 Sami Megdiche
Sami Megdiche Salvatore Mastrangelo
Salvatore Mastrangelo Mohamed Ben Hamouda4
Mohamed Ben Hamouda4 Johannes A. Lenstra
Johannes A. Lenstra Elena Ciani
Elena Ciani- 1Départment des Ressources Animales, Agroalimentaire et Développement Rural, Institut Supérieur Agronomique de Chott-Mariem, Université de Sousse, Sousse, Tunisia
- 2Dipartimento di Bioscienze, Biotecnologie e Biofarmaceutica, University of Bari “Aldo Moro,”Bari, Italy
- 3Dipartimento di Scienze Agrarie, Alimentari e Forestali, University of Palermo, Palermo, Italy
- 4INRA-Tunisie, Institute for Risk Assessment Sciences, Ariana, Tunisia
- 5Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands
Merino sheep represents a valuable genetic resource worldwide. In this study, we investigated selection signatures in Merino (and Merino-derived) sheep breeds using genome-wide SNP data and two different approaches: a classical FST-outlier method and an approach based on the analysis of local ancestry in admixed populations. In order to capture the most reliable signals, we adopted a combined, multi-cohort approach. In particular, scenarios involving four Merino breeds (Spanish Merino, Australian Merino, Chinese Merino, and Sopravissana) were tested via the local ancestry approach, while nine pair-wise breed comparisons contrasting the above breeds, as well as the Gentile di Puglia breed, with non-Merino breeds from the same geographic area were tested via the FST-outlier method. Signals observed using both methods were compared with genome-wide patterns of distribution of runs of homozygosity (ROH) islands. Novel and known selection signatures were detected. The most reliable signals were observed on OAR 3 (MSRB3 and LEMD3), OAR10 (FRY and RXFP2), OAR 13 (RALY), OAR17 (FAM101A), and OAR18 (NFKBIA, SEC23A, and PAX9). All the above overlapped with known QTLs for wool traits, and evidences from the literature of their involvement in skin/hair/wool biology, as well as gene network analysis, further corroborated these results. The signal on OAR10 also contains well known evidence for association with horn morphology and polledness. More elusive biological evidences of association with the Merino phenotype were observed for a number of other genes, notably LOC101120019 and TMEM132B (OAR17), LOC105609948 (OAR3), LOC101110773 (OAR10), and EIF2S2 (OAR17). Taken together, the above results further contribute to decipher the genetic basis underlying the Merino phenotype.
Introduction
Sheep were among the first livestock species to be domesticated (Ryder 1981). Archeological evidences suggest domestication occurred in a region extending from the northern Zagros to southeastern Anatolia ca. 11,000 B.P. (Zeder, 2008). In the last two decades, information from molecular data, as well as discovery and study of novel archaeological sites, has shed new light on the origins and subsequent diffusion of domestic sheep worldwide (Chessa et al., 2009; Meadows et al., 2011; Kijas et al., 2012; Demirci et al., 2013; Singh et al., 2013; Dymova et al., 2017; Ethier et al., 2017; Ivanova et al., 2018a; Ivanova et al., 2018b). Early domesticated sheep are known to have been transported over long distances or even by sea, as early as around 12,000 years BP (Zeder, 2008). They are supposed to have been initially reared mainly for meat and, only during the fifth millennium B.P., specialization for “secondary” products, such as milk and wool, is thought to have occurred (Debono Spiteri et al., 2016). In particular, analysis of viral retro-types combined with archaeological evidence provide support to the hypothesis that specialized wool sheep populations were developed in South-West Asia and then spread throughout Europe, replacing, in the majority of areas, the more primitive domestic stocks (Chessa et al., 2009). Specialization for wool production culminated, in the Middle Ages, with the development of the Merino sheep in Spain. In a recent paper, by analyzing genome-wide SNP data from an intercontinental set of sheep breeds, inclusive of 12 Merino and Merino-derived populations, our group contributed to the reconstruction of the history of Merino development, and the subsequent worldwide merinization process (Ciani et al., 2015).
Well renowned for its premium white fleece and the abundant production of soft, fine, and curly wool, Merino sheep represent a valuable genetic resource worldwide. As such, deciphering the genetic basis underlying the peculiar Merino phenotype is a fundamental aim, and it may further contribute improving wool performances of Merino and Merino-derived breeds. A number of papers have addressed this issue, looking at the genome in search for QTLs (quantitative trait loci) related to wool traits, by using STR markers in Merino (Beh et al., 2001; Bidinost et al., 2008; Roldan et al., 2010), Merino crosses (Rogers et al., 1994; Henry et al., 1998;Zhai et al., 2019), and non-Merino (Allain et al., 1998; Ponz et al., 2001; Allain et al., 2006) sheep populations, or looking at candidate genes (Ling et al., 2014; Rong et al., 2015; Ma et al., 2017; Mu et al., 2017), with keratin genes being among the most studied targets (Parsons et al., 1994; Phuaa et al., 2015; Chai et al., 2017; Li et al., 2017a; Li et al., 2017b; Sulayman et al., 2018; Gong et al., 2019). With the advent of SNP array genotyping technologies, genome-wide association studies (GWAS) using bi-allelic markers have become feasible, and they have been performed in the ovine species to investigate, among others, wool traits (Wang et al., 2014; Bolormaa et al., 2017). An additional approach for connecting DNA to phenotype is the detection of evidence of selective pressure in specific genomic regions by using genome-wide SNP genotype data, also referred to as “selection signatures” analysis. This method has emerged mainly because (i) it does not require the use of phenotypic records, and (ii) unlike GWAS, it can detect selection signatures also when anthropogenic selection has determined complete fixation of the favorable allele (e.g., Qanbari et al., 2014). These features are both relevant in studies addressing genotype–phenotype associations for wool traits, where availability of phenotypic records may represent a limiting issue, and long-term intensive human selection toward wool attributes is likely to have been responsible for the complete prevalence, in the selected populations, of the desired allele. In some of the studies where selection signatures for wool traits have been described, identification of regions affecting wool attributes was not the unique or major goal, with repercussions of this conceptual set-up on the choice of breeds to be contrasted (Zhang et al., 2013; Fariello et al., 2014; Wang et al., 2015; Wei et al., 2015; Seroussi et al., 2017; Rochus et al., 2018). To our knowledge, only two analyses of selection signatures specifically targeting wool attributes have been performed so far (Demars et al., 2017; Gutierrez-Gil et al., 2017). Out of them, only the latter was centered on Merino sheep, which were contrasted, in that study, to the coarse-wool Churra sheep from Spain. Our study follows up on the work by Gutierrez-Gil et al. (2017) to further investigate selection signatures in various Merino (and Merino-derived) sheep breeds under different scenarios, using two different approaches: a classical FST-outlier method and a less usual one based on the analysis of local ancestry in admixed populations (Sankararaman et al., 2008). Signals observed using both methods are also compared with genome-wide patterns of distribution of ROH (runs of homozygosity) islands. We specifically adopted here a multi-cohort approach with the aim of retaining only the most reliable signals.
Materials and Methods
Genotypic Data
A total of 459 unrelated animals arranged in 11 breeds were used in this study (Table S1). Out of them, six were Merino or Merino-derived breeds (Spanish Merino, Australian Merino, Rambouillet, Gentile di Puglia, Sopravissana, and Chinese Merino), and five had no known Merino background (Churra, Ojalada, Bergamasca, Appenninica, and Tibetan) and belonged to the category of “coarse wool” sheep, not purposely selected for wool quality traits (Data Sheet 1). SNP genotypes had been generated in previous published studies (Table S1) by using the Illumina OvineSNP50 Genotyping BeadChip. The whole SNP genotype dataset is available on the WIDDE database (http://widde.toulouse.inra.fr/widde/). The following quality control criteria were applied: (i) individuals with genotyping rate ≤ 90% (command –mind 0.1) were removed; (ii) loci with call rate ≤ 99% (command –geno 0.01), minor allele frequency ≤ 0.005 (command –maf 0.005), and non-autosomal loci were removed; and SNP positions were updated according to the sheep map version Oar_V4. All the above procedures were performed using the PLINK software v. 1.07 (Purcell et al., 2007).
Inference of Local and Global Merino Ancestry
We used the LAMP (Local Ancestry in adMixed Populations) software (Sankararaman et al., 2008) to estimate the individual’s local ancestry of Merino proportion under various scenarios, each contrasting a Merino versus a non-Merino breed, for a total of four cohorts (Table S2). LAMP is a method for estimating ancestries at each locus in a population of admixed individuals i.e., populations formed by the mixing of two or more ancestral populations. The software operates on sliding windows of contiguous SNPs and assigns ancestries by combining the results with a majority vote. The following default settings were adopted: number of generations since admixture (g) = 7 and recombination rate (r) = 1E−08. We opted for adopting default settings in all the tested scenarios since (i) the method was shown to provide robust estimates under different setting configurations in both the literature (Sankararaman et al., 2008) and our preliminary analyses (data not shown). The fraction of global admixture (α) was determined, for each scenario, using the ADMIXTURE software (Alexander et al., 2009). We ran LAMP in the LAMPANC mode, i.e., providing allele frequencies of the two ancestral population proxies. The LAMP analysis provides, among other output results, the marker average ancestry (MAA) related to the two considered ancestral populations. Only MAAs representing the Merino fraction were considered in this study to identify the significant region supposed to be under selection. To this aim, both of the following criteria should be respected by the putative selection signature: (i) local Merino MAA higher than the genome-wide Merino MAA and (ii) being included in the top 5% of SNPs ranked by MAA of Merino proportion.
Detection of FST-Outlier Markers
We adopted the FST-outlier approach implemented in BayeScan (Foll and Gaggiotti, 2008) to detect markers putatively under differential selection pressure in Merino and non-Merino sheep breeds, respectively. To this aim, we performed nine pair-wise comparisons, contrasting each time a Merino versus a non-Merino sheep breed (Table S3). For each cohort, loci that displayed q-val < 0.05 were retained as putatively under selection. Next, we looked for loci that resulted to be putatively under selection in at least four pair-wise comparisons out of nine. For each SNP satisfying the above criteria, we then moved upstream and downstream its position, looking for additional loci with q-val < 0.05 in at least a single pair-wise comparison, and located within 200 kb intervals. We repeated the above process until the next SNP with q-val < 0.05 in at least a single pair-wise comparison was located at a distance higher than 200 kb. Finally, we defined the regions putatively under selection based on the position of the first and the last of the SNPs satisfying the above criteria.
Runs of Homozygosity
ROH were estimated for each animal belonging to the considered breeds using PLINK (Purcell et al., 2007). The following criteria were used to define the ROH: (i) no missing SNP and no heterozygous genotype were allowed in the ROH, (ii) the minimum number of SNPs that constituted the ROH was set to 25, (iii) the minimum SNP density per ROH was set to one SNP every 100 kb, and (iv) the maximum gap between consecutive homozygous SNPs was 250 kb. The minimum length that constituted the ROH was set to 500 Mb. To identify the genomic regions of high homozygosity, the amount of times that each SNP appeared in the ROH was considered and normalized by dividing it by the number of animals included in the analysis. To identify the genomic regions of “high homozygosity,” also called ROH islands, the top 0.999 SNPs of the percentile distribution of the locus homozygosity range within each breed were selected.
Gene and QTL Content of Regions Identified as Under Selection
Annotated genes within the genomic regions putatively under selection were obtained from https://www.ncbi.nlm.nih.gov/genome/gdv/browser/?context=gene&acc=101104604 (NCBI Sheep Genome Data Viewer). The Sheep QTL Database, available at https://www.animalgenome.org/cgi-bin/QTLdb/OA/srchloc?chrom=19&qrange=454178-607539&submit=GO, was interrogated for the presence of QTLs (quantitative trait loci) and significant association signals in the genomic regions identified in this study as putatively under selection. To investigate the biological function and the phenotypes that are known to be affected by each annotated gene, we conducted a comprehensive search in the available literature and public databases, such as NCBI (https://www.ncbi.nlm.nih.gov/), GeneCards (https://www.genecards.org), UniProt (www.uniprot.org), and Amigo2 Gene Ontology database (http://amigo.geneontology.org/amigo). Furthermore, we performed a gene network analysis by using GeneMANIA (Mostafavi et al., 2008). This tool allows to build weighted interaction networks using as a source a very large set of functional association data including protein and genetic interactions, pathways, co-expression, co-localization, and protein domain similarity (see http://pages.genemania.org/help/for a more detailed description of the considered network categories).
Results
Signals of Selection Detected via “Local Ancestry”
Preliminary to the local ancestry analysis, we performed a “global ancestry” analysis using the Bayesian approach implemented in the software ADMIXTURE (Alexander et al., 2009). Individual proportions of global admixture (α) are presented, for the four considered breeds, in Figure S1. The observed patterns support, for all the tested breeds, the formulated scenarios, i.e., that each breed could be considered to derive from the crossbreeding of a given Merino and a given non-Merino breed (breed A and breed B, respectively, in Table S2). Putatively selected regions, identified from LAMP results, are shown, for the four considered breeds, in Table S4–S7. An excess of Merino ancestry was observed at 26, 24, 17, and 22 regions for Australian Merino, Chinese Merino, Sopravissana, and Spanish Merino, respectively. A number of regions were shared by at least three out of the four breeds (Table 1) and, among them, two large regions, on OAR 17 (overlapping signals at 48,474,658–58,410,640 bp) and OAR 18 (overlapping signals at 42,864,163–51,943,741 bp), were shared by all the four breeds.
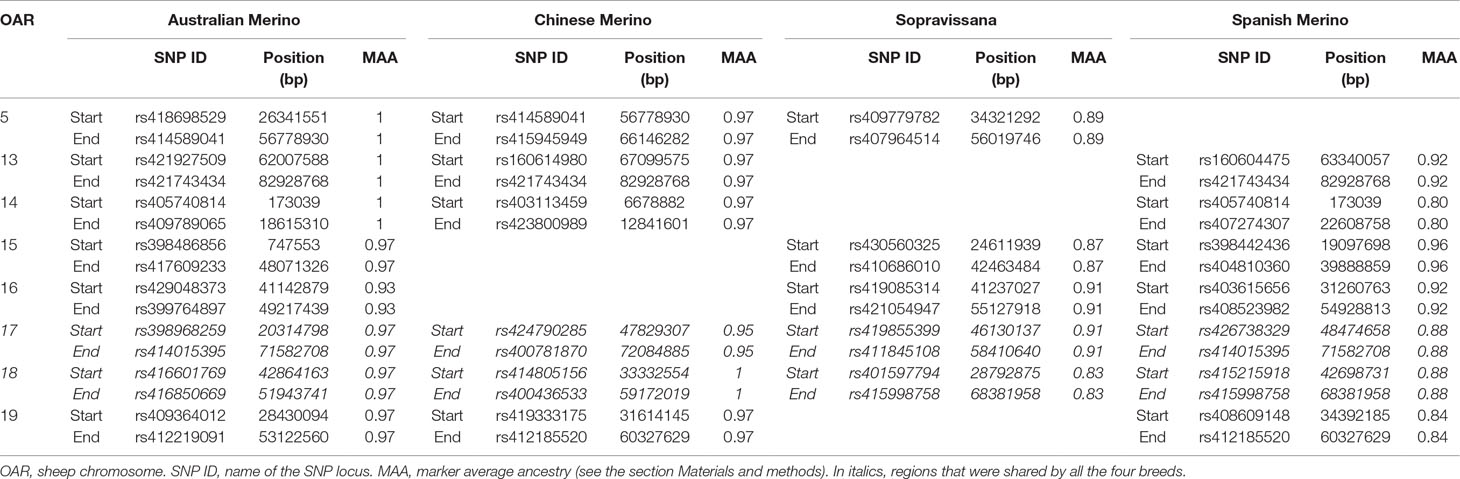
Table 1 Signals of selection detected via local ancestry, shared by at least three of the four considered breeds.
Signals of Selection Detected via the “FST-Outlier” Method
Results of the analyses performed using the FST-outlier approach implemented in BayeScan are presented, for the nine pair-wise comparisons involving Merino and non-Merino breeds, in Table S8. The highest number of significant SNPs (277) was detected for the contrast Chinese Merino vs. Tibetan. A summary of the obtained results is presented in Table 2. Four regions putatively under differential selection pressure were identified, on OAR3 (15,382,6281–154,318,689 bp), OAR10 (29,392,142–29,776,019 bp), OAR13 (62,707,138–62,747,155 bp), and OAR19 (454,178–607,539 bp). Interestingly, in the region on OAR13, one SNP (rs415003205) had q-val < 0.05 in all the nine considered pair-wise comparisons. This is a deep intronic variant (G/A) located at 5,264 bp downstream the end of the first exon of the RALY gene. For the region on OAR3, the locus displaying the highest number of pair-wise comparisons showing signal of selection (six out of nine) was rs423370130 (154,072,493 bp). For the region on OAR19, the highest number of pair-wise comparisons showing signal of selection was five (rs404730996). The large region on OAR10 displayed a maximum of six pair-wise comparisons showing signal of selection, at 29,413,536 bp (rs401979890). In the same region, two loci, out of which one (rs414794714, at 29776019 bp) rather far from rs401979890, had five pair-wise comparisons showing signal of selection. This pattern suggests that the considered region may harbor two different selection signatures.

Table 2 Summary results of the FST-outlier approach for the nine pair-wise comparisons between Merino and non-Merino breeds.
The loci that provided overlapping signals for the same Merino (or Merino-derived) breed with both the “local ancestry” and the “FST-outlier” methods are highlighted in Table S8, while in Tables S4 to S7, the putatively selected regions, detected using the “local ancestry” method, where at least one significant SNP in at least one pair-wise comparison of the “FST-outlier” method involving the corresponding Merino (or Merino-derived) breed was observed, are highlighted in light yellow. In general, the majority of the regions (16/26, Australian Merino; 15/24, Chinese Merino; 9/17 Sopravissana; 12/22 Spanish Merino) showed overlapping signals between the two methods.
Gene and QTL Content of Putatively Selected Regions
The two large regions on OAR 17 and 18, detected as putatively selected by “local ancestry” analysis, contain 148 and 70 genes, respectively (Table S9 and S10). These regions were screened for the presence of known QTLs in sheep (Table S11A). Interestingly, we found two QTLs associated with a wool trait, notably “greasy fleece weight,” at positions 49,606,819–49,606,859 bp and 51,061,367–51,061,407 bp, respectively, in OAR17 (Ebrahimi et al., 2017), and one QTL associated with “staple length,” at position 9,943,363–68,604,602 bp in OAR18 (Allain et al., 2006). The OAR17 QTL at position 49,606,819–49,606,859 bp is located in an inter-genic region, between LOC101120019 (60S ribosomal protein L10a-like, at position 49,486,725–49,520,271 bp) and TMEM132B (transmembrane protein 132B, at position 49,844,529–50,246,808 bp) (data not shown). Similarly, the OAR17 QTL at position 51,061,367–51,061,407 bp is located in an inter-genic region, between LOC101115905 (refilin-A, alias “family with sequence similarity 101, member A” or “filamin-interacting protein FAM101A,” at position 51,021,418–51,035,210 bp) and LOC106991703 (a long non-coding RNA, at position 5,118,8762–51,255,313 bp) (data not shown). The large OAR18 QTL at position 9,943,363–68,604,602 bp includes 693 genes (data not shown) and was hence not useful to refine the signal position. Therefore, the 70 genes detected in the putatively selected region on OAR18 were screened for inclusion in the output of the human GeneCards database using the queries “hair,” “wool,” and “horn,” selected as the most representative of the Merino phenotype. While none of the genes was retrieved when using “wool” or “horn” keywords, a total of 16 out of 70 (23%) genes were retrieved when using the keyword “hair” (Table S10). Notably, three genes displayed particularly high GeneCards relevance scores: NFKBIA (16.1), SEC23A (15.27), and PAX9 (7.17).
The four regions detected as putatively selected by “FST-outlier” analysis contain five (OAR3), four (OAR10), one (OAR13), and two (OAR19) genes (Tables S12A–D). Also, these regions were screened for the presence of known QTLs in sheep (Table S11B). On OAR3, a QTL associated with wool traits (notably, “staple length”) had been previously mapped, within a large chromosome interval (region 1,184,337–224,283,230 bp) encompassing the region detected in this study (Ponz et al., 2001). On OAR10, a genome-wide association study for wool traits in Chinese Merino detected a significant SNP for fiber diameter at position 30 Mb, and several SNPs significant for crimp at 26–27 Mb (Wang et al., 2014). On OAR13, one SNP at 62.9 Mb was associated with wool fiber diameter (Bolormaa et al., 2017).
The results of the gene network analysis for the genes located in the putatively selected regions mentioned above are presented in Figure 1 and Table S13. A total of 57 links are reported for the considered 29 genes, out of which 9 genes had been detected in this study as putatively selected. Interestingly, links were observed not only between genes detected as putatively selected using the same approach, either LAMP or the FST-outlier, but also between genes detected as putatively selected using different approaches (SEC23A/MSRB3, RXFP2/PAX9, EIF2S2/TMEM132B, SEC23A/RXFP2), thus suggesting their complementarity in selection signature detection.
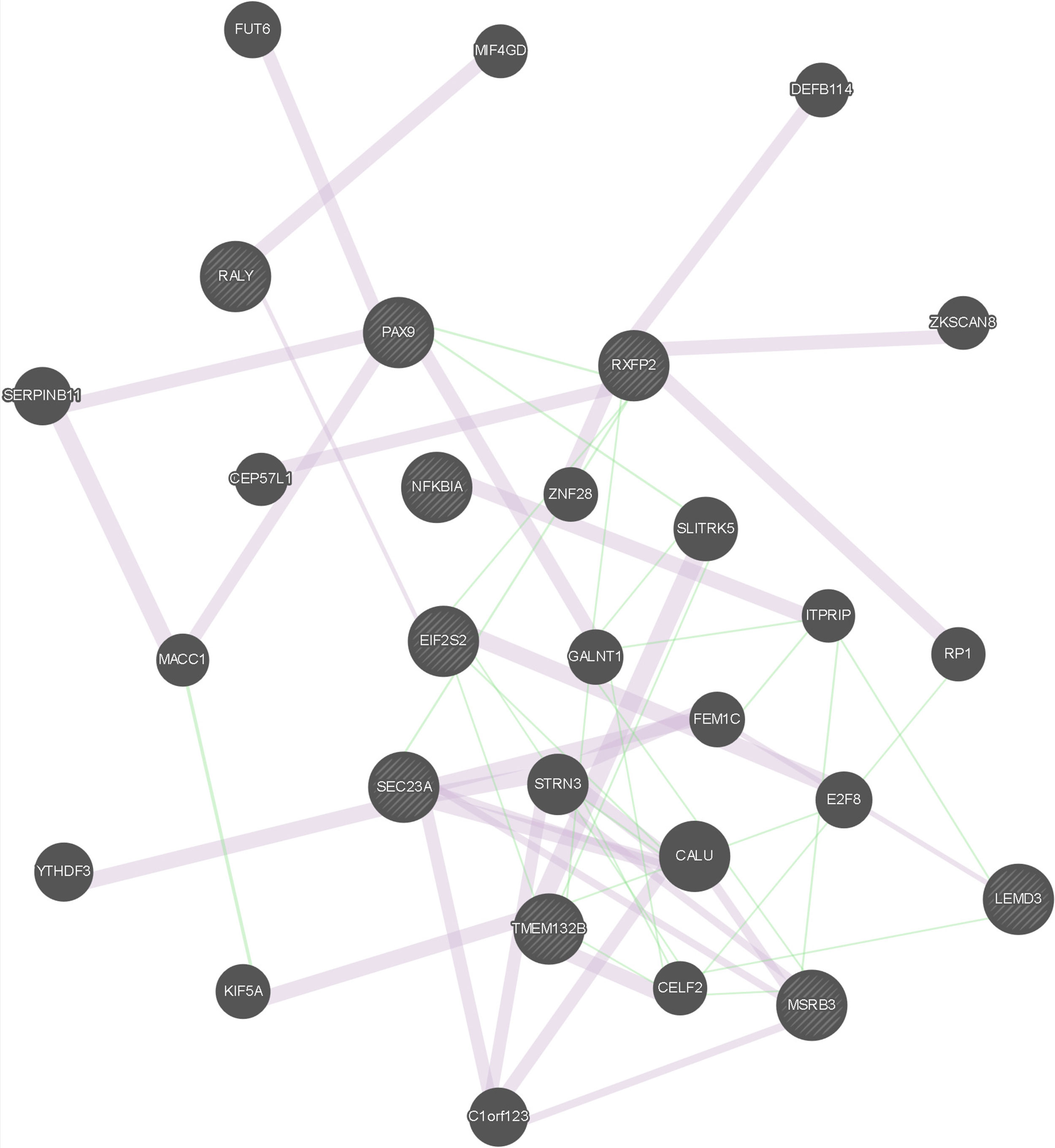
Figure 1 Graphical representation of the gene network analysis. Query genes, i.e., genes detected in this study as putatively selected, are indicated with stripes. Links in light purple indicate the network category co-expression (two genes are linked if their expression levels are similar across conditions in a gene expression study. Most of these data are collected from GEO, the Gene Expression Omnibus); only data associated with a publication are collected. Links in green indicate the network category genetic interaction. (Two genes are functionally associated if the effects of perturbing one gene were found to be modified by perturbations to a second gene. These data are collected from primary studies and BioGRID).
Runs of Homozygosity
Several genomic regions that frequently appeared in a ROH were identified within each breed. Table 3 provides the chromosome position, and the start and end of the detected ROH islands. The top 0.999 SNPs of the percentile distribution of locus homozygosity values led to the use of different thresholds for each breed (from 0.166, in Gentile di Puglia, to 0.261, in Chinese Merino). The genomic distribution of ROH islands was clearly non-uniform among breeds. Gentile di Puglia showed the highest number (21) of ROH islands, followed by the Spanish Merino (12). Gentile di Puglia, together with Sopravissana, also displayed large proportions (33.3% and 50%, respectively) of ROH islands longer than 5 Mb. These results may well reflect the serious bottlenecks experienced by these breeds in the last 70 years. Three overlapping ROH islands were observed between breed pairs. Spanish Merinos and Gentile di Puglia breeds showed a common 5 Mb genomic region on OAR12 (47,013,871 to 52,019,776 bp). Smaller (<1 Mb) genomic regions were shared between Gentile di Puglia and Chinese Merino on OAR2 (99,442,430 to 100,215,565 bp) and between Chinese Merino and Appenninica on OAR16 (30,100,068 to 30,670,323 bp).
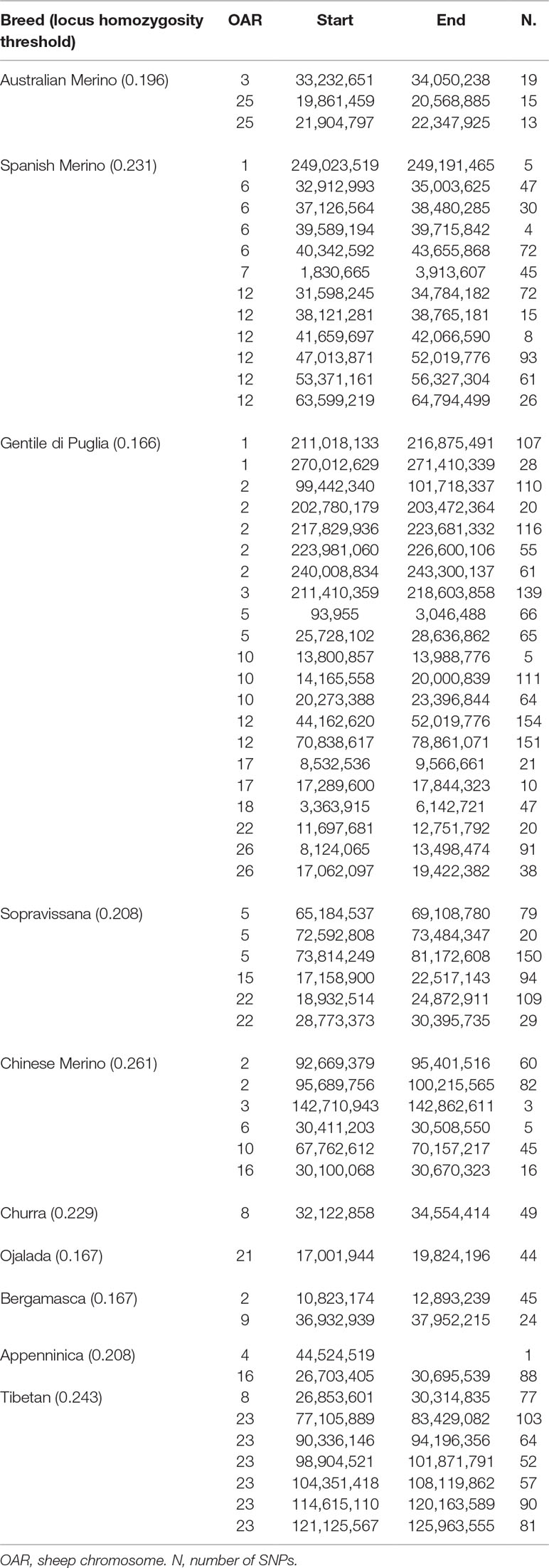
Table 3 List of genomic regions of extended homozygosity (ROH islands) identified in the considered Merino and non-Merino breeds.
When comparing the ROH islands observed within each Merino (or Merino-derived) breed (Table 3) with regions detected by “local ancestry” analysis involving the same Merino (or Merino-derived) breed (Tables S4–S7), we found little overlapping. In particular, the two ROH islands detected on OAR25 in Australian Merino were both included in the LAMP region detected for the same breed on the same chromosome. Similarly, the four ROH islands detected on OAR6 in Spanish Merino were all included in the LAMP region detected for the same breed on the same chromosome. No overlapping was observed for Chinese Merino and Sopravissana.
When comparing the ROH islands observed within each Merino (or Merino-derived) breed (Table 3) with SNPs detected as significantly differentiated in “FST-outlier” pair-wise contrasts involving the same Merino (or Merino-derived) breed (Table S8), some overlapping was observed. In particular, the SNP rs408794746 (34,050,238 bp in OAR3) was significantly differentiated when contrasting Australian Merino with Churra and Ojalada and was also detected within a ROH island in Australian Merino. The significantly differentiated SNP rs425817109 (34,390,603 bp) in the Spanish Merino vs. Churra comparison, and the SNP rs400309388 (34,699,452 bp) in the Spanish Merino vs. Ojalada comparison, were both included within a ROH island detected in Spanish Merino (OAR12). The SNP rs403786137 (215,181,085 bp in OAR1) in the Gentile di Puglia vs. Bergamasca comparison was included within a ROH island detected in Gentile di Puglia. The SNP rs398231484 (216,225,845 bp in OAR3) in the Gentile di Puglia vs. Appenninica comparison was included within a ROH island detected in Gentile di Puglia. The SNP rs407100968 (45,671,005 bp) in the Gentile di Puglia vs. Appenninica comparison and the SNP rs417849493 (48,709,065 bp) in the Gentile di Puglia vs. Bergamasca comparison were both included within a ROH island detected in Gentile di Puglia (OAR12). The SNP rs399908187 (68,477,988 bp in OAR5) in the Sopravissana vs. Bergamasca comparison was included within a ROH island detected in Sopravissana. No significantly differentiated SNP overlapping with ROH islands was detected for the Chinese Merino breed.
Discussion
Comparison Among Approaches for Selection Signatures Detection
In this study, we investigated selection signatures in various Merino (and Merino-derived) sheep breeds using two different approaches. While the “FST-outlier” is considered a classical method for identification of regions putatively under differential selection in pairs of breeds (or group of breeds), the analysis of local ancestry, i.e., the genetic ancestry of an individual at a particular chromosomal location, in admixed populations to detect genomic regions where a significant excess of ancestry from a given parental breed exists (also known as “admixture mapping”) is so far a less popular approach. Among the “admixture mapping” approaches, LAMP has some interesting features that prompted us to opt for this method. Unlike algorithms that are based on reference haplotype frequencies for each of the parental populations, for which larger sample sizes are required to capture haplotypic diversity, LAMP relies on reference allele frequencies (Shriner, 2013) and is consequently less affected by a reduced sample size. Also, it operates on sliding windows of contiguous SNPs, using a “majority vote” for each locus, over all windows that overlap with the SNP, in order to decide the most likely ancestral population at the marker. This simple approach has been shown to provide fast and robust estimates (Sankararaman et al., 2008). Despite LAMP has been developed for estimation of the locus-specific ancestry in recently admixed populations, it has been shown to be robust to inaccuracies in the parameter “number of generations since the admixture.” A critical issue, limiting the widespread use of LAMP, is represented by the choice of the external reference samples to be used as proxies for the true ancestral populations, as the latter are generally not available for sampling (Shriner, 2013). In this study, a set of four hypotheses, each including a test breed and two proxies for the parental populations, were formulated based on historical knowledge on the origin of breeds and the inferred proportions of global admixture. These have to be interpreted with caution given the possible influence of complex patterns of historical admixture known among Merino and Merino-derived sheep populations (Ciani et al., 2015). The four hypotheses were hence tested using the algorithm implemented in LAMP. On the other side, for the “FST-outlier” approach, we were able to define, a set of nine pair-wise comparisons by contrasting (i) Merino populations of Iberian origin (Spanish Merino and Australian Merino, respectively) with non-Merino populations of Iberian origin (Churra and Ojalada, respectively), (ii) Merino populations of Italian origin (Gentile di Puglia and Sopravissana, respectively) with non-Merino populations of Italian origin (Appenninica and Bergamasca, respectively), and (iii) a Merino population of Asian origin (Chinese Merino) with a non-Merino population of Asian origin (Tibetan). The rationale behind the above pairing is that differentially selected loci may be easier to detect when contrasting more homogeneous breeds, such as Merino versus non-Merino breeds from the closest geographical area (Manunza et al., 2016).
Consistently with expectations, the two adopted approaches produced only partly overlapping signals. Indeed, the two methods rely on different algorithms and different assumptions, which also imposed a different organization of the dataset used with the two approaches (four single-breed tests vs. nine pair-wise comparisons, for the “local ancestry” and the FST-outlier” approaches, respectively), thus hampering direct head-to-head comparisons. Notwithstanding, the majority of the regions detected using the “local ancestry” method showed overlapping signals with the “FST-outlier” results. Although we interpreted the above as evidence supporting the robustness of the obtained results, it must be taken into consideration that regions identified by “local ancestry” were generally large, and significant SNPs detected via the “FST-outlier” method may likely occur in there by chance. Indeed, the number of loci identified as putatively under selection pressure using the “FST-outlier” method largely exceeds the number of putatively selected regions identified using the “local ancestry” approach.
In this study, we also investigated genomic regions of high homozygosity (ROH islands), as these have been shown to be abundant in regions under positive selection (Metzger et al., 2015; Mastrangelo et al., 2017; Purfield et al., 2017; Talenti et al., 2017a; Talenti et al., 2017b; Mastrangelo et al., 2018). While we observed little overlapping between ROH islands and regions identified via “local ancestry,” some overlapping was observed between ROH islands and SNPs detected as significant using the “FST-outlier” approach. ROH islands may be the consequence of the genetic hitchhiking phenomenon at loci physically linked to the variant site under direct positive selection pressure. The “local ancestry” approach looks for regions with an excess of ancestry from one of the two parental populations, and not necessarily these regions have to display high homozygosity, although this feature is likely to be observed in case of a strong selective sweep. Similarly, in the “FST-outlier” approach, homozygous genotypes (for different alleles in the two breeds) at loci physically linked to the variant site under direct positive selection pressure may display high frequencies if a strong differential selection existed among the two considered breeds. Also, the argumentation reported above may apply here: the larger number of loci identified as putatively under selection pressure using the “FST-outlier” method may be more likely to occur by chance within large ROH islands compared to the fewer genomic regions identified via “local ancestry.” Moreover, ROH analysis might detect selection related to any trait, while contrasting Merino and non-Merino is more likely to detect signals related to this specific trait. Finally, the existence of ROH islands may be due to non-genetic factors such as demography.
Best Candidate Regions and Putatively Selected Genes
As the aim of this study was to identify loci most likely associated with the Merino phenotype, we arbitrarily identified (i) the best candidate regions detected via the “local ancestry” approach as those being shared by all the four breeds and (ii) the best candidate SNPs detected via the “FST-outlier” approach as those observed in at least 70% of the pair-wise comparisons (six out of nine). Based on the above, two large regions on OAR17 and OAR18 were retained for (i), and three, on OAR3, OAR10, and OAR13, for (ii). The robustness of the adopted procedure was also suggested by the occurrence, in all of the five regions, of QTLs/associations known to be related to wool traits in the ovine species. Moreover, at OAR17, combining analysis of “local ancestry” and inspection of the sheep QTL database allowed to significantly shorten the candidate interval. On the contrary, known QTLs for wool traits described on OAR18 and OAR3 are mapped within extremely large chromosome intervals. These were identified by Allain et al. (2006) and Ponz et al. (2001) who performed whole-genome scans using microsatellite markers on experimental flocks obtained crossing Lacaune with Sarda, and Berrichon du Cher (a Merino-derived breed) with Romanov (a non-Merino breed), respectively. In what follows, the gene content of the best candidate regions is presented by chromosome order.
OAR3
The region detected on OAR3 contains five genes, LOC105609945 (long noncoding RNA), MSRB3 (methionine sulfoxide reductase B3), LOC105609947 (long noncoding RNA), LOC105609948 (a pseudo-gene), and LEMD3 (LEM domain containing 3). Interestingly, the sub-region containing the genes MSRB3, LOC105609947, and LEMD3 was found to harbor a selection signature putatively for tail fat deposition in previous studies contrasting thin- vs. fat-tail sheep breeds, from China (Yuan et al., 2017), and from North Africa and the Chios island (Mastrangelo et al., 2019), for adaptation when contrasting the Red Maasai sheep with the Ethiopian Menz (Fariello et al., 2014), and for ear morphology in Chinese sheep breeds (Wei et al., 2015) and in French Suffolk sheep (Rochus et al., 2018). The latter suggest to consider the genes encoded by the signal on OAR3, notably MSRB3 and LEMD3, as candidates for ear size based on literature showing the possible role of the two genes in ear position in dogs (Vaysse et al., 2011) and ear size in pigs (Wilkinson et al., 2013). A more detailed discussion of each single gene in the OAR3 selection signature is provided below.
LOC105609945—No evidence for involvement of LOC105609945 in any peculiar Merino feature was found.
MSRB3—The methionine sulfoxide reductase B3 (MSRB3, alias DFNB74) catalyzes the reduction of free and protein-bound methionine sulfoxide to methionine. This antioxidant repair enzyme has been described in human epidermal keratinocytes and melanocytes, as well as in hair follicles (Taungjaruwinai et al., 2009). It has been shown to be expressed also in inner and outer hair cells of mouse inner ear (Ahmed et al., 2011). Diseases associated with MSRB3 include deafness (https://www.genecards.org). Down-regulation of MSRB3 has been shown to impair the normal auditory system development through hair cell apoptosis in zebrafish (Shen et al., 2015). The gene has been found in previous selection signatures studies in sheep (Kijas et al., 2012; Fariello et al., 2014; Wei et al., 2015; Manunza et al., 2016; Yuan et al., 2017; Rochus et al., 2018; Mastrangelo et al., 2019). More interestingly, for this study, it has been found within a selection signature observed contrasting fine-wool Merino and coarse-wool Churra sheep breeds (Gutiérrez-Gil et al., 2017). Another line of evidence for the involvement of MSRB3 in hair/wool physiology comes from the observation that actin’s polymerization properties and actin cytoskeletal-mediated events, such as correct bristle development, which are altered by specific oxidation of its conserved methionine (Met)-44 residue on the pointed-end of actin subunits, are rescued by a methionine sulfoxide enzyme reductase (SelR/MsrB) in Drosophila (Hung et al., 2013). In this species, actin plays a role not only in bristle but also in wing hair development (Guild et al., 2005). In mammals, actin has been shown to be one of the major components of both the water-soluble and -insoluble fraction from hair and hair follicles (Vermorken et al., 1981; Lee et al., 2006). Actin bundles in the hair follicle would act as stress fibers and serve as a tensile scaffold for the growth and integrity of the hair follicle (Furumura and Ishikawa, 1996). In Tibetan sheep, microRNAs differentially expressed in wool follicles during anagen, catagen, and telogen phases, thus potentially regulating wool follicle development, targeted, among others, genes in the pathways that regulate the actin cytoskeleton (Liu et al., 2013). MSRB3 also contained the (intronic) SNP that, in this study, displayed the highest number of “FST-outlier” pair-wise comparisons showing signal of selection observed in the OAR3 region.
LOC105609947—No evidence for involvement of LOC105609945 in any peculiar Merino feature was found.
LOC105609948—It’s a pseudo for the ubiquitin-conjugating enzyme E2 D3 gene (UBE2D3), which is part of the bone morphogenic protein (BMP) signaling pathways (gene ontology database accession ID: GO:0030509). BMP ligands (BMP2 and BMP4) when expressed in dermal macro-environment during telogen (resting phase of hair cycle) have been shown to strongly suppress ability of resting hair follicles to be reactivated and grow again (International Patent no. WO2010059861A1 available at https://patents.google.com/patent/WO2010059861A1).
LEMD3—As previously mentioned, the LEM domain containing three gene (alias MAN1) has been found in various selection signatures studies in sheep (Fariello et al., 2014; Wei et al., 2015; Manunza et al., 2016; Yuan et al., 2017; Rochus et al., 2018; Mastrangelo et al., 2019), including the study by Gutierrez-Gil et al. (2017) where fine-wool Merino and coarse-wool Churra sheep breeds were contrasted. Moreover, it has been found associated with the abnormal hair quantity phenotype from the HPO Gene-Disease Associations dataset (Köhler et al., 2014).
OAR10
The region detected on OAR10 includes four genes: LOC106991357 (long noncoding RNA), LOC101110773 (elongation factor 1-alpha 1-like), RXFP2 (relaxin/insulin-like family peptide receptor 2), and LOC106991379 (a pseudo-gene). Despite this region was detected as putatively selected in studies investigating tail fat deposition (Moioli et al., 2015; Yuan et al., 2017; Mastrangelo et al., 2019) and adaptation (Yang et al., 2016; Seroussi et al., 2017), RXFP2 is the most studied gene and is well known for being involved in horn presence/absence and morphology in sheep (Johnston et al., 2011; Kijas et al., 2012; Fariello et al., 2014; Pan et al., 2018). A genome-wide association study for wool traits in Chinese Merino sheep detected, on this chromosome, a significant SNP for “fiber diameter” at position 30 Mb, together with several SNPs significant for “crimp” at 26–27 Mb (Wang et al., 2014). In what follows, a more detailed discussion of the four genes annotated in the OAR10 region is provided.
LOC106991357—No evidence for involvement of this locus in any peculiar Merino feature was found.
LOC101110773—It codes for an elongation factor 1-alpha 1-like. The elongation factor 1-alpha 1 (EF1A1) is a GTP-binding protein which has a primary function as an essential house-keeping gene by delivering aminoacyl-tRNAs to the ribosome during the elongation step of protein translation. EF1A1, together with genes associated to the Usher syndrome, a congenital disease characterized by perturbation of normal organization and growth of hair bundles within the inner ear, is a downstream target of GBX2, which induces EF1A1 activation upon binding to the EF1A1 core promoter. GBX2 has been shown to be expressed in the otic placode, which develops into the inner ear. Loss-of-function and mis-expression studies have shown that GBX2 is essential for development of the inner ear sensory organs. However, neither direct evidence for involvement of EF1A1 in hair bundles organization and growth nor in any peculiar Merino phenotype has been found so far. Another elongation factor type (EF1Bγ) has been proposed to bind to keratin (Kim et al., 2006). The presence of large amounts of EF1Bγ in keratin bundle rich hair fibers would support its biological role in the intermediate filament organization (Sasikumar et al., 2012).
RXFP2—This gene is involved, among others, in the biological process “activation of adenylate cyclase activity” (https://www.uniprot.org/uniprot/Q8WXD0). Adenylate cyclase is responsible for the synthesis of 3’,5’-cyclic adenosine monophosphate (cAMP). Agents that increase cAMP levels have been shown to be potent inhibitors of human and mouse hair follicle growth (Harmon and Nevins, 1997). However, we cannot exclude that, in our tested scenarios, different alleles at this gene may have been differentially selected in the considered breeds as a consequence of selection toward different horn phenotypes. Rochus et al. (2018) highlighted that a number of single nucleotide polymorphisms exist in French sheep in the region extending 100 Kb upstream of RXFP2, with haplotypes in polled sheep being distinct from those observed in horned sheep. From these findings, they suggested that multiple ancient mutations, rather than a single mutation, are likely affecting horn phenotypes. Pan et al. (2018) reported strong association between three SNPs within the RXFP2 gene and horn sizes in a Tibetan population characterized by the presence of animals with heterogeneous horn types.
LOC106991379—No evidence for involvement of this locus in any peculiar Merino feature was found.
It is worth mentioning that, only 0.076 Mb upstream to LOC106991357 on OAR10, the gene FRY is mapped (interval 28,959,450–29,212,913 bp). Looking at our results separately for each tested scenario, we observed that this interval was overlapping with the region detected by the “local ancestry” method in Chinese Merino, as well as with the regions detected by the “FST-outlier” method in the Chinese Merino vs. Tibetan, Australian Merino vs. Churra, and Gentile di Puglia vs. Appenninica comparisons. Moreover, the FRY interval was only slightly upstream to the regions detected by the “FST-outlier” method in the Australian Merino vs. Ojalada (0.2 Mb), Spanish Merino vs. Ojalada (0.18 Mb), Gentile di Puglia vs. Bergamasca (0.18 Mb), Sopravissana vs. Appenninica (0.2 Mb), and Sopravissana vs. Bergamasca (0.18 Mb). In sheep, FRY has been suggested as a key candidate gene for the piebald phenotype in Merino (Garcia-Gamez et al., 2011) and has been suggested to be associated with the black spot phenotype in Valley-type Tibetan sheep (Wei et al., 2015), and with differences in coat color pigmentation distribution between the Awassi and Afec-Assaf sheep (Seroussi et al., 2017). On the contrary, Zhang et al. (2013) detected FRY when contrasting Rambouillet and Suffolk sheep and suggested it to be a candidate gene affecting wool quality. Indeed, FRY encodes a protein furry homolog that, in Drosophila, has been found in growing hairs (He et al., 2005), and whose disruption has been shown to provoke abnormally branched bristles and strong multiple-hair phenotype, with clusters of epidermal hairs and branched hairs (Cong et al., 2001). Fang et al. (2010), following the transgenic FRY protein in vivo, found it to be highly mobile and to accumulate at the distal tip of growing bristles and suggest that it could function in directing/mediating the intracellular transport needed for polarized growth.
OAR13
The region detected on OAR13 contains a single gene (RALY). It encodes a heterogeneous nuclear ribonucleoprotein that binds poly-U-rich elements within several RNAs and regulates the expression of specific transcripts (Cornella et al., 2017; Rossi et al., 2017). In early 90s, the gene was shown to be involved in the pleiotropic lethal yellow phenotype of the mouse due to a deletion of the genes RALY and EIF2S2 (eukaryotic translation initiation factor 2 subunit 2), upstream the ASIP (agouti signaling protein) gene, responsible for the ectopic over-expression of the agouti signaling protein under the control of the RALY promoter (Michaud et al., 1993; Duhl et al., 1994). The gene has been repeatedly detected, often together with the neighbor ASIP gene, in association studies concerning skin pigmentation and skin neoplasms, (https://www.ncbi.nlm.nih.gov/gap/phegeni?tab=1&gene=22913; Jacobs et al., 2015), as well as in several other type of cancers, where it is considered to represent a metastatic marker (Roberts et al., 2019). In 2013, a mutation in this gene has been associated with the saddle tan and black-and-tan phenotypes in Basset Hounds and Pembroke Welsh Corgis (Dreger et al., 2013). Similarly, it has been associated with coat color phenotypes in Chinese and Iranian goats (Guo et al., 2018; Nazari- Ghadikolaei et al., 2018). Worth mentioning that a SNP at OAR13 (position 62.9 Mb) was associated by Bolormaa et al. (2017) with wool fiber diameter in Merino sheep. Although the SNP is not far from the RALY gene, it mapped within the EIF2S2 gene, which has been shown to be involved in protection against chemotherapy-induced alopecia (Nasr et al., 2013).
OAR17
Out of the two pairs of genes flanking the two QTLs on OAR17, one (LOC101120019) is a pseudo-gene related to a 60S ribosomal protein (L10A), one (TMEM132B) codes for a trans-membrane protein, one (LOC101115905) codes for refilin-A (alias FAM101A), and one (LOC106991703) is responsible for the production of a long noncoding RNA. Their possible involvement in the Merino phenotype is discussed here based on evidences from the literature.
LOC101120019—A mutation in a 60S ribosomal protein (L21) has been shown to be involved in hereditary hypotrichosis simplex (HHS), a form of nonsyndromic inherited hair loss disorders (Zhou et al., 2011). 60S ribosomal proteins (L6 and L24) have been shown to be expressed in human anagen hair samples (Carlson et al., 2018). Interestingly, the 60S ribosomal protein L10A has been shown to be expressed in root hairs of Medicago truncatula (Covitz et al., 1998). As is common for genes encoding ribosomal proteins, multiple processed pseudo-genes of the 60S ribosomal protein L10A are dispersed through the genome (https://www.ncbi.nlm.nih.gov/gene/4736). In particular, LOC101120019 on OAR17, being a pseudo-gene, is more likely to play, if any, a regulatory function on the hair physiology.
TMEM132B—The trans-membrane protein 132B is required for normal inner ear hair cell function (https://www.genecards.org/cgi-bin/carddisp.pl?gene=TMEM132E). TMEM132A, but not TMEM132B, TMEM132C, or TMEM132D, was found to be expressed in wool follicle bulb of Tibetan sheep during phase transformation from the middle anagen, to catagen and late telogen/early anagen (Liu et al., 2015). TMEM132E was found to be highly expressed in murine inner hair cells, and a variant in TMEM132E was identified as the most likely cause of autosomal-recessive nonsyndromic hearing loss. Knockdown of the TMEM132E ortholog in zebrafish affected the mechanotransduction of hair cells. (Li et al., 2015).
FAM101A—The gene product is involved in the regulation of the perinuclear actin network and nuclear shape through interaction with filamins. It plays an essential role in the formation of cartilaginous skeletal elements (UniProtKB:Q5SVD0). In addition, it has been shown to be differentially expressed in hair follicle stem cells residing in the bulge of mouse hair follicles versus the epithelial basal cells outside the bulge (Chang, 2014). FAM101A mRNA was detected via next-generation sequencing in wool follicle bulb samples of Tibetan sheep from middle anagen, catagen, and late telogen/early anagen phases (Liu et al., 2015). In a genome-wide association study performed using 50 K SNPs in a Baluchi sheep population, one of the significant SNP markers associated with greasy fleece weight was located within FAM101A (Ebrahimi et al., 2017).
LOC106991703—No evidence for involvement of LOC106991703 in any peculiar Merino feature was found.
OAR18
The region identified on OAR18 included a large number of genes (70) that obviously hampered a detailed analysis of the available literature for each single gene. We hence opted for checking which of the above genes could be retrieved by querying the human GeneCards database using keywords representative of Merino phenotypes. While none of the genes was retrieved when using “wool” or “horn” keywords, 16 genes were retrieved when using the keyword “hair” and, among them, three displayed particularly high GeneCards relevance scores (NFKBIA, SEC23A, and PAX9).
NFKBIA (NFKB inhibitor alpha)—It has been shown to modulate WNT, SHH, and LHX2IS signaling at early stages of hair follicle development in mice. In particular, in the epidermis of mice lacking the transcription factor nuclear factor-kappa B activity, primary hair follicle pre-placode formation is initiated without progression to proper placodes (Tomann et al., 2016). The gene has been also detected as putatively under selection in a Chinese Merino sheep population (Liu et al., 2017).
SEC23A—It is one of the major components and markers of COPII vesicles from endoplasmic reticulum. It has been found associated with the sparse hair phenotype in humans (https://mseqdr.org/hpo_browser.php?8070) Moreover, it may contain causative mutations for an autosomal recessive disease known as cranio–lenticulo–sutural dysplasia, alias Boyadjiev–Jabs syndrome, in which patients have abnormal hair, among other cranio-facial abnormalities. Also, it has been shown to co-localize with the three proteins, transmembrane (Cdh23), scaffold (harmonin), and actin-based motor (Myo7a), whose defect is responsible of various types of the Usher syndrome, a multi-genic congenital disease characterized by perturbation of normal organization and growth of hair bundles within the inner ear (Blanco-Sánchez et al., 2014)
PAX9—It is a member of the paired box (PAX) family of transcription factors. Heterozygous mutations in PAX9 have been associated in humans with non-syndromic tooth agenesis, non-syndromic, and familial oligodontia, with peg-shaped laterals and microdontia incisors. Often, these symptoms are associated with hair defects (Roberts and Chetty, 2018) as the same genes responsible for tooth development are involved in the growth and development of the other tissues derived from the ectoderm, including hair.
In general, biological evidence for the involvement in plausible Merino phenotypes was observed for the vast majority of coding genes in putatively selected regions detected either via “local” ancestry” or the FST-outlier” approach. The above result highlights the power of the multi-cohort approach adopted here. While we cannot exclude that false positive signals may have been retained in this study, still this represents so far the most complete genome-wide study of selection signatures for the Merino phenotype. The selection signatures reported here provide a comprehensive insight into the genetic basis underlining the Merino phenotype in sheep, which appeared here to be mainly represented by wool (and horn) traits. Targeted studies at both physiological and molecular levels will be needed to better understand the biological complexity behind these commercially relevant traits.
Data Availability Statement
The whole SNP genotype dataset is available on the WIDDE database (http://widde.toulouse.inra.fr/widde/).
Author Contributions
Conception of the work: SMe, EC. Data analysis: SMe, SMa, EC. Results interpretation: SMe, SMa, JL, EC. Drafting the article: SMe, EC. Critical revision of the article: SMe, SMa, MH, JL, EC. Final approval of the version to be published: SMe, SMa, MH, JL, EC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors SMa.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01025/full#supplementary-material
Data Sheet 1 | Information on the considered breeds.
Figure S1 | Individual proportions of global admixture for the four considered datasets. Dataset composition: (A) Rambouillet, Chinese Merino, Tibetan; (B) Australian Merino, Sopravissana, Appenninica; (C) Gentile di Puglia, Spanish Merino, Appenninica; (D) Gentile di Puglia, Australian Merino, Appenninica. To estimate admixture proportions of the four test breeds (A, Chinese Merino; B, Sopravissana; C, Spanish Merino; D, Australian Merino), each dataset was assumed to be arranged into two sub-populations. Color codes define the admixture proportions for each animal. Individual proportions of global admixture were averaged within breed to obtain a, the fraction of global admixture, adopted as parameter in the “local ancestry” analyses (see main text).
Table S1 | Sheep breeds considered throughout this study. N, number of genotyped animals.
Table S2 | Sheep breeds considered in the four different scenarios tested using the “local ancestry” approach. N, number of genotyped animals; α, fraction of global admixture.
Table S3 | Sheep breeds considered in the nine pair-wise comparisons tested using the “FST-outlier” approach.
Table S4 | Putatively selected regions identified in Australian Merino using the “local ancestry” approach. Merino genome-wide marker average ancestry (MAA) for this tested scenario was 0.87. For each region, the sheep chromosome (OAR), the name and the position, expressed in base pairs (bp), of the start and end SNPs (SNP ID), together with the MAA values representing the Merino fraction for the start and end SNPs, are provided. In bold and italics the region overlapping with ROHs detected within the Australian Merino breed. Highlighted in light yellow, the regions where at least one significant SNP in at least one pair-wise comparison of the “FST-outlier” method involving the corresponding Merino (or Merino-derived) breed was observed (see main text).
Table S5 | Putatively selected regions identified in Chinese Merino using the “local ancestry” approach. Merino genome-wide marker average ancestry (MAA) for this tested scenario was 0.91. For each region, the sheep chromosome (OAR), the name and the position, expressed in base pairs (bp), of the start and end SNPs (SNP ID), together with the MAA values representing the Merino fraction for the start and end SNPs, are provided. Highlighted in light yellow, the regions where at least one significant SNP in at least one pair-wise comparison of the “FST-outlier” method involving the corresponding Merino (or Merino-derived) breed was observed (see main text).
Table S6 | Putatively selected regions identified in Sopravissana using the “local ancestry” approach. Merino genome-wide marker average ancestry (MAA) for this tested scenario was 0.76. For each region, the sheep chromosome (OAR), the name and the position, expressed in base pairs (bp), of the start and end SNPs (SNP ID), together with the MAA values representing the Merino fraction for the start and end SNPs, are provided. Highlighted in light yellow, the regions where at least one significant SNP in at least one pair-wise comparison of the “FST-outlier” method involving the corresponding Merino (or Merino-derived) breed was observed (see main text).
Table S7 | Putatively selected regions identified in Spanish Merino using the “local ancestry” approach. Merino genome-wide marker average ancestry (MAA) for this tested scenario was 0.77. For each region, the sheep chromosome (OAR), the name and the position, expressed in base pairs (bp), of the start and end SNPs (SNP ID), together with the MAA values representing the Merino fraction for the start and end SNPs, are provided. In bold and italics the region overlapping with ROHs detected within the Spanish Merino breed. Highlighted in light yellow, the regions where at least one significant SNP in at least one pair-wise comparison of the “FST-outlier” method involving the corresponding Merino (or Merino-derived) breed was observed (see main text).
Table S8 | Results of the analyses performed using the “FST-outlier” approach for the nine pair-wise comparisons between Merino and non-Merino breeds. The sheep chromosome (OAR), the name (SNP ID) and the position, expressed in base pairs (bp), of the significant loci displaying qval < 0.05, and the corresponding FST values, are shown. In bold and italics, loci included within ROH islands detected in the same Merino (or Merino-derived) breed (see main text). Highlighted in light yellow, loci located within putatively selected regions identified, for the corresponding four considered breeds (Australian Merino, Spanish Merino, Sopravissana, Chinese Merino) using the “local ancestry” approach.
Table S9 | Gene content of the region on OAR17 detected as putatively selected by “local ancestry” analysis. N., sequential numbers. Gene ID, gene symbol.
Table S10 | Gene content of the region on OAR18 detected as putatively selected by “local ances
Table S11 | Quantitative Trait Loci (QTLs) known in sheep and mapped to regions detected in this study as putatively selected via “local ancestry” (A) and “FST-outlier” (B) analysis. QTL ID, QTL accession code at the NCBI Sheep Genome Data Viewer. OAR, sheep chromosome.
Table S12 | Gene content of the regions on OAR3 (A), OAR10 (B), OAR13 (C) and OAR19 (D) detected as putatively selected by “FST-outlier” analysis. N., sequential number. Gene ID, gene symbol.
Table S13 | Results of the gene network analysis. In italics, interactions between genes detected as putatively selected using the same approach (either LAMP or FST-outlier). In bold, interactions between genes detected as putatively selected using different approaches.
References
Ahmed, Z. M., Yousaf, R., Lee, B. C., Khan, S. N., Lee, S., Lee, K., et al. (2011). Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am. J. Hum. Genet. 88, 19–29. doi: 10.1016/j.ajhg.2010.11.010
Alexander, D. H., Novembre, J., Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Allain, D., Elsen, J. M., Francois, D., Brunei, J. C., Weisbecker, J. L., Schibler, L., et al. (1998). A design aiming at detecting QTL controlling wool traits and other traits in the inra401 sheep line. Proc. 6th World Congr. Genet. Appl. Livest. Prod. 26, 434–436.
Allain, D., Schibler, L., Mura, L., Barillet, F., Sechi, T., Rupp, R., et al. (2006). QTL detection with DNA markers for wool traits in a sheep backcross Sarda × Lacaune resource population. Proc. 8th World Congr. Genet. Appl. Livest. Prod. 2006, 5–7.
Beh, K. J., Callaghan, M. J., Leish, Z., Hulme, D. J., Lenane, I., Maddox, J. F. (2001). A genome scan for QTL affecting fleece and wool traits in Merino sheep. Wool Tech. Sheep Breed. 49, 88–97. doi: 10.1046/j.1365-2052.2002.00829.x
Bidinost, F., Roldan, D. L., Dodero, A. M., Cano, E. M., Taddeo, H. R., Mueller, J. P., et al. (2008). Wool quantitative trait loci in Merino sheep. Small Rumin. Res. 74, 113–118. doi: 10.1016/j.smallrumres.2007.04.005
Blanco-Sánchez, B., Clément, A., Fierro, J., Washbourne, P., Westerfield, M. (2014). Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Model. Mech. 7, 547–559. doi: 10.1242/dmm.014068
Bolormaa, S., Swan, A. A., Brown, D. J., Hatcher, S., Moghaddar, N., van der Werf, J. H., et al. (2017). Multiple-trait QTL mapping and genomic prediction for wool traits in sheep. Genet. Sel. Evol. 49, 62. doi: 10.1186/s12711-017-0337-y
Carlson, T. L., Moini, M., Eckenrode, B. A., Allred, B. M., Donfack, J. (2018). Protein extraction from human anagen head hairs 1-millimeter or less in total length. BioTechniques 64, 170–176. doi: 10.2144/btn-2018-2004
Chai, W., Zhou, H., Forrest, R. H. J., Gong, H., Hodge, S., Hickford, J. G. H. (2017). Polymorphism of KRT83 and its association with selected wool traits in Merino-cross lambs. Small Rumin. Res. 155, 6–11. doi: 10.1016/j.smallrumres.2017.08.019
Chang, C.-Y. (2014) Coordinating Stem Cell Behavior in the Hair Follicle. Student Theses and Dissertations. 217.
Chessa, B., Pereira, F., Arnaud, F., Amorim, A., Goyache, F., Mainland, I., et al. (2009). Revealing the history of sheep domestication using retrovirus integrations. Science 324, 532–536. doi: 10.1126/science.1170587
Ciani, E., Lasagna, E., D’Andrea, M., Alloggio, I., Marroni, F., Ceccobelli, S., et al. (2015). Merino and Merino-derived sheep breeds: a genome-wide intercontinental study. Genet. Sel. Evol. 47, 64. doi: 10.1186/s12711-015-0139-z
Cong, J., Geng, W., He, B., Liu, J., Charlton, J., Adler, P. N. (2001). The furry gene of Drosophila is important for maintaining the integrity of cellular extensions during morphogenesis. Dev. Camb. Engl. 128, 2793–2802.
Cornella, N., Tebaldi, T., Gasperini, L., Singh, J., Padgett, R. A., Rossi, A., et al. (2017). The hnRNP RALY regulates transcription and cell proliferation by modulating the expression of specific factors including the proliferation marker E2F1. J. Biol. Chem. 292, 19674–19692. doi: 10.1074/jbc.M117.795591
Covitz, P. A., Smith, L. S., Long, S. R. (1998). Expressed sequence tags from a root-hair-enriched medicago truncatula cDNA library. Plant Physiol. 117, 1325–1332. doi: 10.1104/pp.117.4.1325
Debono Spiteri, C., Gillis, R. E., Roffet-Salque, M., Castells Navarro, L., Guilaine, J., Manen, C., et al. (2016). Regional asynchronicity in dairy production and processing in early farming communities of the northern Mediterranean. Proc. Natl. Acad. Sci. U. S. A. 113, 13594–13599. doi: 10.1073/pnas.1607810113
Demars, J., Cano, M., Drouilhet, L., Plisson-Petit, F., Bardou, P., Fabre, S., et al. (2017). Genome-wide identification of the mutation underlying fleece variation and discriminating ancestral hairy species from modern woolly sheep. Mol. Biol. Evol. 34, 1722–1729. doi: 10.1093/molbev/msx114
Demirci, S., Baştanlar, E. K., Dağtaş, N. D., Pişkin, E., Engin, A., Özer, F., et al. (2013). Mitochondrial DNA Diversity of Modern, Ancient and Wild Sheep (Ovis gmelinii anatolica) from Turkey: New Insights on the Evolutionary History of Sheep. PLoS ONE 8, e81952. doi: 10.1371/journal.pone.0081952
Dreger, D. L., Parker, H. G., Ostrander, E. A., Schmutz, S. M. (2013). Identification of a mutation that is associated with the saddle tan and black-and-tan phenotypes in Basset Hounds and Pembroke Welsh Corgis. J. Hered. 104, 399–406. doi: 10.1093/jhered/est012
Duhl, D. M., Stevens, M. E., Vrieling, H., Saxon, P. J., Miller, M. W., Epstein, C. J., et al. (1994). Pleiotropic effects of the mouse lethal yellow (Ay) mutation explained by deletion of a maternally expressed gene and the simultaneous production of agouti fusion RNAs. Dev. Camb. Engl. 120, 1695–1708.
Dymova, M. A., Zadorozhny, A. V., Mishukova, O. V., Khrapov, E. A., Druzhkova, A. S., Trifonov, V. A., et al. (2017). Mitochondrial DNA analysis of ancient sheep from Altai. Anim. Genet. 48, 615–618. doi: 10.1111/age.12569
Ebrahimi, F., Gholizadeh, M., Rahimi-Mianji, G., Farhadi, A. (2017). Detection of QTL for greasy fleece weight in sheep using a 50 K single nucleotide polymorphism chip. Trop. Anim. Health Prod. 49, 1657–1662. doi: 10.1007/s11250-017-1373-x
Ethier, J., Bánffy, E., Vuković, J., Leshtakov, K., Bacvarov, K., Roffet-Salque, M., et al. (2017). Earliest expansion of animal husbandry beyond the Mediterranean zone in the sixth millennium BC. Sci. Rep. 7, 7146. doi: 10.1038/s41598-017-07427-x
Fang, X., Lu, Q., Emoto, K., Adler, P. N. (2010). The Drosophila Fry protein interacts with Trc and is highly mobile in vivo. BMC Dev. Biol. 10, 40. doi: 10.1186/1471-213X-10-40
Fariello, M.-I., Servin, B., Tosser-Klopp, G., Rupp, R., Moreno, C., Consortium, I. S. G., et al. (2014). Selection signatures in worldwide sheep populations. PLOS ONE 9, e103813. doi: 10.1371/journal.pone.0103813
Foll, M., Gaggiotti, O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Furumura, M., Ishikawa, H. (1996). Actin bundles in human hair follicles as revealed by confocal laser microscopy. Cell Tissue Res. 283, 425–434. doi: 10.1007/s004410050553
García-Gámez, E., Reverter, A., Whan, V., McWilliam, S. M., Arranz, J., J. International Sheep Genomics Consortium, et al. (2011). Using regulatory and epistatic networks to extend the findings of a genome scan: identifying the gene drivers of pigmentation in merino sheep. PloS One 6, e21158. doi: 10.1371/journal.pone.0021158
Gong, H., Zhou, H., Bai, L., Li, W., Li, S., Wang, J., et al. (2019). Associations between variation in the ovine high glycine-tyrosine keratin-associated protein gene KRTAP20-1 and wool traits. J. Anim. Sci. 97, 587–595. doi: 10.1093/jas/sky465
Guild, G. M., Connelly, P. S., Ruggiero, L., Vranich, K. A., Tilney, L. G. (2005). Actin filament bundles in Drosophila wing hairs: hairs and bristles use different strategies for assembly. Mol. Biol. Cell 16, 3620–3631. doi: 10.1091/mbc.e05-03-0185
Guo, J., Tao, H., Li, P., Li, L., Zhong, T., Wang, L., et al. (2018). Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 8, 10405. doi: 10.1038/s41598-018-28719-w
Gutiérrez-Gil, B., Esteban-Blanco, C., Wiener, P., Chitneedi, P. K., Suarez-Vega, A., Arranz, J.-J. (2017). High-resolution analysis of selection sweeps identified between fine-wool Merino and coarse-wool Churra sheep breeds. Genet. Sel. Evol. 49, 81. doi: 10.1186/s12711-017-0354-x
Harmon, C. S., Nevins, T. D. (1997). Evidence that activation of protein kinase A inhibits human hair follicle growth and hair fibre production in organ culture and DNA synthesis in human and mouse hair follicle organ culture. Br. J. Dermatol. 136 (6), 853–858. doi: 10.1111/j.1365-2133.1997.tb03924.x
He, Y., Fang, X., Emoto, K., Jan, Y.-N., Adler, P. N. (2005). The tricornered Ser/Thr protein kinase is regulated by phosphorylation and interacts with furry during Drosophila wing hair development. Mol. Biol. Cell 16, 689–700. doi: 10.1091/mbc.e04-09-0828
Henry, H., Doods, K., Wuliji, T., Jenkis, Z., Beattie, A., Montgomery, G. (1998). A genome screen for QTL for wool traits in a Merino x Romney backcross flock (Reprinted). Wool Technol. Sheep Breed. 46, 213–217.
Hung, R.-J., Spaeth, C. S., Yesilyurt, H. G., Terman, J. R. (2013). SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 15, 1445–1454. doi: 10.1038/ncb2871
Ivanova, M., De Cupere, B., Ethier, J., Marinova, E. (2018a). Correction: Pioneer farming in southeast Europe during the early sixth millennium BC: Climate-related adaptations in the exploitation of plants and animals. PloS One 13, e0202668. doi: 10.1371/journal.pone.0202668
Ivanova, M., De Cupere, B., Ethier, J., Marinova, E. (2018b). Pioneer farming in southeast Europe during the early sixth millennium BC: Climate-related adaptations in the exploitation of plants and animals. PloS One 13, e0197225. doi: 10.1371/journal.pone.0197225
Jacobs, L. C., Hamer, M. A., Gunn, D. A., Deelen, J., Lall, J. S., van Heemst, D., et al. (2015). A genome-wide association study identifies the skin color genes IRF4, MC1R, ASIP, and BNC2 influencing facial pigmented spots. J. Invest. Dermatol. 135, 1735–1742. doi: 10.1038/jid.2015.62
Johnston, S. E., McEwan, J. C., Pickering, N. K., Kijas, J. W., Beraldi, D., Pilkington, J. G., et al. (2011). Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol. Ecol. 20, 2555–2566. doi: 10.1111/j.1365-294X.2011.05076.x
Kijas, J. W., Lenstra, J. A., Hayes, B., Boitard, S., Neto, L. R. P., Cristobal, M. S., et al. (2012). Genome-Wide Analysis of the World’s Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. PLOS Biol. 10, e1001258. doi: 10.1371/journal.pbio.1001258
Kim, S., Wong, P., Coulombe, P. A. (2006). A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 441, 362–365. doi: 10.1038/nature04659
Köhler, S., Doelken, S. C., Mungall, C. J., Bauer, S., Firth, H. V., Bailleul-Forestier, I., et al. (2014). The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 42, D966–D974. doi: 10.1093/nar/gkt1026
Lee, Y. J., Rice, R. H., Lee, Y. M. (2006). Proteome analysis of human hair shaft: from protein identification to posttranslational modification. Mol. Cell. Proteomics 5, 789–800. doi: 10.1074/mcp.M500278-MCP200
Li, J., Zhao, X., Xin, Q., Shan, S., Jiang, B., Jin, Y., et al. (2015). Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive non syndromic hearing loss DFNB99. Hum. Mutat. 36, 98–105. doi: 10.1002/humu.22712
Li, S., Zhou, H., Gong, H., Zhao, F., Hu, J., Luo, Y., et al. (2017a). Identification of the Ovine Keratin-Associated Protein 26-1 Gene and Its Association with Variation in Wool Traits. Genes 8. doi: 10.3390/genes8090225
Li, S., Zhou, H., Gong, H., Zhao, F., Wang, J., Liu, X., et al. (2017b). Identification of the Ovine Keratin-Associated Protein 22-1 (KAP22-1) Gene and Its Effect on Wool Traits. Genes 8. doi: 10.3390/genes8010027
Ling, Y. H., Xiang, H., Zhang, G., Ding, J. P., Zhang, Z. J., Zhang, Y. H., et al. (2014). Identification of complete linkage disequilibrium in the DSG4 gene and its association with wool length and crimp in Chinese indigenous sheep. Genet. Mol. Res. 13, 5617–5625. doi: 10.4238/2014.July.25.17
Liu, G., Liu, R., Li, Q., Tang, X., Yu, M., Li, X., et al. (2013). Identification of microRNAs in wool follicles during anagen, catagen, and telogen phases in Tibetan sheep. PloS One 8, e77801. doi: 10.1371/journal.pone.0077801
Liu, G., Liu, R., Tang, X., Cao, J., Zhao, S., Yu, M. (2015). Expression profiling reveals genes involved in the regulation of wool follicle bulb regression and regeneration in sheep. Int. J. Mol. Sci. 16, 9152–9166. doi: 10.3390/ijms16059152
Liu, S., He, S., Chen, L., Li, W., Di, J., Liu, M. (2017). Estimates of linkage disequilibrium and effective population sizes in Chinese Merino (Xinjiang type) sheep by genome-wide SNPs. Genes Genomics 39, 733–745. doi: 10.1007/s13258-017-0539-2
Ma, G.-W., Chu, Y.-K., Zhang, W.-J., Qin, F.-Y., Xu, S.-S., Yang, H., et al. (2017). Polymorphisms of FST gene and their association with wool quality traits in Chinese Merino sheep. PloS One 12, e0174868. doi: 10.1371/journal.pone.0174868
Manunza, A., Cardoso, T. F., Noce, A., Martínez, A., Pons, A., Bermejo, L. A., et al. (2016). Population structure of eleven Spanish ovine breeds and detection of selective sweeps with BayeScan and hapFLK. Sci. Rep. 6, 27296. doi: 10.1038/srep27296
Mastrangelo, S., Sardina, M. T., Tolone, M., Di Gerlando, R., Sutera, A. M., Fontanesi, L., et al. (2018). Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds. Animal 12, 2480–2488. doi: 10.1017/S1751731118000629
Mastrangelo, S., Bahbahani, H., Moioli, B., Ahbara, A., Abri, M. A., Almathen, F., et al. (2019). Novel and known signals of selection for fat deposition in domestic sheep breeds from Africa and Eurasia. PLoS One 14, e0209632. doi: 10.1371/journal.pone.0209632
Mastrangelo, S., Tolone, M., Sardina, M. T., Sottile, G., Sutera, A. M., Di Gerlando, R., et al. (2017). Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 49, 84. doi: 10.1186/s12711-017-0360-z
Meadows, J. R. S., Hiendleder, S., Kijas, J. W. (2011). Haplogroup relationships between domestic and wild sheep resolved using a mitogenome panel. Heredity 106, 700–706. doi: 10.1038/hdy.2010.122
Metzger, J., Karwath, M., Tonda, R., Beltran, S., Águeda, L., Gut, M., et al. (2015). Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genomics 16, 764. doi: 10.1186/s12864-015-1977-3
Michaud, E. J., Bultman, S. J., Stubbs, L. J., Woychik, R. P. (1993). The embryonic lethality of homozygous lethal yellow mice (Ay/Ay) is associated with the disruption of a novel RNA-binding protein. Genes Dev. 7, 1203–1213. doi: 10.1101/gad.7.7a.1203
Moioli, B., Pilla, F., Ciani, E. (2015). Signatures of selection identify loci associated with fat tail in sheep. J. Anim. Sci. 93, 4660–4669. doi: 10.2527/jas.2015-9389
Mostafavi, S., Ray, D., Warde-Farley, D., Grouios, C., Morris, Q. (2008). GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biology 9, S4. doi: 10.1186/gb-2008-9-s1-s4
Mu, F., Rong, E., Jing, Y., Yang, H., Ma, G., Yan, X., et al. (2017). Structural characterization and association of ovine Dickkopf-1 gene with wool production and quality traits in Chinese Merino. Genes 8. doi: 10.3390/genes8120400
Nasr, Z., Dow, L. E., Paquet, M., Chu, J., Ravindar, K., Somaiah, R., et al. (2013). Suppression of eukaryotic initiation factor 4E prevents chemotherapy-induced alopecia. BMC Pharmacol. Toxicol. 14, 58. doi: 10.1186/2050-6511-14-58
Nazari-Ghadikolaei, A., Mehrabani-Yeganeh, H., Miarei-Aashtiani, S. R., Staiger, E. A., Rashidi, A., Huson, H. J. (2018). Genome-wide association studies identify candidate genes for coat color and mohair traits in the Iranian Markhoz goat. Front. Genet. 9, 105. doi: 10.3389/fgene.2018.00105
Pan, Z., Li, S., Liu, Q., Wang, Z., Zhou, Z., Di, R., et al. (2018). Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 7. doi: 10.1093/gigascience/giy019
Parsons, Y. M., Cooper, D. W., Piper, L. R. (1994). Evidence of linkage between high-glycine-tyrosine keratin gene loci and wool fibre diameter in a Merino half-sib family. Anim. Genet. 25, 105–108. doi: 10.1111/j.1365-2052.1994.tb00088.x
Phuaa, S. H., Scobieb, D. R., O’Connellb, D., Henrya, H., Doddsa, K. G., Brauninga, R. (2015). Preliminary linkage studies in sheep of keratin and keratin-associated protein genes with fleece weight, wool fibre diameter and fibre curvature. Proc. N. Z. Soc. Anim. Prod. 75, 101–105.
Ponz, R., Moreno, C., Allain, D., Elsen, J. M., Lantier, F., Lantier, I., et al. (2001). Assessment of genetic variation explained by markers for wool traits in sheep via a segment mapping approach. Mamm. Genome 12, 569–572. doi: 10.1007/s003350030007
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Purfield, D. C., McParland, S., Wall, E., Berry, D. P. (2017). The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PloS One 12, e0176780. doi: 10.1371/journal.pone.0176780
Qanbari, S., Pausch, H., Jansen, S., Somel, M., Strom, T. M., Fries, R., et al. (2014). Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 10, e1004148. doi: 10.1371/journal.pgen.1004148
Roberts, M. R., Asgari, M. M., Toland, A. E. (2019). Genome-wide association studies and polygenic risk scores for skin cancer: clinically useful yet? Br. J. Dermatol. doi: 10.1111/bjd.17917
Roberts, T. S., Chetty, M. (2018). Hypohidrotic Ectodermal Dysplasia: Genetic aspects and clinical implications of hypodontia. South Afr. Dent. J. 73, 253–256.
Rochus, C. M., Tortereau, F., Plisson-Petit, F., Restoux, G., Moreno-Romieux, C., Tosser-Klopp, G., et al. (2018). Revealing the selection history of adaptive loci using genome-wide scans for selection: an example from domestic sheep. BMC Genomics 19, 71. doi: 10.1186/s12864-018-4447-x
Rogers, G. R., Hickford, J. G. H., Bickerstaffe, R. (1994). A potential QTL for wool strength located on ovine chromosome 11. Proc. 5th World Congr. Genet. Appl. Livest. Prod. 21, 291–294.
Roldan, D. L., Dodero, A. M., Bidinost, F., Taddeo, H. R., Allain, D., Poli, M. A., et al. (2010). Merino sheep: a further look at quantitative trait loci for wool production. Animal 4, 1330–1340. doi: 10.1017/S1751731110000315
Rong, E. G., Yang, H., Zhang, Z. W., Wang, Z. P., Yan, X. H., Li, H., et al. (2015). Association of methionine synthase gene polymorphisms with wool production and quality traits in Chinese Merino population. J. Anim. Sci. 93, 4601–4609. doi: 10.2527/jas.2015-8963
Rossi, A., Moro, A., Tebaldi, T., Cornella, N., Gasperini, L., Lunelli, L., et al. (2017). Identification and dynamic changes of RNAs isolated from RALY-containing ribonucleoprotein complexes. Nucleic Acids Res. 45, 6775–6792. doi: 10.1093/nar/gkx235
Ryder, M. (1981). A survey of European primitive breeds of sheep. Ann. Genet. Sel. Anim. 13, 381–418. doi: 10.1186/1297-9686-13-4-381
Sankararaman, S., Sridhar, S., Kimmel, G., Halperin, E. (2008). Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82, 290–303. doi: 10.1016/j.ajhg.2007.09.022
Sasikumar, A. N., Perez, W. B., Kinzy, T. G. (2012). The many roles of the eukaryotic elongation factor 1 complex. Wiley Interdiscip. Rev. RNA 3, 543–555. doi: 10.1002/wrna.1118
Seroussi, E., Rosov, A., Shirak, A., Lam, A., Gootwine, E. (2017). Unveiling genomic regions that underlie differences between Afec-Assaf sheep and its parental Awassi breed. Genet. Sel. Evol. 49, 19. doi: 10.1186/s12711-017-0296-3
Shen, X., Liu, F., Wang, Y., Wang, H., Ma, J., Xia, W., et al. (2015). Down-regulation of msrb3 and destruction of normal auditory system development through hair cell apoptosis in zebrafish. Int. J. Dev. Biol. 59, 195–203. doi: 10.1387/ijdb.140200md
Shriner, D. (2013). Overview of admixture mapping. Curr. Protoc. Hum. Genet. 76, 1.23.1–1.23.8. doi: 10.1002/0471142905.hg0123s76
Singh, S., Kumar, S., Kolte, A. P., Kumar, S. (2013). Extensive variation and sub-structuring in lineage A mtDNA in Indian sheep: genetic evidence for domestication of sheep in India. PloS One 8, e77858. doi: 10.1371/journal.pone.0077858
Sulayman, A., Tursun, M., Sulaiman, Y., Huang, X., Tian, K., Tian, Y., et al. (2018). Association analysis of polymovwwrphisms in six keratin genes with wool traits in sheep. Asian-Australas. J. Anim. Sci. 31, 775–783. doi: 10.5713/ajas.17.0349
Talenti, A., Bertolini, F., Pagnacco, G., Pilla, F., Ajmone-Marsan, P., Rothschild, M. F., et al. (2017a). The Valdostana goat: a genome-wide investigation of the distinctiveness of its selective sweep regions. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 28, 114–128. doi: 10.1007/s00335-017-9678-7
Talenti, A., Bertolini, F., Pagnacco, G., Pilla, F., Ajmone-Marsan, P., Rothschild, M. F., et al. (2017b). Erratum to: The Valdostana goat: a genome-wide investigation of the distinctiveness of its selective sweep regions. Mamm. Genome 28, 129. doi: 10.1007/s00335-017-9685-8
Taungjaruwinai, W. M., Bhawan, J., Keady, M., Thiele, J. J. (2009). Differential expression of the antioxidant repair enzyme methionine sulfoxide reductase (MSRA and MSRB) in human skin. Am. J. Dermatopathol. 31, 427–431. doi: 10.1097/DAD.0b013e3181882c21
Tomann, P., Paus, R., Millar, S. E., Scheidereit, C., Schmidt-Ullrich, R. (2016). Lhx2 is a direct NF-κB target gene that promotes primary hair follicle placode down-growth. Development 143, 1512–1522. doi: 10.1242/dev.130898
Vaysse, A., Ratnakumar, A., Derrien, T., Axelsson, E., Rosengren Pielberg, G., Sigurdsson, S., et al. (2011). Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 7, e1002316. doi: 10.1371/journal.pgen.1002316
Vermorken, A. J., Weterings, P. J., Kibbelaar, M. A., Lenstra, J. A., Bloemendal, H. (1981). Isolation and characterization of actin from human hair follicles. FEBS Lett. 127, 105–108. doi: 10.1016/0014-5793(81)80352-3
Wang, H., Zhang, L., Cao, J., Wu, M., Ma, X., Liu, Z., et al. (2015). Genome-wide specific selection in three domestic sheep breeds. PloS One 10, e0128688. doi: 10.1371/journal.pone.0128688
Wang, Z., Zhang, H., Yang, H., Wang, S., Rong, E., Pei, W., et al. (2014). Genome-wide association study for wool production traits in a Chinese Merino sheep population. PloS One 9, e107101. doi: 10.1371/journal.pone.0107101
Wei, C., Wang, H., Liu, G., Wu, M., Cao, J., Liu, Z., et al. (2015). Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genomics 16, 194. doi: 10.1186/s12864-015-1384-9
Wilkinson, S., Lu, Z. H., Megens, H.-J., Archibald, A. L., Haley, C., Jackson, I. J., et al. (2013). Signatures of diversifying selection in European pig breeds. PLoS Genet. 9, e1003453. doi: 10.1371/journal.pgen.1003453
Yang, J., Li, W.-R., Lv, F.-H., He, S.-G., Tian, S.-L., Peng, W.-F., et al. (2016). Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 33, 2576–2592. doi: 10.1093/molbev/msw129
Yuan, Z., Liu, E., Liu, Z., Kijas, J. W., Zhu, C., Hu, S., et al. (2017). Selection signature analysis reveals genes associated with tail type in Chinese indigenous sheep. Anim. Genet. 48, 55–66. doi: 10.1111/age.12477
Zeder, M. A. (2008). Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. 105, 11597–11604. doi: 10.1073/pnas.0801317105
Zhai, M., Xie, Y., Yang, M., Mu, J., Zhao, Z. (2019). Investigation of the relationships between wool quality and microsatellite in hybrids of Australian Merino and Chinese Merino. Kafkas Univ. Vet. Fak. Derg. 25 (2), 163–170. doi: 10.9775/kvfd.2018.2020488
Zhang, L., Mousel, M. R., Wu, X., Michal, J. J., Zhou, X., Ding, B., et al. (2013). Genome-wide genetic diversity and differentially selected regions among Suffolk, Rambouillet, Columbia, Polypay, and Targhee sheep. PloS One 8, e65942. doi: 10.1371/journal.pone.0065942
Keywords: Merino sheep breeds, wool, genome-wide selection signatures, FST-outlier, local ancestry in admixed populations, runs of homozygosity
Citation: Megdiche S, Mastrangelo S, Ben Hamouda M, Lenstra JA and Ciani E (2019) A Combined Multi-Cohort Approach Reveals Novel and Known Genome-Wide Selection Signatures for Wool Traits in Merino and Merino-Derived Sheep Breeds. Front. Genet. 10:1025. doi: 10.3389/fgene.2019.01025
Received: 28 April 2019; Accepted: 24 September 2019;
Published: 25 October 2019.
Edited by:
Francesca Bertolini, Technical University of Denmark, DenmarkReviewed by:
Vincenzo Landi, Universidad de Córdoba,SpainMichelle Mousel, United States Department of Agriculture,United States
Copyright © 2019 Megdiche, Mastrangelo, Ben Hamouda, Lenstra and Ciani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sami Megdiche, bWVnZGljaGVzYW1pQGhvdG1haWwuZnI=; Elena Ciani, ZWxlbmEuY2lhbmlAdW5pYmEuaXQ=