showed slight bronchiectasis or sinusitis, but without situs inversus. These were taken as reference control subjects without KTS. Routine semen analysis and electron microscopic examination showed infertility in adult male patients and multiple ultrastructural abnormalities of cilia in both bronchial and nasal mucosa, respectively (Figure 2). The structural abnormalities of cilia included disarrangement, reduction, and absence of IDA and ODA structures of the subject LEI.
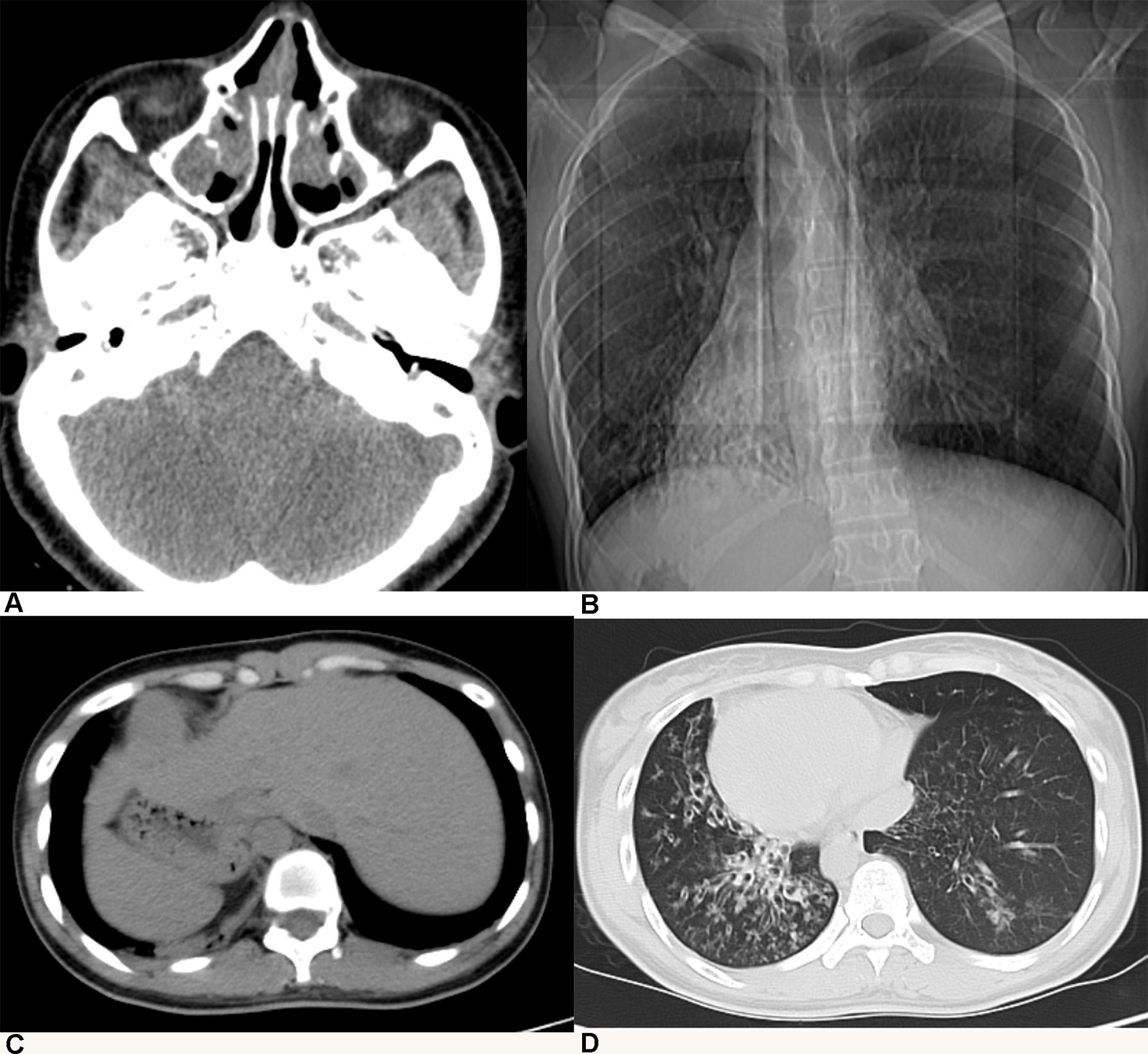
Figure 1 Patients with Kartagener syndrome diagnosed by radiological examination: (A) chronic sinusitis, (B, C) situs inversus, and (D) diffuse bronchiectasis.

Figure 2 Electron microscopic examination of bronchial cilia showed abnormal 9 + 2 structure and deletion of inner or outer dynein arms.
Candidate Gene Rare Variants Identified Using Filtering Analysis
To identify mutations in the Chinese population with KTS, 10 familial normal control subjects were used as annotation controls. We filtered variants to obtain rare mutations with the standard criteria as described in the methodology. With further filtering using integrated bioinformatics pipeline and software, we identified 30 variants of candidate genes (Supplemental Table S3). All KTS subjects carried coding mutations in DNAH1 in the present study. Regarding the known gene variants, we changed the filtering cutoff frequency to 0.01 (gnomAD_all) to obtain all the rare coding variants in the known candidate genes of KTS. In total, 19 variants of the known candidate genes were identified (Tables 2 and 3). The pathogenicity of variants was assessed by four in silico software. All variants were interpreted based on the ACMG guideline (Supplemental Table S3). Nine variants were considered not associated with disease as they were benign or uncertain significance variants (VUS) based on the frequency in the Asian population and ACMG classification (Table 2). Six novel mutations were identified in four genes, which were not included in the dbSNP and gnomAD databases (Table 3). Two novel mutations of DNAH5 were identified in the patient KT4. Only one homozygous variant was identified in LRRC6 (c.64dupT), which is the likely cause for the genetic disorder in KT1 (Table 3). The two heterozygous mutations of DNAH1 in KT1 were compound heterozygous. The variants in DNAI1 and DNAH1 might contribute to the phenotype of KT1. Advanced analysis showed no homozygous mutations in the known candidate genes among patients with KTS (data not shown).

Table 2 The identified likely benign variants of candidate genes in Kartagener syndrome (KTS).
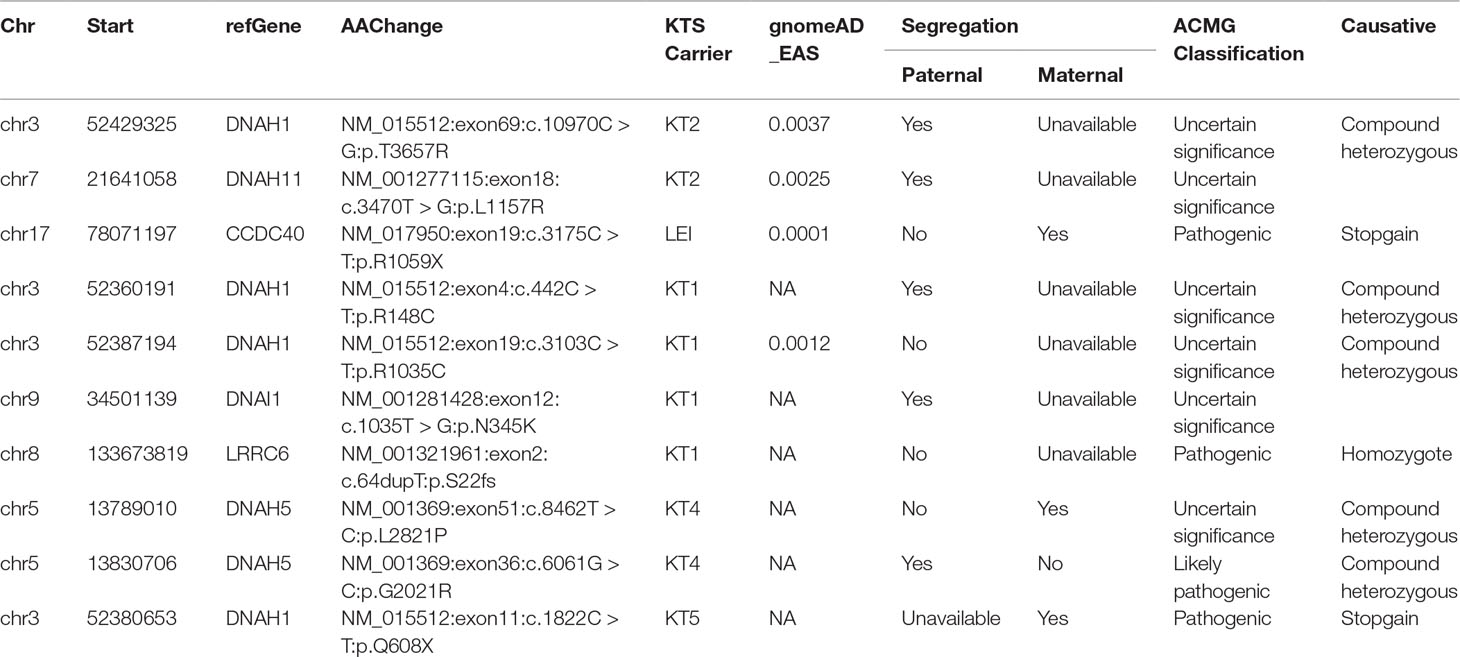
Table 3 Variants pathogenicity assessment and interpretation by the ACMG guideline.
Variants Validation and Pathogenicity Assessment
ACMG prediction and functional analysis showed that four variants are pathogenic, and six variants are of uncertain significance, which were considered KTS-associated variants (Table 3). Sanger sequencing validated all candidate variants including LRRC6, DNAI1, and CCDC40 (Figure 3A). Our study also showed the two rare variants in DNAH5 of KT4 were inherited from the mother and father, respectively, indicating that they are compound heterozygous mutations (Table 3, Supplemental Figure S1). The variant of c.6061G > C in DNAH5 changes the last nucleotide of exon 36 and completely abolishes the splicing signal, which indicated that it is more likely a pathogenic variant than a VUS. Only c.10970C > G of DNAH1 was presented in the father of KT2, indicating that c.4987C > T and c.10970C > G are also highly likely to be compound heterozygous mutations. The variants in DNAH11 might contribute to the phenotype of KT2. Hence, these identified compound heterozygous mutations were the genetic factors, which might cause the KT1, KT2, and KT4 phenotype disorder, but more evidence is needed.
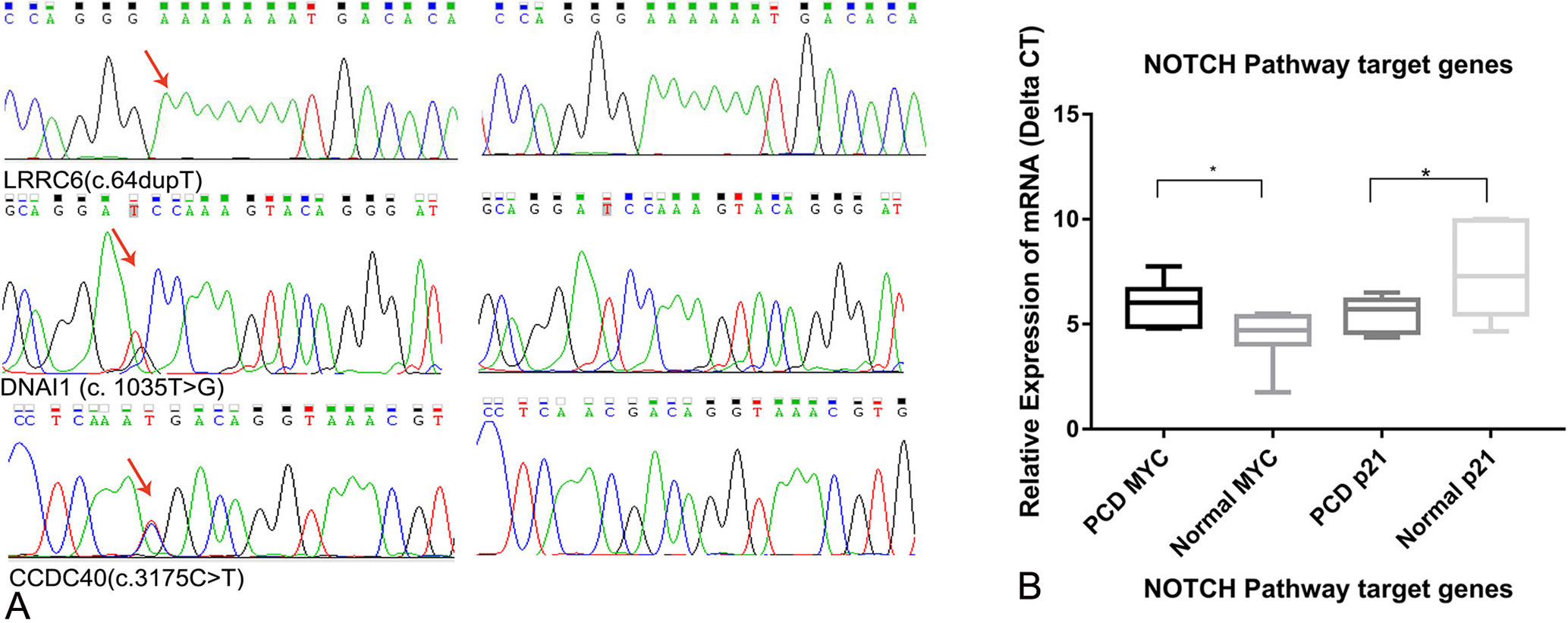
Figure 3 (A) Sanger validation results of the variants DNAI1 (c. 1035T > G), CCDC40 (c. 3175 C > T), and LRRC6 (c.64dupT). (B) Relative expression of the NOTCH pathway target genes MYC and p21 (*p<0.05).
The two heterozygous pathogenic variants were present in LEI and KT5, but this alone does not explain KTS in LEI and KT5, respectively. Since KTS is a recessive genetic disorder, the phenotypic defect in LEI and KT5 may be caused by trans-heterozygous interactions between the carried causative gene and other PCD genes (Li et al., 2016). In summary, in our study, 10 KTS-associated variants were identified among six causative genes of KTS, including four pathogenic variants.
Activation of the NOTCH Signaling Pathway by GALNT11
Real-time expression analysis showed that the expression of MYC and p21 was significantly different among patients with KTS and normal individuals. The expression of p21 was up-regulated by 5.03-fold in patients with KTS compared with that in normal individuals, but that of MYC was down-regulated by 0.29-fold (Figure 3B). Our study demonstrated the NOTCH1 pathway might be abnormally activated in patients with KTS, but the underlying mechanism is still unknown. No significant difference was found in terms of NOTCH1 pathway target gene expression among GALNT11 mutation carriers and controls.
Discussion
In this study, we investigated five Chinese patients with KTS and identified 10 KTS-associated variants that overlapped in six causative genes, namely, CCDC40, DNAH1, DNAH5, DNAH11, DNAI1, and LRRC6. Compound heterozygous mutations were found in DNAH1 and DNAH5. Further analyses showed that the NOTCH pathway might be activated in patients with KTS. Our study showed that patients with KTS carried multiple PCD candidate genes with rare compound heterozygous mutations in causative genes of DNAH1 and DNAH5.
Primary ciliary dyskinesia is remarkably heterogeneous although its inheritance mostly follows an autosomal recessive pattern (Djakow et al., 2016). To date, various gene mutations in over 40 causative genes have been reported, and approximately 35% of PCD remain genetically elusive (Kurkowiak et al., 2015; Yang et al., 2018). A series of monogenic mutations has been reported to contribute to the abnormal structure of motile cilia. The complete or partial deletion of IDAs or ODAs is a common abnormality. Ten KTS-associated variants in six causative genes were identified in our Chinese patients with KST. Defects in the DNAH5 gene could cause ODA deletion (Hornef et al., 2006). Two novel heterozygous mutations in the DNAH5 gene were observed in one patient with PCD, which might be highly deleterious, using in silico prediction methods. One stop–gain mutation was found in CCDC40. Pathogenic mutations in CCDC40 cause IDA defects and microtubular disorganization (Becker-Heck et al., 2011). The frame-shift homozygous mutation of LRRC6 might cause both ODA and IDA defects (Kott et al., 2012). The present study showed that most high-frequency mutations were present in the causative gene DNAH1. Six mutations in DNAH1 were identified in all individuals, indicating that DNAH1 might be a high-risk causative gene in Chinese patients with KTS. The other mutations were identified in PCD causative genes, namely, DNAI2, DNAAF1, DNAAF2, and RPGR, but with uncertain significance (Supplementary Table S3). Further functional investigation would aid in confirming the role of these mutations in the motile cilia structure of KTS.
GALNT11 is a polypeptide N-acetylgalactosaminyl transferase that can catalyze the initiation of protein O-linked glycosylation. O-Glycosylation of NOTCH1 promotes its activation, modulating the essential balance between motile and immotile cilia in the left–right organizer to determine laterality (Boskovski et al., 2013). In the present study, we found a mutation of GALNT11 in three patients with KTS. The missense mutation (c.G71A) was located in the highly conserved domain of GALNT11, and it might disturb the protein function. Therefore, we further analyzed the activation of the NOTCH pathway between patients with KTS and normal controls. Our results showed that the expression of down-stream target genes of p21 was significantly up-regulated in patients with KTS. GALNT11 might contribute to the phenotype of situs inversus in KTS.
We identified compound heterozygous mutations in the causative genes DNAH1 and DNAH5. We only identified one homozygous mutation in the known candidate gene LRRC6 in one patient with KTS. Our study showed that most of the known candidate genes present heterozygous mutations, but each patient with KTS carried multiple gene mutations. One limitation of this study is that DNA samples were not available for all the parents of the recruited KTS patients to confirm the inheritance mode of these mutations. It has been shown that compound heterozygous mutations can also cause extreme phenotypic diversity in the cilia ultrastructure of patients with PCD (Yang et al., 2018). It has also been shown that trans-heterozygous interactions between DNAH6 and other PCD genes can potentially cause heterotaxy and ciliary dysfunction (Li et al., 2016). This indicates that heterotaxy might have a non-monogenic etiology. In summary, KTS is a complex genetic heterogeneity disorder that might be mediated by heterozygous mutations in multiple PCD- or cilia-related genes.
In conclusion, we identified 10 KTS-associated variants in six causative genes by targeted next-generation sequencing, including compound heterozygous mutations in DNAH1 and DNAH5. The potential pathogenic gene GALNT11 activated the NOTCH pathway, which might cause situs inversus in KTS. The various pathogenic mutations in different genes involved in KTS indicate genetic diversity and heterogeneity among patients with KTS. The identification of genetic defects that cause KTS is of great importance for its clinical diagnosis and genetic counseling.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The project was approved by the ethics committee of the Ethics Committee of Shenzhen People’s Hospital. Written informed consent was obtained from the patients for the publication of the patient’s identifiable information. All procedures performed in studies involving human participants in accordance with the 1964 Declaration of Helsinki ethical standards.
Author Contributions
YY, YF, and BL prepared the project proposal and study design. YY and PZ conducted bioinformatics and statistical analysis of sequencing data. QH, PanZ, and ShuL conducted sample collection and Sanger sequencing validation. XT, ShengL, LC, and YF conducted clinical diagnosis of Kartagener syndrome. XH and LC assisted with the prepared and revised manuscript. All the authors have read and approved the final manuscript.
Funding
The study was supported by the Guangdong Provincial Natural Science Foundation (2018A030310674), National Key Research and Development Program of China (2016YFC1304400), the Guangdong Provincial Science and Technology Project (2017A020214016), and Shenzhen Science and Technology Project (JCYJ20170413093032806, JCYJ20180305164128430).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
PCD, primary ciliary dyskinesia; KTS, kartagener syndrome; ODA, outer dynein arm; IDA, inner dynein arm; TEM, transmission electron microscopy; ACMG, American College of Medical Genetics and Genomics; VUS, uncertain significance variants
Acknowledgments
Thanks to laboratory support from the Key Laboratory of Shenzhen Respiratory Diseases (ZDSYS201504301616234) and Shenzhen Public Service Platform on Tumor Precision Medicine and Molecular Diagnosis of Shenzhen People’s Hospital. Thanks to Dr. Liangfei Ao from Renmin Hospital of Wuhan University in samples treatment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00749/full#supplementary-material.
Supplemental Figure S1 | Sanger validation results of the compound heterozygous variants c.8462 > C and c.6061 > C in DNAH5 of KT4
References
Becker-Heck, A., Zohn, I. E., Okabe, N., Pollock, A., Lenhart, K. B., Sullivan-Brown, J., et al. (2011). The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 43 (1), 79–84. doi: 10.1038/ng.727
Boskovski, M. T., Yuan, S., Pedersen, N. B., Goth, C. K., Makova, S., Clausen, H., et al. (2013). The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 504 (7480), 456–459. doi: 10.1038/nature12723
Bush, A., Chodhari, R., Collins, N., Copeland, F., Hall, P., Harcourt, J., et al. (2007). Primary ciliary dyskinesia: current state of the art. Arch. Dis. Child. 92 (12), 1136–1140. doi: 10.1136/adc.2006.096958
Djakow, J., Kramna, L., Dusatkova, L., Uhlik, J., Pursiheimo, J. P., Svobodova, T., et al. (2016). An effective combination of sanger and next generation sequencing in diagnostics of primary ciliary dyskinesia. Pediatr. Pulmonol. 51 (5), 498–509. doi: 10.1002/ppul.23261
Fakhro, K. A., Choi, M., Ware, S. M., Belmont, J. W., Towbin, J. A., Lifton, R. P., et al. (2011). Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. U. S. A. 108 (7), 2915–2920. doi: 10.1073/pnas.1019645108
Guo, T., Tan, Z. P., Chen, H. M., Zheng, D. Y., Liu, L., Huang, X. G., et al. (2017). An effective combination of whole-exome sequencing and runs of homozygosity for the diagnosis of primary ciliary dyskinesia in consanguineous families. Sci. Rep. 7 (1), 7905. doi: 10.1038/s41598-017-08510-z
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T., et al. (2006). DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 174 (2), 120–126. doi: 10.1164/rccm.200601-084OC
Hosie, P., Fitzgerald, D. A., Jaffe, A., Birman, C. S., Morgan, L. (2014). Primary ciliary dyskinesia: overlooked and undertreated in children. J. Paediatr. Child Health 50 (12), 952–958. doi: 10.1111/jpc.12628
Knowles, M. R., Ostrowski, L. E., Loges, N. T., Hurd, T., Leigh, M. W., Huang, L., et al. (2013). Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am. J. Hum. Genet. 93 (4), 711–720. doi: 10.1016/j.ajhg.2013.07.025
Kott, E., Duquesnoy, P., Copin, B., Legendre, M., Dastot-Le Moal, F., Montantin, G., et al. (2012). Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 91 (5), 958–964. doi: 10.1016/j.ajhg.2012.10.003
Kurkowiak, M., Zietkiewicz, E., Witt, M. (2015). Recent advances in primary ciliary dyskinesia genetics. J. Med. Genet. 52 (1), 1–9. doi: 10.1136/jmedgenet-2014-102755
Li, Y., Yagi, H., Onuoha, E. O., Damerla, R. R., Francis, R., Furutani, Y., et al. (2016). DNAH6 and its interactions with PCD genes in heterotaxy and primary ciliary dyskinesia. PLoS Genet. 12 (2), e1005821. doi: 10.1371/journal.pgen.1005821
Lucas, J. S., Burgess, A., Mitchison, H. M., Moya, E., Williamson, M., Hogg, C., et al. (2014). Diagnosis and management of primary ciliary dyskinesia. Arch. Dis. Child. 99 (9), 850–856. doi: 10.1136/archdischild-2013-304831
Lucas, J. S., Paff, T., Goggin, P., Haarman, E. (2016). Diagnostic methods in primary ciliary dyskinesia. Paediatr. Respir. Rev. 18, 8–17. doi: 10.1016/j.prrv.2015.07.017
Merveille, A. C., Davis, E. E., Becker-Heck, A., Legendre, M., Amirav, I., Bataille, G., et al. (2011). CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43 (1), 72–78. doi: 10.1038/ng.726
Olbrich, H., Haffner, K., Kispert, A., Volkel, A., Volz, A., Sasmaz, G., et al. (2002). Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 30 (2), 143–144. doi: 10.1038/ng817
Onoufriadis, A., Shoemark, A., Munye, M. M., James, C. T., Schmidts, M., Patel, M., et al. (2014). Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 51 (1), 61–67. doi: 10.1136/jmedgenet-2013-101938
Paff, T., Kooi, I. E., Moutaouakil, Y., Riesebos, E., Sistermans, E. A., Daniels, H., et al. (2018). Diagnostic yield of a targeted gene panel in primary ciliary dyskinesia patients. Hum. Mutat. 39 (5), 653–665. doi: 10.1002/humu.23403
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038/gim.2015.30
Takeuchi, K., Kitano, M., Kiyotoshi, H., Ikegami, K., Ogawa, S., Ikejiri, M., et al. (2018). A targeted next-generation sequencing panel reveals novel mutations in Japanese patients with primary ciliary dyskinesia. Auris Nasus Larynx 45 (3), 585–591. doi: 10.1016/j.anl.2017.09.007
Tarkar, A., Loges, N. T., Slagle, C. E., Francis, R., Dougherty, G. W., Tamayo, J. V., et al. (2013). DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 45 (9), 995–1003. doi: 10.1038/ng.2707
Wallmeier, J., Al-Mutairi, D. A., Chen, C. T., Loges, N. T., Pennekamp, P., Menchen, T., et al. (2014). Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Genet. 46 (6), 646–651. doi: 10.1038/ng.2961
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi: 10.1093/nar/gkq603
Wilson, C. W., Nguyen, C. T., Chen, M. H., Yang, J. H., Gacayan, R., Huang, J., et al. (2009). Fused has evolved divergent roles in vertebrate hedgehog signalling and motile ciliogenesis. Nature 459 (7243), 98–102. doi: 10.1038/nature07883
Yang, L., Banerjee, S., Cao, J., Bai, X., Peng, Z., Chen, H., et al. (2018). Compound heterozygous variants in the coiled-coil domain containing 40 gene in a Chinese family with primary ciliary dyskinesia cause extreme phenotypic diversity in cilia ultrastructure. Front. Genet. 9, 23. doi: 10.3389/fgene.2018.00023
Keywords: Kartagener syndrome, compound heterozygous, CCDC40, DNAH1, DNAH5, DNAI1, NOTCH pathway
Citation: Yue Y, Huang Q, Zhu P, Zhao P, Tan X, Liu S, Li S, Han X, Cheng L, Li B and Fu Y (2019) Identification of Pathogenic Mutations and Investigation of the NOTCH Pathway Activation in Kartagener Syndrome. Front. Genet. 10:749. doi: 10.3389/fgene.2019.00749
Received: 02 May 2019; Accepted: 17 July 2019;
Published: 22 August 2019.
Edited by:
Merlin G. Butler, University of Kansas Medical Center, United StatesReviewed by:
Fan Jin, Zhejiang University, ChinaMaria Paola Lombardi, University of Amsterdam, Netherlands
Copyright © 2019 Yue, Huang, Zhu, Zhao, Tan, Liu, Li, Han, Cheng, Li and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingyun Fu, eWluZ3l1bmZ1MjAxN0AxNjMuY29t; Bo Li, MTMzNTI5OTU2NTZAMTYzLmNvbQ==