 Ting Wang
Ting Wang Jun Ma
Jun Ma Qin Zhang
Qin Zhang Ang Gao2
Ang Gao2 Hong Li
Hong Li Jingjing Xiang
Jingjing Xiang Benjing Wang
Benjing Wang- 1Newborn Screening Laboratory, Center for Reproduction and Genetics, the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, China
- 2Genetic Clinic, Center for Reproduction and Genetics, the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, China
- 3Infertility Clinic, Center for Reproduction and Genetics, the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, China
- 4Genetic Laboratory, Center for Reproduction and Genetics, the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, China
Expanded newborn screening for inborn errors of metabolism (IEMs) by tandem mass spectrometry (MS/MS) could simultaneously analyze more than 40 metabolites and identify about 50 kinds of IEMs. Next generation sequencing (NGS) targeting hundreds of IMEs-associated genes as a follow-up test in expanded newborn screening has been used for genetic analysis of patients. The spectrum, prevalence, and genetic characteristic of IEMs vary dramatically in different populations. To determine the spectrum, prevalence, and gene mutations of IEMs in newborns in Suzhou, China, 401,660 newborns were screened by MS/MS and 138 patients were referred to genetic analysis by NGS. The spectrum of 22 IEMs were observed in Suzhou population of newborns, and the overall incidence (excluding short chain acyl-CoA dehydrogenase deficiency (SCADD) and 3-Methylcrotonyl-CoA carboxylase deficiency (3-MCCD)) was 1/3,163. The prevalence of each IEM ranged from 1/401,660 to 1/19,128, while phenylketonuria (PKU) (1/19,128) and Mild hyperphenylalaninemia (M-HPA) (1/19,128) were the most common IEMs, followed by primary carnitine uptake defect (PCUD) (1/26,777), SCADD (1/28,690), hypermethioninemia (H-MET) (1/30,893), 3-MCCD (1/33,412) and methylmalonic acidemia (MMA) (1/40,166). Moreover, 89 reported mutations and 51 novel mutations in 25 IMEs-associated genes were detected in 138 patients with one of 22 IEMs. Some hotspot mutations were observed for ten IEMs, including PAH gene c.728G > A, c.611A > G, and c.721C > T for Phenylketonuria, PAH gene c.158G > A, c.1238G > C, c.728G > A, and c.1315+6T > A for M-HPA, SLC22A5 gene c.1400C > G, c.51C > G, and c.760C > T for PCUD, ACADS gene c.1031A > G, c.164C > T, and c.1130C > T for SCAD deficiency, MAT1A gene c.791G > A for H-MET, MCCC1 gene c.639+2T > A and c.863A > G for 3-MCCD, MMUT gene c.1663G > A for MMA, SLC25A13 gene c.IVS16ins3Kb and c.852_855delTATG for cittrullinemia II, PTS gene c.259C > T and c.166G > A for Tetrahydrobiopterin deficiency, and ACAD8 gene c.1000C > T and c.286C > A for Isobutyryl coa dehydrogenase deficiency. All these hotspot mutations were reported to be pathogenic or likely pathogenic, except a novel mutation of ACAD8 gene c.286C > A. These mutational hotspots could be potential candidates for gene screening and these novel mutations expanded the mutational spectrum of IEMs. Therefore, our findings could be of value for genetic counseling and genetic diagnosis of IEMs.
Introduction
Inborn errors of metabolism (IMEs) are a large group of monogenic diseases resulting in death and abnormalities of physical and neurological development at almost all stages of life. IMEs are always caused by the defect of an enzyme, its coenzyme, or a transporter leading to the accumulation of its substrate and/or the insufficiency of its downstream products. Nowadays, the introduction of tandem mass spectrometry (TMS) allows screening for more than 50 IMEs using dried blood spot in the neonatal period (Therrell et al., 2015). For the neonates screened to have IMEs, some serious clinical consequence could be prevented, including mild to severe irreversible mental retardation, lifelong disability, physical handicaps, coma, and early death, if early diagnosis and treatment were implemented (Therrell et al., 2015). Therefore, expanded newborn screening program covering dozens of diseases has been implemented in the majority of developed countries. For example, population-based data are available in the United States (Centers for Disease Control and Prevention, 2008; Gallant et al., 2012; Hsu et al., 2013), Canada (Karaceper et al., 2016), United Kingdom (Sanderson et al., 2006), Germany (Lindner et al., 2008), France (Dhondt, 2010), Egypt (Hassan et al., 2016), Greece (Loukas et al., 2010), Saudi Arabia (Alfadhel et al., 2017), Australia (Wiley et al., 1999; Webster et al., 2003), South Korea (Yoon et al., 2005), Singapore (Lim et al., 2014), and Japan (Shibata et al., 2018).
The advent of target capture and next generation sequencing (NGS) enables simultaneously sequence a large group of targeted genes accounting for numerous diseases, which has become the best choice for identification of genetic etiology of IMEs following expanded newborn screening program. The utility of NGS in expanded newborn screening has enriched our understanding of genetic etiology, genetic characteristics, and phenotype-genotype correlation of IMEs. Some hotspot variants resulting in the defect of enzymes have been identified in patients with IMEs, such as ACADS variants c.511C > T and c.625G > A for short chain acyl-CoA dehydrogenase deficiency (SCADD; MIM# 201470) (Tonin et al., 2016; Nochi et al., 2017), PAH variant c.728C > A for phenylketonuria (PKU; MIM# 261600) (Liu et al., 2017), and so on. Also, many IMEs have a dramatic variation of symptoms and the outcome of the affected patients was correlated with genotype, such as medium chain acyl-CoA dehydrogenase deficiency (MCADD; MIM# 201450) (Ensenauer et al., 2005; Maier et al., 2005), very long chain acyl-CoA dehydrogenase deficiency (VLCADD; MIM# 609016) (Andresen et al., 1999; Obaid et al., 2018), and so on. In addition, the spectrum, the incidence, and the genetic characteristics of IMEs vary dramatically in different regions and populations.
Expanded newborn screening was introduced in China in 2004, later than developed countries. In the milestone pilot study, a total of 371,942 newborns were screened in four centers, and the collective estimated incidence of overall IMEs was 1/3,795 in live births, with a sensitivity of 98.99% and a specificity of 99.83% (Shi et al., 2012). Recently, targeted sequencing of genes associated to more than 50 IMEs by NGS was used as a follow-up test for genetic diagnosis after the expanded newborn screening, and some novel variants were found in Chinese patients. In Suzhou, the expanded newborn screening program targeting 27 IMEs started in 2014. Until now, its screening rate is closed to 100% of live births and more than 400,000 newborns have been referred to expand newborn screening. A total of 22 kinds of IMEs were identified in Suzhou population and 153 infants were diagnosed with one of these IMEs. Almost all these patients were referred to genetic analysis via targeted NGS. 140 variants in 25 IMEs-associated genes were found in 138 patients. Some hotspot variants were also observed in Suzhou patients, including c.791G > A in MATA1 gene for hypermethioninemia (MIM# 250850), c.158G > A in PAH gene for mild hyperphenylalaninemia (M-HPA; MIM# 261600), c.721C > T in PAH gene for PKU (MIM# 261600), c.852_855delTATG in SLC25A13 gene for citrullinemia type II (CTLN 2; MIM# 605814), c.639+2T > A in MCCC1 gene for 3- methylcroton acyl coenzyme A carboxylase deficiency (3-MCCD; MIM# 210200 and 210210), c.1400C > G in SLC22A5 gene for primary carnitine uptake defect (PCUD; MIM# 212140), and c1031A > G in ACADS gene for SCADD. These hotspot mutations could explain the relative high incidence of associated IMEs. As a result, it is critical to screen these mutations and prenatal genetic consulting for Suzhou population. These mutations are good candidates for further research on genetic characteristics in other Chinese populations.
Material and Methods
Subjects
A total of 401,660 newborns were referred to expand newborn screening. Informed and written consent was obtained from the parents of all screened newborns. Our screened protocol is consistent with other newborn screening centers in China, and was shown in Figure 1. The protocol was reviewed and approved by Ethic committee of the Affiliated Suzhou Hospital of Nanjing Medical University.
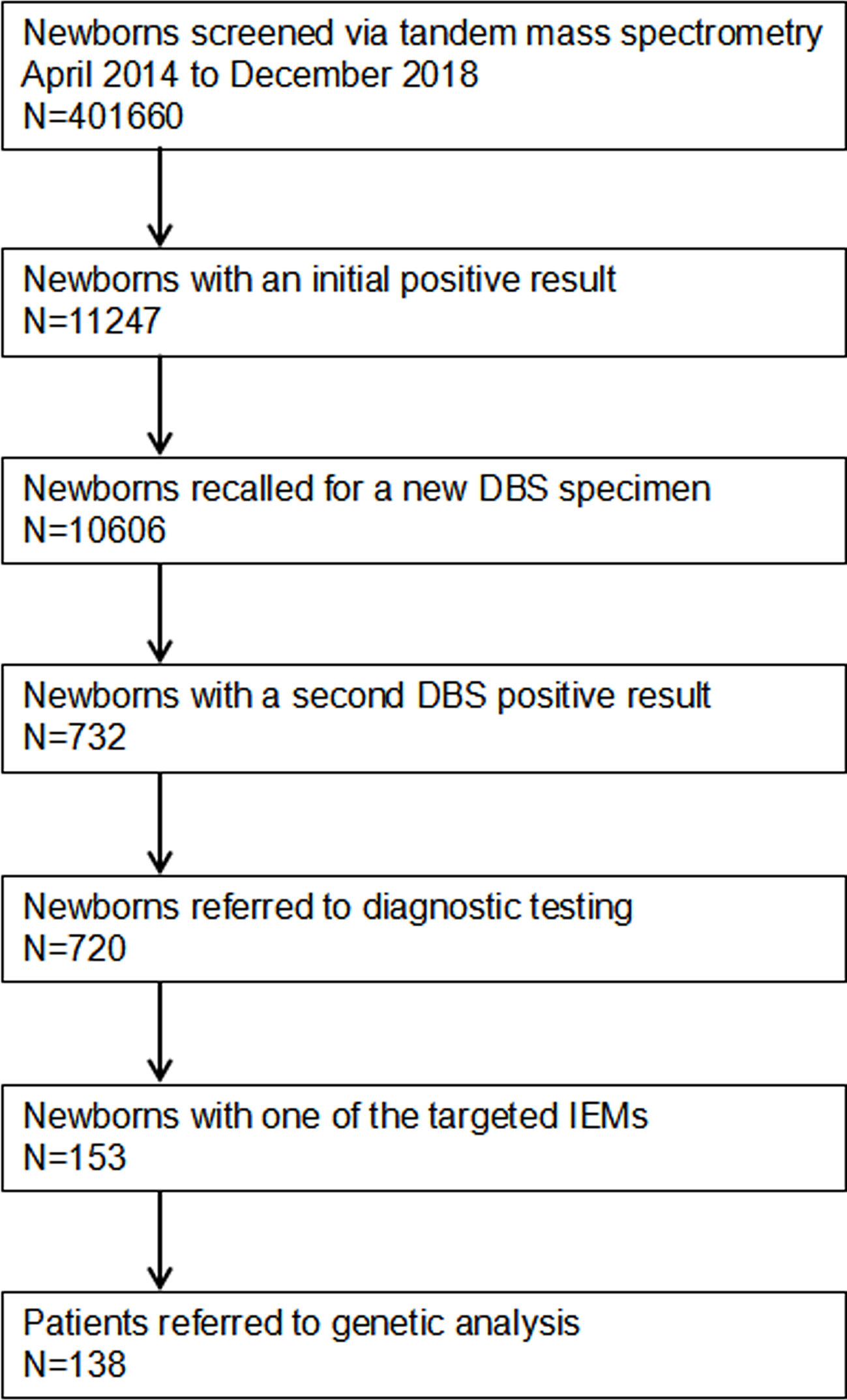
Figure 1 Flowchart of expanded newborn screening for inborn errors of metabolism and genetic analysis of patients. DBS, dried blood spot; IEMs, inborn errors of metabolisms.
Expanded Newborn Screening Assay
Eleven amino acids, 30 acylcarnitines, free carnitine, and succinylacetone were tested using tandem mass spectrometry (Supplementary Table 1). Assays for screening inborn metabolism disorders were performed using screening kit (PerkinElmer, USA) and Waters HPLC-tandem mass spectrometry (TQD, Waters, USA). In brief, 100-ul extract liquor containing internal standards was added into U bottom plates. After incubating for 45 min at 45°C, 75-ul extract liquor was transferred into V bottom plates. After 2 h standing at ambient temperature, 25-ul liquor was injected into tandem mass spectrometry for metabolites analyses. Three levels of internal quality controls including blank, low, and high were used for quality control.
Positive Results for IMEs
In our screening panel, 26 kinds of IEMs were included. Each IEM had two or more indicators including metabolites and ratios, and their cut-off values. When DBS results met the positive rules of IEMs, they were considered as positive. All the positive rules of IEMs were shown in Table 1.

Table 1 Conditions and their positive rules in expanded newborn screening panel.
Genetic Analysis
High throughput sequencing was performed on all patients diagnosed with one kind of IEMs using the expanded edition panel of IMEs (Genuine Diagnostic, Hangzhou, China) including 306 genes related to IEMs. In brief, the target sequences were enriched using Agilent SureSelect Human Exon Sequence Capture Kit (Agilent Technologies, Inc, California, USA). Next, the captured products were purified using Agencourt AMPure XP beads (Beckman Coulter, Inc, Miami, USA). Then, the sequencing library was established using TruePrepTM DNA Library Prep Kit V2 (Vazyme Biotech, New Jersey, USA) and TruePrepTM Index Kit V2 (Vazyme Biotech, New Jersey, USA) and was examined by Agilent High Sensitivity DNA Kit (Agilent Technologies, Inc, California, USA). Finally, the sequencing library was quantified by Illumina DNA Standards and Primer Premix Kit (KAPA Biosystems, Boston, USA), and massively parallel sequenced on Illumina HiSeq 2500 system.
Statistical Analysis
Statistical analysis was performed using SPSS17.0 version. The difference of categorical data was compared using Chi-square test. The difference of measurement data was compared by analysis of variance. p < 0.05 was considered to be statistical significance.
Results
A total of 401,660 newborns were screened by expanded newborn screening program (Figure 1). After initial screening, 11,247 (2.80%) newborns, who had positive results, were recalled for a new specimen. However, only 10,606 (94.30%) newborns with an initial positive result were collected a new specimen. After a repeated test, 732 (6.90%) newborns with a second positive result were determined to be suspect positive, and 720 (98.36%) of them were referred to diagnostic testing. Finally, 153 infants were diagnosed with one of IMEs and treated, and 138 of them were referred to genetic analysis. As SCADD and 3-MCCD were diseases with questionable phenotype, the overall incidence (excluding SCADD and 3-MCCD) was 1/3163. The comparison of all characteristics between normal newborns and patients did not reach at significant difference, including age at testing (p = 0.574), gender (p = 0.260), gestational age (p = 0.691), birth weight (p = 0.795), number of fetus (p = 0.988), register region (p = 0.571), and household registration (p = 0.166). The characteristics of newborns screened by expanded newborn screening program were shown in Table 2.
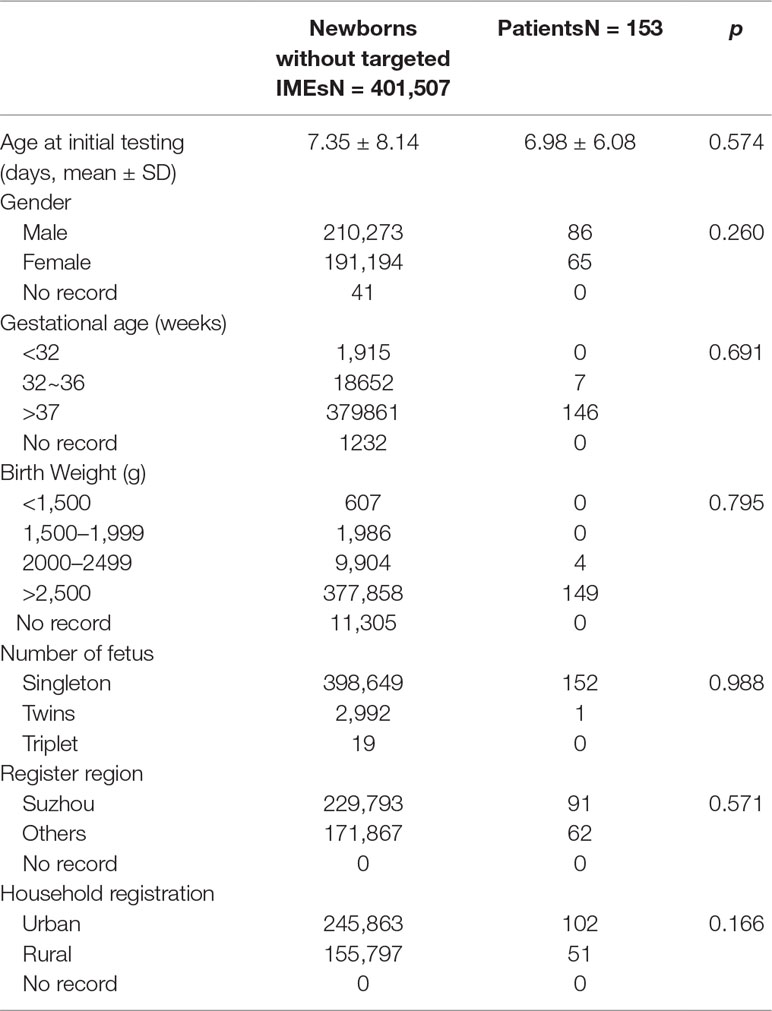
Table 2 Characteristics of newborns screened by expanded newborn screening program.
Of 22 IEMs, 10 were amino acid metabolic disorders (AAMDs), 7 were organic acid metabolic disorders (OAMDs), and 5 were fatty acid metabolic disorders (FAMDs). The AAMDs were the most common diseases, accounting for 51.63% of patients, followed by FAMDs (19.61%) and OAMDs (28.76%). The overall prevalence of AAMDs, FAMDs, and OAMDs was 1/5,084, 1/11,814, and 1/10,041, respectively. HPA may be induced by PAH defect or tetrahydrobiopterin deficiency. A total of 48 infants with HPA were found, including 42 (87.5%) infants with PAH defect and 6 (12.5%) infants with tetrahydrobiopterin deficiency caused by PTPS (MIM* 612719) defect. The incidence of HPA, PAH defect, and PTPS defect were 1/8,368, 1/9,563, and 1/66,943, respectively. Furthermore, 42 infants with PAH defect were classified into two groups: 21 (50%) infants with PKU (≥360 μmol/L Phe) and 21 (50%) infants with M-HPA (120 μmol/L to 360 μmol/L Phe) (Chen et al., 2015). Of the 10 AAMDs, PKU and M-HPA were the most common diseases, accounting for 26.58% of patients, respectively, followed by hypermethioninemia (16.46%). The prevalence of single AAMD ranged from 1/401,660 to 1/19,127. Of the 7 OAMDs, 3-MCCD was the most common disease, accounting for 40.00% of patients, followed by methylmalonic acidemia (MMA; MIM# 251000) (33.33%). The prevalence of single OAMD ranged from 1/401,660 to 1/33,412. Of the 5 FAMDs, PCUD was the most common disease, accounting for 34.09% of patients, followed by SCADD (31.82%), VLCADD (13.64%), and MCADD (11.36%). The prevalence of single FAMD ranged from 1/100,411 to 1/26,777. All the above data were shown in Figure 2 and Table 3.
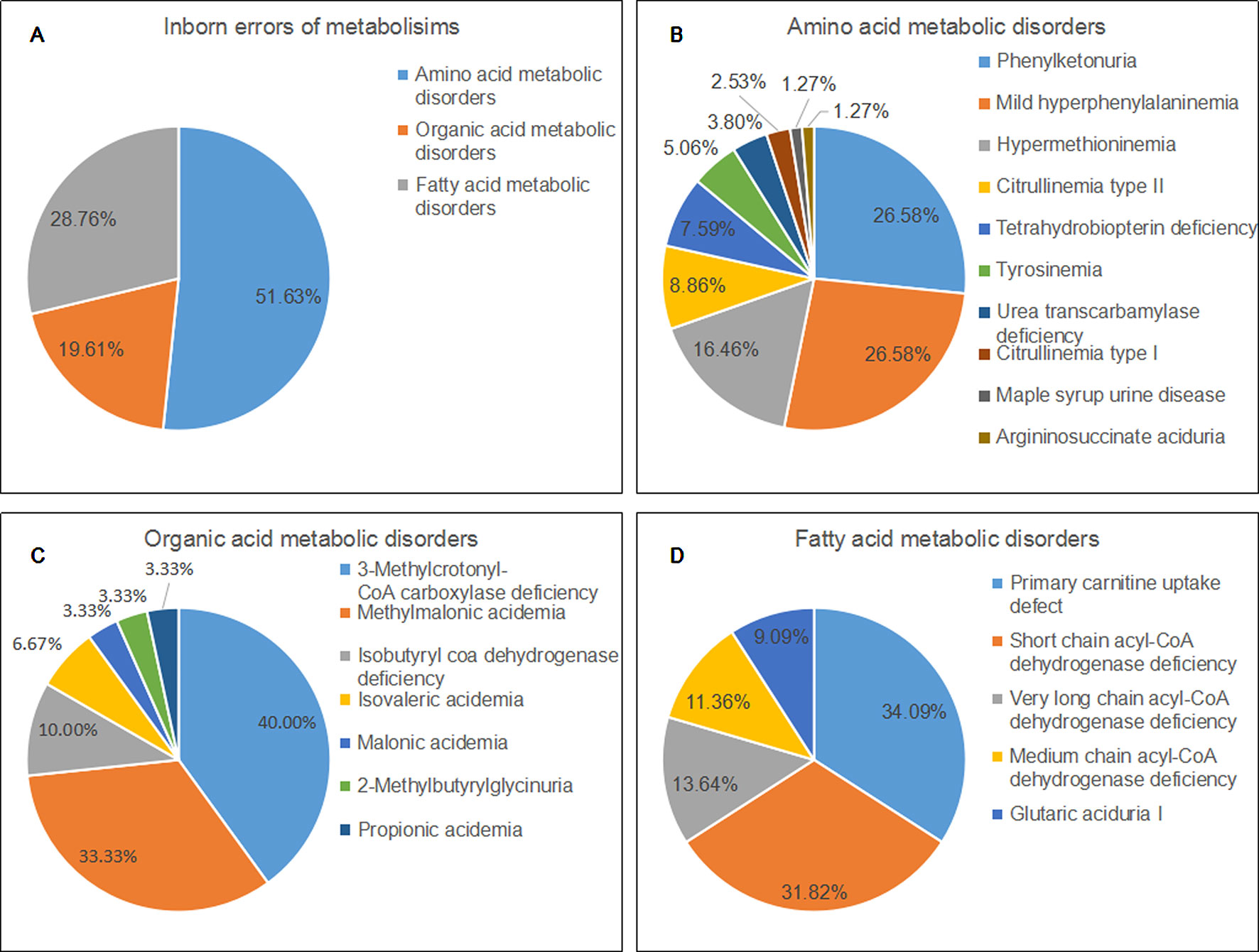
Figure 2 Disease spectrum and distribution of inborn errors of metabolisms. (A) The percentage of three categories of inborn errors of metabolisms. (B) the percentage of different kinds of amino acid metabolic disorders. (C) the percentage of different kinds of organic acid metabolic disorders. (D) the percentage of different kinds of fatty acid metabolic disorders.
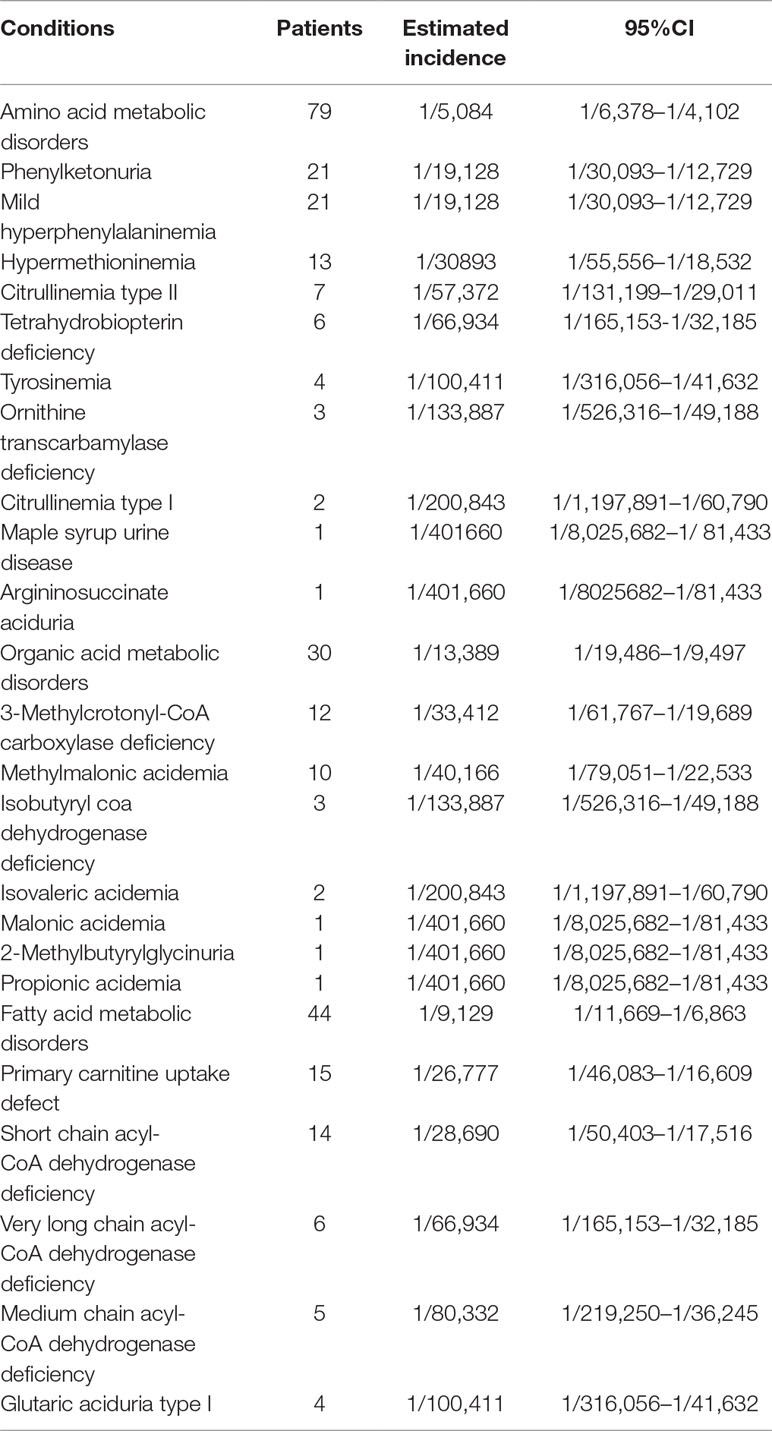
Table 3 The spectrum and incidence of conditions from 401660 newborns screened by expanded newborn screening program.
With regard to genetic analysis, 89 reported mutations and 51 novel mutations were detected in 138 patients with one of IEMs (Table 4). All common IEMs affecting more than 10 patients had mutational hotspots. In 12 patients with PKU, 17 mutations were detected and the c.728G > A was the most common mutations in PAH gene, accounting for 20.8% of mutational alleles and 41.7% of patients, followed by c.611A > G (8.3% and 16.7%), c.721C > T (8.3% and 16.7%), and c.498C > G (8.3% and 8.3%). All the four hot mutations are pathogenic (www.ncbi.nlm.nih.gov/clinvar). In 18 patients with M-HPA, 19 mutations were detected and the c.158G > A was the most common mutations in PAH gene, accounting for 25.0% of mutational alleles and 50.0% of patients, followed by c.1238G > C (11.1% and 22.2%), c.728G > A (8.3% and 16.7%), and c.1315+6T > A (8.3% and 16.7%). However, the c.158G > A has a uncertain significance of pathogenicity (www.ncbi.nlm.nih.gov/clinvar). In 15 patients with PCUD, nine mutations were detected and the c.1400C > G was the most common mutation in SLC22A5 (MIM* 603377) gene, accounting for 50.0% of mutational alleles and 80% of patients, followed by c.51C > G (13.3% and 26.7%) and c.760C > T (13.3% and 26.7%). All the three mutations are pathogenic and/or likely pathogenic (). In 14 patients with SCAD deficiency, nine mutations were detected and the c.1031A > G was the most common mutation in ACADS (MIM* 606885) gene, accounting for 42.9% of mutational alleles and 71.4% of patients, followed by c.164C > T (17.9% and 28.6%) and c.1130C > T (10.7% and 21.4%). The two mutation c.1031A > G and c.164C > T are likely pathogenic, and the c.1130C > T had conflicting interpretations of pathogenicity (www.ncbi.nlm.nih.gov/clinvar). Hypermethioninemia is able to be inherited by dominant transmission of MAT1A (MIM* 610550) gene. In 13 patients with hypermethioninemia, five mutations were detected and the c.791G > A was the most common mutation in MAT1A gene, accounting for 71.4% of mutational alleles and 76.9% of patients. The c.791G > A is pathogenic (www.ncbi.nlm.nih.gov/clinvar) and dominantly inherited. In 12 patients with 3-Methylcrotonyl-CoA carboxylase deficiency, 13 mutations and 2 mutations were detected in MCCC1 (MIM* 609010) gene and MCCC2 (MIM* 609014) gene. The most common mutation is c.639+2T > A of MCCC1 gene, accounting for 16.7% of mutational alleles and 33.3% of patients, followed by c.863A > G of MCCC1 gene (12.5% and 25.0%). The c.639+2T > A is pathogenic, but the c.863A > G has uncertain significance of pathogenicity (). Of 10 patients with MMA, 7 carried MMUT (MIM* 609058) gene mutations and 3 carried MMACHC (MIM* 609831) gene mutations. In all patients with MMA, the most common mutation was c.1663G > A of MMUT gene, accounting for 15.0% of mutational alleles and 30% of patients, followed by c.729_730insTT of MMUT gene (10.0% and 20.0%) and c.609G > A of MMACHC gene (10.0% and 20.0%).
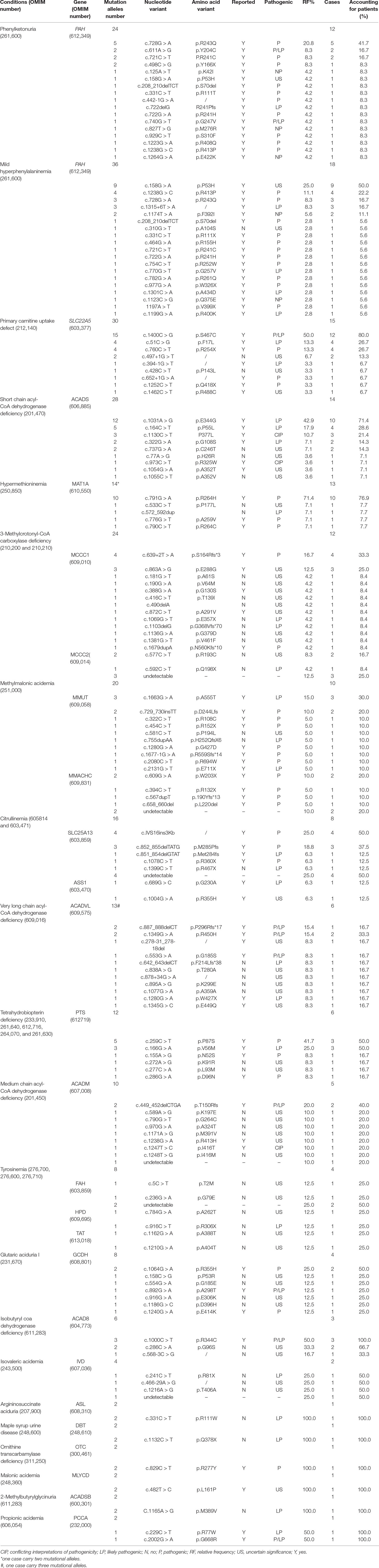
Table 4 Mutations detected in patients with inborn error of metabolism identified by expanded newborn screening.
Other IEMs including citrullinemia (MIM# 605814 and 603471), Tetrahydrobiopterin deficiency (MIM# 233910, 261640, 612716, 264070, and 261630), and Isobutyryl coa dehydrogenase deficiency (IBD; MIM# 611283) were also observed to have mutational hotspots. Of eight patients with Citrullinemia, seven were confirmed CTLN2 caused by mutations in SLC25A13 (MIM* 603859) gene, and only one cittrullinemia I (CTLN 1; MIM# 215700) caused by mutations in ASS1 (MIM* 603470) gene. In all patients with citrullinemia, the most common mutation was c.IVS16ins3Kb of SLC25A13 gene, accounting for 25.0% of mutational alleles and 50.0% of patients, followed by c.852_855delTATG of SLC25A13 gene (18.8% and 37.5%). Both c.IVS16ins3Kb and c.852_855delTATG of SLC25A13 gene are pathogenic for CTLN2 (www.ncbi.nlm.nih.gov/clinvar). In six patients with Tetrahydrobiopterin deficiency, six mutations in PTS (MIM* 612719) gene were detected and the c.259C > T was the most common mutation, accounting for 41.7% of mutational alleles and 50.0% of patients, followed by c.166G > A (25.0% and 50.0%). Furthermore, the c.259C > T is pathogenic and the c.166G > A is likely pathogenic (www.ncbi.nlm.nih.gov/clinvar), and the two mutations account for 83.3% of patients. Interestingly, all the three patients with Isobutyryl coa dehydrogenase deficiency were heterozygous for the c.1000C > T variant of ACAD8 (MIM* 604773) gene, and two patients were heterozygous for the c.286C > A variant, which has not been reported. The c.1000C > T of the ACAD8 gene was reported to be pathogenic and likely pathogenic in patients with IBD (www.ncbi.nlm.nih.gov/clinvar). Obviously, the two mutations are hotspots and main causes for IBD.
Discussion
PKU is an autosomal recessive genetic AAMD caused by deficiency of phenylalanine hydroxylase (PAH) (Blau et al., 2010). They were the most common IMEs identified by expanded newborn screening program, and the incidence of both were about 1/20,000. Up to now, more than 800 PAH mutations have been identified in patients with deficiency of PAH (Zhang et al., 2018). Some hotspot mutations exist in PAH gene and vary in different populations. For example, the most common PAH mutation is c.1222C > T in American (Kaul et al., 1994), IVS10-11G > A in Iranian (Zamanfar et al., 2017; Esfahani and Vallian, 2018; Rastegar Moghadam et al., 2018) and Spanish (Bueno et al., 2013; Aldámiz-Echevarría et al., 2016), c.168+5G > C in western Iranian (Alibakhshi et al., 2014), c.1238G > C in Japanese (Okano et al., 2011; Dateki et al., 2016), c.728G > A in Chinese (Zhou et al., 2012; Li et al., 2015; Zhang et al, 2018), c.781C > T in Karachays (Gundorova et al., 2018), c.1068C > A and c.728G > A in south Korean (Lee et al., 2008), c.1162G > A in Brazilian (Vieira Neto et al., 2018), c.782G > A in Syrian (Murad et al., 2013), and c.1222C > T in Australian (Ho et al., 2014). These different hotspot mutations suggested different origins. Similar to other Chinese populations, in our cohort, the c.728G > A is the most common PAH mutation that account for 20.8% of mutational alleles and 41.7% of classical PKU patients. However, in patients with M-HPA, the c.158C > A is the most common mutation that account for 25.0% of mutational alleles and 50.0% of patients. In a Japanese population, the c.158C > A also exhibited a relative higher prevalence in patients with hyperphenylalaninemia compared with PKU (Dateki et al., 2016). It appears that PAH deficiency has a correlation between genotype and clinical phenotype, and the c.158C > A could be considered as a marker for differentiating hyperphenylalaninemia from classical PKU.
PCUD is the second common IME. It shows a large variation of prevalence in different populations. For example, the prevalence of PCUD is 1/297 in Faroese (Rasmussen et al., 2014), 1/120,000 in Australian (Wilcken et al., 2003), 1/40,000 in Japanese (Koizumi et al., 1999), and 1/20,000–70,000 in American (Magoulas and El-Hattab, 2012). According to the available data, the prevalence of PCUD ranges from 1/45,000 to 1/8,000 in different areas of China (Han et al., 2012; Sun et al., 2017; Zheng et al., 2017; Guo et al., 2018), while the incidence of PCUD is 1/26,777 in Suzhou population. PCUD is caused by deficiency of organic cation transporter 2 (OCTN2) that results from variants in SLC22A5 gene. The symptomatic patients presented a variety of clinical symptoms, including muscle weakness, dilated cardiomyopathy, hepatomegaly, encephalopathy, sudden infant death, feeding difficulty, recurrent pneumonia, vomiting, abdominal pain, and diarrhea (Han et al., 2014). These symptoms might be caused by different genotypes of PCUD (Rose et al., 2012; Chen et al., 2013). To date, more than 110 SLC22A5 mutations have been reported and hotspot mutations vary in different population (Han et al., 2014). For example, C.844T > C is always observed in Caucasian PCUD patients (Burwinkel et al., 1999; Vaz et al., 1999; Wang et al., 1999), and c.1400C > G is the hotspot mutation in Southeast Asian (Koizumi et al., 1999; Nezu et al., 1999; Tang et al., 1999). However, in California patients, no obvious hotspot mutation was observed in PCUD patients (Gallant et al., 2017). This could be explained by the fact that those patients are multi-ethnic. With regard to Chinese, the hotspot mutations are similar in different regions. Chen et al. found the most common mutations were c.760C > T (32.9%), c.1400C > G (21.1%), and c.51C > G (14.5%) in Taiwan PCD patients (Chen et al., 2013). Han et al. reported that c.760C > T is the most common mutation in patients with symptomatic, and c.51C > G in patients with asymptomatic in Shanghai (Han et al., 2014). Guo et al. observed that c.1400C > G were the most common mutation in five Jining PCUD patients (Guo et al., 2018). Sun et al. also noted that the c.1400C > G was the most common mutation in seven Nanjing PCUG patients (Sun et al., 2017). Tan et al. found the c.51C > G is the most common mutations in Liuzhou PCUD patients (Tan et al., 2017). In agreement with most studies, the c.1400C > G is the most common mutation, with a relative frequency of 50% and accounting for 80% of Suzhou PCUD patients.
SCADD is the third prevalent disease of IMEs and the most prevalent disease of fatty acid metabolic errors in Suzhou population. SCADD had a wide spectrum of symptoms, including hepatic dysfunction, bilateral optic atrophy, vomiting, dysmorphic facial features, feeding difficulties, metabolic acidosis, epilepsy, ketotic hypoglycemia, developmental delay, lethargy, seizures, dystonia, myopathy, and hypotonia (Kılıç et al., 2017; Nochi et al., 2017). However, almost all patients with SCADD identified by newborn screening present no symptom or significant health tissue (Jethva et al., 2008; Waisbren et al., 2008; Huang et al., 2016; Zheng et al., 2017). Therefore, SCADD was not included in expanded newborn screening panels in many newborn screening centers (Dietzen et al., 2009; Mak et al., 2013; Smon et al., 2018). The reported incidence of SCADD is 1/25,000∼1/45,000 worldwide (Zytkovicz et al., 2001; Loukas et al., 2010; Lim et al., 2014). In consistence with the above reports, the incidence of SCADD in Suzhou population is 1/28,690. SCADD is caused by the deficiency of SCAD that is encoded by ACADS gene. Until now, about 70 variants have been reported to be pathogenic or likely pathogenic in ACADS gene, including two common variants, c.511C > T and c.625G > A (Tonin et al., 2016; Nochi et al., 2017). Most patients with SCAD deficiency carry two mutation alleles of the two common variants, or harbor one of them in combination with a rare variant in ACADS gene (van Maldegem et al., 2010), and the hotspot in Ashkenazi Jewish patients is a pathogenic c.319C > T mutation (Tein et al., 2008). However, 71.4% of Suzhou patients with SCAD deficiency carried the pathogenic mutation c.1031A > G, similar to Zhejiang SCADD patients (Huang et al., 2016), but different from Jining SCADD patients (Guo et al., 2018).
Several conditions, including deficiency in cystathionine β-synthase activity, tyrosinemia type I, and liver disease, could result in abnormal elevation of serum methionine. In this study, hypermethioninemia specially refers to abnormal elevated methionine caused by the abolished or reduced activity of hepatic methionine adenosyltransferase (MAT) I/III that are encoded by MATA1 gene. More than 37 mutations described previously range from truncating mutations with no residual enzyme activity to mild missense mutations (Mudd., 2011; Chien et al., 2015). The prevalence of MAT I/III deficiency was reported to range from 1/110,000 to 1/20,000 in different newborn populations (Chien et al., 2005; Couce et al., 2008; Martins et al., 2012; Couce et al., 2013; Nagao et al., 2013). However, in mainland of China, the incidence is unreported. Our study reported a prevalence of 1/30,893 in Suzhou population of newborns. Of 13 Suzhou hypermethioninemia patients, 10 cases carried the dominant mutation c.791G > A (Pérez Mato et al., 2001; Muriello et al., 2017). Previous studies reported that the c.791G > A was the most prevalent mutation in Asian populations, such as Japanese, Chinese in Taiwan, and so on (Chien et al., 2005; Nagao et al., 2013). MATA1 deficiency is inherited either as autosomal-recessive or autosomal-dominant. Most MAT1A mutations give rise to autosomal recessive phenotypes, but several autosomal dominant mutations have also been observed, including c.776C > T, c.791G > A (Muriello et al., 2017), c.746G > A, and c.838G > A (Kim et al., 2016). With the exception of a few individuals with hypermethioninemia who present with abnormal neurological symptoms, most patients generally are free of major clinical manifestation. Hypermethioninemia shows clinical symptoms correlated to genotypes (Chou, 2000), while the c.791G > A could lead to mild hypermethioninemia. Of all MATA1 mutations related to hypermethioninemia, the c.791G > A was the most common mutation identified in patients screened by expanded newborn screening (Couce et al., 2008; Martins et al., 2012; Couce et al., 2013). The c.791G > A mutation was the most prevalent mutation in Asian populations, including Japanese (Nagao et al., 2013) and Taiwan population (Chien et al., 2005). As expected, the c.791G > A was the most prevalent (80%) mutation in Suzhou newborns. In addition, another autosomal dominant mutation c.776G > T was found in one patient, and t one patient carried a novel heterozygous c.533C > T mutation. It appears that MATA1 deficiency is mainly inherited via autosomal dominant mode in Suzhou population. Furthermore, we found no Suzhou patients exhibit obvious clinical abnormality. There is a wide range of clinical manifestations in individuals with mutations in MAT1A gene, from completely asymptomatic to neurological problems associated with brain demyelination (Furujo et al., 2012). As a result, we speculated that the extent of clinical manifestations is associated with the inherited mode, which needs further research.
3-MCCD is an autosomal recessive inborn error of leucine metabolism, resulting in leukodystrophy, developmental delays, hypoglycemia, acidosis, failure to thrive, lactic acidosis, and hyperammonemia (Elpeleg et al., 1992; de Kremer et al., 2002; Forsyth et al., 2016). Despite cases with 3-MCCD identified by expanded newborn screening are more than previous expected, a growing number of reports have shown that the majority of cases are in fact asymptomatic (Stadler et al., 2006; Arnold et al., 2008; Arnold et al., 2012; Lam et al., 2013; Ye et al., 2014; Rips et al., 2016). This suggests this condition might represent a biochemical phenotype, but not a disease, and therefore should be excluded from newborn screening panels (Wilcken, 2008; Forsyth et al., 2016; Rips et al., 2016). The 3-MCCD is classified into type I (MIM# 210200) and type II (MIM# 210210), caused by MCCC1 gene and MCCC2 gene, respectively. Until now, at least 66 MCCC1 and 83 MCCC2 mutations have been reported (Yang et al., 2015). The 3-MCCD was the most prevalent organic acid metabolic error and showed a large variation of incidence from 1/27,000 to 1/110,000 in different countries (Yang et al., 2015; Fonseca et al., 2016). Similar to another Chinese population (Yang et al., 2015), the incidence of 3-MCCD is about 1/33,412 in Suzhou population. Some previous reports revealed that MCCC2 mutations were the main etiology of 3-MCCD (Uematsu et al., 2007; Cho et al., 2012; Grünert et al., 2012; Fonseca et al., 2016). However, in Zhejiang population, almost all (5/6) 3-MCCD patients carried one or two MCCC1 mutations (Yang et al., 2015). Similarly, of 12 Suzhou patients with 3-MCCD, 75% (9/12) were caused by MCCC1 mutations, which suggested the MCCC1 mutations might be prevalent in China (Yang et al., 2015). It is worth mentioning that further genetic testing for more Chinese patients should be conducted to confirm the above conclusion. Several mutations were observed to have a relative high frequency in 3-MCCD patients, including c.838G > T (4/12), c.295G > A (3/56), c.1574+1G > A (3/56) in MCCC2 gene and c.1155A > C (4/56) in MCCC1 gene (Dantas et al., 2005; Cho et al., 2012). However, most studies did not observe mutational hotspot of the two genes (Stadler et al., 2006; Yang et al., 2015; Smon et al., 2018). Contrary to a previous report on Zhejiang patients, a mutational hotspot c.639+2T > A, that was observed in only one Zhejiang patient and predicted to be pathogenic (Yang et al., 2015), had a high prevalence (4/12) in Suzhou patients. This inconsistency could be caused by the diversity of races or a small sample size of patients. As a result, the mutational hotspot should be confirmed by further research based on a large number of patients.
MMA is a family of lethal, severe, and multisystems organic acid metabolic errors, which has a wide clinical spectrum, including anorexia, failure to thrive, hypotonia, developmental delay, progressive renal failure, functional immune impairment, optic nerve atrophy, and hematologic abnormalities. MMA is classified into two main forms according to phenotype, including isolated methylmalonic acidurias and combined methylmalonic aciduria and homocystinuria, and caused by the defects of 10 genes, including MUT, MMAA, MMAB, CD320, MMADHC, LMBRD1, HCFC1, ABCD4, MCEE, and SUCLA2. According to previous reports, the incidence of MMA was 1/50,000 in Japan (Shigematsu et al., 2002), 1/85,000 in Taiwan of China (Cheng et al., 2010), and 1/250,000 in Germany (Schulze et al., 2003). However, in mainland China, the incidence of MMA ranged from 1/3,920 to 1/26,000 (Tu, 2011; Han et al., 2016; Yin, 2016; Zhao et al., 2016). In Suzhou, the incidence of MMA is about 1/40,000, obviously higher than that in the above countries, but lower than that in Shandong, Henan, Beijing, Shanghai, and Taiwan. Recently, a study with large sample size, containing 1,003 MMA patients derived from 26 provinces or cities of China, demonstrated that MMA cblC and MMA mut were the two major types in China. Similar to the report by Liu et al., the MMA cblC and the MMA mut were also the most prevalent types in Suzhou. However, contrary to Liu et al. report, in Suzhou population, the MMA mut (six patients) was more prevalent than MMA cblC (three patients). Several hotspot mutations were reported in MUT gene and MMADHC gene. Han et al. reported that the c.729_730insTT of the MMUT gene was the most common mutation in Shanghai patients (Han et al., 2015). Liu et al. reported that the c.609G > A and the c.658_660delAAG of the MMACHC gene were the most common mutations in 70 unrelated MMA cblC patients (Liu et al., 2010). Yu et al. reported that the c.609G > A and the c.658_660delAAG were the most common mutations detected in 13 (81%) out of 16 MMA cblC patients (Yu et al., 2015). However, in Suzhou patients with MMA, the mutations c.1663G > A and c.729_730insTT of the MMUT gene and c.609G > A of the MMACHC gene were the most common mutations. Therefore, the hotspot mutations in Chinese patients with MMA might be c.609G > A and c.658_660delAAG of the MMACHC gene and might be c.1663G > A and c.729_730insTT of the MMUT gene.
Citrullinemia is an autosomal recessive disorder and a urea cycle disease leading to a wide spectrum of phenotypes, from life-threatening neonatal hyperammonemia to adult onset with mild symptoms, and even no manifestation (Saheki and Kobayashi, 2002; Gao et al., 2003; Häberle et al., 2003; Enns et al., 2005; Dimmock et al., 2008; Komatsu et al., 2008; Häberle et al., 2009; Salek et al., 2010). This disease is classified into CTLN 1 and CTLN 2, caused by mutations of ASS1 gene and SLC25A13 gene, respectively. The estimated prevalence of CTLN 1 and CTLN 2 is 1 in 44,300–200,000 (Kasper et al., 2010; Niu et al., 2010) and 1 in 7,100–230,000 (Yamaguchi et al., 2002; Kobayashi et al., 2003; Lu et al., 2005; Tabata et al., 2008; Kikuchi et al., 2012) based on expanded newborn screening, respectively. However, most CTLN2 cases were identified in countries of East Asia, especially in Japan. More than 137 mutations in ASS1 gene have been identified in worldwide patients (Diez-Fernandez et al., 2017). The c.1168G > A mutation is the most common mutation in several ethnic groups, including Germans, Spaniards, and Turks, but rare in Asians (Gao et al., 2003; Engel et al., 2009; Diez-Fernandez et al., 2017). Whereas, the c.421-2A > G is the most frequent mutation in East Asians (Kobayashi et al., 1995; Lee et al., 2013; Woo et al., 2013). However, in this study, only one CTLN 1 patient and two mutations of the ASS1 gene were identified in Suzhou citurillinemia patients, which could be caused by ethnic specificity. With regard to CTLN2, a higher prevalence is observed in Suzhou patients compared to CTLN1. In previous studies, mutation detection of the SLC25A13 gene was very high, greater than 90% of CTLN2 in East Asians (Yasuda et al., 2000; Saheki and Kobayashi, 2002; Yamaguchi et al., 2002; Kobayashi et al., 2003; Saheki et al., 2004; Lu et al., 2005). In contrast to CTLN1, the CTLN2 had a narrow spectrum of mutations in SLC25A13 gene and highly clustered mutations (Woo et al., 2014). The c.851_854del mutation in SLC25A13, which was suspected to have a founder effect, was identified in CTLN2 patients throughout East Asian countries, such as China, Japan, Korea, and so on (Kobayashi et al., 1999; Tanaka et al., 2002; Imamura et al., 2003; Tabata et al., 2008). According to targeted mutation analysis, the frequency of overall mutations in CTLN2 is estimated to be 1/65–79 in Chinese, 1/69–73 in Japanese, 1/50–112 in Korean, and 1/70–97 in Taiwanese (Yamaguchi et al., 2002; Saheki and Kobayashi, 2002; Kobayashi et al., 2003; Saheki et al., 2004; Lu et al., 2005). In above countries, c.1177+1G>A and c.851_854del are the most common mutations in CTLN 2 patients. In Japanese patients, the c.1177+1G>A mutation (up to 43.1% of detected alleles) had the highest frequency, followed by c.851_854del mutation (up to 38.9%) (Saheki and Kobayashi, 2002; Yamaguchi et al., 2002). In Korea patients, the IVS16ins3kb and the c.851_854del were the most common mutations and were found at very high frequencies (100%) (Ko et al., 2007). In Hong Kong CTLN2 patients, the c.851_854del GTAT, IVS16ins3kb and c.852_855delTATG were the most common mutations of SLC25A13 gene (Hui et al., 2014; Chong et al., 2018). In another Chinese population, the c.851_854del GTAT was the most common mutation and was observed in 100% CTLN2 patients (Lin et al., 2017). Some other studies also observed c.851_854del GTAT was the most common mutation in Chinese populations (Xing et al., 2010; Fu et al., 2011; Song et al., 2011). In our study, we found the c.852_855delTATG and the IVS16ins3kb were the most common mutations and accounted for 75.0% of cases. In addition, another reported hotspot mutation c.851_854del GTAT was also observed in one Suzhou patient. As a result, c.851_854del, c.852_855delTATG, and IVS16ins3kb might be the most common mutations and should draw more attention in genetic analysis of Chinese CTLN2 patients.
Tetrahydrobiopterin deficiency (or BH4 deficiency) is a rare inborn metabolic disorder characterized by the deficiency of tetrahydrobiopterin or BH4 and caused by mutations in one of the four genes, including GCH1, PCBD1, PTS, and QDPR. This condition is inherited by autosomal recessive pattern and has a wide spectrum of symptoms, including intellectual disability, progressive problems with development, movement disorders, difficulty swallowing, seizures, behavioral problems, and inability to control body temperature. The total prevalence of this condition is estimated 1/500,000 to 1/1,000,000 worldwide and was relative high in Asian populations. In mainland of China, BH4 deficiency accounted for 8.55% of patients with HPA (Ye et al., 2009), significantly higher than 1%–3% of HPA worldwide (Blau et al., 1996). In our study, the prevalence of BH4 deficiency in Suzhou newborns was about 1/67,000, higher than 1/140,000 in the mainland of China (Ye et al., 2009; Li et al., 2018). Two teams reported the mutations in PTS gene were the main cause of BH4 deficiency, accounting for more than 95% of Chinese patients (Ye et al., 2009; Ye et al., 2013; Li et al., 2018). Similar to the above reports, all these six Suzhou patients with BH4 deficiency were caused by mutations in PTS gene. To date, more than 90 mutations in PTS gene have been reported in different populations. There are several mutational hotspots in different regions. Wang and coworkers investigated 204 PTPS deficiency patients and found the c.259C > T (38.2%) in PTS gene was the most common mutation, followed by c.84-291A > G (11%) (Wang et al., 2018). Ye and coworkers investigated 136 Chinese patients with PTPS deficiency and found c.259C > T (42.9%) in PTS gene was the most common mutation, followed by c.286G > A (13.4%) (Ye et al., 2013). Similar to the report by Ye et al., Li and coworkers reported that the c.259C > T (31.82%) in PTS gene was the most common mutation, followed by c.286G > A (13.64%) (Li et al., 2018). In Suzhou BH4 deficiency patients, we also found the c.259C > T was the most common mutation, accounting for 41.7% of mutation alleles. However, the second common mutation was c.166G > A, accounting for 25% of mutation alleles. The two most common mutations accounted for 83.3% of BH4-deficiency patients. The remaining four mutation alleles were c.155A > G, c.272A > G, c.277C > A, and c.286G > A, respectively, and all of these mutations were reported previously. However, c.155A > G, c.272A > G, and c.286G > A were reported as common mutations in Chinese patients with BH4 deficiency (Ye et al., 2013; Li et al., 2018; Wang et al., 2018). This difference could be caused by a small sample size and different populations. As a result, these mutational hotspots are potential candidates for genetic analysis of Chinese patients with BH4 deficiency.
IBD deficiency is a very rare disorder characterized by disrupting the breakdown of Val. This condition is an autosomal recessively inherited disease and caused by mutations in the ACAD8 gene. To the best of our knowledge, only 27 patients with IBD deficiency were reported in literature, and 28 mutations in the ACAD8 gene were detected in these patients (Yun et al., 2015; Santra et al., 2016; Lin et al., 2018). Most patients with IBD deficiency were asymptomatic in neonatal period, and a few had developed features such as dilated cardiomyopathy, hypotonia, developmental delay, and speech delay (Yun et al., 2015; Lin et al., 2018). In Suzhou population of newborns, three patients with IBD deficiency were identified from more than 400,000 newborns, and all patients remained asymptomatic during treatment and follow-up. Two reported common mutations were detected, including c.1000C > T and c.286C > A in the ACAD8 gene. Recently, Lin and coworker reported six Chinese patients with IBD deficiency and found the c.286C > A (7/14) was the most common mutation (Lin et al., 2018), followed by c.1000C > T. However, we found that the c.1000C > T was the most common mutation accounting for 50.0% (3/6) of mutational alleles in Suzhou patients with IBD deficiency, followed by c.286C > A (33.3%). As a result, the two mutations c.286C > A and c.1000C > T in the ACAD8 gene could be considered as mutational hotspots resulting in IBD deficiency in Chinese population. In addition, a novel heterozygous mutation c.568-3C > G was found in one patient with IBD deficiency. Our results characterized the mutational hotspots in the ACAD8 gene in Chinese patients with IBD deficiency and broaden the mutational spectrum of the ACAD8 gene.
There were five patients who were affected with Argininosuccinic aciduria (ASA; MIM# 207900), Maple syrup urine disease (MSUD; MIM# 248600), ornithine transcarbamylase deficiency (OTD; MIM# 311250), 2-methylbutyrylglycinuria (MBG; MIM# 248360), and Malonic acidemia (MA; MIM# 248360), respectively. Despite these five kinds of IEMs are extremely rare in Suzhou population, the cases affected with one of these IEMs are homozygous for one of the mutations, including c.331C > T in ASL, c.1132C > T in DBT, c.829C > T in OTC, 1165A > G in ACADSB, and c.482T > C in MLYCD. Therefore, these mutations might be hotspots causing the above five IEMs. In Suzhou patients, the remaining six IEMs were not observed to have mutational hotspots, including MCAD deficiency, VLCAD deficiency, glutaric acidemia type I (GA-I; MIM# 231670), tyrosinemia (MIM# 276700, 276600, and 276710), isovaleric acidemia (IVA; MIM# 243500), and propionic acidemia (PROP; MIM# 606054). In 32 mutation alleles of 16 patients with one of above six IEMs, 24 reported mutations and 6 novel mutations were detected. This might be caused by a small sample size and further research is needed.
In summary, we have detected a few mutational hotspots and some novel mutations that account for most Suzhou patients with IEMs identified by expanded newborn screening that might be pathogenic. These mutational hotspots could be potential candidates for gene screening and these novel mutations expanded the mutational spectrum of IEMs. Our findings could be of value for genetic counseling and genetic diagnosis of IEMs.
Data Availability Statement
The datasets generated for this study can be found in Sequence Read Archive using the accession number PRJNA566217.
Ethics Statement
The protocol was reviewed and approved by Ethic committee of the affiliated Suzhou hospital of Nanjing Medical University.
Author Contributions
BW and JX conceived and designed the research, analyzed data, and wrote the manuscript. TW, JM, and BW conducted experiments and reviewed the manuscript. QW conducted experiments. HL analyzed data and reviewed the manuscript. QZ and AG took part in diagnosis and treatment of infants with IME. All authors read and approved the manuscript.
Funding
This study was supported by grants from the Jiangsu Maternal and Children Health Care Research Project (F201603 and F201715), the Jiangsu Provincial Medical Innovation Team (CXTDB2017013), the Suzhou Clinical Medical Expert Team (SZYJTD201708), the Jiangsu Maternal and Children Health Care Key Discipline (FXK201748), the Suzhou Science and Technology Support Program (SYS201649), the Suzhou Key Medical Center (SZZX201505), and Suzhou Industry Technology Innovation Project (SYS201770).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all colleagues for their contributions, and Pin Zhu for technical supports in genetic analysis.
Abbreviations
ASA, Argininosuccinate aciduria; BH4 deficiency, Tetrahydrobiopterin deficiency; BKT, beta-Ketothiolase deficiency; CACT, Carnitine-acylcarnitine translocase deficiency; CPT-Ia, Carnitine palmitoyltransferase I deficiency; CPY-II, Carnitine palmitoyltransferase II deficiency; EMA, Ethylmalonic encephalopathy; GA-I, Glutaric aciduria type I; H-ARG, Arginemia; HCY, Homocystinuria; H-MET, Hypermethioninemia; HMG, 3-Hydroxy-3-methylglutaric aciduria; H-ORN, Hyperornithinemia; H-PRO, Hyperprolinemia; H-TYR, Tyrosinemia; IBG, Isobutyrylglycinuria; IVA, Isovaleric acidemia; LCHADD, long-chain L-3-Hydroxy acyl-CoA dehydrogenase deficiency; MADD, mutiple Acyl-coa dehydrogenases deficiency; MAL, Malonic acidemia; MCADD, Medium chain acyl-CoA dehydrogenase deficiency; MCD, multiple Carboxylase deficiency (MCD); M-HPA, mild Hyperphenylalaninemia; MSUD, Maple syrup urine disease; MUT, Methylmalonic acidemia; NKHG, Nonketotic hyperglycinemia; OTC, Ornithine transcarbamylase deficiency; PCUD, Primary carnitine uptake defect; PKU, Phenylketonuria; PROP, Propionic acidemia; SCADD, Short chain acyl-CoA dehydrogenase deficiency; TFP, Trifunctional protein deficiency; TYR-I, Tyrosinemia type I; VLCADD, very long chain acyl-CoA dehydrogenase deficiency; 2M3HBA, 2-Methyl-3-hydroxybutyric aciduria; 2MBG, 2-Methylbutyrylglycinuria; 3-MCC, 3-Methylcrotonyl-CoA carboxylase deficiency; 3MGA, 3-Methylglutaconic aciduria.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01052/full#supplementary-material
References
Aldámiz-Echevarría, L., Llarena, M., Bueno, M. A., Dalmau, J., Vitoria, I., Fernández-Marmiesse, A., et al. (2016). Molecular epidemiology, genotype-phenotype correlation and BH4 responsiveness in Spanish patients with phenylketonuria. J. Hum. Genet. 61, 731–744. doi: 10.1038/jhg.2016.38
Alfadhel, M., Al Othaim, A., Al Saif, S., Al Mutairi, F., Alsayed, M., Rahbeeni, Z., et al. (2017). Expanded newborn screening program in saudi arabia: incidence of screened disorders. J. Paediatr. Child. Health 53, 585–591. doi: 10.1111/jpc.13469
Alibakhshi, R., Moradi, K., Mohebbi, Z., Ghadiri, K. (2014). Mutation analysis of PAH gene in patients with PKU in western Iran and its association with polymorphisms: identification of four novel mutations. Metab. Brain. Dis. 29, 131–138. doi: 10.1007/s11011-013-9432-0
Andresen, B. S., Olpin, S., Poorthuis, B. J., Scholte, H. R., Vianey-Saban, C., Wanders, R., et al. (1999). Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 64, 479–494. doi: 10.1086/302261
Arnold, G. L., Koeberl, D. D., Matern, D., Barshop, B., Braverman, N., Burton, B., et al. (2008). A Delphi-based consensus clinical practice protocol for the diagnosis and management of 3-methylcrotonyl CoA carboxylase deficiency. Mol. Genet. Metab. 93, 363–370. doi: 10.1016/j.ymgme.2007.11.002
Arnold, G. L., Salazar, D., Neidich, J. A., Suwannarat, P., Graham, B. H., Lichter-Konecki, U., et al. (2012). Outcome of infants diagnosed with 3-methyl-crotonyl-CoA-carboxylase deficiency by newborn screening. Mol. Genet. Metab. 106, 439–441. doi: 10.1016/j.ymgme.2012.04.006
Blau, N., Barnes, I., Dhondt, J. L. (1996). International database of tetrahydrobiopterin deficiencies. J. Inherit. Metab. Dis. 19, 8–14. doi: 10.1007/BF01799342
Blau, N., van Spronsen, F. J., Levy, H. L. (2010). Phenylketonuria. Lancet. 376, 1417–1427. doi: 10.1016/S0140-6736(10)60961-0
Bueno, M. A., González-Lamuño, D., Delgado-Pecellín, C., Aldámiz-Echevarría, L., Pérez, B., Desviat, L. R., et al. (2013). Molecular epidemiology and genotype-phenotype correlation in phenylketonuria patients from South Spain. J. Hum. Genet. 58, 279–284. doi: 10.1038/jhg.2013.16
Burwinkel, B., Kreuder, J., Schweitzer, S., Vorgerd, M., Gempel, K., Gerbitz, K. D., et al. (1999). Carnitine transporter OCTN2 mutations in systemic primary carnitine deficiency: a novel Arg169Gln mutation and a recurrent Arg282ter mutation associated with an unconventional splicing abnormality. Biochem. Biophys. Res. Commun. 261, 484–487. doi: 10.1006/bbrc.1999.1060
Centers for Disease Control and Prevention (CDC). (2008). Impact of expanded newborn screening–United States, 2006. MMWR Morb. Mortal. Wkly. Rep. 57, 1012–1015.
Chen, Y. C., Chien, Y. H., Chen, P. W., Leung-Sang Tang, N., Chiu, P. C., Hwu, W. L., et al. (2013). Carnitine uptake defect (primary carnitine deficiency): risk in genotype-phenotype correlation. Hum. Mutat. 34, 655. doi: 10.1002/humu.22286
Chen, Y. F., Jia, H. T., Chen, Z. H., Song, J. P., Liang, Y., Pei, J. J., et al. (2015). Mutational spectrum of phenylketonuria in Jiangsu province. Eur. J. Pediatr. 174, 1333–1338. doi: 10.1007/s00431-015-2539-z
Cheng, K. H., Liu, M. Y., Kao, C. H., Chen, Y. J., Hsiao, K. J., Liu, T. T., et al. (2010). Newborn screening for methylmalonic aciduria by tandem mass spectrometry: 7 years’ experience from two centers in Taiwan. J. Chin. Med. Assoc. 73, 314–318. doi: 10.1016/S1726-4901(10)70067-8
Chien, Y. H., Abdenur, J. E., Baronio, F., Bannick, A. A., Corrales, F., Couce, M., et al. (2015). Mudd's disease (MAT I/III deficiency): a survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet. J. Rare Dis. 10, 99. doi: 10.1186/s13023-015-0321-y
Chien, Y. H., Chiang, S. C., Huang, A., Hwu, W. L. (2005). Spectrum of hypermethioninemia in neonatal screening. Early. Hum. Dev. 81, 529–533. doi: 10.1016/j.earlhumdev.2004.11.005
Cho, S. Y., Park, H. D., Lee, Y. W., Ki, C. S., Lee, S. Y., Sohn, Y. B., et al. (2012). Mutational spectrum in eight Korean patients with 3-methylcrotonyl-CoA carboxylase deficiency. Clin.Genet. 81, 96–98. doi: 10.1111/j.1399-0004.2011.01704.x
Chong, S. C., Lo, P., Chow, C. W., Yuen, L., Chu, W. C. W., Leung, T. Y., et al. (2018). Molecular and clinical characterization of citrin deficiency in a cohort of Chinese patients in Hong Kong. Mol. Genet. Metab. Rep. 17, 3–8. doi: 10.1016/j.ymgmr.2018.08.002
Chou, J. Y. (2000). Molecular genetics of hepatic methionine adenosyltransferase deficiency. Pharmacol. Ther. 85, 1–9. doi: 10.1016/S0163-7258(99)00047-9
Couce, M. L., Bóveda, M. D., Castiñeiras, D. E., Corrales, F. J., Mora, M. I., Fraga, J. M., et al. (2008). Hypermethioninaemia due to methionine adenosyltransferase I/III (MATI/III) deficiency: diagnosis in an expanded neonatal screening programme. J. Inherit. Metab. Dis. 31, S233–S239. doi: 10.1007/s10545-008-0811-3
Couce, M. L., Bóveda, M. D., García-Jimémez, C., Balmaseda, E., Vives, I., Castiñeiras, D. E., et al. (2013). Clinical and metabolic findings in patients with methionine adenosyltransferase I/III deficiency detected by newborn screening. Mol. Genet. Metab. 110, 218–221. doi: 10.1016/j.ymgme.2013.08.003
Dantas, M. F., Suormala, T., Randolph, A., Coelho, D., Fowler, B., Valle, D., et al. (2005). 3-Methylcrotonyl-CoA carboxylase deficiency: mutation analysis in 28 probands, 9 symptomatic and 19 detected by newborn screening. Hum. Mutat. 26, 164. doi: 10.1002/humu.9352
Dateki, S., Watanabe, S., Nakatomi, A., Kinoshita, E., Matsumoto, T., Yoshiura, K., et al. (2016). Genetic background of hyperphenylalaninemia in Nagasaki, Japan. Pediatr. Int. 58, 431–433. doi: 10.1111/ped.12924
de Kremer, R. D., Latini, A., Suormala, T., Baumgartner, E. R., Laróvere, L., Civallero, G., et al. (2002). Leukodystrophy and CSF purine abnormalities associated with isolated 3-methylcrotonyl-CoA carboxylase deficiency. Metab. Brain. Dis. 17, 13–18. doi: 10.1023/A:1014096112916
Dhondt, J. L. (2010). Expanded newborn screening: social and ethical issues. J. Inherit. Metab. Dis. 33, S211–S217. doi: 10.1007/s10545-010-9138-y
Dietzen, D. J., Rinaldo, P., Whitley, R. J., Rhead, W. J., Hannon, W. H., Garg, U. C., et al. (2009). National academy of clinical biochemistry laboratory medicine practice guidelines: follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry; executive summary. Clin. Chem. 55, 1615–1626. doi: 10.1373/clinchem.2009.131300
Diez-Fernandez, C., Rüfenacht, V., Häberle, J. (2017). Mutations in the human argininosuccinate synthetase (ASS1) gene, impact on patients, common changes, and structural considerations. Hum. Mutat. 38, 471–484. doi: 10.1002/humu.23184
Dimmock, D. P., Trapane, P., Feigenbaum, A., Keegan, C. E., Cederbaum, S., Gibson, J., et al. (2008). The role of molecular testing and enzyme analysis in the management of hypomorphic citrullinemia. Am. J. Med. Genet. A. 146A, 2885–2890. doi: 10.1002/ajmg.a.32527
Elpeleg, O. N., Havkin, S., Barash, V., Jakobs, C., Glick, B., Shalev, R. S. (1992). Familial hypotonia of childhood caused by isolated 3-methylcrotonyl-coenzyme A carboxylase deficiency. J. Pediatr. 121, 407–410. doi: 10.1016/S0022-3476(05)81796-2
Engel, K., Höhne, W., Häberle, J. (2009). Mutations and polymorphisms in the human argininosuccinate synthetase (ASS1) gene. Hum. Mutat. 30, 300–307. doi: 10.1002/humu.20847
Enns, G. M., O'Brien, W. E., Kobayashi, K., Shinzawa, H., Pellegrino, J. E. (2005). Postpartum "psychosis" in mild argininosuccinate synthetase deficiency. Obstet. Gynecol. 105, 1244–1246. doi: 10.1097/01.AOG.0000157769.90230.24
Ensenauer, R., Winters, J. L., Parton, P. A., Kronn, D. F., Kim, J. W., Matern, D., et al. (2005). Genotypic differences of MCAD deficiency in the Asian population: novel genotype and clinical symptoms preceding newborn screening notification. Genet. Med. 7, 339–343. doi: 10.1097/01.GIM.0000164548.54482.9D
Esfahani, M. S., Vallian, S. (2018). A comprehensive study of phenylalanine hydroxylase gene mutations in the Iranian phenylketonuria patients. Eur. J. Med. Genet. S1769–7212 (18), 30562–30567. doi: 10.1016/j.ejmg.2018.10.011
Fonseca, H., Azevedo, L., Serrano, C., Sousa, C., Marcão, A., Vilarinho, L. (2016). 3-Methylcrotonyl-CoA carboxylase deficiency: Mutational spectrum derived from comprehensive newborn screening. Gene 594, 203–210. doi: 10.1016/j.gene.2016.09.003
Forsyth, R., Vockley, C. W., Edick, M. J., Cameron, C. A., Hiner, S. J., Berry, S. A., et al. (2016). Outcomes of cases with 3-methylcrotonyl-CoA carboxylase (3-MCC) deficiency- Report from the Inborn Errors of Metabolism Information System. Mol. Genet. Metab. 118, 15–20. doi: 10.1016/j.ymgme.2016.02.002
Fu, H. Y., Zhang, S. R., Wang, X. H., Saheki, T., Kobayashi, K., Wang, J. S. (2011). The mutation spectrum of the SLC25A13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia. J. Gastroenterol. 46, 510–518. doi: 10.1007/s00535-010-0329-y
Furujo, M., Kinoshita, M., Nagao, M., Kubo, T. (2012). Methionine adenosyltransferase I/III deficiency: neurological manifestations and relevance of S-adenosylmethionine. Mol. Genet. Metab. 107, 253–256. doi: 10.1016/j.ymgme.2012.08.002
Gallant, N. M., Leydiker, K., Tang, H., Feuchtbaum, L., Lorey, F., Puckett, R., et al. (2012). Biochemical, molecular, and clinical characteristics of children with short chain acyl-CoA dehydrogenase deficiency detected by newborn screening in California. Mol. Genet. Metab. 106, 55–61. doi: 10.1016/j.ymgme.2012.02.007
Gallant, N. M., Leydiker, K., Wilnai, Y., Lee, C., Lorey, F., Feuchtbaum, L., et al. (2017). Biochemical characteristics of newborns with carnitine transporter defect identified by newborn screening in California. Mol. Genet. Metab. 122, 76–84. doi: 10.1016/j.ymgme.2017.06.015
Gao, H. Z., Kobayashi, K., Tabata, A., Tsuge, H., Iijima, M., Yasuda, T., et al. (2003). Identification of 16 novel mutations in the argininosuccinate synthetase gene and genotype-phenotype correlation in 38 classical citrullinemia patients. Hum. Mutat. 22, 24–34. doi: 10.1002/humu.10230
Grünert, S. C., Stucki, M., Morscher, R. J., Suormala, T., Bürer, C., Burda, P., et al. (2012). 3-methylcrotonyl-CoA carboxylase deficiency: clinical, biochemical, enzymatic and molecular studies in 88 individuals. Orphanet. J. Rare Dis. 7, 31. doi: 10.1186/1750-1172-7-31
Gundorova, P., Zinchenko, R. A., Kuznetsova, I. A., Bliznetz, E. A., Stepanova, A. A., Polyakov, A. V. (2018). Molecular-genetic causes for the high frequency of phenylketonuria in the population from the North Caucasus. PLoS One 13, e0201489. doi: 10.1371/journal.pone.0201489
Guo, K., Zhou, X., Chen, X., Wu, Y., Liu, C., Kong, Q. (2018). Expanded newborn screening for inborn errors of metabolism and genetic characteristics in a chinese population. Front. Genet. 9, 122. doi: 10.3389/fgene.2018.00122
Häberle, J., Pauli, S., Schmidt, E., Schulze-Eilfing, B., Berning, C., Koch, H. G. (2003). Mild citrullinemia in Caucasians is an allelic variant of argininosuccinate synthetase deficiency (citrullinemia type 1). Mol. Genet. Metab. 80, 302–306. doi: 10.1016/j.ymgme.2003.08.002
Häberle, J., Vilaseca, M. A., Meli, C., Rigoldi, M., Jara, F., Vecchio, I., et al. (2009). First manifestation of citrullinemia type I as differential diagnosis to postpartum psychosis in the puerperal period. Eur. J. Obstet. Gynecol. Reprod. Biol. 149, 228–229. doi: 10.1016/j.ejogrb.2009.11.014
Han, B., Cao, Z., Tian, L., Zou, H., Yang, L., Zhu, W., et al. (2016). Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic acidemia and homocysteinemia (cblCtype) in Shandong province, China. Brain. Dev. 38, 491–497. doi: 10.1016/j.braindev.2015.10.016
Han, L., Wang, F., Wang, Y., Ye, J., Qiu, W., Zhang, H., et al. (2014). Analysis of genetic mutations in Chinese patients with systemic primary carnitine deficiency. Eur. J. Med. Genet. 57, 571–575. doi: 10.1016/j.ejmg.2014.08.001
Han, L. S., Huang, Z., Han, F., Ye, J., Qiu, W. J., Zhang, H. W., et al. (2015). Clinical features and MUT gene mutation spectrum in Chinese patients with isolated methylmalonic acidemia: identification of ten novel allelic variants. World J. Pediatr. 11, 358–365. doi: 10.1007/s12519-015-0043-1
Han, L. S., Ye, J., Qiu, W. J., Zhang, H. W., Wang, Y., Ji, W. J., et al. (2012). Primary carnitine deficiency in 17 patients: diagnosis, treatment and follow up [Article in Chinese]. Zhonghua Er Ke Za Zhi 50, 405–409.
Hassan, F. A., El-Mougy, F., Sharaf, S. A., Mandour, I., Morgan, M. F., Selim, L. A., et al. (2016). Inborn errors of metabolism detectable by tandem mass spectrometry in Egypt: the first newborn screening pilot study. J. Med. Screen. 23, 124–129. doi: 10.1177/0969141315618229
Ho, G., Alexander, I., Bhattacharya, K., Dennison, B., Ellaway, C., Thompson, S., et al. (2014). The molecular bases of phenylketonuria (PKU) in New South Wales, Australia: mutation profile and correlation with tetrahydrobiopterin (BH4) responsiveness. JIMD Rep. 14, 55–65. doi: 10.1007/8904_2013_284
Huang, X. W., Zhang, Y., Yang, J. B., Hong, F., Qian, G. L., Tong, F., et al. (2016). Clinical, biochemical and gene mutation characteristics of short chain acyl-coenzyme A dehydrogenase deficiency by neonatal screening [Article in Chinese]. Zhonghua Er Ke Za Zhi 54, 927–930.
Hui, J., Tang, N. L., Li, C. K., Law, L. K., To, K. F., Yau, P., et al. (2014). Inherited metabolic diseases in the Southern Chinese population: spectrum of diseases and estimated incidence from recurrent mutations. Pathology 46, 375–382. doi: 10.1097/PAT.0000000000000140
Hsu, C. F., Kang, K. T., Leo Lee, Y., Chie, W. C. (2013). Cost-effectiveness of expanded newborn screening in Texas. Value Health 16, 1103–1104. doi: 10.1016/j.jval.2013.06.019
Imamura, Y., Kobayashi, K., Shibatou, T., Aburada, S., Tahara, K., Kubozono., et al. (2003). Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type IIcitrullinemia: a case report of siblings showing homozygous SLC25A13 mutation with and without the disease. Hepatol. Res. 26, 68–72. doi: 10.1016/S1386-6346(02)00331-5
Jethva, R., Bennett, M. J., Vockley, J. (2008). Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol. Genet. Metab. 95, 195–200. doi: 10.1016/j.ymgme.2008.09.007
Karaceper, M. D., Chakraborty, P., Coyle, D., Wilson, K., Kronick, J. B., Hawken, S., et al. (2016). The health system impact of false positive newborn screening results for medium-chain acyl-CoA dehydrogenase deficiency: a cohort study. Orphanet. J. Rare Dis. 11, 12. doi: 10.1186/s13023-016-0391-5
Kasper, D. C., Ratschmann, R., Metz, T. F., Mechtler, T. P., Möslinger, D., Konstantopoulou, V., et al. (2010). The national Austrian newborn screening program - eight years experience with massspectrometry. past, present, and future goals. Wien. Klin. Wochenschr. 122, 607–613. doi: 10.1007/s00508-010-1457-3
Kaul, R., Matalon, R., Allen, R., Fisch, R. O., Michals, K., Petrosky, A., et al. (1994). Frequency of 12 mutations in 114 children with phenylketonuria in the Midwest region of the USA. J. Inherit. Metab. Dis. 17, 356–358. doi: 10.1007/BF00711829
Kılıç, M., Şenel, S., Karaer, K., Ceylaner, S. (2017). Microcephaly and developmental delay caused by short-chain acyl-CoA dehydrogenase deficiency. Turk. J. Pediatr. 59, 708–710. doi: 10.24953/turkjped.2017.06.016
Kikuchi, A., Arai-Ichinoi, N., Sakamoto, O., Matsubara, Y., Saheki, T., Kobayashi, K., et al. (2012). Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13. Mol. Genet. Metab. 105, 553–558. doi: 10.1016/j.ymgme.2011.12.024
Kim, Y. M., Kim, J. H., Choi, J., Gu-Hwan, K., Kim, J. M., Kang, M., et al. (2016). Determination of autosomal dominant or recessive methionine adenosyltransferase I/III deficiencies based on clinical and molecular studies. Int. J. Mol. Med. 22, 147–155. doi:10.2119/molmed.2015.00254
Ko, J. M., Kim, G. H., Kim, J. H., Kim, J. Y., Choi, J. H., Ushikai, M., et al. (2007). Six cases of citrin deficiency in Korea. Int. J. Mol. Med. 20, 809–815. doi: 10.3892/ijmm.20.6.809
Kobayashi, K., Bang Lu, Y., Xian Li, M., Nishi, I., Hsiao, K. J., Choeh, K., et al. (2003). Screening of nine SLC25A13 mutations: their frequency in patients with citrin deficiency and high carrier rates in Asian populations. Mol. Genet. Metab. 80, 356–359. doi: 10.1016/S1096-7192(03)00140-9
Kobayashi, K., Kakinoki, H., Fukushige, T., Shaheen, N., Terazono, H., Saheki, T. (1995). Nature and frequency of mutations in the argininosuccinate synthetase gene that cause classical citrullinemia. Hum. Genet. 96, 454–463. doi: 10.1007/BF00191806
Kobayashi, K., Sinasac, D. S., Iijima, M., Boright, A. P., Begum, L., Lee, J. R., et al. (1999). The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 22, 159–163. doi: 10.1038/9667
Koizumi, A., Nozaki, J., Ohura, T., Kayo, T., Wada, Y., Nezu, J., et al. (1999). Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum. Mol. Genet. 8, 2247–2254. doi: 10.1093/hmg/8.12.2247
Komatsu, M., Yazaki, M., Tanaka, N., Sano, K., Hashimoto, E., Takei, Y., et al. (2008). Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liverdisease. J. Hepatol. 49, 810–820. doi: 10.1016/j.jhep.2008.05.016
Lam, C., Carter, J. M., Cederbaum, S. D., Neidich, J., Gallant, N. M., Lorey, F., et al. (2013). Analysis of cases of 3-methylcrotonyl CoA carboxylase deficiency (3-MCCD) in the California newborn screening program reported in the state database. Mol. Genet. Metab. 110, 477–483. doi: 10.1016/j.ymgme.2013.09.006
Lee, B. H., Kim, Y. M., Heo, S. H., Kim, G. H., Choi, I. H., Lee, B. S., et al. (2013). High prevalence of neonatal presentation in Korean patients with citrullinemia type 1, and their shared mutations. Mol. Genet. Metab. 108, 18–24. doi: 10.1016/j.ymgme.2012.11.011
Lee, Y. W., Lee, D. H., Kim, N. D., Lee, S. T., Ahn, J. Y., Choi, T. Y., et al. (2008). Mutation analysis of PAH gene and characterization of a recurrent deletion mutation in Korean patients with phenylketonuria. Exp. Mol. Med. 40, 533–540. doi: 10.3858/emm.2008.40.5.533
Li, N., Jia, H., Liu, Z., Tao, J., Chen, S., Li, X., et al. (2015). Molecular characterisation of phenylketonuria in a Chinese mainland population using next-generation sequencing. Sci. Rep. 5, 15769. doi: 10.1038/srep15769
Li, N., Yu, P., Rao, B., Deng, Y., Guo, Y., Huang, Y., et al. (2018). Molecular genetics of tetrahydrobiopterin deficiency in Chinese patients. J. Pediatr. Endocrinol. Metab. 31, 911–916. doi: 10.1515/jpem-2018-0037
Lim, J. S., Tan, E. S., John, C. M., Poh, S., Yeo, S. J., Ang, J. S., et al. (2014). Inborn Error of Metabolism (IEM) screening in Singapore by electrospray ionization-tandem mass spectrometry (ESI/MS/MS): An 8 year journey from pilot to current program. Mol. Genet. Metab. 113, 53–61. doi: 10.1016/j.ymgme.2014.07.018
Lin, Y., Peng, W., Jiang, M., Lin, C., Lin, W., Zheng, Z., et al. (2018). Clinical, biochemical and genetic analysis of Chinese patients with isobutyryl-CoA dehydrogenase deficiency. Clin. Chim. Acta. 487, 133–138. doi: 10.1016/j.cca.2018.09.033
Lin, Y., Yu, K., Li, L., Zheng, Z., Lin, W., Fu, Q. (2017). Mutational analysis of ASS1, ASL and SLC25A13 genes in six Chinese patients with citrullinemia [Article in Chinese]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 34, 676–679.
Lindner, M., Gramer, G., Haege, G., Fang-Hoffmann, J., Schwab, K. O., Tacke, U., et al. (2008). Efficacy and outcome of expanded newborn screening for metabolic diseases–report of 10 years from South-West Germany. Orphanet. J. Rare Dis. 6, 44. doi:10.1186/1750-1172-6-44
Liu, M. Y., Yang, Y. L., Chang, Y. C., Chiang, S. H., Lin, S. P., Han, L. S., et al. (2010). Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria. J. Hum. Genet. 55, 621–626. doi:10.1038/jhg.2010.81
Liu, N., Huang, Q., Li, Q., Zhao, D., Li, X., Cui, L., et al. (2017). Spectrum of PAH gene variants among a population of Han Chinese patients with phenylketonuria from northern China. BMC Med. Genet. 18, 108. doi: 10.1186/s12881-017-0467-7
Loukas, Y. L., Soumelas, G. S., Dotsikas, Y., Georgiou, V., Molou, E., Thodi, G., et al. (2010). Expanded newborn screening in Greece: 30 months of experience. J. Inherit. Metab. Dis. 33, S341–S348. doi: 10.1007/s10545-010-9181-8
Lu, Y. B., Kobayashi, K., Ushikai, M., Tabata, A., Iijima, M., Li, M. X., et al. (2005). Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J. Hum. Genet. 50, 338–346. doi: 10.1007/s10038-005-0262-8
Magoulas, P. L., El-Hattab, A. W. (2012). Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet. J. Rare Dis. 7, 68. doi: 10.1186/1750-1172-7-68
Maier, E. M., Liebl, B., Röschinger, W., Nennstiel-Ratzel, U., Fingerhut, R., Olgemöller, B., et al. (2005). Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum. Mutat. 25, 443–452. doi: 10.1002/humu.20163
Mak, C. M., Lee, H. C., Chan, A. Y., Lam, C. W. (2013). Inborn errors of metabolism and expanded newborn screening: review and update. Crit. Rev. Clin. Lab. Sci. 50, 142–162. doi: 10.3109/10408363.2013.847896
Martins, E., Marcão, A., Bandeira, A., Fonseca, H., Nogueira, C., Vilarinho, L. (2012). Methionine adenosyltransferase I/III deficiency in portugal: high frequency of a dominantly inherited form in a small area of douro high lands. JIMD Rep. 6, 107–112. doi: 10.1007/8904_2011_124
Mudd, S. H. (2011). Hypermethioninemias of genetic and non-genetic origin: A review. Am. J. Med. Genet. C. Semin. Med. Genet. 157C, 3–32. doi: 10.1002/ajmg.c.30293
Murad, H., Dabboul, A., Moassas, F., Alasmar, D., Al-Achkar, W. (2013). Mutation spectrum of phenylketonuria in Syrian population: genotype-phenotype correlation. Gene 528, 241–247. doi: 10.1016/j.gene.2013.07.001
Muriello, M. J., Viall, S., Bottiglieri, T., Cusmano-Ozog, K., Ferreira, C. R. (2017). Confirmation that MAT1A p.Ala259Val mutation causes autosomal dominant hypermethioninemia. Mol. Genet. Metab. Rep. 13, 9–12. doi: 10.1016/j.ymgmr.2017.07.004
Nagao, M., Tanaka, T., Furujo, M. (2013). Spectrum of mutations associated with methionine adenosyltransferase I/IIIdeficiency among individuals identified during newborn screening in Japan. Mol. Genet. Metab. 110, 460–464. doi: 10.1016/j.ymgme.2013.10.013
Nezu, J., Tamai, I., Oku, A., Ohashi, R., Yabuuchi, H., Hashimoto, N., et al. (1999). Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 21, 91–94. doi: 10.1038/5030
Niu, D. M., Chien, Y. H., Chiang, C. C., Ho, H. C., Hwu, W. L., Kao, S. M., et al. (2010). Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J. Inherit. Metab. Dis. 33, S295–S305. doi: 10.1007/s10545-010-9129-z
Nochi, Z., Olsen, R. K. J., Gregersen, N. (2017). Short-chain acyl-CoA dehydrogenase deficiency: from gene to cell pathology and possible disease mechanisms. J. Inherit. Metab. Dis. 40, 641–655. doi: 10.1007/s10545-017-0047-1
Obaid, A., Nashabat, M., Alfadhel, M., Alasmari, A., Al Mutairi, F., Alswaid, A., et al. (2018). Clinical, biochemical, and molecular features in 37 saudi patients with very long chain acyl coa dehydrogenase deficiency. JIMD Rep. 40, 47–53. doi: 10.1007/8904_2017_58
Okano, Y., Kudo, S., Nishi, Y., Sakaguchi, T., Aso, K. (2011). Molecular characterization of phenylketonuria and tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency in Japan. J. Hum. Genet. 56, 306–312. doi: 10.1038/jhg.2011.10
Pérez Mato, I., Sanchez del Pino, M. M., Chamberlin, M. E., Mudd, S. H., Mato, J. M., Corrales, F. J. (2001). Biochemical basis for the dominant inheritance of hypermethioninemia associated with the R264H mutation of the MAT1A gene. A monomeric methionine adenosyltransferase with tripolyphosphatase activity. J. Biol. Chem. 276, 13803–13809. doi: 10.1074/jbc.M009017200
Rasmussen, J., Nielsen, O. W., Janzen, N., Duno, M., Gislason, H., Køber, L., et al. (2014). Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J. Inherit. Metab. Dis. 37, 215–222. doi: 10.1007/s10545-013-9606-2
Rastegar Moghadam, M., Shojaei, A., Babaei, V., Rohani, F., Ghazi, F. (2018). Mutation analysis of Phenylalanine hydroxylase gene in Iranian patients with Phenylketonuria. Med. J. Islam Repub. Iran 32, 21. doi: 10.14196/mjiri.32.21
Rips, J., Almashanu, S., Mandel, H., Josephsberg, S., Lerman-Sagie, T., Zerem, A., et al. (2016). Primary and maternal 3-methylcrotonyl-CoA carboxylase deficiency: insights from the Israel newborn screening program. J. Inherit. Metab. Dis. 39, 211–217. doi: 10.1007/s10545-015-9899-4
Rose, E. C., di San Filippo, C. A., Ndukwe Erlingsson, U. C., Ardon, O., Pasquali, M., Longo, N. (2012). Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 33, 118–123. doi: 10.1002/humu.21607
Saheki, T., Kobayashi, K. (2002). Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 47, 333–341. doi: 10.1007/s100380200046
Saheki, T., Kobayashi, K., Iijima, M., Horiuchi, M., Begum, L., Jalil, M. A., et al. (2004). Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the ureacycle. Mol. Genet. Metab. 81 Suppl 1, S20–S26. doi: 10.1016/j.ymgme.2004.01.006
Salek, J., Byrne, J., Box, T., Longo, N., Sussman, N. (2010). Recurrent liver failure in a 25-year-old female. Liver Transpl. 16, 1049–1053. doi: 10.1002/lt.22118
Sanderson, S., Green, A., Preece, M. A., Burton, H. (2006). The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child. 91, 896–899. doi: 10.1136/adc.2005.091637
Santra, S., Macdonald, A., Preece, M. A., Olsen, R. K., Andresen, B. S. (2016). Long-term outcome of isobutyryl-CoA dehydrogenase deficiency diagnosed following an episode of ketotic hypoglycaemia. Mol. Genet. Metab. Rep. 10, 28–30. doi: 10.1016/j.ymgmr.2016.11.005
Schulze, A., Lindner, M., Kohlmüller, D., Olgemöller, K., Mayatepek, E., Hoffmann, G. F. (2003). Expanded newborn screening for inborn errors of metabolism by electrosprayionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics 111, 1399–1406. doi: 10.1542/peds.111.6.1399
Shi, X. T., Cai, J., Wang, Y. Y., Tu, W. J., Wang, W. P., Gong, L. M., et al. (2012). Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep. 6, 79–83. doi: 10.1007/8904_2011_119
Shibata, N., Hasegawa, Y., Yamada, K., Kobayashi, H., Purevsuren, J., Yang, Y., et al. (2018). Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective screening vs. Expanded newborn screening. Mol. Genet. Metab. Rep. 16, 5–10. doi: 10.1016/j.ymgmr.2018.05.003
Shigematsu, Y., Hirano, S., Hata, I., Tanaka, Y., Sudo, M., Sakura, N., et al. (2002). Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 776, 39–48. doi: 10.1016/S1570-0232(02)00077-6
Smon, A., Repic Lampret, B., Groselj, U., Zerjav Tansek, M., Kovac, J., Perko, D., et al. (2018). Next generation sequencing as a follow-up test in an expanded new born screening programme. Clin. Biochem. 52, 48–55. doi: 10.1016/j.clinbiochem.2017.10.016
Song, Y. Z., Deng, M., Chen, F. P., Wen, F., Guo, L., Cao, S. L., et al. (2011). Genotypic and phenotypic features of citrin deficiency: five-year experience in a Chinese pediatric center. Int. J. Mol. Med. 28, 33–40. doi: 10.3892/ijmm.2011.653
Stadler, S. C., Polanetz, R., Maier, E. M., Heidenreich, S. C., Niederer, B., Mayerhofer, P. U., et al. (2006). Newborn screening for 3-methylcrotonyl-CoA carboxylase deficiency: population heterogeneity of MCCA and MCCB mutations and impact on risk assessment. Hum. Mutat. 27, 748–759. doi: 10.1002/humu.20349
Sun, Y., Wang, Y. Y., Jiang, T. (2017). Clinical features and genotyping of patients with primary carnitine deficiency identified by newborn screening. J. Pediatr. Endocrinol. Metab. 30, 879–883. doi: 10.1515/jpem-2017-0002
Tabata, A., Sheng, J. S., Ushikai, M., Song, Y. Z., Gao, H. Z., Lu, Y. B., et al. (2008). Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J. Hum. Genet. 53, 534–545. doi: 10.1007/s10038-008-0282-2
Tan, J. Q., Chen, D. Y., Li, Z. T., Yan, T. Z., Huang, J. W., Cai, R. (2017). Genetic diagnosis of 10 neonates with primary carnitine deficiency [Article in Chinese]. Zhongguo Dang Dai Er Ke Za Zhi 19, 1150–1154.
Tanaka, T., Nagao, M., Tsutsumi, H. (2002). Application of mutation analysis for the previously uncertain cases of adult-onset type IIcitrullinemia (CTLN2) and their clinical profiles. Tohoku J. Exp. Med. 198, 89–97. doi: 10.1620/tjem.198.89
Tang, N. L., Ganapathy, V., Wu, X., Hui, J., Seth, P., Yuen, P. M., et al. (1999). Mutations of OCTN2, an organic cation/carnitine transporter, lead to deficient cellular carnitine uptake in primary carnitine deficiency. Hum. Mol. Genet. 8, 655–660. doi: 10.1093/hmg/8.4.655
Tein, I., Elpeleg, O., Ben-Zeev, B., Korman, S. H., Lossos, A., Lev, D., et al. (2008). Short-chain acyl-CoA dehydrogenase gene mutation (c.319C>T) presents with clinical heterogeneity and is candidate founder mutation in individuals of Ashkenazi Jewish origin. Mol. Genet. Metab. 93, 179–189. doi: 10.1016/j.ymgme.2007.09.021
Therrell, B. L., Padilla, C. D., Loeber, J. G., Kneisser, I., Saadallah, A., Borrajo, G. J., et al. (2015). Current status of newborn screening worldwide: 2015. Semin. Perinatol. 39, 171–187. doi: 10.1053/j.semperi.2015.03.002
Tonin, R., Caciotti, A., Funghini, S., Pasquini, E., Mooney, S. D., Cai, B., et al. (2016). Clinical relevance of short-chain acyl-CoA dehydrogenase (SCAD) deficiency: exploring the role of new variants including the first SCAD-disease-causing allele carrying a synonymous mutation. BBA Clin. 5, 114–119. doi: 10.1016/j.bbacli.2016.03.004
Tu, W. J. (2011). Methylmalonic acidemia in mainland China. Ann. Nutr. Metab. 58, 281. doi: 10.1159/000331469
Uematsu, M., Sakamoto, O., Sugawara, N., Kumagai, N., Morimoto, T., Yamaguchi, S., et al. (2007). Novel mutations in five Japanese patients with 3-methylcrotonyl-CoA carboxylase deficiency. J. Hum. Genet. 52, 1040–1043. doi: 10.1007/s10038-007-0211-9
van Maldegem, B. T., Wanders, R. J., Wijburg, F. A. (2010). Clinical aspects of short-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 33, 507–511. doi: 10.1007/s10545-010-9080-z
Vaz, F. M., Scholte, H. R., Ruiter, J., Hussaarts-Odijk, L. M., Pereira, R. R., Schweitzer, S., et al. (1999). Identification of two novel mutations in OCTN2 of three patients with systemic carnitine deficiency. Hum. Genet. 105, 157–161. doi: 10.1007/s004399900105
Vieira Neto, E., Laranjeira, F., Quelhas, D., Ribeiro, I., Seabra, A., Mineiro, N., et al. (2018). Mutation analysis of the PAH gene in phenylketonuria patients from Rio de Janeiro, Southeast Brazil. Mol. Genet. Genomic Med. 6, 575–591. doi: 10.1002/mgg3.408
Waisbren, S. E., Levy, H. L., Noble, M., Matern, D., Gregersen, N., Pasley, K., et al. (2008). Short-chain acyl-CoA dehydrogenase (SCAD) deficiency: an examination of the medical and neurodevelopmental characteristics of 14 cases identified through newborn screening or clinical symptoms. Mol. Genet. Metab. 95, 39–45. doi: 10.1016/j.ymgme.2008.06.002
Wang, R., Shen, N., Ye, J., Han, L., Qiu, W., Zhang, H., et al. (2018). Mutation spectrum of hyperphenylalaninemia candidate genes and the genotype-phenotype correlation in the Chinese population. Clin. Chim. Acta. 481, 132–138. doi: 10.1016/j.cca.2018.02.035
Wang, Y., Ye, J., Ganapathy, V., Longo, N. (1999). Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc. Natl. Acad. Sci. U. S. A. 96, 2356–2360. doi: 10.1073/pnas.96.5.2356
Webster, D., Joint Newborn Screening Committee; Human Genetics Society of Australasia; Royal Australasian College of Physicians. (2003). Newborn screening in Australia and New Zealand. Southeast Asian J. Trop. Med. Public Health 34, 69–70.
Wilcken, B., Wiley, V., Hammond, J., Carpenter, K. (2003). Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N. Engl. J. Med. 348, 2304–2312. doi: 10.1056/NEJMoa025225
Wilcken, B. (2008). The consequences of extended newborn screening programmes: do we know who needs treatment? J. Inherit. Metab. Dis. 31, 173–177. doi: 10.1007/s10545-008-0843-8
Wiley, V., Carpenter, K., Wilcken, B. (1999). Newborn screening with tandem mass spectrometry: 12 months’ experience in NSW Australia. Acta. Paediatr. Suppl. 88, 48–51. doi: 10.1111/j.1651-2227.1999.tb01157.x
Woo, H. I., Ki, C. S., Lee, S. Y., Kim, J. W., Song, J., Jin, D. K., et al. (2013). Mutation spectrum of the ASS1 gene in Korean patients with citrullinemia type I. Clin. Biochem. 46, 209–213. doi: 10.1016/j.clinbiochem.2012.10.008
Woo, H. I., Park, H. D., Lee, Y. W. (2014). Molecular genetics of citrullinemia types I and II. Clin. Chim. Acta. 431, 1–8. doi: 10.1016/j.cca.2014.01.032
Xing, Y. Z., Qiu, W. J., Ye, J., Han, L. S., Xu, S. S., Zhang, H. W., et al. (2010). Studies on the clinical manifestation and SLC25A13 gene mutation of Chinese patients with neonatal intrahepatic cholestasis caused by citrin deficiency [Article in Chinese]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 27, 180–185.
Yamaguchi, N., Kobayashi, K., Yasuda, T., Nishi, I., Iijima, M., Nakagawa, M., et al. (2002). Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations. Hum. Mutat. 19, 122–130. doi: 10.1002/humu.10022
Yang, L., Yang, J., Zhang, T., Weng, C., Hong, F., Tong, F., et al. (2015). Identification of eight novel mutations and transcript analysis of two splicing mutations in Chinese newborns with MCC deficiency. Clin. Genet. 88, 484–488. doi: 10.1111/cge.12535
Yasuda, T., Yamaguchi, N., Kobayashi, K., Nishi, I., Horinouchi, H., Jalil, M. A., et al. (2000). Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum. Genet. 107, 537–545. doi: 10.1007/s004390000430
Ye, J., Yang, Y., Yu, W., Zou, H., Jiang, J., Yang, R., et al. (2013). Demographics, diagnosis and treatment of 256 patients with tetrahydrobiopterin deficiency in mainland China: results of a retrospective, multicentre study. J. Inherit. Metab. Dis. 36, 893–901. doi: 10.1007/s10545-012-9550-6
Ye, J., Qiu, W. J., Han, L. S., Zhou, J. D., Gao, X. L., Gu, X. F. (2009). The investigation of differential diagnostic development and incidence of tetrahydrobiopterin deficiency[Article in Chinese]. Zhonghua Yu Fang Yi Xue Za Zhi 43, 128–131.
Ye, J., Gong, L., Han, L., Qiu, W., Zhang, H., Gao, X., et al. (2014). [Follow up and gene mutation analysis in cases suspected as 3-methylcrotonyl-coenzyme A carboxylase deficiency by neonatal screening]. Zhonghua Er Ke Za Zhi. 52, 409–414.
Yin, F. (2016). Analysis of genetic mebolism error screening results for 44639 newborns using tandem mass spectrum. Maternal Child Health Care China 31, 3098–3101.
Yoon, H. R., Lee, K. R., Kang, S., Lee, D. H., Yoo, H. W., Min, W. K., et al. (2005). Screening of newborns and high-risk group of children for inborn metabolic disorders using tandem mass spectrometry in South Korea: a three-year report. Clin. Chim.Acta. 354, 167–180. doi: 10.1016/j.cccn.2004.11.032
Yu, Y. F., Li, F., Ma, H. W. (2015). Relationship of genotypes with clinical phenotypes and outcomes in children with cobalamin C type combined methylmalonic aciduria and homocystinuria [Article in Chinese]. Zhongguo Dang Dai Er Ke Za Zhi 17, 769–774.
Yun, J. W., Jo, K. I., Woo, H. I., Lee, S. Y., Ki, C. S., Kim, J. W., et al. (2015). A novel ACAD8 mutation in asymptomatic patients with isobutyryl-CoA dehydrogenase deficiency and a review of the ACAD8 mutation spectrum. Clin. Genet. 87, 196–198. doi: 10.1111/cge.12350
Zamanfar, D., Jalali, H., Mahdavi, M. R., Maadanisani, M., Zaeri, H., Asadpoor, E. (2017). Investigation of five common mutations on phenylalanine hydroxylase gene of phenylketonuria patients from two provinces in North of Iran. Int. J. Prev. Med. 8, 89. doi: 10.4103/ijpvm.IJPVM_378_16. eCollection 2017
Zhang, Z., Gao, J. J., Feng, Y., Zhu, L. L., Yan, H., Shi, X. F., et al. (2018). Mutational spectrum of the phenylalanine hydroxylase gene in patients with phenylketonuria in the central region of China. Scand. J. Clin. Lab. Invest. 78, 211–218. doi: 10.1080/00365513.2018.1434898
Zhao, D. H., Zhu, X. Y., Li, X. L., Ni, M., JIA, C. L., Zhang, J. (2016). Analysis of methylmalonic academia screening results of 349858 newborns in Henan Province (in Chinese). Chin. J. Birth Health Heredity 24, 86–87. doi: 10.13404/j.cnki.cjbhh.2016.08.039
Zheng, J., Zhang, Y., Hong, F., Yang, J., Tong, F., Mao, H., et al. (2017). Screening for fatty acid oxidation disorders of newborns in Zhejiang province: prevalence, outcome and follow-up [Article in Chinese]. Zhejiang Da Xue Xue Bao Yi Xue Ban. 46, 248–255.
Zhou, Y. A., Ma, Y. X., Zhang, Q. B., Gao, W. H., Liu, J. P., Yang, J. P., et al. (2012). Mutations of the phenylalanine hydroxylase gene in patients with phenylketonuria in Shanxi, China. Genet. Mol. Biol. 35, 709–713. doi: 10.1590/S1415-47572012005000069
Zytkovicz, T. H., Fitzgerald, E. F., Marsden, D., Larson, C. A., Shih, V. E., Johnson, D. M., et al. (2001). Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin. Chem. 47, 1945–1955.
Keywords: expanded newborn screening, inborn errors of metabolism, tandem mass spectrometry, disease spectrum, prevalence, genetic characteristics, hotspot mutation
Citation: Wang T, Ma J, Zhang Q, Gao A, Wang Q, Li H, Xiang J and Wang B (2019) Expanded Newborn Screening for Inborn Errors of Metabolism by Tandem Mass Spectrometry in Suzhou, China: Disease Spectrum, Prevalence, Genetic Characteristics in a Chinese Population. Front. Genet. 10:1052. doi: 10.3389/fgene.2019.01052
Received: 18 March 2019; Accepted: 01 October 2019;
Published: 29 October 2019.
Edited by:
Merlin G. Butler, University of Kansas Medical Center, United StatesReviewed by:
Xusheng Wang, University of North Dakota, United StatesNelson L. S. Tang, The Chinese University of Hong Kong, China
Copyright © 2019 Wang, Ma, Zhang, Gao, Wang, Li, Xiang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjing Wang, d2FuZ2JqODUwMTEzQDE2My5jb20=; Jingjing Xiang, eGlhbmdqaW5namluZzIwMTNAMTYzLmNvbQ==
†These authors have contributed equally to this work