 Ye Xu1Yunqiao Dong2Yazhou Xu1
Ye Xu1Yunqiao Dong2Yazhou Xu1 Zetian Lai1Binbin Jin1Yanqiang Hao3Yonghui Gao1
Zetian Lai1Binbin Jin1Yanqiang Hao3Yonghui Gao1 Yan Sun1
Yan Sun1 Xiao-guang Chen1
Xiao-guang Chen1 Jinbao Gu1*
Jinbao Gu1*- 1Guangdong Provincial Key Laboratory of Tropical Disease Research, Department of Pathogen Biology, School of Public Health, Southern Medical University, Guangzhou, China
- 2Reproductive Medical Center of Guangdong Women and Children Hospital, Guangdong Women and Children Hospital, Guangzhou, China
- 3Department of Laboratory Medicine, Guangdong Women and Children Hospital, Guangzhou, China
The Asia tiger mosquito (Aedes albopictus) is an important vector of arboviruses, and females can transmit pathogens such as the dengue, zika, and chikungunya viruses. Understanding sex-related differences in this mosquito is fundamental for vector control. However, there are no reports of systematic characterization of long non-coding RNAs (lncRNAs) in male and female Ae. albopictus. To investigate the roles of coding and non-coding RNAs in both sexes of Ae. albopictus, RNA sequencing was performed on male and female samples. The results showed 305 differentially expressed protein-coding genes (DEGs) between males and females, of which 198 were highly expressed in males and 125 were highly expressed in females. Sex-associated gene ontology terms were enriched. Analysis with the FEELnc software identified 2,623 novel lncRNAs, of which 26 showed male high expression and 11 showed female high expression. Quantitative real-time PCR of randomly selected DEGs and lncRNAs supported the validity of the RNA sequencing results. Knocking down male high-expressed gene AALF000433 in male adults reduced the egg hatching rate. This study provides valuable data on sex-specific expression of protein-coding genes and lncRNAs in adult Ae. albopictus, which will guide further studies aimed at understanding sex development and determination mechanisms in this species.
Introduction
Aedes albopictus, an important vector of arboviruses such as dengue (Paupy et al., 2010), zika (Liu et al., 2017), and chikungunya (Vega-Rúa et al., 2014), is an aggressive, invasive mosquito species with a worldwide distribution. Consequently, it is a significant threat to human health. Vector control is a useful way to prevent epidemics of mosquito-borne infectious diseases. Gene drive systems have been proposed as a means of vector control, by either inhibiting the target population or spreading effector genes that make the population refractory to the relevant disease pathogens (Adelman and Tu, 2016). As only the adult female Ae. albopictus feeds on animal blood and transmits pathogens, a possible approach to reduce disease transmission is the release of only male Ae. albopictus. Hence, it is important to understand the sex determination mechanism(s) used by this species.
In recent years, advances in next-generation sequencing technology and bioinformatic tools have afforded a better understanding of the pathways underlying different biological processes and of the key factors related to such processes, e.g., mRNAs, long non-coding RNAs (lncRNAs), and microRNAs. Access to the genomic sequence from Ae. albopictus (Chen et al., 2015; Miller et al., 2018) has also facilitated the development of various omics technologies (Tsujimoto et al., 2017) in this species. More recently, RNA sequencing (RNA-Seq) studies in Ae. albopictus have explored differentially expressed mosquito genes in chikungunya virus-infected and uninfected mosquitoes; the transcriptomic changes after dengue virus infection have also been examined (Tsujimoto et al., 2017). Clearly, the use of omics technologies has strong potential to enhance our understanding of the interactions between viruses and their hosts.
lncRNAs form a class of transcripts that are longer than 200 nucleotides but do not encode proteins (Esteller, 2011). Strong evidence is accumulating that lncRNAs have regulatory roles in some major biological processes, including transcriptional and post-transcriptional gene regulation, regulation of genomic stability, and epigenetic regulation. lncRNAs are transcribed from various regions, including promoters and intragenic, intergenic, exonic, and intronic regions, as well as enhancer sequences and antisense strands, based on their genome positions (Jarroux et al., 2017). Most lncRNAs are retained in the nucleus, presumably to target the transcription machinery and for nuclear genome surveillance. lncRNAs have also been reported to be differentially expressed under diverse conditions (Hung et al., 2011). Thus, lncRNAs are currently of great interest in mosquito research and so far, 2,949 putative lncRNAs have been found in Anopheles gambiae (Jenkins et al., 2015).
In Ae. albopictus, the potential relationships of lncRNAs with sex- or sexual reproduction-associated processes are beginning to attract research attention. Here, we analyzed in detail the mRNA and lncRNA profiles of male and female Ae. albopictus.
Materials and Methods
Mosquitoes
The Ae. albopictus Foshan (Guangdong, China) strain used in this research (established in the laboratory in 1981) was provided by the Center for Disease Control of Guangdong, China. Ae. albopictus mosquitoes were reared in an artificial climate box at 25 ± 1°C, with a 10:14 h daily light–dark cycle, and 70–80% relative humidity. Larvae were fed on fish food (Yee®, Shandong, China) and the adults were fed on a 10% glucose solution. Three replicons of both male or female Ae. albopictus were collected two days after emergence. Each pool contained 15 male or female Ae. albopictus individuals.
RNA Extraction
RNA was extracted from each pool using TRIzol (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s recommendations. RNA quality was determined using a 2100 Bioanalyzer (Agilent), and the concentration was determined using an ND-2000 instrument (NanoDrop Technologies). Ribosomal RNA (rRNA) was removed from the RNA preparations using an Epicentre Ribo-Zero rRNA Removal Kit (Epicentre, Madison, WI, USA). The rRNA-depleted RNA was used to prepare sequencing libraries with the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA), according to the manufacturer’s instructions. The prepared libraries were then sequenced on the Illumina HiSeq™ 4000 platform (2 × 150-bp read lengths) by Gene Denovo Technologies (Guangzhou, China).
Sequencing Data Analysis
An analysis pipeline was employed to explore the transcriptome profile and identify novel lncRNAs (Figure S1). High-quality clean reads were obtained by processing the raw data to remove adapter sequences, low-quality reads, and poly-N sequences using Trimmomatic version 0.35 (Bolger et al., 2014). Next, the clean reads were mapped to the genome of Ae. albopictus strain Foshan (Chen et al., 2015; Miller et al., 2018) using Hisat2 version 2.1.1 (Pertea et al., 2016) with the default parameters. StringTie version 1.3.6 (Pertea et al., 2015) was employed to assemble and quantify transcripts with Ae. albopictus Foshan strain reference annotation (AaloF1.2, VectorBase, https://www.vectorbase.org) (Lawson et al., 2008). The Ballgown package version 2.16.0 (Frazee et al., 2015) was used to analyze the differentially expressed protein-coding genes (DEGs) in the R (version 3.6) environment (R Core Team, 2013). Fragments per kilobase per million reads was used to measure expression levels of protein-coding genes (Mortazavi et al., 2008). Low-abundance transcripts with a variance across samples of less than one were filtered out. DEGs were defined as genes with two-fold or greater changes between samples from male and female mosquitoes with false discovery rate (FDR) < 0.05. All assembled transcript sequences were generated and reannotated with the eggNOG-mapper (Huerta-Cepas et al., 2015), based on the eggNOG 4.5.1 database (Huerta-Cepas et al., 2015). For enrichment analysis, the R package ClusterProfiler version 3.12.0 (Yu et al., 2012) was used for gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. FDR <0.05 was regarded as the cutoff criterion for both GO and KEGG enrichment analysis.
Identification of Novel lncRNAs, Data Analysis, and Functional Enrichment Analyses for lncRNAs
Ae. albopictus lncRNA loci were detected using FEELnc version 0.1.1 (Wucher et al., 2017), a program that predicts lncRNA loci using a random forest model trained with multi k-mer frequencies and relaxed open reading frames. First, the FEELnc filter module was used to identify non-lncRNA transcripts from reconstructed transcripts of StringTie version 1.3.6, as described in the previous section. The FEELnc filter module also filters out short transcripts (default 200 nucleotides) and can deal with single-exon transcripts. Second, the FEELnc coding potential module (FEELnc codpot) was used to compute a coding potation score for each of the candidate transcripts in the output of the FEELnc filter module. Third, predicted lncRNA transcript sequences were searched against the National Center for Biotechnology Information (NCBI) nonredundant protein database by BLASTX (McGinnis and Madden, 2004). Transcripts with significant homology to known proteins (e.g., e-value < 1e-10, target coverage > 80%, and identity > 90%) were removed. Last, the FEELnc classifier module (FEELnc classifier) was employed to classify Ae. albopictus lncRNAs into two major types (“genic” and “intergenic”) and six subtypes. The FEELnc classifier module was used for possible function prediction of differentially expressed lncRNAs based on their nearest-neighbor protein-coding genes. Functional annotation enrichment analyses of differentially expressed lncRNAs were performed as described in the previous section.
Quantitative Real-Time PCR (qRT-PCR) Validation
Five significantly differentially expressed lncRNAs and mRNAs were randomly selected. Expression levels of selected lncRNAs and mRNAs in the whole body, midgut (MG), and salivary gland (SG) of both males and females, and in the male testis and female ovary were validated by qRT-PCR with three biological replicates. RNA and cDNA were synthesized from RNA isolated from each organism using M-MLV reverse transcriptase (Invitrogen). qRT-PCR was performed using SYBR Green assays with specific primers (Table S1), each in a total reaction volume of 20 μl, as described previously (Huang et al., 2016), with a program of 95°C (10 min), 40 cycles of 95°C (10 sec), and 60°C (1 min). β-actin was used for normalization, and the 2−ΔΔCT method (Livak and Schmittgen, 2001) was used to estimate the relative expression of each lncRNA and mRNA. The results were analyzed using student’s t-tests (Prescott, 2004), with P <0.05 considered to be statistically significant.
Production of Double-Stranded RNA (dsRNA)
The total RNA of male adult was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. cDNA was synthesized with oligo-d(T) using a SuperScript™ III First-Strand Synthesis System (Invitrogen, Carlsbad, USA) as described in the manufacturer’s protocol. Sense and antisense segments primers (Table S4) with T7 promoter were designed to amplify the templates of a 794 bp segment of AALF000433 (Figure S3). PCR amplification was performed using PrimeSTAR® HS DNA Polymerase (Takara Bio, Beijing, China) with conditions as follows: 95°C for 5 min, followed by 30 cycles of 30 s 95°C, 30 s at 58°C, and 60 s at 72°C, with a final extension step of 72°C for 10 min. Amplified PCR products were electrophoresed on 1.2% agarose gels and target fragments were extracted using MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0 (Takara Bio, Beijing, China). Sense and antisense RNAs were synthesized in vitro using T7 RiboMAX™ Express RNAi System (Promaga, USA) with 1 μg of DNA template in a total volume of 20 μL, respectively. To anneal the RNA strands, mix equal volumes of complementary RNA reactions together and incubate at 70°C for 10 minutes, then slowly cool to room temperature (∼20 minutes) according to the manufacturer’s protocol. Remaining single-strand RNAs and DNA templates in the reactions were digested by RNase and DNase. dsRNAs were purified by phenol chloroform extraction, ethanol precipitated and re-suspended in nuclease free water. A green fluorescent protein (GFP)-derived dsRNA was used as a control.
Adult Injection, Mosquito Mating, and Egg Collection
Injections of dsRNAs into 2-day-old single reared adult Ae. albopictus males were performed under a dissecting microscope. Doses of approximately 30 ng of AALF000433 dsRNAs or gfp dsRNAs were injected into the thorax and immediately transferred to small plastic cups (900 ml, 11 cm top diameter) and provided with 10% sucrose solution. The relative expression levels of AALF000433 were quantified by qRT-PCR five days after injection. Gene expression levels were analyzed by the 2−ΔΔCT method (Livak and Schmittgen, 2001). For each treatment described above, three independent mosquitoes were used as biological replicates. Five days after injection, each injected individual male was transferred to a small cup (250 ml, 8 cm top diameter) with one female (one female crossed with one male per cage) for mating for one day. After fully mating, each single female was placed in an oviposition site to lay eggs. The hatching rate was measured. For each treatment described above, ten independent mosquitoes were used as biological replicates. The results were analyzed using student’s t-tests (Prescott, 2004), with P < 0.05 considered to be statistically significant.
Results
Sequencing Data Overview
High-throughput sequencing was performed on an Illumina HiSeq™ 4000 platform with female and male mosquito samples. After adaptor removal and quality trimming, 69,027,078, 83,886,978, and 76,075,782 clean reads from female samples, and 78,871,518, 91,141,086, and 78,543,476 clean reads from male samples were obtained. In addition, nearly 60% of trimmed reads could be mapped to the Ae. albopictus Foshan strain genome (Table 1).
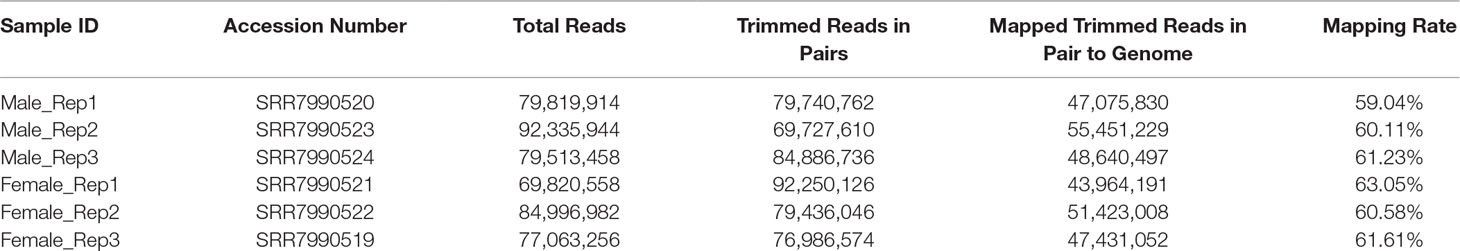
Table 1 Summary of the sequence data.
Overall transcript expression datasets of samples were used for principal component analysis. The overall transcript profiles of the male and female libraries showed the highest similarity, indicating that genome-wide variance of the biological replicates was negligible, and remarkable differences were observed between the sexes (Figure S2).
Gene Expression Analysis and DEGs Identification
StringTie version 1.3.6 was used for transcript assembly and detection of expression. A total of 39,492 transcripts from 29,862 protein-coding genes were assembled. A total of 305 protein-coding genes were considered to be differentially expressed between the male and female mosquito samples (Table S2). Among these DEGs, 198 had male high-expression and 125 female high-expression. Volcano plot analysis (Figure 1A) was used to depict the gene expression distribution. Hierarchical clustering analysis (Figure 1B) of 305 DEGs was performed between the different libraries; in the figure, red and green lines indicate male high-expression and female high-expression levels for each library.
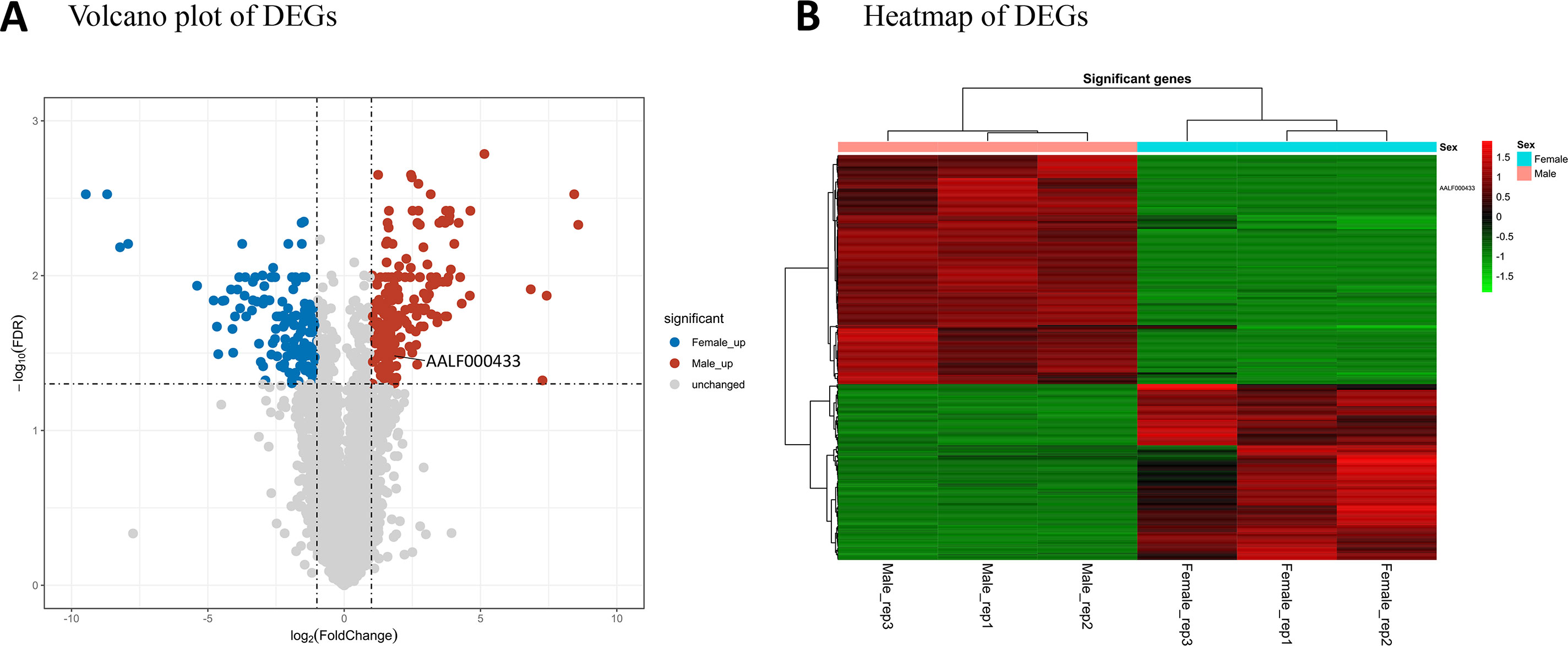
Figure 1 mRNA expression profiles of male and female Ae. albopictus adults. (A) Volcano plot analysis of gene expression variation. Red and blue points correspond to 2.0-fold changes in male and female high-expression mRNAs, respectively, and indicate FDR < 0.05. (B) Hierarchical clustering of the 305 DEGs in the six samples. Green represents low expression levels and red represents high expression levels.
Functional Enrichment of DEGs
To further elucidate the functional roles of the DEGs, we used the eggNOG database and ClustalProfile R package for GO and KEGG pathway enrichment analysis. The top 15 significantly enriched (FDR < 0.05) GO terms in the categories of biological process (BP), cellular component (CC), and molecular function (MF) were obtained.
Among the BP terms for male high-expression protein-coding genes, the most significantly enriched GO terms were related to male sex function (Figure 2A): spermatogenesis, spermatid development, spermatid differentiation, and skeletal myofibril assembly (movement in host). The most enriched GO CC terms of male samples were sperm flagellum, 9 + 2 motile cilium, motile cilium, cilium, and sperm part. The MF terms for the male samples indicated that motor activity was very important; representative terms were microfilament motor activity, motor activity, structural constituent of cytoskeleton, actin-dependent ATPase activity, and adenine nucleotide transmembrane transporter activity. Fifteen protein-coding genes, including 13 novel and two known protein-coding genes, were associated with sperm-related terms. (Figures 2B, C). KEGG pathway analysis showed that nine overrepresented pathways with FDR of <0.05 were enriched in male samples (Figures 2D–F).
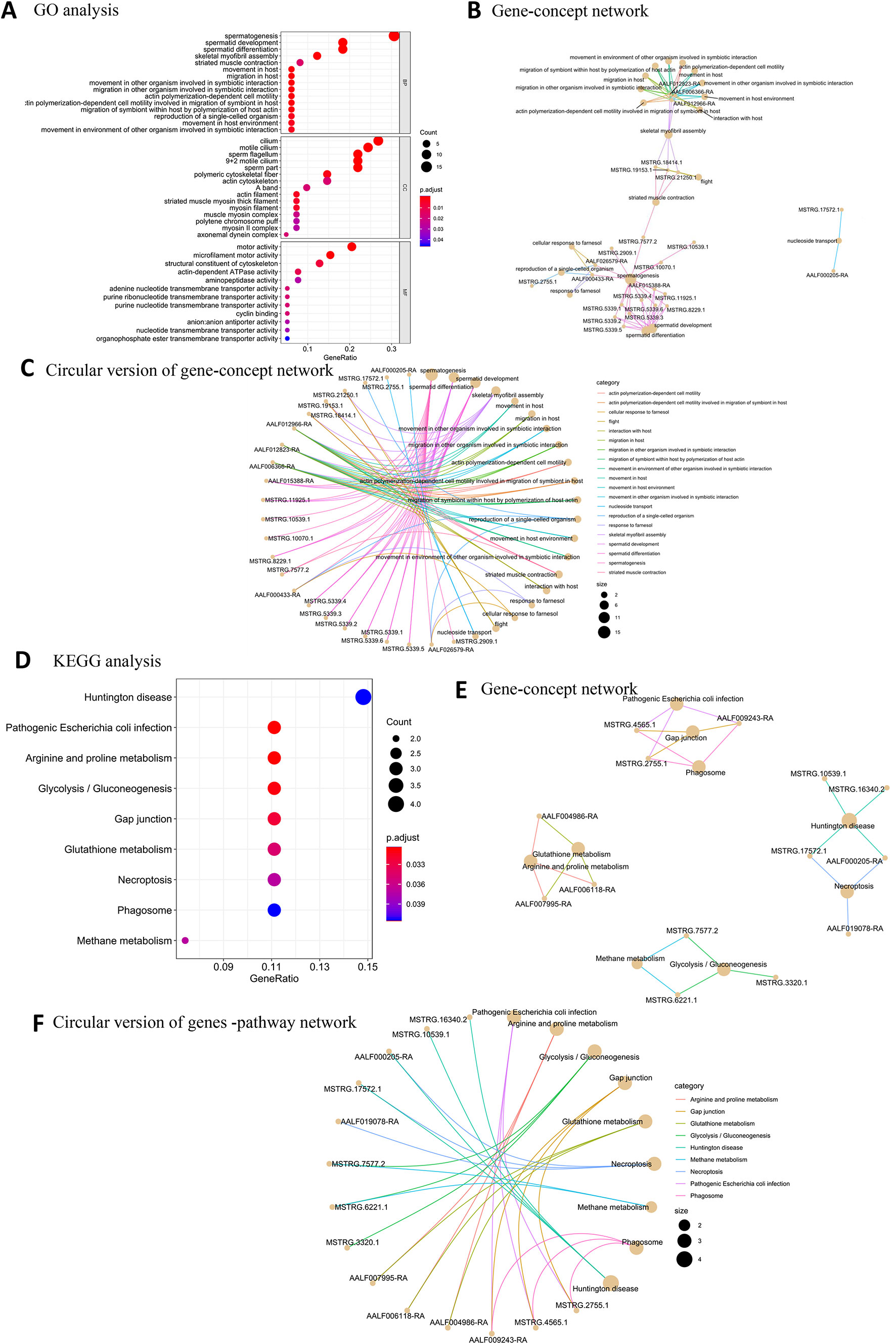
Figure 2 GO and KEGG pathway enrichment analysis of male high-expression protein-coding genes. (A) The top 20 GO terms for BP, CC, and MF for male high-expression protein-coding genes. (B) Gene-concept network depicting the linkages of male high-expression protein-coding genes and biological concepts as a network. (C) Circular version of gene-concept network for male high-expression protein-coding genes. (D) Top KEGG pathway terms for male high-expression protein-coding genes. (E) Protein-coding gene-pathway network depicting the linkages of male high-expression protein-coding genes and KEGG pathways as a network. (F) Circular version of male high-expression protein-coding gene-pathway network.
Among the BP terms for female high-expression protein-coding genes, the most significantly enriched GO terms were related to chromatin assembly or disassembly, chromatin organization, development of primary female sexual characteristics, female sex differentiation, and connective tissue development. The most enriched CC terms were nucleosome, DNA packaging complex, protein-DNA complex, chromatin, and myofilament. The significantly enriched MF terms were proximal promoter DNA-binding transcription repressor activity, RNA polymerase II-specific, serine-type endopeptidase activity, serine-type peptidase activity, and serine hydrolase activity (Figure 3A). Four new protein-coding genes (MSTRG.14455.2, MSTRG.14455.2, MSTRG.16151.1, and MSTRG.5389.3) were associated with sex determination (Figures 3B, C). Six overrepresented KEGG pathway terms were enriched in the female samples (Figures 3D–F).
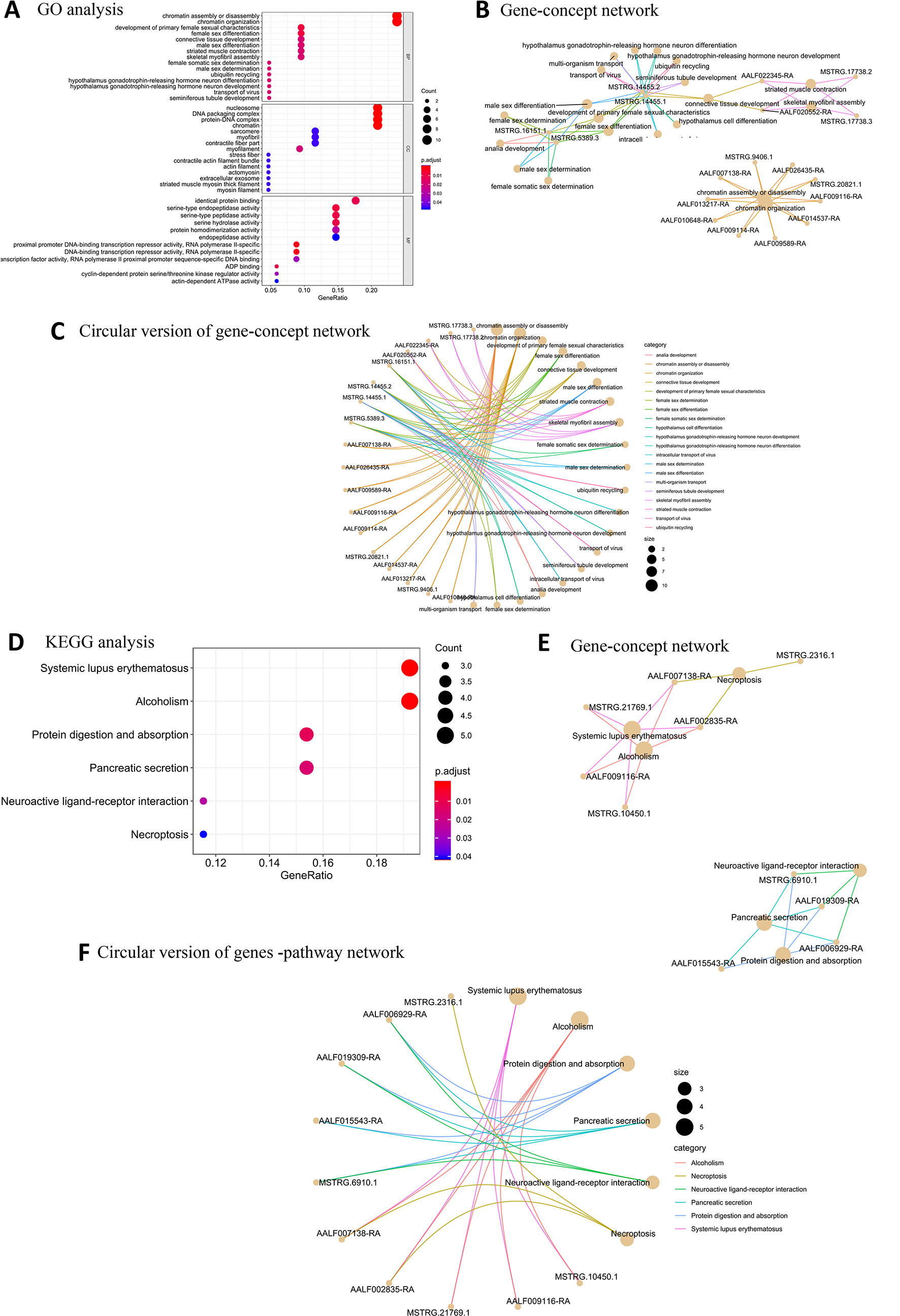
Figure 3 GO and KEGG pathway enrichment analysis of female high-expression protein-coding genes. (A) Top 20 GO terms for BP, CC, and MF for female high-expression protein-coding genes. (B) Gene-concept network depicting the linkages of female high-expression protein-coding genes and biological concepts as a network. (C) Circular version of female high-expression protein-coding gene-concept network. (D) Top KEGG pathway terms for female high-expression protein-coding genes. (E) Protein-coding gene-pathway network depicting the linkages of female high-expression protein-coding genes and KEGG pathways as a network. (F) Circular version of female high-expression protein-coding gene-pathway network.
The GO terms skeletal myofibril assembly (Figure 4A), actin filament, striated muscle myosin thick filament, myosin filament, myofilament (Figure 4B), and actin-dependent ATPase activity (Figure 4C) were enriched in both male and female samples with different protein-coding genes. The glutathione metabolism pathway was enriched in both male (AALF004986-RA, AALF006118-RA, AALF007995-RA) and female samples (AALF019309-RA, AALF006929-RA) with different protein-coding genes (Figure 4D).
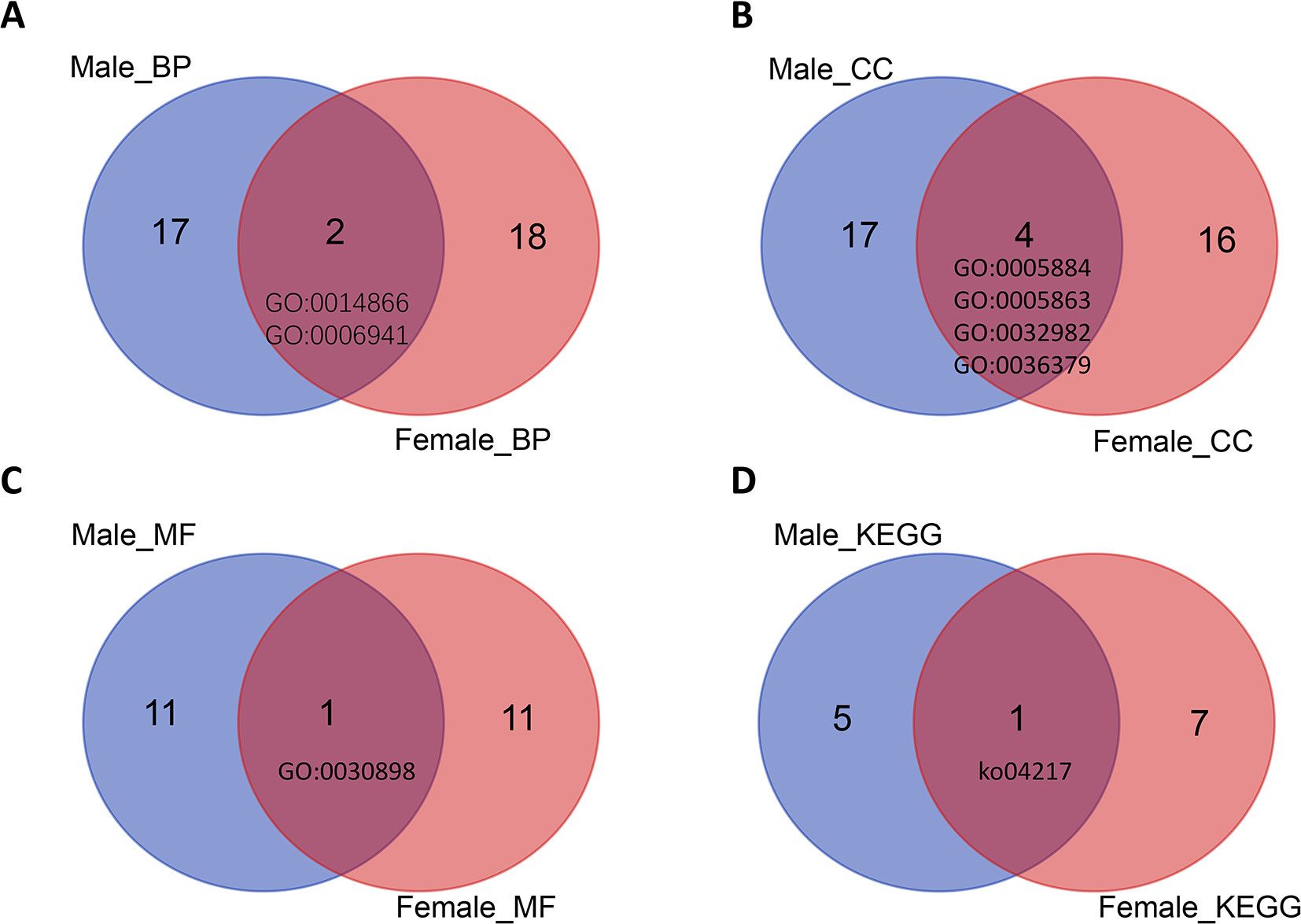
Figure 4 Venn plots of GO terms and KEGG pathway terms for both male and female high-expression protein-coding genes. (A) Two BP term was enriched in male and female samples. (B) Four CC terms were enriched in male and female samples. (C) One MF term was enriched in male and female samples. (D) One KEGG term was enriched in male and female samples.
Ae. albopictus lncRNAs Predicted by FEELnc
The optimal coding potential score (CPS) was used to determine the coding status of the RNA gene models. FEELnc automatically computes the CPS cut-off that maximizes classification performance and provides users with a two-graph receiver operating characteristic (ROC) curve to display the performance of the model and visualize the CPS. The results showed a CPS cut-off of 0.383 (Figure 5A). In total, 2,623 novel lncRNAs were identified. Using the FEELclassifier module, the class distributions of the 1484 lncRNAs were classified by comparison with annotated protein-coding genes from VectorBase. We found 325 genic and 1,159 intergenic lncRNAs (Figure 5B).
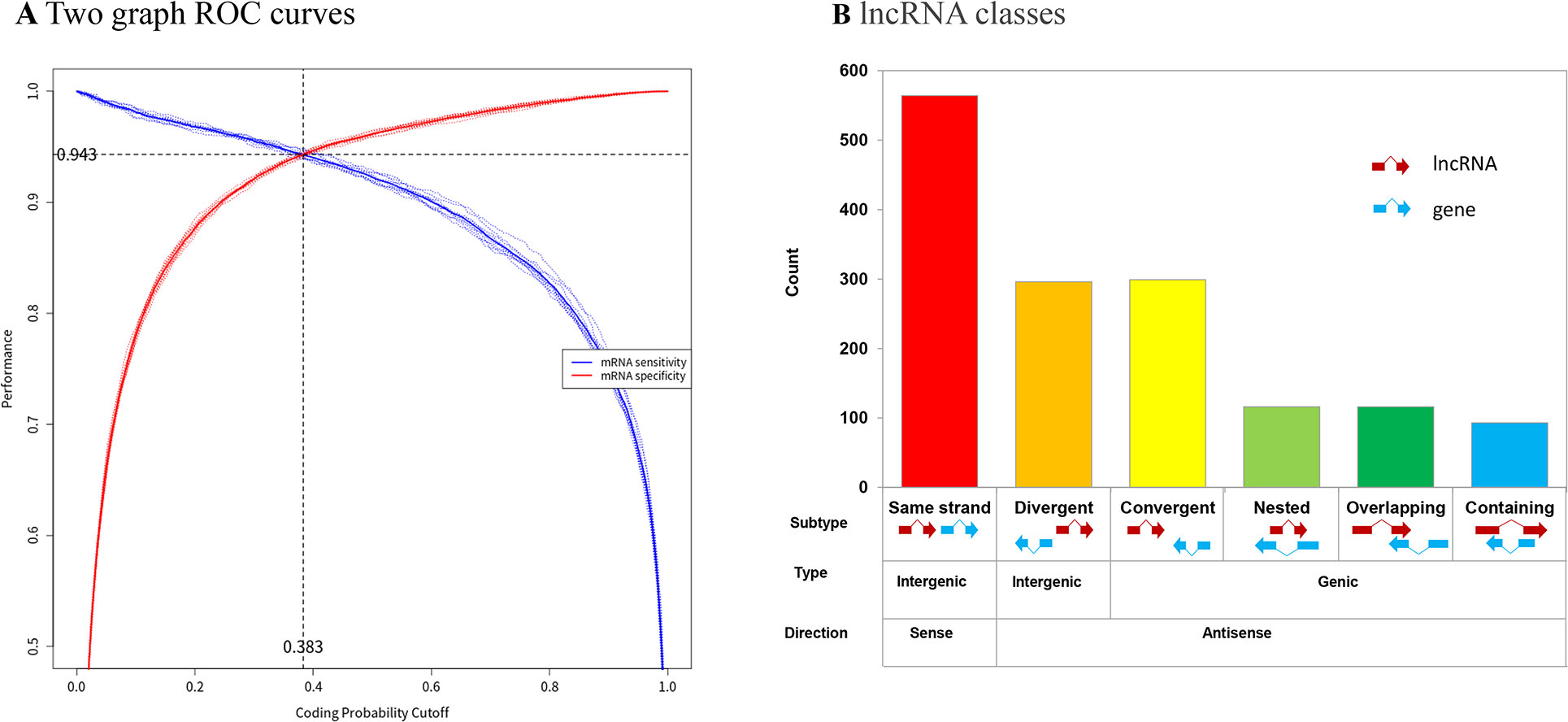
Figure 5 Identification of novel lncRNAs in Ae. albopictus. (A) Two ROC curves for automatic detection of optimized CPS threshold and user specificity threshold. Red line indicates mRNA specificity. Blue line indicates lncRNA specificity. (B) lncRNA classes based on RNA partners from reference annotations. Types and counts of novel lncRNA molecules in Ae. albopictus.
Identification of Differentially Expressed lncRNAs Between Females and Males
Differentially expressed lncRNAs between the male and female samples were also analyzed. A total of 37 lncRNAs were considered to be differentially transcribed between the male and female samples (Table S3). Among these, 26 male high-expression lncRNAs and 11 female high-expression lncRNAs were detected (Figure 6).
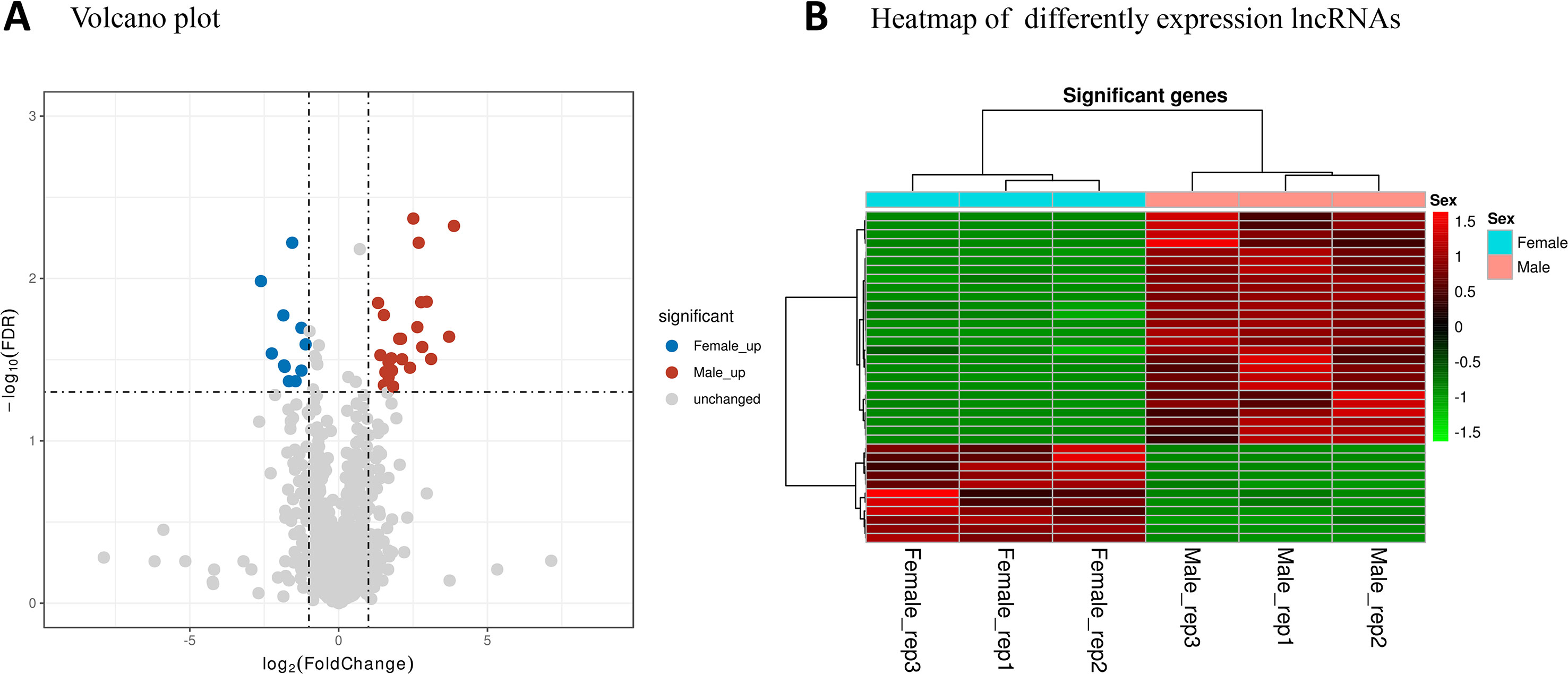
Figure 6 lncRNA expression profiles in male and female Ae. albopictus mosquitoes. (A) Volcano plot analysis of lncRNA expression variation between males and females. Red and blue points correspond to 2.0-fold changes in male/female high-expression mRNAs, respectively, and indicate FDR < 0.05. (B) Hierarchical clustering of the differentially expressed lncRNAs. Green represents low expression levels and red represents high expression levels.
The expression relationships between differentially expressed lncRNAs and their target protein-coding genes were also analyzed. The results showed co-high expression of two significant male high-expression lncRNAs and their target genes using relaxed criteria (P <0.05 for both lncRNAs and protein-coding genes). Two female high-expression lncRNAs showed co-high-expression with their target genes (Figure 7).
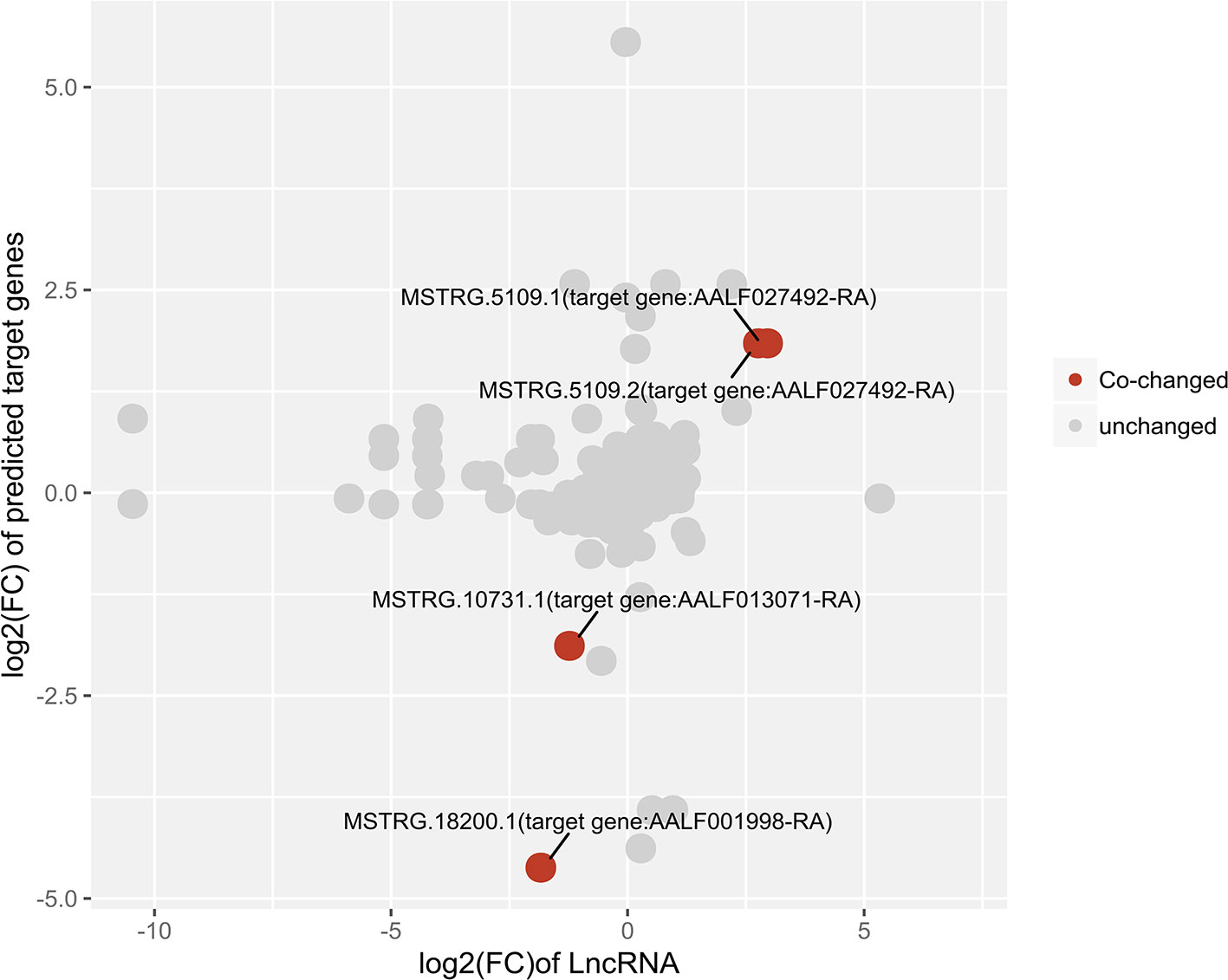
Figure 7 Correlation plot of lncRNA:mRNA pairs of differentially expressed lncRNAs and their predicted target protein-coding genes.
GO and KEGG Pathway Analyses of Protein-Coding Genes Targeted by Differentially Expressed lncRNAs
As lncRNAs are known to regulate their nearest-neighbor protein-coding genes (Rinn et al., 2007; Faghihi et al., 2008; Guttman et al., 2009; Villegas et al., 2014), we performed GO term and KEGG pathway enrichment analysis on nearest-neighbor protein-coding genes to predict the potential function of the differentially expressed lncRNAs. In male samples, seven GO terms of MF were significantly enriched (FDR < 0.05) (Figures 8A, B). No overrepresented KEGG pathway was enriched in the male samples. In the female samples, 30 GO terms were significantly enriched (FDR < 0.05) (Figures 8C, D). The KEGG pathway analysis revealed that the glutathione metabolism [ko00480] pathway was enriched in the female samples (Figures 8E, F).
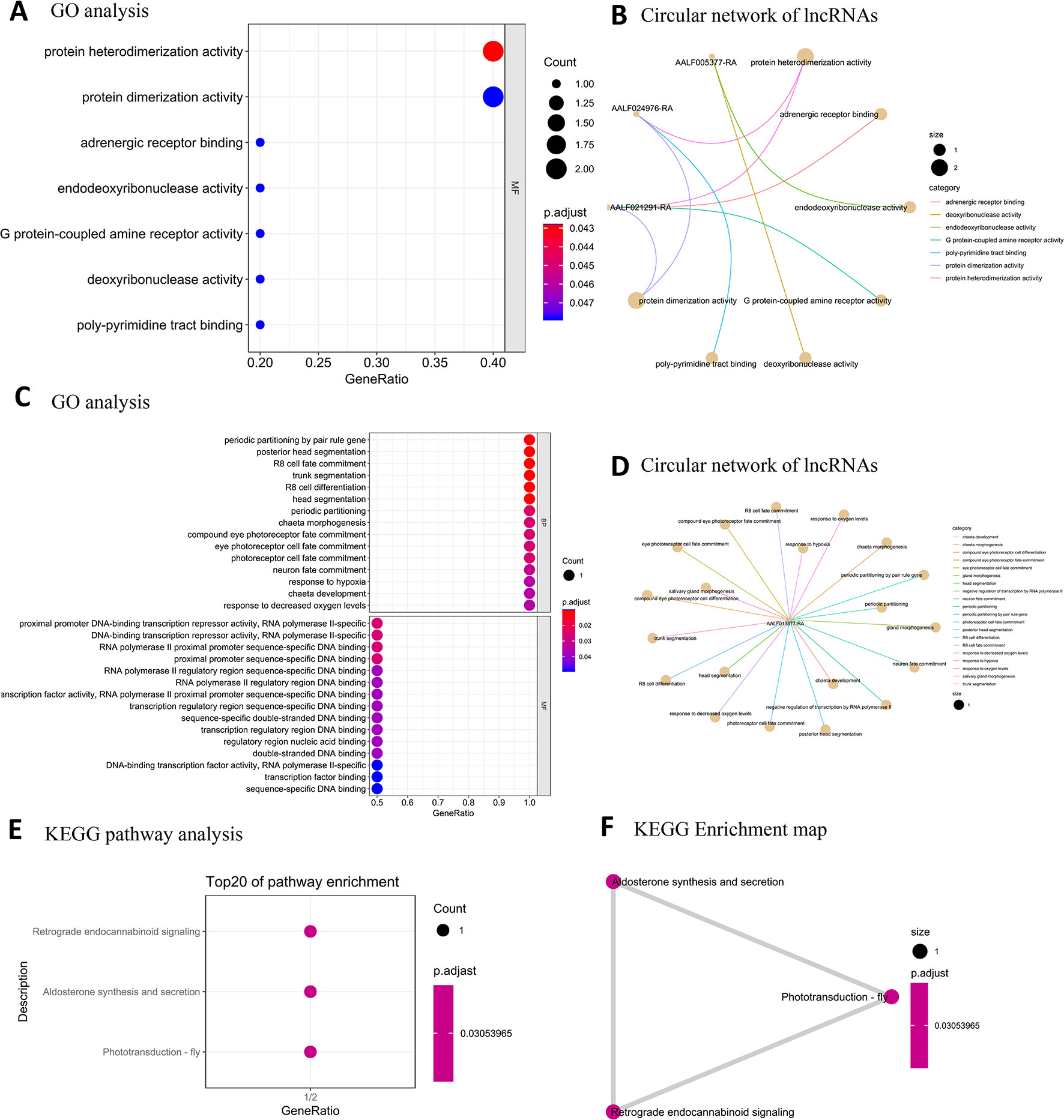
Figure 8 GO and KEGG pathway enrichment analysis of male/female high-expression lncRNA target protein-coding genes. (A) Top GO terms for MF for male high-expression lncRNA target protein-coding genes. (B) Circular network of male high-expression lncRNA target protein-coding genes and BCs. (C) Top GO terms for female high-expression lncRNA target protein-coding genes. (D) Circular network of female high-expression lncRNA target protein-coding genes and BCs (E) Top KEGG pathway terms for female high-expression lncRNA expression. (F) Enrichment map organizing enriched terms into a network with edges weighted by the ratio of overlapping gene sets.
Validation of Differentially Expressed lncRNAs and mRNAs
Five significantly differentially expressed lncRNAs and mRNAs were randomly selected and validated using qRT-PCR. According to the results, three selected lncRNAs (MSTRG.883.1, MSTRG.9253.1, and MSTRG.8012.1) (Figure 9A) and all five selected protein-coding genes (MSTRG.14385, MSTRG8850, AALF009243, MSTRG.10018, and MSTRG.10539) (Figure 9B) showed significant low-expression in male adults, consistent with the RNA-Seq data. These lncRNAs were also detected in the mosquito sex organs, MG, and SG. Three selected lncRNAs (MSTRG.883.1, MSTRG.9253.1, and MSTRG.1188.1) and two selected protein-coding genes (MSTRG.14385 and MSTRG.10539) were significantly differentially expressed between male testis and female ovary. Five selected lncRNAs (MSTRG.883.1, MSTRG.9253.1, MSTRG.5389.3, MSTRG.8012.1, and MSTRG.1188.1) and three selected protein-coding genes (MSTRG.14385, MSTRG.10018, and MSTRG.10539) were significantly differentially expressed between male MG and female MG. One selected lncRNA (MSTRG. 8012.1) and one selected protein-coding gene (MSTRG.10018) were significantly differentially expressed between male SG and female SG. Therefore, the qRT-PCR results support the validity of the RNA-Seq results, indicating that these lncRNAs and mRNAs may be involved in sex differences in Aedes mosquitoes.
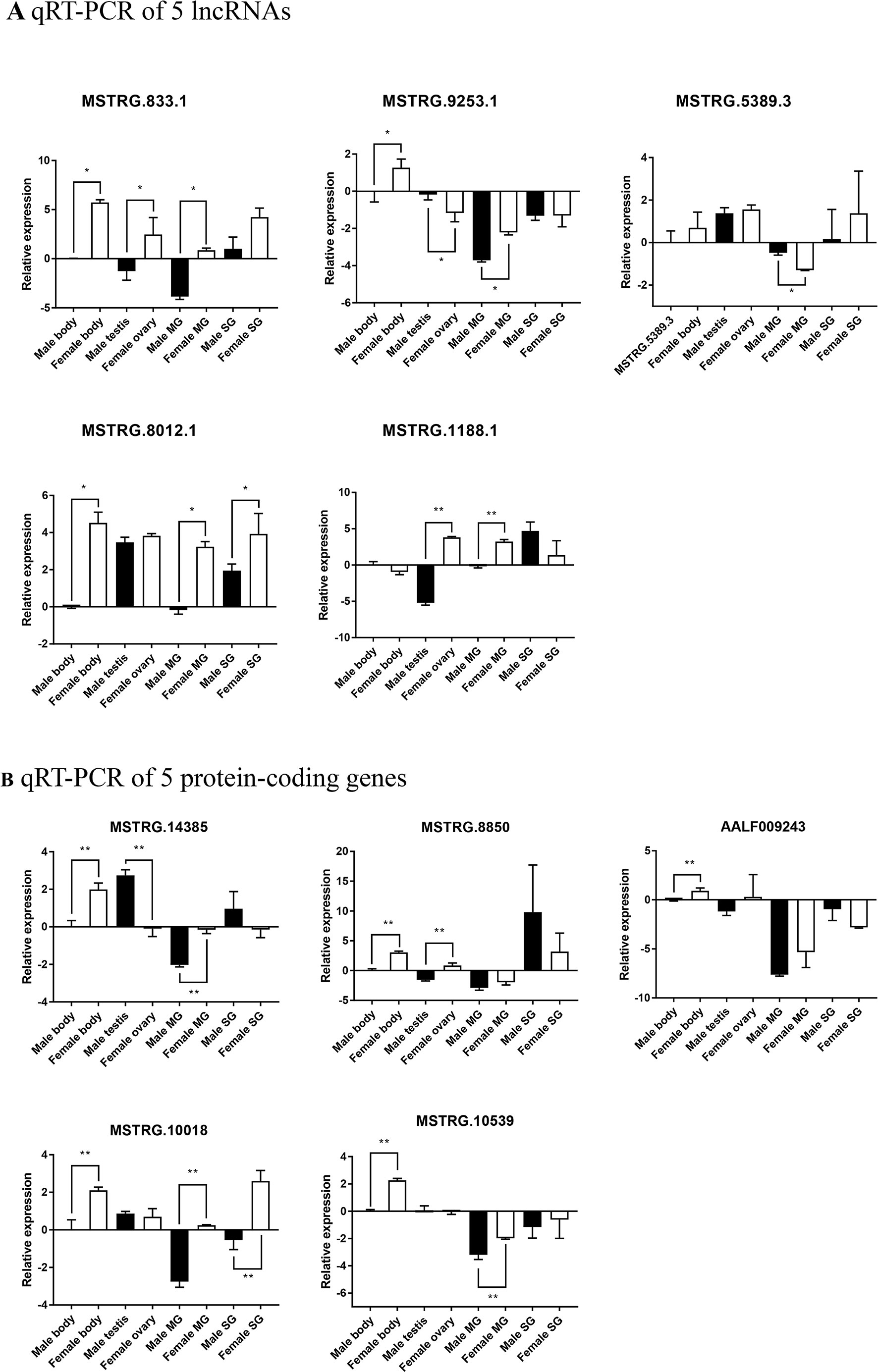
Figure 9 qRT-PCR-based quantitation of male and female lncRNAs and protein-coding genes with significant differential expression levels in whole body, sex organs, MG, and SG. Relative expression levels of (A) five lncRNAs and (B) five protein-coding genes. Data represent mean ± standard deviation, *P < 0.05, **P < 0.01.
Knocking Down AALF000433 in Male Adults Could Reduce Egg Hatching Rate
As mentioned in GO enrichment analysis, a male up-regulated gene, the AALF00043 gene, was enriched in the GO category of cellular response to farnesol, reproduction of a single-celled organism, and spermatogenesis (Figure 2B). Thus, it was selected for knockdown experiments to verify its association with reproductive activity in Ae. albopictus. The gene expression level of AALF00043 was reduced to 29.5% of the normal level five days after injection (Figure 10A). Treated male adults were mated with single reared females individually. Hatching rates of single females were measured. The hatching rate after treatment (62.1 ± 14%, n = 10) was significantly lower than that of untreated controls (84.2 ± 16%, n = 10) (P <0.05) (Figure 10B).
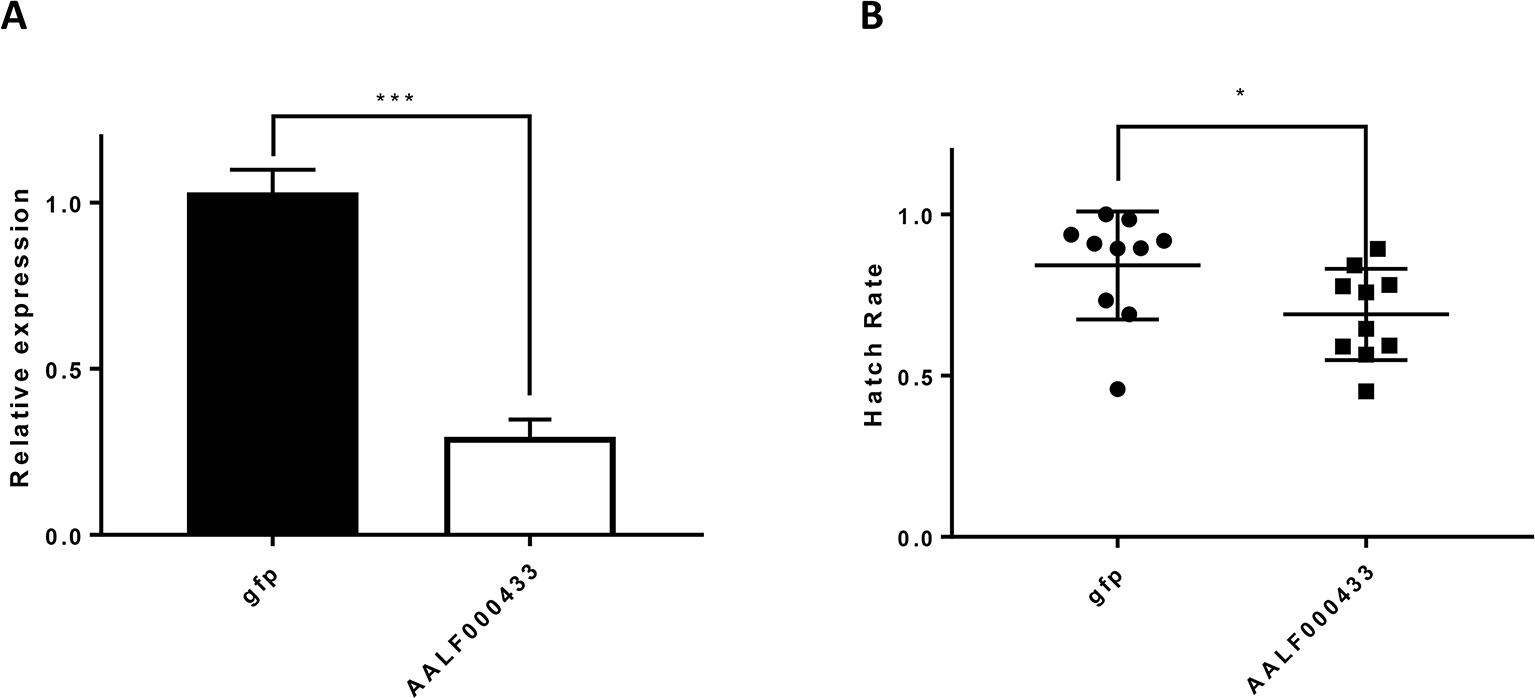
Figure 10 Knockdown of AALF000433 in male adults. (A) Analysis of the effects of AALF000433 interference in male adults by qRT-PCR. Bars represent the standard error of the mean (SEM) (n = 3). The x-axis indicates the groups. (B) The hatch rate of eggs from each female mate in each treatment group. Data are shown as mean ± SEM (*P < 0.05, ***P < 0.001).
Discussion
Various sex determination mechanisms are present in insects. The mechanisms of sex determination in mosquitoes are of particular interest, as only the adult females can bite and transmit diseases. For the past three decades, Ae. albopictus has been monitored as the vector for dengue transmission in south China (Jing et al., 2012). With the aim of obtaining further insight into the sex determination mechanisms and sexual reproduction processes in Ae. albopictus, we conducted a transcriptomic analysis of coding and non-coding gene changes occurring in males and females of this species. This is the first study to provide data on sex-specific protein-coding genes and lncRNAs expression in adult Ae. albopictus.
lncRNAs have regulatory roles in various important biological processes. lncRNA profiles have been studied in several mosquito species (Padron et al., 2014; Jenkins et al., 2015; Etebari et al., 2016), showing associations of lncRNA function with aspects of reproduction such as sex determination (Zhang et al., 2010; Mulvey et al., 2014), meiosis (Shichino et al., 2014), and spermatogenesis (Arun et al., 2012; Nolasco et al., 2012). In 2014, Mulvery et al. showed that transgenic lines ectopically expressing certain lncRNAs could affect the process of Drosophila sex determination and alter levels of other lncRNAs (Mulvey et al., 2014). Our study identified 37 lncRNAs that were differentially transcribed between male and female samples; these are promising candidates for exploring the function of lncRNAs in sex determination and development.
FEELnc, an alignment-free program that accurately annotates lncRNAs based on a random forest model trained with general features, and BLASTX (McGinnis and Madden, 2004) were used to identify and classify lncRNAs in this study. FEELnc (Wucher et al., 2017) has been shown to achieve similar or better classification performance on GENCODE (Harrow et al., 2012) and NONCODE (Bu et al., 2011) data sets when compared with five programs [PhyloCSF (Lin et al., 2011), CPC (Kong et al., 2007), CPAT (Wang et al., 2013), PLEK (Li et al., 2014), and CNCI (Sun et al., 2013)], and has previously been used to identify lncRNAs (Che et al., 2018; Donato et al., 2018; Wang et al., 2018). In this study, 2,623 novel lncRNAs were identified, of which 1,484 were classified. Various studies have suggested that lncRNAs can regulate the expression of neighboring genes in cis (Rinn et al., 2007; Faghihi et al., 2008; Guttman et al., 2009; Villegas et al., 2014).
The Ae. albopictus Foshan genome (Chen et al., 2015) and the corresponding protein-coding gene set annotations facilitate assembly and quantification of transcripts for further lncRNA identification. Using Hisat2-StringTie-Ballgown pipeline (Pertea et al., 2016), 305 DEGs and 37 differentially expressed lncRNAs between male and female samples were identified (|log2(fold change)| > 1, FDR < 0.05). As expected, several sperm development BP terms were enriched. Importantly, MSTRG.7577.2, MSTRG.19153.1, MSTRG.18414.1, and MSTRG.21250.1 putatively connect the biological processes of spermatogenesis and cell movement. These DEGs could play important parts in the reproductive development of male sperm. In this study, male high-expression gene AALF000433 was knocked down in male adults using dsRNA. The results showed a strong relationship between knockdown of AALF000433 and a decreased egg hatching rate. The BLAST results showed that AALF000433 encodes the nucleosome assembly protein (Nap1). In 2013, Kimura et al. showed that Drosophila Nap1 is localized at the apical tip of the sperm head, and the nap1 mutant exhibits disruption of the nuclear bundle (Kimura, 2013). This implies that knocking down the expression of nap1 in Ae. albopictus might affect sperm activity, resulting in the low egg hatching rate. Sex determination BP terms were also enriched in female samples (Doublesex [dsx] gene isoform, MSTRG.16151.1, and MSTRG.5389.3). The dsx gene could have an ultimate discriminatory role for sex determination in insects. The dsx pre-messenger RNA produces male- and female-specific splicing patterns that encode the male-DSXM and female-DSXF proteins. These proteins determine male and female development, respectively. The function of dsx has been explored in several insects, including Drosophila (Shirangi et al., 2016), beetles (Ledón-Rettig et al., 2017), Bombyx mori (Xu et al., 2017), A. gambiae (Kyrou et al., 2018), and honey bees (McAfee et al., 2019). Previous studies have suggested that lncRNAs can regulate neighboring coding genes (Villegas and Zaphiropoulos, 2015). In the current study, the functions of differentially expressed lncRNAs were predicted by exploring their partner protein-coding genes. Enriched terms indicated that potential target protein-coding genes were associated with DNA-binding molecular functions.
The accuracy of the RNA-Seq analysis was verified by qRT-PCR using five randomly selected significantly expressed protein-coding genes and lncRNAs. All of the results were consistent with the RNA-Seq data, supporting the reliability of the RNA-Seq data. The present research findings expand our current knowledge about lncRNAs and their relationships with sex development protein-coding genes in Ae. albopictus.
Conclusion
We have presented the first genome-wide analysis of lncRNAs from adult male and female Ae. albopictus, and identified the specifically expressed lncRNAs and mRNAs in males and females. These results will be of use in further studies to obtain a deeper understanding of Ae. albopictus sex determination mechanisms.
Data Availability Statement
Sequencing data were deposited in the NCBI Sequence Read Archive under the accession numbers SRR7990520, SRR7990523, SRR7990524, SRR7990521, SRR7990522, and SRR7990519.
Author Contributions
JG and YX conceived, designed, and supervised this study. YX performed data analysis and wrote the first draft. BJ prepared the samples. YD, ZL, YH, YG, YS, and X-g C participated in discussions and provided valuable advice and practical contributions. YX, and YZX participated in knockdown experiment. All authors reviewed, edited, and approved the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81672054), the Natural Science Foundation of Guangdong Province (2017A030313120), the Research Team Program of the Natural Science Foundation of Guangdong (2014A030312016), and the China Postdoctoral Science Foundation (2018M633078).
Conflict of Interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00975/full#supplementary-material
Figure S1 | Overview of transcriptome analysis and lncRNA identification.
Figure S2 | Principal component analysis (PCA) of the general transcriptome characteristics. The first principal component (PC1) accounted for 62.3% and the second principal component (PC2) for 23% of the total variance in the dataset. The plot indicates that the transcriptome data are highly reproducible.
Figure S3 | Location of dsRNA in AALF000433 and the sequence. Light Blue indicate the sequence of dsRNA.
Table S1 | Primers used for qRT-PCR.
Table S2 | The detail information of all DEGs.
Table S3 | Detailed information for all differentially expressed lncRNAs.
Table S4 | Primers for AALF000433 dsRNA and qRT-PCR.
References
Adelman, Z. N., Tu, Z. (2016). Control of mosquito-borne infectious diseases: sex and gene drive. Trends Parasitol. 32, 219–229. doi: 10.1016/j.pt.2015.12.003
Arun, G., Akhade, V. S., Donakonda, S., Rao, M. R. S. (2012). mrhl RNA, a long noncoding RNA, negatively regulates Wnt signaling through its protein partner Ddx5/p68 in mouse spermatogonial cells. Mol. Cell. Biol. 32, 3140–3152. doi: 10.1128/MCB.00006-12
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bu, D., Yu, K., Sun, S., Xie, C., Skogerbø, G., Miao, R., et al. (2011). NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 40, D210–D215. doi: 10.1093/nar/gkr1175
Che, T., Li, D., Jin, L., Fu, Y., Liu, Y., Liu, P., et al. (2018). Long non-coding RNAs and mRNAs profiling during spleen development in pig. PloS One 13, e0193552. doi: 10.1371/journal.pone.0193552
Chen, X.-G., Jiang, X., Gu, J., Xu, M., Wu, Y., Deng, Y., et al. (2015). Genome sequence of the Asian Tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc. Natl. Acad. Sci. U.S.A. 112, E5907–E5915. doi: 10.1073/pnas.1516410112
Donato, L., Scimone, C., Rinaldi, C., D’angelo, R., Sidoti, A. (2018). Non-coding RNAome of RPE cells under oxidative stress suggests unknown regulative aspects of Retinitis pigmentosa etiopathogenesis. Sci. Rep. 8, 16638. doi: 10.1038/s41598-018-35086-z
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861. doi: 10.1038/nrg3074
Etebari, K., Asad, S., Zhang, G., Asgari, S. (2016). Identification of Aedes aegypti long intergenic non-coding RNAs and their association with Wolbachia and dengue virus infection. PLoS Negl. Trop. Dis. 10, e0005069. doi: 10.1371/journal.pntd.0005069
Faghihi, M. A., Modarresi, F., Khalil, A. M., Wood, D. E., Sahagan, B. G., Morgan, T. E., et al. (2008). Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 14, 723. doi: 10.1038/nm1784
Frazee, A. C., Pertea, G., Jaffe, A. E., Langmead, B., Salzberg, S. L., Leek, J. T. (2015). Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 33, 243. doi: 10.1038/nbt.3172
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223. doi: 10.1038/nature07672
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F., et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. doi: 10.1101/gr.135350.111
Huang, M., Zhong, Z., Lv, M., Shu, J., Tian, Q., Chen, J. (2016). Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget 7, 47186. doi: 10.18632/oncotarget.9706
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2015). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Hung, T., Wang, Y., Lin, M. F., Koegel, A. K., Kotake, Y., Grant, G. D., et al. (2011). Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 43, 621. doi: 10.1038/ng.848
Jarroux, J., Morillon, A., Pinskaya, M. (2017). “History, discovery, and classification of lncRNAs,” in Long Non Coding RNA Biology. Ed. Rao, M. (Singapore: Springer), 1–46. doi: 10.1007/978-981-10-5203-3_1
Jenkins, A. M., Waterhouse, R. M., Muskavitch, M. A. (2015). Long non-coding RNA discovery across the genus anopheles reveals conserved secondary structures within and beyond the Gambiae complex. BMC Genomics 16, 337. doi: 10.1186/s12864-015-1507-3
Jing, Q.-L., Yang, Z.-C., Luo, L., Xiao, X.-C., Di, B., He, P., et al. (2012). Emergence of dengue virus 4 genotype II in Guangzhou, China, 2010: survey and molecular epidemiology of one community outbreak. BMC Infect. Dis. 12, 87. doi: 10.1186/1471-2334-12-87
Kimura, S. (2013). The Nap family proteins, CG5017/Hanabi and Nap1, are essential for Drosophila spermiogenesis. FEBS Lett. 587, 922–929. doi: 10.1016/j.febslet.2013.02.019
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Kyrou, K., Hammond, A. M., Galizi, R., Kranjc, N., Burt, A., Beaghton, A. K., et al. (2018). A CRISPR–Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol. 36, 1062. doi: 10.1038/nbt.4245
Lawson, D., Arensburger, P., Atkinson, P., Besansky, N. J., Bruggner, R. V., Butler, R., et al. (2008). VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res. 37, D583–D587. doi: 10.1093/nar/gkn857
Ledón-Rettig, C., Zattara, E., Moczek, A. (2017). Asymmetric interactions between doublesex and tissue-and sex-specific target genes mediate sexual dimorphism in beetles. Nat. Commun. 8, 14593. doi: 10.1038/ncomms14593
Li, A., Zhang, J., Zhou, Z. (2014). PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinf. 15, 311. doi: 10.1186/1471-2105-15-311
Lin, M. F., Jungreis, I., Kellis, M. (2011). PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–i282. doi: 10.1093/bioinformatics/btr209
Liu, Z., Zhou, T., Lai, Z., Zhang, Z., Jia, Z., Zhou, G., et al. (2017). Competence of Aedes aegypti, Ae. albopictus, and Culex quinquefasciatus mosquitoes as Zika virus vectors, China. Emerging Infect. Dis. 23, 1085. doi: 10.3201/eid2307.161528
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– DDCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
McAfee, A., Pettis, J. S., Tarpy, D. R., Foster, L. J. (2019). Feminizer and doublesex knock-outs cause honey bees to switch sexes. PLoS Biol. 17, e3000256. doi: 10.1371/journal.pbio.3000256
McGinnis, S., Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
Miller, J. R., Koren, S., Dilley, K. A., Puri, V., Brown, D. M., Harkins, D. M., et al. (2018). Analysis of the Aedes albopictus C6/36 genome provides insight into cell line utility for viral propagation. GigaScience 7, gix135. doi: 10.1093/gigascience/gix135
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621. doi: 10.1038/nmeth.1226
Mulvey, B. B., Olcese, U., Cabrera, J. R., Horabin, J. I. (2014). An interactive network of long non-coding RNAs facilitates the Drosophila sex determination decision. Biochim. Biophys. Acta Gene Regul. Mech. 1839, 773–784. doi: 10.1016/j.bbagrm.2014.06.007
Nolasco, S., Bellido, J., Gonçalves, J., Tavares, A., Zabala, J. C., Soares, H. (2012). The expression of tubulin cofactor A (TBCA) is regulated by a noncoding antisense Tbca RNA during testis maturation. PloS One 7, e42536. doi: 10.1371/journal.pone.0042536
Padron, A., Molina-Cruz, A., Quinones, M., Ribeiro, J. M., Ramphul, U., Rodrigues, J., et al. (2014). In depth annotation of the Anopheles gambiae mosquito midgut transcriptome. BMC Genomics 15, 636. doi: 10.1186/1471-2164-15-636
Paupy, C., Ollomo, B., Kamgang, B., Moutailler, S., Rousset, D., Demanou, M., et al. (2010). Comparative role of Aedes albopictus and Aedes aegypti in the emergence of Dengue and Chikungunya in central Africa. Vector-Borne Zoonotic Dis. 10, 259–266. doi: 10.1089/vbz.2009.0005
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650. doi: 10.1038/nprot.2016.095
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290. doi: 10.1038/nbt.3122
Prescott, P. (2004). “Student’s t-Tests,” in Encyclopedia of Statistical Sciences. Eds. Kotz, S., Read, C. B., Balakrishnan, N., Vidakovic, B., Johnson, N. L. (USA: Wiley-Interscience), 8372–8377.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. doi: 10.1016/j.cell.2007.05.022
Shichino, Y., Yamashita, A., Yamamoto, M. (2014). Meiotic long non-coding meiRNA accumulates as a dot at its genetic locus facilitated by Mmi1 and plays as a decoy to lure Mmi1. Open Biol. 4, 140022. doi: 10.1098/rsob.140022
Shirangi, T. R., Wong, A. M., Truman, J. W., Stern, D. L. (2016). Doublesex regulates the connectivity of a neural circuit controlling Drosophila male courtship song. Dev. Cell 37, 533–544. doi: 10.1016/j.devcel.2016.05.012
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41, e166–e166. doi: 10.1093/nar/gkt646
R Core Team. (2013). R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Tsujimoto, H., Hanley, K. A., Sundararajan, A., Devitt, N. P., Schilkey, F. D., Hansen, I. A. (2017). Dengue virus serotype 2 infection alters midgut and carcass gene expression in the Asian tiger mosquito, Aedes albopictus. PloS One 12, e0171345. doi: 10.1371/journal.pone.0171345
Vega-Rúa, A., Zouache, K., Girod, R., Failloux, A.-B., Lourenço-De-Oliveira, R. (2014). High vector competence of Aedes aegypti and Aedes albopictus from ten American countries as a crucial factor of the spread of Chikungunya. J. Virol 88, 6294–6306. doi: 10.1128/JVI.00370-14
Villegas, V., Zaphiropoulos, P. (2015). Neighboring gene regulation by antisense long non-coding RNAs. Int. J. Mol. Sci. 16, 3251–3266. doi: 10.3390/ijms16023251
Villegas, V. E., Rahman, M. F.-U., Fernandez-Barrena, M. G., Diao, Y., Liapi, E., Sonkoly, E., et al. (2014). Identification of novel non-coding RNA-based negative feedback regulating the expression of the oncogenic transcription factor GLI1. Mol. Oncol. 8, 912–926. doi: 10.1016/j.molonc.2014.03.009
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., Li, W. (2013). CPAT: coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74–e74. doi: 10.1093/nar/gkt006
Wang, Y., Xu, T., He, W., Shen, X., Zhao, Q., Bai, J., et al. (2018). Genome-wide identification and characterization of putative lncRNAs in the diamondback moth, Plutella xylostella (L.). Genomics 110, 35–42. doi: 10.1016/j.ygeno.2017.08.003
Wucher, V., Legeai, F., Hedan, B., Rizk, G., Lagoutte, L., Leeb, T., et al. (2017). FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 45, e57–e57. doi: 10.1093/nar/gkw1306
Xu, J., Zhan, S., Chen, S., Zeng, B., Li, Z., James, A. A., et al. (2017). Sexually dimorphic traits in the silkworm, Bombyx mori, are regulated by doublesex. Insect Biochem. Mol. Biol. 80, 42–51. doi: 10.1016/j.ibmb.2016.11.005
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: J. Integrative Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
Keywords: lncRNAs, Aedes albopictus, transcriptome, sex, mosquitoes
Citation: Xu Y, Dong Y, Xu Y, Lai Z, Jin B, Hao Y, Gao Y, Sun Y, Chen X-g and Gu J (2019) Differentiation of Long Non-Coding RNA and mRNA Expression Profiles in Male and Female Aedes albopictus. Front. Genet. 10:975. doi: 10.3389/fgene.2019.00975
Received: 15 May 2019; Accepted: 13 September 2019;
Published: 14 October 2019.
Edited by:
Naomi Lynne Forrester, Keele University, United KingdomCopyright © 2019 Xu, Dong, Xu, Lai, Jin, Hao, Gao, Sun, Chen and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinbao Gu, Z3VqaW5iYW9Ac211LmVkdS5jbg==